Back to Journals » International Journal of General Medicine » Volume 15
Epigenetic Modifications in the Pathogenesis of Systemic Sclerosis
Received 3 January 2022
Accepted for publication 4 March 2022
Published 19 March 2022 Volume 2022:15 Pages 3155—3166
DOI https://doi.org/10.2147/IJGM.S356877
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Jiangfan Yu,1 Rui Tang,2 Ke Ding3
1Department of Dermatology, Second Xiangya Hospital of Central South University, Changsha, 410011, People’s Republic of China; 2Department of Rheumatology and Immunology, Second Xiangya Hospital of Central South University, Changsha, 410011, People’s Republic of China; 3Department of Urology, Xiangya Hospital of Central South University, Changsha, 410008, People’s Republic of China
Correspondence: Ke Ding, Department of Urology, Xiangya Hospital of Central South University, Changsha, 410008, People’s Republic of China, Email [email protected]
Abstract: Systemic sclerosis is a rare chronic autoimmune disease, which mainly manifests as immune disorders, vascular damage, and progressive fibrosis. The etiology of SSc is complex and involves multiple factors. Both genetic and environmental factors are involved in its pathogenesis. As one of the molecular mechanisms of environmental factors, epigenetic regulation plays an important role in the occurrence and development of systemic sclerosis, which involves DNA methylation, histone modification and non-coding RNA regulation. This review summarizes research advances in epigenetics, including exosomes, lncRNA, and mentions possible biomarkers and therapeutic targets among them.
Keywords: systemic sclerosis, pathogenesis, epigenetics, biomarkers
Introduction
Systemic sclerosis (SSc) is an chronic autoimmune disease involving immune system disorders, microvascular injury, and increasing organ fibrosis. The cause of SSc is still unclear, and may be related to factors such as genetics,1 environmental factors (air pollution, viral infection, chemical substances such as silicon, etc.),2 female hormones,3 cellular and humoral immune abnormalities.4 The pathogenesis of SSc remains ambiguous, and main processes currently recognized includes: immune disorders cause the body to produce a large amount of autoantibodies and cytokines to exacerbate autoimmunity; abnormal structure and function of arterioles or microcirculation, leading to fibrosis of the tube wall; fibroblasts dysfunction leads to excessive accumulation of collagen and other matrix components. These three processes are related to each other.5 SSc is divided into three subtypes: limited cutaneous SSc (lcSSc), diffuse cutaneous SSc (dcSSc) and SSc without cutaneous involvement.6 This disease can affects multiple organs throughout the body, and once severe pulmonary hypertension and scleroderma renal crisis occur, the situation is life-threatening. Because the current treatments are still limited, the prognosis and quality of life of SSc patients are not optimistic. And early diagnosis of SSc without skin lesions is challenging. Therefore, finding suitable biomarkers to assist in diagnosis or targeted therapy is particularly important.
Studies have shown that if a person has a positive family history of SSc, then his/her incidence rate of SSc will be higher than others, with the relative risk of siblings increases by 15–19 times, and first-degree relatives increases by 13–15 times.7 Compared with the general population, certain ethnic groups have an increased prevalence of SSc and different clinical manifestations.8 All of the above have confirmed that genetic factors play a part in the pathogenesis of SSc. However, it cannot fully explain the onset of this complex disease, as well illustrated by the largely incomplete concordance among identical twins.9 Besides, environmental factors can cause phenotypic changes, indicating that it may be involved in the pathogenesis of SSc. As one of the hot research mechanisms of environmental factors, the growing evidence implicates that epigenetics may provide additional explanations for immune system disorders including SSc (Figure 1).
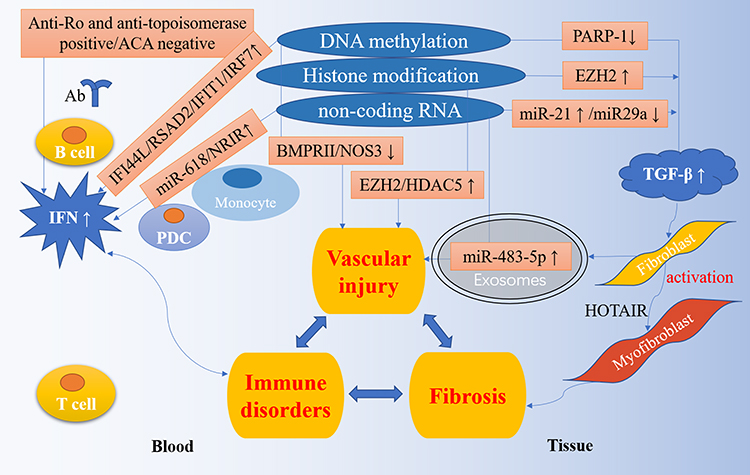 |
Figure 1 Epigenetic regulation in the pathogenesis of SSc. The pathogenesis of SSc is characterized by three processes: immune disorders, vascular damage and fibrosis. A large number of studies have shown that epigenetics plays a vital role in the pathogenesis of SSc. IFI44L, RSAD2, IFIT1, IRF7 and other IFN related genes are highly expressed in the blood due to hypomethylation; anti-Ro and anti-topoisomerase positive and ACA negative are related with IFN highly expression; the up-regulation of miR-618 and NRIR are also associated with IFN production. These controls have led to the high IFN expression pattern of SSc. The down-regulation of PARP-1 and miR-29a, and the up-regulation of EZH2 and miR-21 participate in the TGF-β pathway and activate fibroblasts into myofibroblasts. In addition, the decrease of BMPRII and NOS3, the increase of EZH2, HDAC5, and miR-483-5p are involved in vascular injury. |
Epigenetics
Epigenetics is the subject of studying the heritable changes in genes without changing their nucleotide sequences. There are a lot of research in various disciplines, and it has developed into an increasingly diverse field of scientific inquiry. It is mainly composed of three parts: DNA methylation, histone modification and non-coding RNA-mediated regulation. It typically involves changes in gene expression levels, chromatin structural stability and other functions.10 More and more studies have shown that epigenetics is a biological process, and its disorder is related to the occurrence of some autoimmune diseases.11
DNA methylation is an important research content in epigenetics. In this biological process, the role of DNA methyltransferase (DNMT) is crucial.12 Since this process mainly occurs in the promoter region of the gene CpG island, its effect will inhibit gene transcription.13 DNMTs mainly includes DNMT1, DNMT3a and DNMT3b. Different DNMTs have different functions. For example, DNMT1 is ubiquitous and maintains DNA methylation level, while DNMT3a and DNMT3b promote the re-methylation process, thus ensuring methylation during embryonic development.14
Besides DNA methylation, histone modification is another epigenetic mechanism, which is a covalent post-translational regulation, including ubiquitination, phosphorylation, methylation, acetylation, ADP ribosylation, sulfonylation, etc.15 By changing the electric charge of histones and the conformation of chromatin, the expression of genes can be regulated. Among them, acetylation and methylation are the most studied.16 Histone acetylases (HATs) and histone deacetylases (HDACs) are the two main enzymes involved in the process of histone acetylation. By recruiting or removing acetyl groups on histones, histone acetylation is at different levels, thereby activating or inhibiting gene transcription.17 While histone methyltransferase (HMT) and demethylase (HDM) are the other two main enzymes that catalyze histone methylation. How histone methylation affects gene expression depends on the position of amino acid residues and the number of methyl groups.18 The results of histone modifications are affected by many aspects, and there are also interrelationships among various modifications.
