Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Epithelial cell adhesion molecule aptamer functionalized PLGA-lecithin-curcumin-PEG nanoparticles for targeted drug delivery to human colorectal adenocarcinoma cells
Authors Li L, Xiang D, Shigdar S, Yang W, Li Q, Lin J, Liu K, Duan W
Received 27 December 2013
Accepted for publication 17 January 2014
Published 21 February 2014 Volume 2014:9(1) Pages 1083—1096
DOI https://doi.org/10.2147/IJN.S59779
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Lei Li,1,* Dongxi Xiang,2,* Sarah Shigdar,2 Wenrong Yang,3 Qiong Li,2 Jia Lin,4 Kexin Liu,1 Wei Duan2
1College of Pharmacy, Dalian Medical University, Dalian, People's Republic of China; 2School of Medicine, Faculty of Health, Deakin University, Waurn Ponds, VIC, Australia; 3School of Life and Environmental Sciences, Faculty of Science, Engineering and Built Environment, Deakin University, Waurn Ponds, VIC, Australia; 4Department of Biochemistry and Molecular Biology, West China School of Preclinical and Forensic Medicine, Sichuan University, Chengdu, People's Republic of China
*These authors contributed equally to this work
Abstract: To improve the efficacy of drug delivery, active targeted nanotechnology-based drug delivery systems are gaining considerable attention as they have the potential to reduce side effects, minimize toxicity, and improve efficacy of anticancer treatment. In this work CUR-NPs (curcumin-loaded lipid-polymer-lecithin hybrid nanoparticles) were synthesized and functionalized with ribonucleic acid (RNA) Aptamers (Apts) against epithelial cell adhesion molecule (EpCAM) for targeted delivery to colorectal adenocarcinoma cells. These CUR-encapsulated bioconjugates (Apt-CUR-NPs) were characterized for particle size, zeta potential, drug encapsulation, stability, and release. The in vitro specific cell binding, cellular uptake, and cytotoxicity of Apt-CUR-NPs were also studied. The Apt-CUR-NP bioconjugates exhibited increased binding to HT29 colon cancer cells and enhancement in cellular uptake when compared to CUR-NPs functionalized with a control Apt (P<0.01). Furthermore, a substantial improvement in cytotoxicity was achieved toward HT29 cells with Apt-CUR-NP bioconjugates. The encapsulation of CUR in Apt-CUR-NPs resulted in the increased bioavailability of delivered CUR over a period of 24 hours compared to that of free CUR in vivo. These results show that the EpCAM Apt-functionalized CUR-NPs enhance the targeting and drug delivery of CUR to colorectal cancer cells. Further development of CUR-encapsulated, nanosized carriers will lead to improved targeted delivery of novel chemotherapeutic agents to colorectal cancer cells.
Keywords: PLGA-lecithin-PEG nanoparticles, curcumin, EpCAM, aptamer, targeted drug delivery
Introduction
One of the main goals of modern cancer chemotherapy is to deliver a safe and effective dose of therapeutic agents preferentially to disease site(s) while sparing normal tissues.1 To improve the efficacy of drug delivery, nanotechnology-based drug-delivery systems are gaining considerable attention, as they have the potential to reduce side effects, minimize toxicity, and improve anticancer treatment efficacy. Among these delivery systems, liposome and polymer nanoparticles (NPs) are the two most promising classes owing to their biocompatible and biodegradable properties, and they have been approved for clinical use by the US Food and Drug Administration.2,3 However, these two first-generation nanomedicines utilize a “passive targeting” principle for cancer-drug delivery. The new generation of cancer nanodrug delivery systems seeks to adopt an “active targeting” approach, in addition to the passive targeting.
Recently, biodegradable lipid-polymer hybrid nanoparticles that take advantage of the positive attributes of polymeric nanoparticles have emerged as a more attractive class of drug delivery carriers.3,4 The hybrid nanoparticles can be a robust drug-delivery platform using a single-step strategy of nanoprecipitation and self-assembly to achieve high drug-encapsulation efficiency, ease of manufacturing, tunable and sustained drug release, and good serum stability.4 These hybrid nanoparticles are mainly composed of 1) poly (D, L-lactide-co-glycolide) (PLGA) as a hydrophobic core for encapsulating hydrophobic drugs; 2) lecithin as a monolayer around the PLGA core; and 3) 1,2-distearoyl-sn-glycero- 3-phosphoethanolamine-N-carboxy(polyethylene glycol) 2000 (DSPE-PEG2000-COOH) as a polyethylene glycol (PEG) shell that intersperses into the lipid shell to prolong the circulation half-life in vivo and to provide functionalization groups for the modification of targeting ligands.
A desirable attribute of a targeted cancer treatment is to deliver chemotherapeutic drugs to cancer cells over a sufficient time without affecting the surrounding noncancerous tissues. This can be accomplished by targeting ligands that recognize tumor-specific or tumor-associated surface markers.5 Aptamers (Apts), single-stranded oligonucleotides selected via an in vitro process of systematic evolution of ligands by exponential enrichment, can be considered as the nucleic-acid analogs of antibodies.6 Apts have some favorable characteristics that could rival traditional antibodies, including small size, low immunogenicity, great stability over extreme conditions, and high affinity and specificity toward their targets.5,7
Curcumin (CUR) is a natural polyphenol present in turmeric (Curcuma longa) with low intrinsic toxicity but a wide range of biological activities, including antitumor, antioxidant, and anti-inflammatory properties.8 From in vivo studies, it has been demonstrated that CUR is capable of inhibiting the growth of implanted human tumors and carcinogen-induced tumors.9 CUR mediates its antiproliferative, anti-invasive, and proapoptotic effects on cancer cells, including cancer stem/progenitor cells and their progenies, through multiple molecular mechanisms. However, CUR has low solubility (approximately 0.0004 mg/mL at pH 7.3) and limited bioavailability in the blood circulation when administered orally in humans, and it is degraded within 30 minutes at basic pH in aqueous solution, which greatly limits its utility in cancer treatment as a free drug.10,11 Various studies have shown that encapsulation of CUR in PLGA nanoparticles and liposomes can overcome such limitations.9,12,13 Most of these targeted CUR-PLGA nanoparticles tested are characterized by a size of about 200 nm.14 However, for cancer treatment, nanoparticles with a diameter of 30–200 nm are desirable, depending on the type of tumor in question, in order to promote better tissue penetration and cellular uptake and longer blood circulation.15
To date, no study has encapsulated CUR with PLGA-lecithin-PEG nanoparticles. Being a good dispersing agent, lecithin has promising traits as a nanomedicine carrier due to its ability to increase the stability and drug-loading beyond that which PLGA nanoparticles have achieved.16 In this study, we developed CUR-encapsulated small PLGA-lecithin-PEG nanoparticles (CUR-NPs) with a size of around 100 nm for addressing the solubility and low bioavailability of CUR and optimizing the particle size for better antitumor traits. Second, we functionalized the CUR-NPs with an Apt against epithelial cell adhesion molecule (EpCAM), a protein that is overexpressed on colorectal adenocarcinoma cells, to develop EpCAM Apt-CUR-NP bioconjugates. We then assessed whether these nanoparticle–Apt bioconjugates could deliver drugs to colon cancer cells with specific targeting and enhancement of cellular uptake. We showed that these Apt-CUR-NP conjugates exhibited a higher drug-encapsulating yield and a more-sustained drug-release profile than did free CUR. In addition, good cellular targeting toward colon cancer cells was observed for the conjugate, resulting in enhancement of cellular uptake and cytotoxicity. Our data suggest that the EpCAM Apt-functionalized CUR-NP is a promising drug-delivery carrier for colon cancer therapy.