In addition to the above two processes, non-coding RNA is also one of the most studied epigenetic mechanisms, consisting of microRNA (miRNA) and long non-coding RNA (lncRNA). Among them, miRNA are about 22 nucleotides, while lncRNA is more than 200 nucleotides.19 miRNA usually binds to the 3’-UTR of target mRNA, thereby regulating gene expression after transcription, and then preventing gene translation through mRNA cleavage and degradation.20 It regulates a variety of key pathways, and dysregulation can lead to inflammation, fibrosis, angiogenesis, etc.21 lncRNA is a group of newly identified molecules and current research on it is still at an early stage. Relevant studies have shown that lncRNA may participate in various gene regulatory mechanisms, including gene transcription and may act a pivotal part in regulating the development of the disease.22
The aim of this review is to summarizing the research results of predecessors in epigenetics and the latest research progress in recent years, and strive to further understand the pathogenesis of SSc and find the most possible biomarkers.
Abnormal DNA Methylation
DNA Methylation and Immune-Related Cells
Previous studies have confirmed that IFI44L was hypomethylated and together with its overexpression in the blood in immune-related diseases such as systemic lupus erythematosus (SLE) and Sjogren’s syndrome (SS), suggesting that IFI44L methylation differences may serve as a serum biomarker for these common autoimmune diseases.23 Similarly, IFI44L was overexpressed in SSc blood, as were RSAD2 and TYROBP.24–26 More recently, Ramos et al found 27 differentially expressed genes by detecting the whole genome DNA methylation in whole blood, and newly discovered CYFIP1 gene hypomethylation, STAT3, TNFRSF1A, TNXB, TLE3 gene hypermethylation. Correspondingly, TNXB expression was low in SSc blood.27 Another research by Chen et al also newly found that IFIT1, IRF7, MX1, OAS1 and USP18 in CD4+T cells of SSc patients were significantly hypomethylated.28 The above IFI44L, RSAD2, IFIT1, IRF7 and other genes were all type I interferon (IFN) related genes. Dysfunction of type I IFN has been studied in various immune-related diseases such as SSc and SLE.29 Further study has reported that some SSc patients had high expression of IFN-induced genes,30 and one of the possible explanations is that the serum of most anti-topoisomerase positive SSc patients could induce IFN production.31 SSc serum antibodies might have a gradient relationship with IFN markers,32 which will provide favorable evidence for the research of the pathogenesis and therapeutic targets of SSc.
Wang et al has observed that hypermethylation and decreased expression of FOXP3 in CD4+T cells in patients with SSc, the promoter methylation status and expression level of FOXP3 were closely related to disease activity.33 Subsequent studies also confirmed that FOXP3 was hypermethylated in the peripheral blood of SSc, besides, PRF1, ITGAL hypermethylation and CDKN2A and CD70 hypomethylation were also found.34 Compared with healthy women, women with SSc had lower DNA regulatory sequence methylation levels and significantly higher CD40L mRNA expression in CD4+T cells.35 This might be an explanation for female susceptibility. Moreover, abnormal methylation changes might cause the differential expression of genes in peripheral blood mononuclear cells (PBMC) and also participated in the aberrant activation, proliferation and migration of immune cells in SSc patients. Simultaneously, four methylation-regulated genes F2R, FYN, PAG1 and PRKCH were differentially expressed between SSc with and without interstitial lung disease (ILD).36 These differentially methylated sites further revealed epigenetic alterations in the pathogenesis of SSc.
DNA Methylation and Fibroblasts
Altorok et al found that COL8A1, COL16A1, COL29A1 in dcSSc fibroblasts and COL1A1, COL6A3, COL12A1 in lcSSc fibroblasts were all hypomethylated by detecting genome-wide DNA methylation in skin fibroblasts from SSc patients. Furthermore, PAX9 and TNXB were hypomethylated in both dcSSc and lcSSc, which are related to collagen synthesis and extracellular matrix (ECM) glycoproteins.37 Similarly, the methylation level of SSc fibroblasts decreased, and the level of DNA demethylases TET1 mRNA increased; in the hypoxic environment, TET1 expression in SSc fibroblasts was abnormally regulated and was accompanied by overall DNA hypomethylation.38 Recently, studies demonstrated that the enzyme poly(ADP-ribose) polymerase-1 (PARP-1) negatively regulated transforming growth factor β (TGF-β) signal in cutaneous fibrosis.39 The decreased expression of PARP-1 was due to DNA hypermethylation in its promoter region, and decreased PARP-1 might be directly participate in the excessive activation of TGF-β signaling and the sustained activation of fibroblasts.39 Recent studies have reported hypomethylation of DLX5 and TMEM140 in skin fibroblasts from African-American SSc patients, accompanied by overexpression of these genes.40 It also provided further strong evidence that people of African ancestry were more likely to develop SSc, but the specific factors behind racial differences remained elusive, and epigenetics may provide a more scientifically plausible platform for unraveling this mystery.
MeCP2 protein is the main component of the methylated CpG binding domain (MBD) family. It inhibits the transcription of its downstream target genes through specific binding to methylated DNA and is an important transcription inhibitor. Compared with the control group, the levels of MeCP2, MBD-1, MBD-2, DNMT1 of SSc fibroblasts increased significantly, and CpG islands in the promoter region of the collagen suppressor gene FLI1 of SSc skin fibroblasts were hypermethylated.41 Besides the overall DNA methylation differences in SSc dermal fibroblasts, the transcription factors genes RUNX1, RUNX2 and RUNX3 of dcSSc and lcSSc were also hypomethylated,37,42 and DNA methylation was also involved in the effect of the TGF-β pathway.42 The study of Henderson et al showed that MeCP2 highly expressed in SSc fibroblasts enhanced Wnt signal transduction and collagen synthesis.43 However, the study of He et al unveiled that the overexpression of MeCP2 in skin fibroblasts inhibited the differentiation, proliferation and migration of myofibroblasts. Genes such as PLAU, NID2, and ADA partially mediated the anti-fibrotic effect of MeCP2.44 Although the reasons for the inconsistency between the two studies were not clear, these results all illustrated that abnormal DNA methylation was closely related to the pathogenesis of SSc, and its specific molecular mechanisms deserved further study.
DNA Methylation and Endothelial Cells
Microvascular endothelial cells (MVEC) played a critical role in regulating the contraction and expansion of normal blood vessels, and many studies have confirmed the damage of endothelial cells in SSc.45–48 Wang et al have demonstrated that extensive methylation of CpG sites in the promoter region of bone morphogenetic protein receptor type II receptor (BMPRII) leads to its reduced expression, which resulted in MVECs of SSc being more prone to apoptosis than healthy controls. Corresponding, adding 2-deoxy-5-azacytidine and trichostatin A (TSA) to SSC MVECs normalized BMPRII expression level.49 Nitric oxide (NO) has been previously reported to be one of the most important vasodilators, synthesized by endothelial nitric oxide synthase (eNOS) encoded by the NOS3 gene. A significant down-regulation of NOS3 expression was observed in SSc MVECs and the CpG site in the promoter region of NOS3 was extensively methylated.50 Recent studies have pointed out that there were significant differences in genome-wide DNA methylation in SSc MVEC, mainly at 2455 CpG sites. The hypermethylated genes identified in SSc MVEC included NOS1, DNMT3A, DNMT3B, HDAC4 and ANGPT2.51 This further proved that the methylation of genes related to angiogenesis was involved in the occurrence and development of scleroderma.
DNA Methylation and Serum Cytokine
In a study by Zhu et al, the DNA methylation changes of serum cytokines were analyzed in combination with the genome-wide methylation expression profile, and it was displayed that the transcription initiation regions of IL-10, IL-12P70, IL-1β, IL-1ra and VEGF all have DNA hypomethylation, suggesting that they may all be associated with DNA methylation.52
Histone Modification
Histone Modification and Immune-Related Cells
Previous research by Wang et al has shown that the overall histone H4 hyperacetylation and the overall histone H3K9 hypomethylation in the B cells of SSc patients. Overall histone H4 acetylation was negatively correlated with HDAC2 expression. At the same time, histone modifications in SSc B cells were related to skin thickness and disease activity, indicating that it might become a valuable epigenetic marker.53 Ciechomska et al have proven that under certain conditions, histone demethylation in monocytes promoted transdifferentiation of fibroblasts in SSc.54 In addition to B cells, histone modifications at the whole genome level were also associated with changes in the phenotype of SSc monocytes. Van der Kroef et al have reported that there were abnormal histone methylation modifications at multiple gene sites. The expression of some genes was directly related to the level of histone methylation near the transcription start point. Genes related to histone modification were mainly involved in immune, IFN and antiviral pathways.55
Histone Modification and Fibroblasts
Histone modifications have a role in regulating the expression of genes associated with fibroblast activation of myofibroblasts, governing their fate, as there are persistent histone DNA methylation and deacetylation in SSc fibroblasts.56 The study by Wang et al has shown that adding HDAC inhibitors (TSA) significantly reduced the expression of type I collagen in SSc fibroblasts; compared with the control group, the levels of HDAC1 and HDAC6 of SSc fibroblasts increased significantly. The acetylation of histones H3 and H4 in the promoter region of FLI1 gene of SSc fibroblasts was significantly reduced besides.41 Recently, the effect of the histone methyltransferase enhancer of zeste homolog 2 (EZH2) has been studied in SSc fibroblasts. EZH2 mediated the trimethylation of histone H3 lysine 27 (H3K27me3) to inhibit transcription. In a previous study, Kramer et al demonstrated that inhibition of H3K27me3 stimulated collagen release from fibroblasts in a time and dose-dependent manner, and treatment with DZNep (an inhibitor of EZH2 and H3K27me3) exacerbated fibrosis induced by bleomycin or by overexpression of TGF-βI.57 Moreover, inhibition of H3K27me3 induced the expression of the profibrotic transcription factor Fra-2 both in vitro and in vivo, and addition of Fra-2 siRNA reduced the profibrotic effect of DZNep.57 These all suggested inhibiting H3K27me3 could induce fibrosis. However, another study by Tsou et al showed the opposite, DZNep dose-dependently reduced the expression of COL1A1, TGF-β, LRRC16A, Fra-2 and other fibrosis-related genes in SSc fibroblasts, and inhibited the migration activity of SSc fibroblasts.58 In addition, EZH2 inhibitors prevented bleomycin-induced skin fibrosis, and overexpression of EZH2 accelerated wound healing, while high-dose GSK126 (another EZH2 inhibitor) significantly inhibited wound healing; furthermore, hypermethylation and low expression of LRRC16A gene were associated with EZH2-mediated cell migration.58 The above showed that EZH2 inhibitors could prevent fibrosis in vitro and in vivo. Meanwhile, the study by Wasson et al found that inhibiting EZH2 with GSK126 also inhibited the increase in collagen and α-SMA expression in SSc dermal fibroblasts, confirming former work of Tsou et al.59 One possible explanation for this inconsistent result is that the upregulation of H3K27me3 in SSc fibroblasts may be a compensatory attempt to neutralize the increased expression of profibrotic genes in SSc fibroblasts under different disease conditions. The regulation of fibrosis by H3K27me3 may be related to the diversity of histone methylases and the complexity of the molecular mechanisms of transcriptional regulation. H3K27me3 may be a central regulator of SSc fibrosis, and the exact role requires further studies to clarify.