Material and methods
Cells and reagents
All cell lines used were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA), including HT29 (human colon cancer, EpCAM+) cells and HEK293T (human embryonic kidney, EpCAM−) cells. CUR was purchased from Henan Zhongda Biological Engineering Co, Ltd (Zhengzhou City, People’s Republic of China). PLGA (lactide to glycolide ratio 50/50) with ester-terminated was obtained from Sigma-Aldrich (St Louis, MO, USA). Pegylated phospholipid (DSPE-PEG2000-COOH) was obtained from Avanti Polar Lipids, Inc. (Alabaster, AL, USA). Soybean lecithin consisting of 90%–95% phosphatidylcholine was obtained from MP Biomedicals (Solon, OH, USA). Amicon Ultra-4 centrifugal filters with a molecular weight cut-off of 10 kDa were obtained from EMD Millipore (Billerica, MA, USA). N-hydroxysuccinimide (NHS), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC), and penicillin/streptomycin solution were obtained from Sigma-Aldrich. All reagents were of analytical grade. Trypan blue, trypsin (0.25%) and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay kit were purchased from Sigma-Aldrich. EpCAM Apt (sequence: 5′-Cy5 – A(2′-F-C)G(2′-F-U)A(2′-F-U) (2′-F-C) (2′-F-C) (2′-F-C) (2′-F-U) (2′-F-U) (2′-F-U) (2′-F-U) (2′-F-C)G(2′-F-C)G(2′-F-U)-C6-amino linker-3′; where F =2′-fluoro, molecular weight: 6345) and negative control Apt, (sequence: 5′-Cy5 – A(2′-O-Me-C)G(2′-O-Me-U)A(2′-O-Me-U) (2′-O-Me-C) (2′-O-Me-C) (2′-O-Me-C) (2′-O-Me-U) (2′-O-Me-U) (2′-O-Me-U) (2′-O-Me-U) (2′-O-Me-C)G(2′-O-Me-C)G(2′-O-Me-U)- C6-amino linker-3′; where 2′-O-Me = 2′-methoxy, molecular weight: 6502) were synthesized by Biospring GmbH (Frankfurt, Germany). All other reagents were of analytical or chromatographic grade and purchased from Sigma-Aldrich.
Synthesizing of Apt-CUR-NPs
Preparation of CUR-NPs
CUR-NPs were prepared from PLGA, soybean lecithin, and DSPE-PEG2000-COOH (or DSPE-PEG2000-Apt) using a nanoprecipitation technique with a slight modification.17 PLGA (10 mg/mL) and CUR (1 mg/mL) were dissolved in acetonitrile and mixed together at a final concentration of 5 mg/mL and 125 μg/mL, respectively. Lecithin and DSPE-PEG2000-COOH (4:1, molar ratio) were dissolved in 4% ethanol aqueous at 20% of the PLGA polymer weight and heated at 65°C for 3 minutes. The PLGA/acetonitrile solution was then added to the lipid aqueous solution in a dropwise manner to allow self-assembly for 20 minutes. The organic solvent was eliminated at low pressure and at 37°C via rotary evaporation. The remaining organic solvent and free drug molecules were removed by washing the nanoparticle solution three times using a Microsep™ Advance Centrifugal Device (Pall Corporation, Ann Arbor, MI, USA; molecular weight cutoff of 10 kDa) and then suspended in distilled water. The nanoparticles were used immediately or stored at −4°C for later use.
Conjugation of Apt-CUR-NPs
The conjugation of Apt to nanoparticles was achieved through an amide bond between the carboxylic groups of DSPE-PEG2000-COOH and an amino group at the 3′ end of the Apt with two steps. Initially, amine-terminated Apt was conjugated to the carboxyl-functionalized DSPE-PEG using EDC and NHS activation chemistry. Briefly, DSPE-PEG2000-COOH was incubated with 100 μL of phosphate-buffered saline (PBS) (pH 6.0) containing 400 mmol/L EDC and 100 mmol/L NHS for 30 minutes at room temperature with gentle shaking. The resulting NHS-activated DSPE-PEG2000-COOH was covalently linked to prefolded amine-modified Apt with a molar ratio of Apt to DSPE-PEG2000-COOH of 1:10 over 2 hours at room temperature. The reaction mixture was then washed with water three times using a Microsep™ Advance Centrifugal device (Pall Corporation, Ann Arbor, MI, USA; molecular weight cutoff of 3 kDa) for removing unbound Apts. The resulting DSPE-PEG2000-Apt conjugates were then used in the preparation of lipid–polymer combinational nanoparticles according to the synthesis protocol described above.
Determination of characteristics of the NPs
Particle size, zeta potential, and surface morphology
Dynamic light scattering analysis using a Zetasizer Nano-ZS instrument (Malvern Instruments Ltd, Malvern, Worcestershire, UK) was employed to analyze the size distribution of CUR-NPs and Apt-CUR-NPs in dispersion and the zeta potential of the particles. The shape and particle morphology of CUR-NPs and Apt-CUR-NPs were analyzed using a field emission transmission electron micrograph (JEM-2200-FS; JEOL, Peabody, MA, USA). One drop of the sample solution was deposited onto a carbon-coated copper grid that had been previously hydrophilized under ultraviolet (UV) light and air-dried at room temperature prior to examination under transmission electron microscopy (TEM).
Determination of the successful bioconjugates of Apt to CUR-NPs
Agarose gel electrophoresis
For confirmation of Apt conjugation, nanoparticles prepared as described above were subjected to separation via electrophoresis using a 0.8% agarose gel. The electrophoresis was performed at 90 V for 90 minutes, followed by imaging and analysis using an Image Quant LAS 4000 (GE Healthcare Life Sciences, Pittsburgh, PA, USA).
Surface chemistry evaluation
The Apt conjugated to the surface of nanoparticles was confirmed on the basis of the surface chemistry measured by X-ray photoelectron spectroscopy (XPS). XPS spectra were obtained using an EscaLab 250XI spectrometer with a monochromated Al Kα source (1486.68 eV), hemispherical analyzer, and multichannel detector (Thermo Fisher Scientific, Waltham, MA, USA). The pass energy was fixed at 20 eV or 100 eV to ensure sufficient resolution and sensitivity. Binding energies of elements were corrected with reference to silicon wafer SiSi2p (102 eV). The XPS spectrum was analyzed with the curve-fitting program XPSPEAK 4.1 (Informer Technologies, Inc., Pittsburgh, PA, USA), and involved background subtraction using Shirley routines and subsequent nonlinear least-squares fitting to mix Gaussian–Lorentzian functions.
Determination of drug encapsulation efficiency and loading capacity
The drug concentration was calculated using the linear portion of the calibration curve obtained by the UV spectrophotometer at 427 nm for serial drug concentrations. The entrapment efficiency (E [%]) of CUR loaded in PLGA-lipid nanoparticles was determined as follows: the nonencapsulated CUR was separated from nanoparticles by filtration with a Microsep™ Advance Centrifugal Device (Pall Corporation; molecular weight cutoff of 10 kDa), followed by centrifugation at 21,000× g for 5 minutes. The cleared supernatant was used for the determination of the nonencapsulated CUR by UV spectrophotometry. The E (%) was calculated by the formula:

The yield corresponds to the ratio of CUR in nanoparticles recovered to the total amount of dried nanoparticles. The Y (%) was calculated by:

The stability of CUR in solution and nanoparticles in different PBS solutions (0.01 M phosphate, pH 7.4), with or without 10% fetal bovine serum (FBS) was estimated by the UV spectrophotometry method. Free CUR and CUR-NPs were prepared to a final volume of 5 mL with a fixed concentration of 20 μg/mL and incubated in a shaker rotating at 100 rpm at 37°C for 8 hours. Free CUR was dissolved in PBS with the help of dimethyl sulfoxide (DMSO; final concentration 0.1% volume/volume [v/v]). At predetermined time points (0 hours, 0.5 hour, 1 hour, 2 hours, 3 hours, 4 hours, 5 hours, 6 hours, 7 hours, and 8 hours), 100 μL of solutions containing free CUR or CUR-NPs were removed and mixed with 400 μL of acetonitrile to determine the concentration of CUR, as described above.
In vitro drug release
The release of CUR from CUR-NPs was analyzed in PBS (pH 7.4) containing 0.1% weight/volume (w/v) Tween-80 with continuous magnetic stirring at 100 rpm. Two mL of CUR-NPs was added into a Slide-A-Lyzer Dialysis Cassette (Thermo Fisher Scientific, Rockford, IL, USA; molecular weight cutoff of 2 kDa) and the cassette was placed into a beaker containing 200 mL of the phosphate–Tween buffer as described above. The CUR-NPs were dialyzed with magnetic stirring for 72 hours at 37°C. At specific times, a given volume of nanoparticles was withdrawn from the dialysis bag for analysis and the entire buffer solution in the beaker was removed and replaced with fresh buffer solution to maintain a sink condition. CUR was extracted from nanoparticles by acetonitrile, and concentrations in nanoparticles were determined as described above. A standard curve of CUR was prepared under identical conditions.
Targeted binding abilities of Apt-CUR-NPs
HT29 cells or HEK293T cells were seeded in 35 mm glass-bottom dishes and incubated at 37°C in 5% CO2 for 24 hours. The Dulbecco’s Modified Eagle’s Medium for HEK293T cells and McCoy’s 5A Medium for HT29 cells were then replaced with full culture medium (without phenol red) containing 4 μg/mL free CUR or equivalent amount of CUR-NPs, Apt-CUR-NPs, or control-Apt-CUR-NPs. One hour later, cells were washed twice with PBS and imaged for cellular uptake studies using a FluoviewFV10i fluorescence laser scanning confocal microscope (Olympus Corporation, Tokyo, Japan) at 37°C. The binding abilities obtained from confocal images were quantitatively analysed by the Image Pro program (Media Cybernetics, Inc., Rockville, MD, USA).
Cellular uptake study
CUR content in cells and plasma was determined using a high-performance liquid chromatography (HPLC) system consisting of an e2695 Separation Module and a 2475 Multi λ Fluorescence Detector (Waters Corporation, Milford, MA, USA) with the mobile phase composed of acetonitrile–2% glacial acetic acid (48:52, v/v) at a flow rate of 1.0 mL/minute at 25°C. The excitation and emission wavelengths were set at 425 nm and 475 nm, respectively. Chromatographic separation was performed using a Nova-Pak® C18 column (Waters Corporation; 3.9×150 mm 4 μm) with a Nova-Pak® C18 guard column. All solvents for HPLC procedures were prepared freshly and filtered with 0.22 μm membrane before using.
The stock solution of CUR (10 mM) was prepared in DMSO and diluted with the culture medium to obtain the desired concentrations. The final concentration of DMSO was less than 0.1% in the working culture medium. For loading of CUR, HT29 and HEK293T cells (1×106 cells/mL) were incubated with CUR at 20 nM for 30 minutes or 60 minutes, and washed thrice with cold PBS. For the estimation of cellular uptake, the pellet of washed and CUR-loaded cells was suspended in 200 μL of acetonitrile and sonicated for 20 minutes, followed by centrifugation at 21,000×g for 10 minutes, and the concentration of CUR in the supernatant was determined by HPLC assay with the fluorescence detector. The estimated uptake at each point was expressed as pmoL/million cells and the vehicle control was performed for each cell line.
In vitro cell viability assay
The viability of cells was determined by the MTT assay, which measures the mitochondrial conversion of MTT to formazan as detected by the change of optical density at 570 nm.12 After 24 hours of incubation, HT29 and HEK293T cells were exposed to free CUR, CUR-NPs, Apt-CUR-NPs, or control-Apt-CUR-NPs for 2 hours at different concentrations (4 μg/mL and 8 μg/mL). The drug was then replaced with full culture medium (without phenol red) containing 1% penicillin/streptomycin and further incubated for 48 hours. Following the incubation, the medium was removed and 20 μL of MTT reagent was added into each well and incubated for another 4 hours. The reaction was terminated by removing MTT prior to the addition of 150 μL/well DMSO. The absorbance of the wells, including the blanks, was measured at 570 nm using a VICTOR TM X5 Multilabel HTS Reader (PerkinElmer, Waltham, MA, USA). All experiments were performed in triplicate and repeated thrice.
Pharmacokinetics study in vivo
To investigate the pharmacokinetic properties of free CUR and CUR-NPs in vivo, male Sprague Dawley rats with body weights ranging from 250–300 g were randomly divided into two experimental groups (5–6 rats per group) with intravenous administration of drugs at 4 mg/kg. The blood from the tail was serially collected into heparinized tubes at 0 minutes, 5 minutes, 15 minutes, 30 minutes and 1 hour, 1.5 hours, 2 hours, 3 hours, 4 hours, 6 hours, 8 hours, 10 hours, and 24 hours. To separate the plasma and blood cells, blood samples (200 μL) were centrifuged at 10,000× g at 4°C for 10 minutes and the supernatant was stored at −20°C until the determination of CUR by HPLC.
Statistical analysis
All data are shown as the mean ± standard deviation. One-way analysis of variance was used to identify statistical significance among groups. Statistical significance was set at P<0.05.
Results
Generation and characterization of the nanoparticles
To generate the nanoparticle-Apt bioconjugates for targeted drug delivery to cancer cells, we used the nanoprecipitation method to encapsulate CUR within PLGA-PEG-COOH nanoparticles followed by the conjugation of the EpCAM Apt (Figure 1A). Structurally, this vehicle is composed of a hydrophobic polymeric core made of PLGA and CUR, a lipid layer composed of lecithin and DSPE-PEG2000-COOH with the link of Apt. The polymeric core and the lipid shell are associated through hydrophobic interactions, van der Waals forces, electrostatic interactions, or other noncovalent forces; the hydrophilic polymer stealth layer is often conjugated to the lipid shell through covalent bonds.17 The surface morphology of CUR-NPs and Apt-CUR-NPs was determined by TEM, and these nanoparticles showed narrow size distribution (polydispersity index: P<0.2) (Figure 1B). The physicochemical characteristics of formed nanoparticles were determined by their size, surface charge (zeta potential), CUR encapsulation, and surface morphology, which are summarized in Table 1. Bioconjugation of Apts to CUR-NPs led to a slight increase in particle size (from 86.11±1.4 nm to 90±1.9 nm), and more negative zeta potential (−26.9±2.7 mV to −36.3±4.2 mV and −39.9±3.7 mV, EpCAM Apt and negative control Apt bioconjugates respectively). The CUR encapsulated efficiency of CUR-NPs and Apt-CUR-NPs were 90.13%±4.2% and 89.98%±3.8%, respectively (n=3). Unlike free CUR, which has poor solubility in aqueous media, the aqueous solubility of CUR-NPs was improved greatly; the increased surface area due to the reduction of particle size, together with advanced biocompatibility of PLGA and PEG may contribute to the remarkable enhancement of solubility.