Histone Modification and Endothelial Cells
In another study by Tsou et al, it was reported that the expression of HDAC5 in endothelial cells of SSc patients was significantly higher than that of healthy controls. Silencing HDAC5 in SSc endothelial cells restored normal angiogenesis. CYR61, PVRL2 and FSTL1 were key genes involved in angiogenesis and fibrosis regulated by HDAC5. Knockdown of HDAC5 and CYR61, PVRL2 or FSTL1 at the same time inhibited the angiogenesis of SSC endothelial cells. On the contrary, overexpression of these genes alone resulted in an increase in angiogenesis in SSc, indicating that these genes regulated by HDAC5 played a functional role in the angiogenesis disorder of SSc.60 The use of HDAC inhibitors has also shown promising anti-fibrosis effects both in vivo and in vitro.41,61 Previous studies have proven that EZH2 affected the regulation of genes related to endothelial cells function, especially genes related to cell adhesion and migration.62 The overexpression of EZH2 in normal dermal endothelial cells inhibited angiogenesis, while the decrease of EZH2 led to an increase in angiogenesis. EZH2 was elevated in SSc endothelial cells, knocking down EZH2 in SSc endothelial cells significantly increased angiogenesis compared to the control. Similarly, treatment of SSc endothelial cells with DZNep reduced the expression of EZH2 in SSc endothelial cells by approximately 50%, thereby significantly enhancing angiogenesis. These data indicated that EZH2 might inhibit angiogenesis in SSc endothelial cells by inhibiting pro-angiogenic genes or activating anti-angiogenic genes. Further research reported that inhibition of EZH2 in endothelial cells restored the normal angiogenesis of SSc by activating the NOTCH pathway, especially by up-regulating NOTCH ligand DLL4.58
Non-Coding RNA-Mediated Regulation
miRNAs in the Blood
The expression of miR-483-5p increased in the serum, fibroblasts and endothelial cells of SSc patients, and regulated the expression of fibrosis-related genes. It was worth noting that miR-483-5p was also up-regulated in localized scleroderma, but it was not up-regulated in other autoimmune diseases (such as SLE and SS), suggesting that it has a potential role as a unique and precise biomarker and mediator of SSc fibrosis.63 In addition, Koba et al reported that combined detection of serum miR-206 and miR-21 levels was more helpful to distinguish scleroderma patients from normal people than detection of miR-206 or miR-21 alone.64 Furthermore, miR-5196 in monocytes and serum of SSc patients increased significantly, and was positively correlated with CRP levels. Exogenous transfection of miR-5196 reduced the expression of TIMP-1 that promoted fibrosis, suggesting that it may be a potential regulator of SSc fibrosis, and miR-5196 was considered to be a potential biomarker for SSc.65 Plasmacytoid dendritic cells (PDCs) were an important source of I type IFNs and participated in the occurrence and maintenance of autoimmunity. The expression of miR-618 in peripheral blood PDCs of SSc patients was up-regulated, including those with early disease without skin fibrosis. IFN regulatory factor 8 was a key transcription factor for PDC formation and activation, and it has been identified as a target of miR-618. The up-regulation of miR-618 inhibited the development of PDCs and enhanced their ability to secrete IFN-α, which may be one of the reasons for the type I IFN characteristics of SSc patients. Therefore, miR-618 may be an important epigenetic target in SSc immune regulation.66 In PBMC of SSc-ILD, the expression of miR-155 was closely related to lung function.67 Moreover, the level of miR-200C in PBMC of SSc-ILD patients was negatively correlated with forced vital capacity and forced expiratory volume in 1 second, and the level of miR-200C was positively correlated with the severity of SSc-ILD. Thus miR-200C could be used as a biomarker of the severity of ILD in SSc patients.68 And circulating miRNA profiles were different between lcSSc and dcSSc patients and between patients with different autoantibodies.69
miRNAs in the Fibroblasts
The study by Liakouli et al found that EGFL7 (Epidermal Growth Factor Like-domain 7), which played an important role in angiogenesis, and its specific negative regulator miR-126 might be related to the pathogenesis of SSc vascular disease and fibrosis.70 In addition, the serum expression level of miR-126 could distinguish RA patients from SSc patients in the study of Stypinska et al.71 In both dcSSc and TGF-β treated fibroblasts, miR-21 increased while miR-29a decreased. MiR-21 and TGF-β had a synergistic effect in inducing collagen production, while miR-29a reduced the collagen production of dermal fibroblasts induced by TGF-β.72 Other studies have also confirmed the pro-fibrotic effect of miR-21 and the anti-fibrotic effect of miR-29a.73–75 Besides being involved in the production of collagen, miR-21 and miR-29a also played a role in the apoptosis of myofibroblasts. Previous studies showed that the overexpression and inhibition of miR-29a and miR-21 respectively induced apoptosis of dermal fibroblasts.76,77 These results suggested that miR-21 and miR-29a may be potential therapeutic targets or useful prognostic or diagnostic markers. Several other related miRNAs in SSc fibroblasts were miR-145, miR-155, miR-196a, let-7a, miR-29b-3p, miR-138-5p and miR-146b-5p, which were associated with immune responses and fibroblast activation.78–85
miRNAs in the Lungs
Previous studies have reported that miR-155 and miR-143 in lung tissue of SSC-ILD patients were closely related to the high-resolution CT score.67 Moreover, He et al identified 26 differentially expressed miRNAs by comparing SSc-ILD samples with healthy control lung tissue samples. Among them, 2 miRNAs (hsa-let-7e and hsa-miR-455) in SSc-ILD samples were significantly up-regulated, while the other 24 miRNAs were down-regulated. The top 10 dysregulated miRNAs were hsa-miR-4459, hsa-miR-4507, hsa-miR-4417, hsa-miR-3687, hsa-miR-1246, hsa-miR4668, hsa-miR-4530, hsa-miR-4299, hsa-miR-4516 and has-miR-663b, mainly associated with inflammation and fibroblast regulation related signaling pathways, such as ECM-receptor interaction, TGF-β signaling pathway, Wnt signaling pathway, and cell adhesion connection. CX3CL1, FOSL1 and FOSB were the target genes predicted by these miRNAs.86 A large cohort study showed that CX3XL1 was associated with anti-topoisomerase I antibodies and pulmonary fibrosis, as well as the progression of ILD.87 The expression of FOSL1 in the lung mediated anti-fibrosis effects by regulating the expression of proinflammatory, profibrotic and fibrotic genes, providing a potential biomarker for poor prognosis and treatment options for pulmonary fibrosis.88 However, these studies have some limitations due to the limited number of samples and lack of sufficient in vivo and in vitro experimental verification. Therefore, its clinical significance needs to be further studied.
lncRNAs
So far, only a few lncRNAs that regulate immune response have been reported in SSc. NRIR in monocytes of patients with SSc was closely related to IFN response, which further supports the abnormal phenomenon of IFN in SSc.89 Abd-Elmawla et al analyzed the plasma expression profile of lncRNA, and studied its relationship with disease types and related clinical indicators, and proposed plasma SPRY4-IT1, HOTTIP, ANCR and TINCR as new candidate biomarkers for SSc.90 Further, the study by Takata et al unveiled that OTUD6B-AS1 regulated cell proliferation and apoptosis through the expression of cyclinD1 independent of the sense gene, revealing its potential value in regulating the apoptosis of fibroblasts and vascular smooth muscle cells, and may participate in the pathogenesis of SSc.91 Another study by Pachera et al showed that the expression of H19X was dose-dependent with the concentration of TGF-β and could effectively induce fibroblast differentiation and survival, enhance ECM production and tissue progressive fibrosis.92 Of note, the study by Wang et al demonstrated that intrinsic TGF-β activation may result in increased expression of TSIX in the serum of patients with SSc, and that TSIX was able to regulate collagen production.93 Since increased levels of TSIX appear to correlate with levels of skin fibrosis, it has the potential to act as a novel disease biomarker that may also serve as a potential therapeutic target.94 Recently, HOTAIR was found to induce skin fibroblasts differentiation in vitro, driving EZH2-dependent myofibroblast activation in SSc through miRNA 34a-dependent activation of NOTCH.59 Apart from Notch signaling, HOTAIR can also induce fibrosis through Hedgehog signaling.95 The differentially expressed ncRNA00201 and PSMB8-AS1 in blood and MGC12916 in fibroblasts were also confirmed to be important players in the pathogenesis of SSc.40,96,97 The involvement of lncRNAs in the pathogenesis of scleroderma is intricate and complicated, which may be through the epigenetic regulation of the gene itself, interacting with miRNAs, or affecting the secretion of important cytokines such as IL-6 and TNFα,97 and participating in regulating the expression of fibrosis-related genes. At present, the field of lncRNA is still in its infancy, and related research is still limited but proved to be effective. Therefore, this is a promising prospect and it is worthwhile to further research the epigenetic mechanism of SSc.
Exosomes
Exosomes are thought to mediate cell-to-cell communication by transferring their contents (protein, RNA, miRNA, lncRNA, circRNA and DNA) to target cells. The study by Wermuth et al have shown that compared with the normal control group, 6 fibrotic miRNAs in serum exosomes isolated from SSc patients increased, and 10 anti-fibrotic miRNAs decreased. In addition, serum exosomes isolated from SSc patients stimulated normal human skin fibroblasts to express fibrosis genes and type I collagen, and produced and secreted fibronectin in a dose-dependent manner.98
Interestingly, many miRNAs were encapsulated in exosomes for circulation, such as miR-483-5p mentioned earlier. These findings suggest that miRNAs in exosomes may be involved in the transmission of fibrotic signals, and related study has further confirmed this view.99
Conclusion
Epigenetics has been in development for decades and has been implicated in many autoimmune diseases. In recent years, with the in-depth understanding of various pathway mechanisms, it has been found that epigenetics plays a pivotal role in the occurrence and development of systemic scleroderma. This review summarizes the recent advances in epigenetics in the pathogenesis of SSc and attempts to find suitable disease-related biomarkers and therapeutic targets (Table 1). However, up to now, the etiology of SSc is still under research, and studies on the specific mechanisms of tissue fibrosis, vascular lesions, and immune disorders in SSc are limited. Moreover, the results of some experiments are inconsistent, which also shows that the epigenetic mechanism of SSc is inscrutable, which requires further exploration.
 |
Table 1 Epigenetic Regulation in the Pathogenesis of SSc |
With the progress of research, more and more epigenetically related molecules have been excavated for their medical value, and agonists or inhibitors of the above molecules are also a promising choice for future targeted therapy of SSc. If the non-invasive detection of the above-mentioned biological molecules can achieve the effects of judging the condition, assisting the treatment, and evaluating the prognosis, this will become more scientifically meaningful, but many obstacles need to be overcome. As mentioned earlier in the text, many genes related to the interferon pathway undergo epigenetic changes and appear to be associated with different antibodies in SSc, which provides the possibility of targeted therapy in the clinic. If the above-mentioned epigenetically related molecules can be developed into novel dressings with percutaneous absorption or micro sustained release, it may also be a future direction for the treatment of localized scleroderma, but it also requires a lot of follow-up and research.
Ethics Statement
The authors confirm to have complied with ethical standards.
Funding
This work was supported by the National Natural Science Foundation of China (No. 82003363 and No. 82001738), the Changsha Municipal Natural Science Foundation (kq2007059), and Hunan Provincial Natural Science Foundation of China (2021JJ40820).
Disclosure
The authors declare that they have no conflicts of interest for this work.
References
1. Orvain C, Assassi S, Avouac J, et al. Systemic sclerosis pathogenesis: contribution of recent advances in genetics. Curr Opin Rheumatol. 2020;32(6):505–514. doi:10.1097/BOR.0000000000000735
2. Ferri C, Arcangeletti M-C, Caselli E, et al. Insights into the knowledge of complex diseases: environmental infectious/toxic agents as potential etiopathogenetic factors of systemic sclerosis. J Autoimmun. 2021;124:102727. doi:10.1016/j.jaut.2021.102727
3. Hughes M, Pauling JD, Armstrong-James L, et al. Gender-related differences in systemic sclerosis. Autoimmun Rev. 2020;19(4):102494. doi:10.1016/j.autrev.2020.102494
4. Zuber JP, Spertini F. Immunological basis of systemic sclerosis. Rheumatology (Oxford). 2006;45 Suppl 3:iii23–25. doi:10.1093/rheumatology/kel285
5. Pattanaik D, Brown M, Postlethwaite BC, et al. Pathogenesis of systemic sclerosis. Front Immunol. 2015;6:272. doi:10.3389/fimmu.2015.00272
6. van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American college of rheumatology/European league against rheumatism collaborative initiative. Ann Rheum Dis. 2013;72(11):1747–1755. doi:10.1136/annrheumdis-2013-204424
7. Arnett FC, Cho M, Chatterjee S, et al. Familial occurrence frequencies and relative risks for systemic sclerosis (scleroderma) in three United States cohorts. Arthritis Rheum. 2001;44(6):1359–1362. doi:10.1002/1529-0131(200106)44:6<1359::AID-ART228>3.0.CO;2-S
8. Barnes J, Mayes MD. Epidemiology of systemic sclerosis: incidence, prevalence, survival, risk factors, malignancy, and environmental triggers. Curr Opin Rheumatol. 2012;24(2):165–170. doi:10.1097/BOR.0b013e32834ff2e8
9. Feghali-Bostwick C, Medsger TJ, Wright TM. Analysis of systemic sclerosis in twins reveals low concordance for disease and high concordance for the presence of antinuclear antibodies. Arthritis Rheum. 2003;48(7):1956–1963. doi:10.1002/art.11173
10. Luo Y, Wang Y, Shu Y, et al. Epigenetic mechanisms: an emerging role in pathogenesis and its therapeutic potential in systemic sclerosis. Int J Biochem Cell Biol. 2015;67:92–100. doi:10.1016/j.biocel.2015.05.023
11. Wu H, Chen Y, Zhu H, et al. The pathogenic role of dysregulated epigenetic modifications in autoimmune diseases. Front Immunol. 2019;10:2305. doi:10.3389/fimmu.2019.02305
12. Schubeler D. Function and information content of DNA methylation. Nature. 2015;517(7534):321–326. doi:10.1038/nature14192
13. Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128(4):669–681. doi:10.1016/j.cell.2007.01.033
14. Jurkowska RZ, Jeltsch A. Mechanisms and biological roles of DNA methyltransferases and DNA methylation: from past achievements to future challenges. Adv Exp Med Biol. 2016;945:1–17.