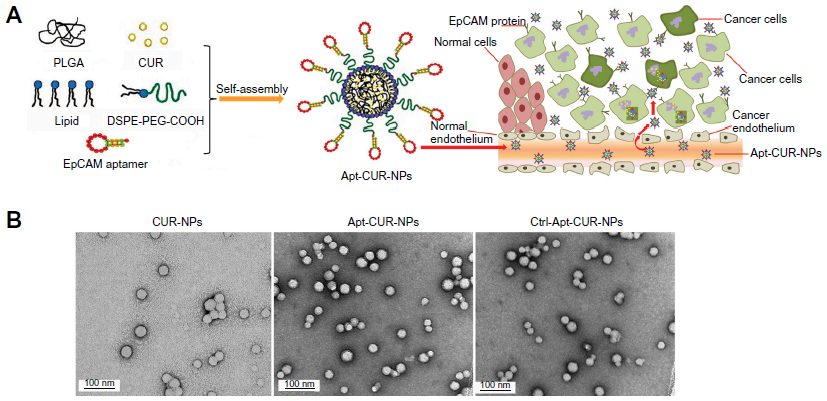 | Figure 1 Synthesization and characterization of Apt-CUR-PLGA-lecithin-PEG NPs. |
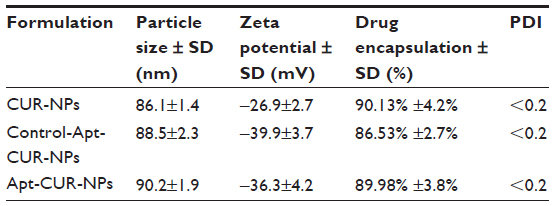 | Table 1 Particle size, zeta potential, drug encapsulation efficacy, and polydispersity index for CUR-NPs, control-Apt-CUR-NPs, and Apt-CUR-NPs |
Confirmation of the NP–Apt bioconjugates
The conjugation of the EpCAM Apt to CUR-NPs was determined by agarose gel electrophoresis. As shown in Figure 2A, the migration of the free EpCAM Apts matched that of the 19 base pairs (bp) size marker, while both of the positive and control Apt-CUR-NP bioconjugates almost stayed at the origin. To confirm the solid conjugation of Apt-CUR-NPs, free Apt and CUR-NPs were mixed together without any conjugation process and subjected to gel electrophoresis; these two elements were well separated on the gel. Taken together, the data reveal the efficient conjugation between the CUR-NPs and EpCAM Apt.
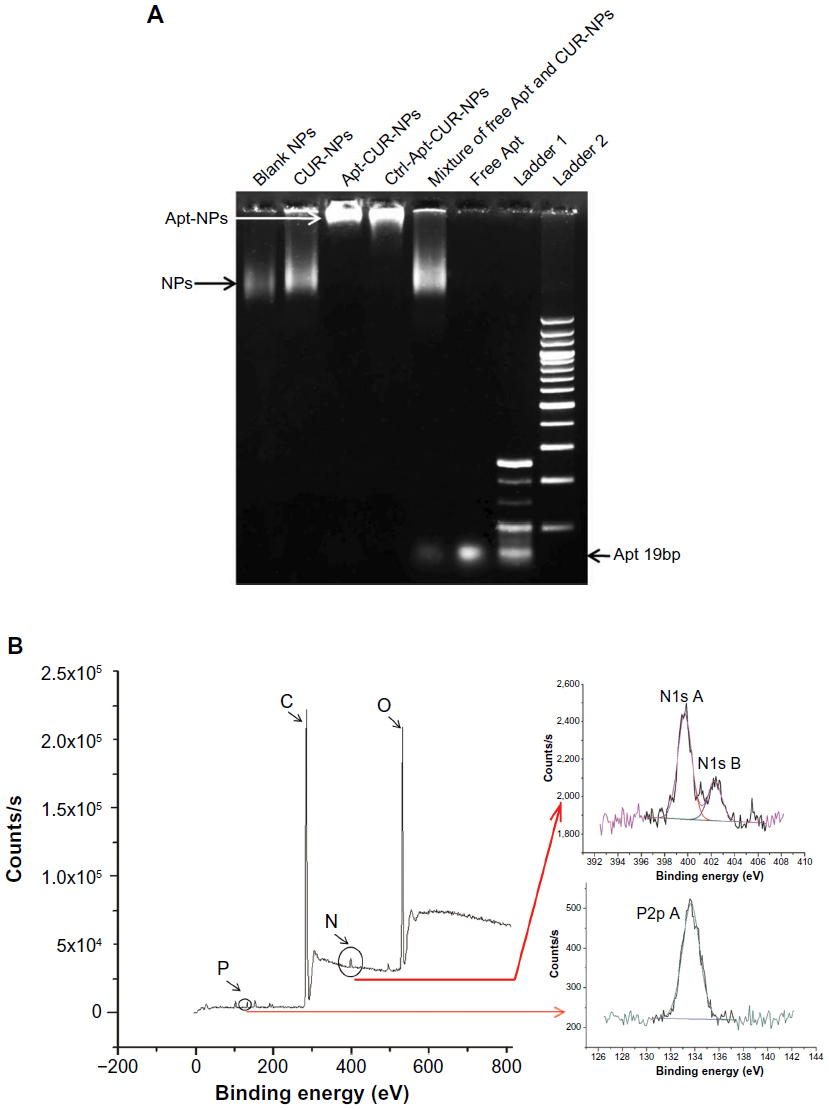 | Figure 2 Determination of successful bioconjugates of apt to CUR-PLGA-lecithin-PEG NPs. |
The successful attachment of Apt (-NH2) to PLGA-CUR-PEG nanoparticles (-COOH) was further confirmed by N1 peak in XPS for identification of chemical species on particle surfaces (Figure 2B). The spectrum of nitrogen (N) indicates the formation of an amide bond by the EDC/NHS activation and attachment of Apt to nanoparticles, confirming the surface structure of Apt-modified CUR-NPs. A prominent peak (399.7 eV) of nitrogen originating from the C=N double bond nitrogen of the imidazole ring in ribonucleic acid (RNA) was obtained, demonstrating the attachment of NH2-Apt to CUR-NPs. The peak at 133.6 eV was attributed to P2p envelope, which could only be ascribed to Apt according to the chemical composition of the sample. These data clearly confirm that the Apts have been successfully conjugated to CUR-NPs.
Drug stability in nanoparticles and in vitro release study
The stability of anticancer drugs for avoiding biodegradation in vivo is a major challenge for targeted delivery of therapeutic agents, especially for CUR. To study the stability of CUR in our nanoparticles, free CUR and CUR-NPs were incubated at physiological pH 7.4 with or without 10% FBS followed by determination of CUR retention with a UV spectrophotometer. As shown in Figure 3A, more than 95% of free CUR underwent rapid degradation in PBS after 6 hours of incubation. In contrast, the majority of CUR in CUR-NPs remained in PBS with and without 10% FBS, and approximately 75% of CUR remained stable. Therefore, encapsulation of CUR into PLGA-lecithin-PEG nanoparticles significantly improved its stability by protecting the encapsulated CUR against biodegradation.
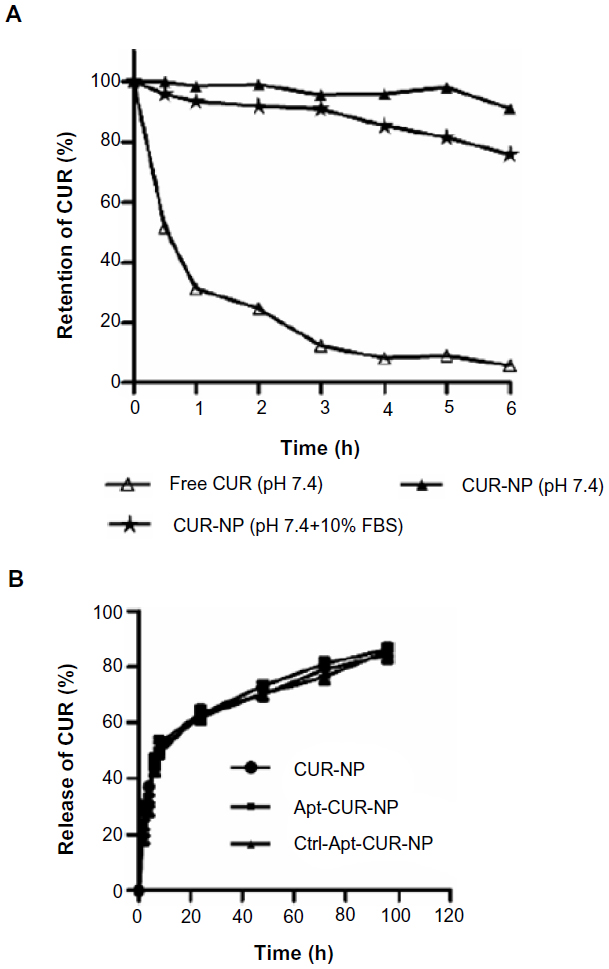 | Figure 3 Stability and release profile of free CUR and CUR-NPs. |
Following determination of the CUR retention, we proceeded to investigate the in vitro release of CUR from CUR-NPs and Apt-CUR-NPs in PBS (pH 7.4, containing 0.1% Tween-80). We added 0.1% Tween-80 to the dialysis buffer to generate a sink condition for CUR, as it is poorly soluble in PBS. A similar rapid release of about 50% CUR from all nanoparticles was observed after 8 hours of incubation, followed by a steady continued release pattern afterward (Figure 3B). The initial burst release might be due to the dissociation of surface-absorbed drugs present in the amphipathic lipid fence, while the sustained release was likely due to the slow release of CUR entrapped inside the polymer matrix. Thus, the results indicate that the encapsulation of CUR in PLGA-lecithin-PEG nanoparticles affords its continuous and prolonged release.
Apt-CUR-NPS selectively and effectively deliver drugs to targeted cells
The binding of Apt-CUR-NP bioconjugates to HT29 cells and the subsequent internalization was studied using confocal microscopy. As shown in Figure 4A, the conjugation of Apt to CUR-NPs led to significantly enhanced internalization of the EpCAM Apt CUR-NP conjugates to HT29 cells compared with the conjugates using the negative control Apt, which has the same nucleic acid sequence but different chemical modification and thus abolished its ability to bind to EpCAM. There was no gross difference in the binding of EpCAM-negative HEK293T cells between the bioconjugates and the control group. The cell binding and internalization of Apt-CUR-NPs and control were further quantified using the Image Pro program. As shown in Figure 4B, the data demonstrates a 64-fold enhancement in the binding and/or internalization of Apt-CUR-NP bioconjugates to HT29 cells compared with CUR-NPs functionalized with the negative control Apt. However, there was a remarkably low binding efficiency of nanoparticles in nontargeted HEK293T cells, which was most likely due to nonspecific phagocytosis, a phenomenon known to exist in many cells toward nanoparticles. The binding of bioconjugates to HT29 cells was observed at an early incubation stage (5 minutes), while the differential binding and internalization of Apt-CUR-NP bioconjugates became markedly pronounced after 60 minutes (data not shown). These results demonstrate that Apt-CUR-NP bioconjugates can enhance the delivery of CUR to EpCAM-expressing HT29 colorectal cancer cells.
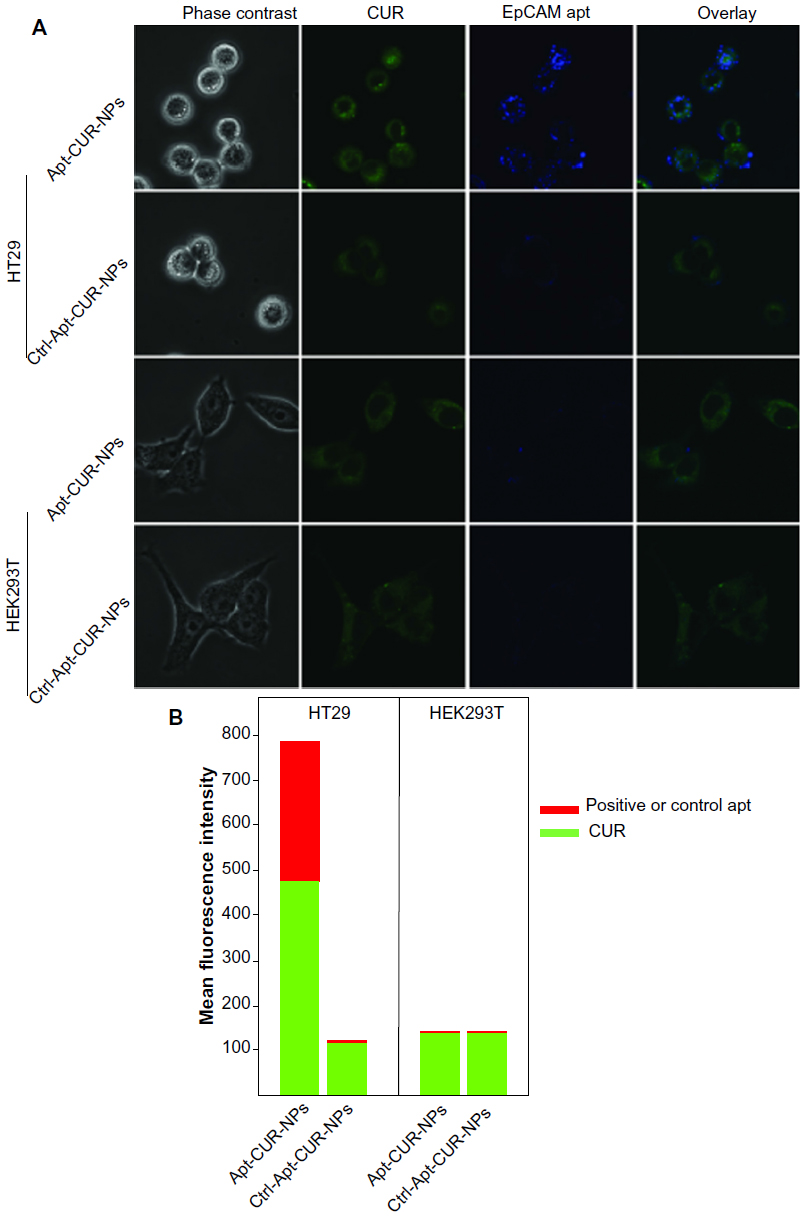 | Figure 4 Selective internalization of Apt-functionalized CUR-PLGA-lecithin-PEG nanoparticles to colon cancer cells. |
Confocal microscopy revealed a particulate pattern for both CUR fluorescence and Apt fluorescence in HT29 cells treated with Apt-CUR-NP bioconjugates (Figure 4). In contrast, in HT29 cells treated with CUR-NPs conjugated with the negative control Apt and HEK293T cells, the fluorescence for CUR lacked such a particulate pattern. This particulate pattern is indicative of endocytosis, as we previously showed for the EpCAM Apt upon binding to HT29 cells.18 Following the demonstration of Apt-CUR-NPs binding to HT29 cells qualitatively using microscopy, we next sought to confirm the differential uptake of the Apt-CUR-NP bioconjugate quantitatively using HPLC. In a 30-minute time course study, the relative cellular uptake of Apt-CUR-NPs was found to be 138.0 pmol per 1×106 cells, compared with 86.6 pmol, 107.2 pmol, and 113.6 pmol per 1×106 cells for free CUR, control-CUR-NPs, and CUR-NPs, respectively (Figure 5). There were statistically significant differences between Apt-CUR-NPs and free CUR groups (P<0.01, n=5), while the differences between Apt-CUR-NPs and the other two groups (control-CUR-NP, and CUR-NP) were also statistically significant (P<0.05, n=5). Similar results were observed after 60-minutes incubation of cells with different groups in which cellular uptakes in the free CUR, control- CUR-NPs, and CUR-NPs were all statistically significantly different compared with Apt-CUR-NPs. This differential uptake of bioconjugates by HT29 cells was reproducibly observed with cells of different passage numbers. Thus, these results suggest that Apt-CUR-NPs are capable of efficiently targeting HT29 cells and delivering CUR inside HT29 cells via endocytosis.