15. Rothbart SB, Strahl BD. Interpreting the language of histone and DNA modifications. Biochim Biophys Acta. 2014;1839(8):627–643. doi:10.1016/j.bbagrm.2014.03.001
16. Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi:10.1016/j.cell.2007.02.005
17. Wapenaar H, Dekker FJ. Histone acetyltransferases: challenges in targeting bi-substrate enzymes. Clin Epigenetics. 2016;8:59. doi:10.1186/s13148-016-0225-2
18. Song Y, Wu F, Wu J. Targeting histone methylation for cancer therapy: enzymes, inhibitors, biological activity and perspectives. J Hematol Oncol. 2016;9(1):49. doi:10.1186/s13045-016-0279-9
19. Zhao C-N, Mao Y-M, Liu L-N, et al. Emerging role of lncRNAs in systemic lupus erythematosus. Biomed Pharmacother. 2018;106:584–592. doi:10.1016/j.biopha.2018.06.175
20. Winter J, Jung S, Keller S, et al. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009;11(3):228–234. doi:10.1038/ncb0309-228
21. Zhang T, Hu J, Wang X, et al. MicroRNA-378 promotes hepatic inflammation and fibrosis via modulation of the NF-kappaB-TNFalpha pathway. J Hepatol. 2019;70(1):87–96. doi:10.1016/j.jhep.2018.08.026
22. Robinson EK, Covarrubias S, Carpenter S. The how and why of lncRNA function: an innate immune perspective. Biochim Biophys Acta Gene Regul Mech. 2020;1863(4):194419. doi:10.1016/j.bbagrm.2019.194419
23. Zhao M, Zhou Y, Zhu B, et al. IFI44L promoter methylation as a blood biomarker for systemic lupus erythematosus. Ann Rheum Dis. 2016;75(11):1998–2006. doi:10.1136/annrheumdis-2015-208410
24. Christmann RB, Hayes E, Pendergrass S, et al. Interferon and alternative activation of monocyte/macrophages in systemic sclerosis-associated pulmonary arterial hypertension. Arthritis Rheum. 2011;63(6):1718–1728. doi:10.1002/art.30318
25. Bos CL, van Baarsen LGM, Timmer TCG, et al. Molecular subtypes of systemic sclerosis in association with anti-centromere antibodies and digital ulcers. Genes Immun. 2009;10(3):210–218. doi:10.1038/gene.2008.98
26. Tan FK, Zhou X, Mayes MD, et al. Signatures of differentially regulated interferon gene expression and vasculotrophism in the peripheral blood cells of systemic sclerosis patients. Rheumatology (Oxford). 2006;45(6):694–702. doi:10.1093/rheumatology/kei244
27. Ramos PS, Zimmerman KD, Haddad S, et al. Integrative analysis of DNA methylation in discordant twins unveils distinct architectures of systemic sclerosis subsets. Clin Epigenetics. 2019;11(1):58. doi:10.1186/s13148-019-0652-y
28. Chen S, Pu W, Guo S, et al. Genome-wide DNA methylation profiles reveal common epigenetic patterns of interferon-related genes in multiple autoimmune diseases. Front Genet. 2019;10:223. doi:10.3389/fgene.2019.00223
29. Chen K, Liu J, Cao X. Regulation of type I interferon signaling in immunity and inflammation: a comprehensive review. J Autoimmun. 2017;83:1–11. doi:10.1016/j.jaut.2017.03.008
30. Bujor AM, El Adili F, Parvez A, et al. Fli1 downregulation in scleroderma myeloid cells has profibrotic and proinflammatory effects. Front Immunol. 2020;11:800. doi:10.3389/fimmu.2020.00800
31. Kim D, Peck A, Santer D, et al. Induction of interferon-alpha by scleroderma sera containing autoantibodies to topoisomerase I: association of higher interferon-alpha activity with lung fibrosis. Arthritis Rheum. 2008;58(7):2163–2173. doi:10.1002/art.23486
32. Assassi S, Mayes MD, Arnett FC, et al. Systemic sclerosis and lupus: points in an interferon-mediated continuum. Arthritis Rheum. 2010;62(2):589–598. doi:10.1002/art.27224
33. Wang YY, Wang Q, Sun XH, et al. DNA hypermethylation of the forkhead box protein 3 (FOXP3) promoter in CD4+ T cells of patients with systemic sclerosis. Br J Dermatol. 2014;171(1):39–47. doi:10.1111/bjd.12913
34. Matatiele P, Tikly M, Tarr G, et al. DNA methylation similarities in genes of black South Africans with systemic lupus erythematosus and systemic sclerosis. J Biomed Sci. 2015;22(1):34. doi:10.1186/s12929-015-0142-2
35. Lian X, Xiao R, Hu X, et al. DNA demethylation of CD40l in CD4+ T cells from women with systemic sclerosis: a possible explanation for female susceptibility. Arthritis Rheum. 2012;64(7):2338–2345. doi:10.1002/art.34376
36. Zhu H, Zhu C, Mi W, et al. Integration of genome-wide DNA methylation and transcription uncovered aberrant methylation-regulated genes and pathways in the peripheral blood mononuclear cells of systemic sclerosis. Int J Rheumatol. 2018;2018:7342472. doi:10.1155/2018/7342472
37. Altorok N, Tsou P-S, Coit P, et al. Genome-wide DNA methylation analysis in dermal fibroblasts from patients with diffuse and limited systemic sclerosis reveals common and subset-specific DNA methylation aberrancies. Ann Rheum Dis. 2015;74(8):1612–1620. doi:10.1136/annrheumdis-2014-205303
38. Hattori M, Yokoyama Y, Hattori T, et al. Global DNA hypomethylation and hypoxia-induced expression of the ten eleven translocation (TET) family, TET1, in scleroderma fibroblasts. Exp Dermatol. 2015;24(11):841–846. doi:10.1111/exd.12767
39. Zhang Y, Pötter S, Chen C-W, et al. Poly(ADP-ribose) polymerase-1 regulates fibroblast activation in systemic sclerosis. Ann Rheum Dis. 2018;77(5):744–751. doi:10.1136/annrheumdis-2017-212265
40. Baker FD, da Silveira W, Hazard ES, et al. Differential DNA methylation landscape in skin fibroblasts from African Americans with systemic sclerosis. Genes (Basel). 2021;12(2):129.