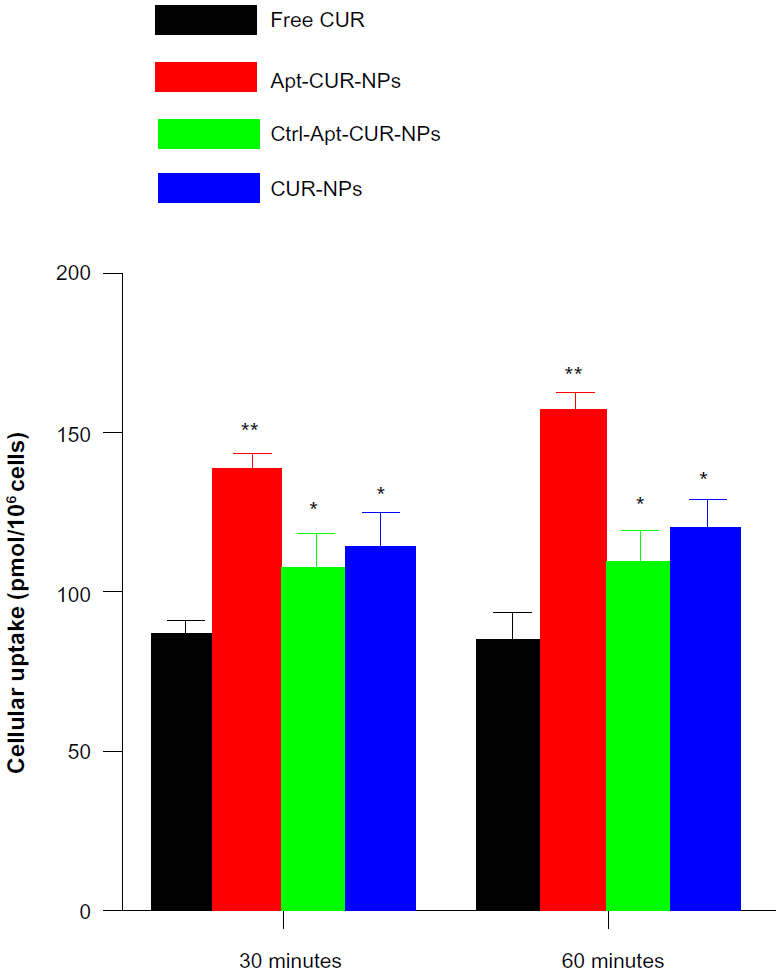 | Figure 5 Apt-CUR-NPs selectively and effectively deliver drugs to targeted cells. |
In vitro cell viability assay
After the confirmation of the ability of the Apt-NP bioconjugates to deliver CUR to target cells, we further evaluated the in vitro antiproliferation activity of free CUR, NPs alone, CUR-NPs, Apt-CUR-NPs, and control-Apt-CUR-NPs against both HT29 and HEK293T cells using a cell viability assay (MTT). Cells were treated with the respective reagent for 2 hours followed by 48-hour incubation in drug-free media, and the cell viability was determined. As shown in Figure 6, Apt-CUR-NPs were more cytotoxic to colorectal cancer HT29 cells than all controls at the concentration equivalent of 4 μg/mL CUR. While free CUR was toxic against both HT29 and HEK293T cells, the Apt-CUR-NP bioconjugate was significantly more potent against the EpCAM-expressing HT29 cells relative to the EpCAM-negative HEK293T cells (cell viabilities 58.9%±2.6% and 72.4%±1.3%, respectively, P<0.05). The data indicated that the cytotoxicity of Apt-CUR-NPs was more enhanced than that of free CUR. The observed cytotoxicity of Apt-CUR-NPs to HEK293T cells was likely due to uptake of CUR released after the dissociation of CUR from CUR-NP bioconjugates. In addition, similar inhibitory effects were obtained when the CUR concentration increased to 8 μg/mL in these groups (data not shown). Taken together, these results indicate that CUR-NPs bioconjugated with EpCAM Apt could effectively deliver drugs to EpCAM-expression colorectal cancer cells in vitro.
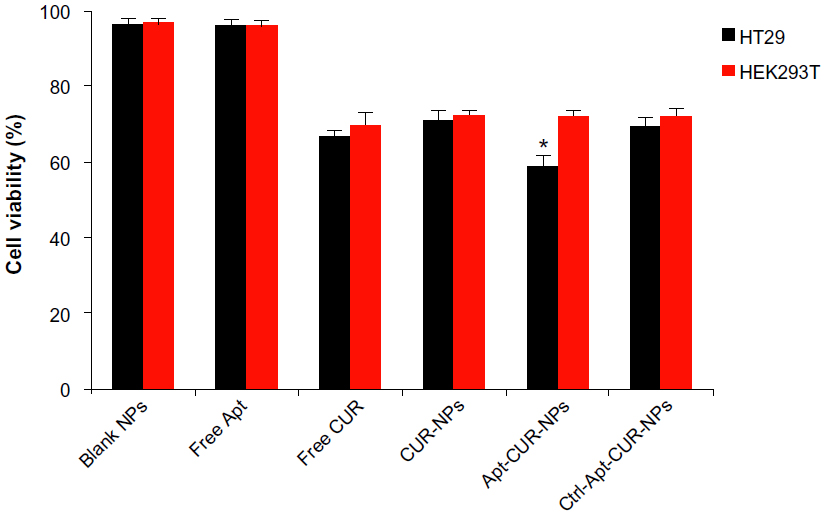 | Figure 6 Measurement of the cytotoxicity effects of Apt-CUR-NPs to cancer cells by MTT assay. |
Pharmacokinetics study in vivo
The systemic pharmacokinetics and bioavailability after intravenous administrations of 4 mg/kg of free CUR and an equivalent dose of CUR-NPs are summarized in Table 2. After the intravenous administration of free CUR suspension, a maximum plasma concentration of approximately 1,502.66±559.55 ng/mL was observed in 5 minutes. Thereafter, the free CUR was diminished abruptly and became undetectable in the serum 6 hours after administration, as the drug was distributed and rapidly metabolized, resulting in a short elimination half-time of approximately 1.07 hours. In a sharp contrast, a sustained release of CUR over 24 hours was observed when it was delivered by CUR-NPs; the maximum plasma concentration reached 1487.51±503.49 ng/mL even after 2-hour administration, contributing to a significant increase of t1/2 (about sixfold) compared with that of free CUR (P<0.01). There was a statistically significant difference in both area under the curve (AUC)(0–t) and AUC(0–∞) between free CUR and CUR-NPs (P<0.05). In addition, the CUR from CUR-NPs presented a relative bioavailability threefold superior to that of free CUR.
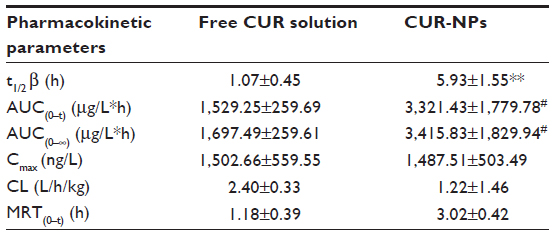 | Table 2 Summary of pharmacokinetic parameters for free CUR and CUR-NPs |
Discussion
Chemotherapy agents used for cancer therapy have limited efficacy mainly due to their low specificity toward cancer cells and poor pharmacobioavailability in vivo, resulting in incidence of normal tissue toxicity and major side effects.19–21 To address the problem, there is a high demand to explore therapeutic modalities with no or minimal side effects to normal cells. In this case, the study of using targeting molecules for cancer-specific therapy, linking to nanosized drug carriers, has been a promising strategy for targeted drug delivery and controlled drug release in the battle against cancer.5,22 In this study, we synthesized a bioconjugate composed of an RNA Apt specifically targeting the EpCAM protein of colorectal adenocarcinoma cells and CUR-encapsulated PLGA-lecithin-PEG-nanoparticles, and we performed a series of in vitro studies to demonstrate that the Apt-CUR-NP bioconjugate can efficiently deliver CUR to colon cancer cells.
A variety of elements should be taken into consideration in order to synthesize a drug-encapsulated nanoparticle with high efficiency, including selecting the right polymer composition, a reliable solvent for good drug solubility, and a solid technique.19,23 We chose the nanoprecipitation method to synthesize nanoparticles with lecithin-surrounded PLGA and its derivatives for the purpose of highly efficient encapsulation of CUR (90.13% ±4.2%); the reaction condition for nanoprecipitation was gentle, which could minimize the probability of structural damage of Apt.19,24 Since we plan to explore a functional drug nanocarrier using Apts for targeted delivery of drug-released nanoparticles to specific cells, along with the fact that EpCAM represents a well-characterized target of cancer stem cells in colon cancer, we selected the RNA Apt against EpCAM proteins of colorectal adenocarcinoma cells to generate the nanoparticle–Apt bioconjugates.
The mean size of generated bioconjugate nanoparticles including both CUR-NPs and Apt-CUR-NPs was smaller than 100 nm (about 90 nm), which is consistent with the notion that particle size less than 150 nm is desirable for promising extravasation of tumor microvasculature and tumor accumulation.25 Zeta potential is another important index for the stability of nanoparticle formulations, because high values can prevent the aggregation of nanoparticles in buffer due to strong repellent forces.26 Particles that are generated from PLGA are expected to have a slightly negative surface charge, a desirable characteristic as particles with a negative surface charge may specifically interact with the negatively charged Apts and increase their binding characteristics.27,28 In this study, the zeta potentials of CUR-NPs and Apt-CUR-NPs were −26.9 mV and −36.3 mV, respectively. It is possible that the conjugation of carboxylic acid functional groups in the terminal ends of polymers with amino groups in Apts resulted in additional negative charge on the surface of the nanoparticles, which could minimize the interaction between Apts and nanoparticles.17,29
Surface composition of nanoparticles is a key determinant of the drug bioavailability and other pharmacokinetic parameters.30,31 In this study, CUR was encapsulated into PLGA-lecithin-PEG. The formed CUR-NPs had a significantly increased half-life and mean retention time compared with that of free CUR (approximately sixfold and threefold, respectively). The approach of PEGylation has been used to prolong the circulation half-life, and it is useful for minimizing nanoparticle aggregation and beneficial in preventing the clogging of small vasculature and improving size-based targeting, which may also minimize the interaction between the conjugated Apts with the nanoparticle surface.32–34 The drug release from PLGA nanoparticles is a complicated process and many factors can affect the process, including the nanoparticle diffusion, degradation speed, stability of the protected layer, and physicochemical properties of drugs.19,23 Our CUR-NPs exhibited an initial release of CUR, which is understandable because of the burst release of deposited or weakly attached drugs on the nanoparticle surface.19,35 The sustained release of CUR from CUR-NPs was consistent with previous studies for the release process of encapsulated anticancer drugs from PLGA-NPs.35,36 Also, consistent with the literature,17,29 we demonstrated that the attachment of Apt to nanoparticles does not affect the drug release from the lipid–polymer, as the payload release profile was not significantly different between CUR-NPs and Apt-CUR-NPs.
To study whether the Apt-functionalized bioconjugate could selectively and effectively deliver drugs to colon cancer cells, the HT29 cell line with abundant EpCAM proteins on cell surfaces was chosen as a model, with HEK293T cells that do not express EpCAM as a negative control.18 Indeed, the binding of Apt-CUR-NPs to HT29 cells was increased when compared with that of control-Apt-CUR-NPs as well as with the EpCAM-negative cell line HEK293T. To complement the results achieved from confocal microscopy, we further quantitatively evaluated the cellular uptake of CUR-NPs and Apt-CUR-NPs in HT29 cells using an HPLC assay to verify the enhancement uptake of Apt-CUR-NPs by colon cancer cells. The data demonstrate that the average cellular uptake of CUR in Apt-CUR-NPs by HT29 cells was statistically significantly higher than that in all control groups. It is considered that the targeted therapeutic effect of nanoparticles is a strategy of increased internalization and enhanced retention of the drug-loaded nanoparticles within targeted cells.37 In this study, the targeted drug delivery of Apt-CUR-NP bioconjugate offers increased drug sensitivity and enhancement of drug cellular uptake. In order to further study whether the Apt–nanoparticle bioconjugate can be useful for targeted drug delivery, we compared in vitro cytotoxicity induced by Apt-CUR-NPs, free CUR, and other controls in the HT29 cell line. Cells treated with Apt-CUR-NPs produced more cytotoxicity than the control-Apt-CUR-NPs and free CUR; these results were consistent with cellular uptake results. The enhanced toxicity of Apt-CUR-NPs in colon cancer cells was likely due to EpCAM Apts attached to the surface of nanoparticles that facilitate the targeting of Apt-CUR-NPs to HT29 cells, followed by the endocytosis of nanoparticles and drug release inside of targeted cells. Of note, in in vitro cellular assays, cells are displayed as a monolayer and exposed to a constant drug concentration; this pattern cannot offer a reliable prediction of therapeutic effects in vivo because of that cells are exposed to different drug levels after the drug absorption. Therefore, it is important to evaluate the pharmacokinetic characteristic in vivo after the intravenous administration of CUR-NPs and free CUR. The results demonstrate that the half-life of CUR-NPs was 5 hours longer than that of free CUR. It is likely that solid, larger-size lipid–polymer nanoparticles could prolong drug circulation in vivo and thus facilitate the effective delivery of the payload to tumors due to extended systemic exposure.
In conclusion, we developed a targeted drug delivery carrier composed of curcumin-encapsulated nanoparticles and EpCAM RNA Apts. These multifunctional Apt-CUR-NP bioconjugates can be used as effective therapeutic modalities to deliver drugs to colon cancer cells, resulting in the selective and effective delivery of CUR to EpCAM-expressing cancer cells with enhancement of cellular uptake. This bioconjugate system could be further developed as a potential approach for both the detection and killing of colorectal adenocarcinoma cells. Further validation of Apt-CUR-NP bioconjugates in additional in vivo studies will lead to improvement of CUR delivery after systemic administration for targeted cancer treatment.
Acknowledgments
We thank Dr Yongbai Yin from the School of Physics at the University of Sydney for technical assistance. Wei Duan and his work were supported by grants from the National Health and Medical Research Council of Australia, Australia–India Strategic Research Fund, and The CASS Foundation. Lei Li was supported by a grant from National Natural Science Foundation of China (#81202484).