41. Wang Y, Fan PS, Kahaleh B. Association between enhanced type I collagen expression and epigenetic repression of the FLI1 gene in scleroderma fibroblasts. Arthritis Rheum. 2006;54(7):2271–2279. doi:10.1002/art.21948
42. Noda S, Asano Y, Nishimura S, et al. Simultaneous downregulation of KLF5 and Fli1 is a key feature underlying systemic sclerosis. Nat Commun. 2014;5(1):5797. doi:10.1038/ncomms6797
43. Henderson J, Brown M, Horsburgh S, et al. Methyl cap binding protein 2: a key epigenetic protein in systemic sclerosis. Rheumatology (Oxford). 2019;58(3):527–535. doi:10.1093/rheumatology/key327
44. He Y, Tsou P-S, Khanna D, et al. Methyl-CpG-binding protein 2 mediates antifibrotic effects in scleroderma fibroblasts. Ann Rheum Dis. 2018;77(8):1208–1218. doi:10.1136/annrheumdis-2018-213022
45. Marden G, Wan Q, Wilks J, et al. The role of the oncostatin M/OSM receptor beta axis in activating dermal microvascular endothelial cells in systemic sclerosis. Arthritis Res Ther. 2020;22(1):179. doi:10.1186/s13075-020-02266-0
46. Takagi K, Kawamoto M, Higuchi T, et al. Single nucleotide polymorphisms of the HIF1A gene are associated with susceptibility to pulmonary arterial hypertension in systemic sclerosis and contribute to SSc-PAH disease severity. Int J Rheum Dis. 2020;23(5):674–680. doi:10.1111/1756-185X.13822
47. Nakamura K, Taniguchi T, Hirabayashi M, et al. Altered properties of endothelial cells and mesenchymal stem cells underlying the development of scleroderma-like vasculopathy in KLF5 +/−;Fli-1 +/− Mice. Arthritis Rheumatol. 2020;72(12):2136–2146. doi:10.1002/art.41423
48. Zhang Y, Zhu H, Layritz F, et al. Recombinant adenosine deaminase ameliorates inflammation, vascular disease, and fibrosis in preclinical models of systemic sclerosis. Arthritis Rheumatol. 2020;72(8):1385–1395. doi:10.1002/art.41259
49. Wang Y, Kahaleh B. Epigenetic repression of bone morphogenetic protein receptor II expression in scleroderma. J Cell Mol Med. 2013;17(10):1291–1299. doi:10.1111/jcmm.12105
50. Fish JE, Marsden PA. Endothelial nitric oxide synthase: insight into cell-specific gene regulation in the vascular endothelium. Cell Mol Life Sci. 2006;63(2):144–162. doi:10.1007/s00018-005-5421-8
51. Ramahi A, Altorok N, Kahaleh B. Epigenetics and systemic sclerosis: an answer to disease onset and evolution? Eur J Rheumatol. 2020;7(Suppl 3):S147–S156. doi:10.5152/eurjrheum.2020.19112
52. Zhu HL, Du Q, Chen WL, et al. [Altered serum cytokine expression profile in systemic sclerosis and its regulatory mechanisms]. Beijing Da Xue Xue Bao Yi Xue Ban. 2019;51(4):716–722. Chinese. doi:10.19723/j.issn.1671-167X.2019.04.021
53. Wang Y, Yang Y, Luo Y, et al. Aberrant histone modification in peripheral blood B cells from patients with systemic sclerosis. Clin Immunol. 2013;149(1):46–54. doi:10.1016/j.clim.2013.06.006
54. Ciechomska M, O’Reilly S, Przyborski S, et al. Histone demethylation and toll-like receptor 8-Dependent cross-talk in monocytes promotes transdifferentiation of fibroblasts in systemic sclerosis Via Fra-2. Arthritis Rheumatol. 2016;68(6):1493–1504. doi:10.1002/art.39602
55. van der Kroef M, Castellucci M, Mokry M, et al. Histone modifications underlie monocyte dysregulation in patients with systemic sclerosis, underlining the treatment potential of epigenetic targeting. Ann Rheum Dis. 2019;78(4):529–538. doi:10.1136/annrheumdis-2018-214295
56. Tsou PS, Varga J, O’Reilly S. Advances in epigenetics in systemic sclerosis: molecular mechanisms and therapeutic potential. Nat Rev Rheumatol. 2021;17(10):596–607. doi:10.1038/s41584-021-00683-2
57. Kramer M, Dees C, Huang J, et al. Inhibition of H3K27 histone trimethylation activates fibroblasts and induces fibrosis. Ann Rheum Dis. 2013;72(4):614–620. doi:10.1136/annrheumdis-2012-201615
58. Tsou PS, Campbell P, Amin MA, et al. Inhibition of EZH2 prevents fibrosis and restores normal angiogenesis in scleroderma. Proc Natl Acad Sci U S A. 2019;116(9):3695–3702. doi:10.1073/pnas.1813006116
59. Wasson CW, Abignano G, Hermes H, et al. Long non-coding RNA HOTAIR drives EZH2-dependent myofibroblast activation in systemic sclerosis through miRNA 34a-dependent activation of NOTCH. Ann Rheum Dis. 2020;79(4):507–517. doi:10.1136/annrheumdis-2019-216542
60. Tsou P-S, Wren JD, Amin MA, et al. Histone deacetylase 5 is overexpressed in scleroderma endothelial cells and impairs angiogenesis via repression of proangiogenic factors. Arthritis Rheumatol. 2016;68(12):2975–2985. doi:10.1002/art.39828
61. Huber LC, Distler JHW, Moritz F, et al. Trichostatin A prevents the accumulation of extracellular matrix in a mouse model of bleomycin-induced skin fibrosis. Arthritis Rheum. 2007;56(8):2755–2764. doi:10.1002/art.22759
62. Maleszewska M, Vanchin B, Harmsen MC, et al. The decrease in histone methyltransferase EZH2 in response to fluid shear stress alters endothelial gene expression and promotes quiescence. Angiogenesis. 2016;19(1):9–24. doi:10.1007/s10456-015-9485-2
63. Chouri E, Servaas NH, Bekker CPJ, et al. Serum microRNA screening and functional studies reveal miR-483-5p as a potential driver of fibrosis in systemic sclerosis. J Autoimmun. 2018;89:162–170. doi:10.1016/j.jaut.2017.12.015
64. Koba S, Jinnin M, Inoue K, et al. Expression analysis of multiple microRNAs in each patient with scleroderma. Exp Dermatol. 2013;22(7):489–491. doi:10.1111/exd.12173