Disclosure
The authors report no conflicts of interest in this work.
References
Gullotti E, Yeo Y. Extracellularly activated nanocarriers: a new paradigm of tumor targeted drug delivery. Mol Pharm. 2009;6(4):1041–1051. | |
Cheng J, Teply BA, Sherifi I, et al. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials. 2007;28(5):869–876. | |
Chan JM, Zhang L, Yuet KP, et al. PLGA-lecithin-PEG core-shell nanoparticles for controlled drug delivery. Biomaterials. 2009;30(8):1627–1634. | |
Zheng Y, Yu B, Weecharangsan W, et al. Transferrin-conjugated lipid-coated PLGA nanoparticles for targeted delivery of aromatase inhibitor 7alpha-APTADD to breast cancer cells. Int J Pharm. 2010;390(2):234–241. | |
Farokhzad OC, Jon S, Khademhosseini A, Tran TN, Lavan DA, Langer R. Nanoparticle-aptamer bioconjugates: a new approach for targeting prostate cancer cells. Cancer Res. 2004;64(21):7668–7672. | |
Shigdar S, Ward AC, De A, Yang CJ, Wei M, Duan W. Clinical applications of aptamers and nucleic acid therapeutics in haematological malignancies. Br J Haematol. 2011;155(1):3–13. | |
Shigdar S, Lin J, Li Y, et al. Cancer stem cell targeting: the next generation of cancer therapy and molecular imaging. Ther Deliv. 2012;3(2):227–244. | |
Maheshwari RK, Singh AK, Gaddipati J, Srimal RC. Multiple biological activities of curcumin: a short review. Life Sci. 2006;78(18):2081–2087. | |
Anand P, Nair HB, Sung B, et al. Design of curcumin-loaded PLGA nanoparticles formulation with enhanced cellular uptake, and increased bioactivity in vitro and superior bioavailability in vivo. Biochem Pharmacol. 2010;79(3):330–338. | |
Yallapu MM, Jaggi M, Chauhan SC. Curcumin nanoformulations: a future nanomedicine for cancer. Drug Discov Today. 2012; 17(1–2):71–80. | |
Sharma RA, Gescher AJ, Steward WP. Curcumin: the story so far. Eur J Cancer. 2005;41(13):1955–1968. | |
Jung SH, Jung SH, Seong H, Cho SH, Jeong KS, Shin BC. Polyethylene glycol-complexed cationic liposome for enhanced cellular uptake and anticancer activity. Int J Pharm. 2009;382(1–2):254–261. | |
Nair KL, Thulasidasan AK, Deepa G, Anto RJ, Kumar GS. Purely aqueous PLGA nanoparticulate formulations of curcumin exhibit enhanced anticancer activity with dependence on the combination of the carrier. Int J Pharm. 2012;425(1–2):44–52. | |
Mathew A, Fukuda T, Nagaoka Y, et al. Curcumin loaded-PLGA nanoparticles conjugated with Tet-1 peptide for potential use in Alzheimer’s disease. PLoS One. 2012;7(3):e32616. | |
Albanese A, Tang PS, Chan WC. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu Rev Biomed Eng. 2012;14:1–16. | |
Bouarab L, Maherani B, Kheirolomoom A, et al. Influence of lecithin-lipid composition on physico-chemical properties of nanoliposomes loaded with a hydrophobic molecule. Colloids Surf B Biointerfaces. 2013;115C:197–204. | |
Aravind A, Jeyamohan P, Nair R, et al. AS1411 aptamer tagged PLGA-lecithin-PEG nanoparticles for tumor cell targeting and drug delivery. Biotechnol Bioeng. 2012;109(11):2920–2931. | |
Shigdar S, Qiao L, Zhou SF, et al. RNA aptamers targeting cancer stem cell marker CD133. Cancer Lett. 2013;330(1):84–95. | |
Yallapu MM, Gupta BK, Jaggi M, Chauhan SC. Fabrication of curcumin encapsulated PLGA nanoparticles for improved therapeutic effects in metastatic cancer cells. J Colloid Interface Sci. 2010;351(1):19–29. | |
Markman M. Pharmaceutical management of ovarian cancer: current status. Drugs. 2008;68(6):771–789. | |
Herzog TJ, Pothuri B. Ovarian cancer: a focus on management of recurrent disease. Nat Clin Pract Oncol. 2006;3(11):604–611. | |
Langer R. Drug delivery. Drugs on target. Science. 2001; 293(5527):58–59. | |
Bala I, Hariharan S, Kumar MN. PLGA nanoparticles in drug delivery: the state of the art. Crit Rev Ther Drug Carrier Syst. 2004;21(5):387–422. | |
Chan VS. Nanomedicine: An unresolved regulatory issue. Regul Toxicol Pharmacol. 2006;46(3):218–224. | |
Farokhzad OC, Karp JM, Langer R. Nanoparticle-aptamer bioconjugates for cancer targeting. Expert Opin Drug Deliv. 2006;3(3):311–324. | |
Kim JY, Kim JK, Park JS, Byun Y, Kim CK. The use of PEGylated liposomes to prolong circulation lifetimes of tissue plasminogen activator. Biomaterials. 2009;30(29):5751–5756. | |
Shaikh J, Ankola DD, Beniwal V, Singh D, Kumar MN. Nanoparticle encapsulation improves oral bioavailability of curcumin by at least 9-fold when compared to curcumin administered with piperine as absorption enhancer. Eur J Pharm Sci. 2009;37(3–4):223–230. | |
Grabovac V, Bernkop-Schnürch A. Development and in vitro evaluation of surface modified poly(lactide-co-glycolide) nanoparticles with chitosan-4-thiobutylamidine. Drug Dev Ind Pharm. 2007;33(7):767–774. | |
Guo J, Gao X, Su L, et al. Aptamer-functionalized PEG-PLGA nanoparticles for enhanced anti-glioma drug delivery. Biomaterials. 2011;32(31):8010–8020. | |
Hoffart V, Lamprecht A, Maincent P, Lecompte T, Vigneron C, Ubrich N. Oral bioavailability of a low molecular weight heparin using a polymeric delivery system. J Control Release. 2006;113(1):38–42. | |
Khalil NM, do Nascimento TC, Casa DM, et al. Pharmacokinetics of curcumin-loaded PLGA and PLGA-PEG blend nanoparticles after oral administration in rats. Colloids Surf B Biointerfaces. 2013;101:353–360. | |
Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R. Biodegradable long-circulating polymeric nanospheres. Science. 1994;263(5153):1600–1603. | |
Fenske DB, MacLachlan I, Cullis PR. Stabilized plasmid-lipid particles: a systemic gene therapy vector. Methods Enzymol. 2002;346:36–71. | |
Fang RH, Hu CM, Zhang L. Nanoparticles disguised as red blood cells to evade the immune system. Expert Opin Biol Ther. 2012;12(4):385–389. | |
Patil YB, Toti US, Khdair A, Ma L, Panyam J. Single-step surface functionalization of polymeric nanoparticles for targeted drug delivery. Biomaterials. 2009;30(5):859–866. | |
Sahu A, Bora U, Kasoju N, Goswami P. Synthesis of novel biodegradable and self-assembling methoxy poly(ethylene glycol)-palmitate nanocarrier for curcumin delivery to cancer cells. Acta Biomater. 2008;4(6):1752–1761. | |
Punfa W, Yodkeeree S, Pitchakarn P, Ampasavate C, Limtrakul P. Enhancement of cellular uptake and cytotoxicity of curcumin-loaded PLGA nanoparticles by conjugation with anti-P-glycoprotein in drug resistance cancer cells. Acta Pharmacol Sin. 2012;33(6):823–831. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.