65. Ciechomska M, Zarecki P, Merdas M, et al. The role of microRNA-5196 in the pathogenesis of systemic sclerosis. Eur J Clin Invest. 2017;47(8):555–564. doi:10.1111/eci.12776
66. Rossato M, Affandi AJ, Thordardottir S, et al. Association of MicroRNA-618 expression with altered frequency and activation of plasmacytoid dendritic cells in patients with systemic sclerosis. Arthritis Rheumatol. 2017;69(9):1891–1902. doi:10.1002/art.40163
67. Christmann RB, Wooten A, Sampaio-Barros P, et al. miR-155 in the progression of lung fibrosis in systemic sclerosis. Arthritis Res Ther. 2016;18(1):155. doi:10.1186/s13075-016-1054-6
68. Jiang Z, Tao J-H, Zuo T, et al. The correlation between miR-200c and the severity of interstitial lung disease associated with different connective tissue diseases. Scand J Rheumatol. 2017;46(2):122–129. doi:10.3109/03009742.2016.1167950
69. Wuttge DM, Carlsen AL, Teku G, et al. Specific autoantibody profiles and disease subgroups correlate with circulating micro-RNA in systemic sclerosis. Rheumatology (Oxford). 2015;54(11):2100–2107. doi:10.1093/rheumatology/kev234
70. Liakouli V, Cipriani P, Di Benedetto P, et al. Epidermal growth factor like-domain 7 and miR-126 are abnormally expressed in diffuse systemic sclerosis fibroblasts. Sci Rep. 2019;9(1):4589. doi:10.1038/s41598-019-39485-8
71. Stypinska B, Wajda A, Walczuk E, et al. The serum cell-free microRNA expression profile in MCTD, SLE, SSc, and RA patients. J Clin Med. 2020;9(1):161. doi:10.3390/jcm9010161
72. Jafarinejad-Farsangi S, Gharibdoost F, Farazmand A, et al. MicroRNA-21 and microRNA-29a modulate the expression of collagen in dermal fibroblasts of patients with systemic sclerosis. Autoimmunity. 2019;52(3):108–116. doi:10.1080/08916934.2019.1621856
73. Zhu H, Luo H, Li Y, et al. MicroRNA-21 in scleroderma fibrosis and its function in TGF-beta-regulated fibrosis-related genes expression. J Clin Immunol. 2013;33(6):1100–1109. doi:10.1007/s10875-013-9896-z
74. O’Reilly S. miRNA-29a in systemic sclerosis: a valid target. Autoimmunity. 2015;48(8):511–512. doi:10.3109/08916934.2015.1077232
75. Maurer B, Stanczyk J, Jüngel A, et al. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheum. 2010;62(6):1733–1743. doi:10.1002/art.27443
76. Jafarinejad-Farsangi S, Farazmand A, Mahmoudi M, et al. MicroRNA-29a induces apoptosis via increasing the Bax: bcl-2 ratio in dermal fibroblasts of patients with systemic sclerosis. Autoimmunity. 2015;48(6):369–378. doi:10.3109/08916934.2015.1030616
77. Jafarinejad-Farsangi S, Farazmand A, Gharibdoost F, et al. Inhibition of MicroRNA-21 induces apoptosis in dermal fibroblasts of patients with systemic sclerosis. Int J Dermatol. 2016;55(11):1259–1267. doi:10.1111/ijd.13308
78. Mullenbrock S, Liu F, Szak S, et al. Systems analysis of transcriptomic and proteomic profiles identifies novel regulation of fibrotic programs by miRNAs in pulmonary fibrosis fibroblasts. Genes (Basel). 2018;9(12):588. doi:10.3390/genes9120588
79. Henderson J, Distler J, O’Reilly S. The role of epigenetic modifications in systemic sclerosis: a druggable target. Trends Mol Med. 2019;25(5):395–411. doi:10.1016/j.molmed.2019.02.001
80. Long H, Wang X, Chen Y, et al. Dysregulation of microRNAs in autoimmune diseases: pathogenesis, biomarkers and potential therapeutic targets. Cancer Lett. 2018;428:90–103. doi:10.1016/j.canlet.2018.04.016
81. Wermuth PJ, Piera-Velazquez S, Rosenbloom J, et al. Existing and novel biomarkers for precision medicine in systemic sclerosis. Nat Rev Rheumatol. 2018;14(7):421–432. doi:10.1038/s41584-018-0021-9
82. Bagnato G, Roberts WN, Roman J, et al. A systematic review of overlapping microRNA patterns in systemic sclerosis and idiopathic pulmonary fibrosis. Eur Respir Rev. 2017;26(144):160125. doi:10.1183/16000617.0125-2016
83. Zuo X, Zhang L, Luo H, et al. Systematic approach to understanding the pathogenesis of systemic sclerosis. Clin Genet. 2017;92(4):365–371. doi:10.1111/cge.12946
84. Artlett CM, Sassi-Gaha S, Hope JL, et al. Mir-155 is overexpressed in systemic sclerosis fibroblasts and is required for NLRP3 inflammasome-mediated collagen synthesis during fibrosis. Arthritis Res Ther. 2017;19(1):144. doi:10.1186/s13075-017-1331-z
85. Zhang L, Wu H, Zhao M, et al. Meta-analysis of differentially expressed microRNAs in systemic sclerosis. Int J Rheum Dis. 2020;23(10):1297–1304. doi:10.1111/1756-185X.13924
86. He Y, Liu H, Wang S, Chen Y. In silico detection and characterization of microRNAs and their target genes in microRNA microarray datasets from patients with systemic sclerosis-interstitial lung disease. DNA Cell Biol. 2019;38(9):933–944. doi:10.1089/dna.2019.4780
87. Hoffmann-Vold AM, Weigt SS, Palchevskiy V, et al. Augmented concentrations of CX3CL1 are associated with interstitial lung disease in systemic sclerosis. PLoS One. 2018;13(11):e0206545. doi:10.1371/journal.pone.0206545
88. Rajasekaran S, Vaz M, Reddy SP. Fra-1/AP-1 transcription factor negatively regulates pulmonary fibrosis in vivo. PLoS One. 2012;7(7):e41611. doi:10.1371/journal.pone.0041611
89. Mariotti B, Servaas NH, Rossato M, et al. The long non-coding RNA NRIR drives IFN-response in monocytes: implication for systemic sclerosis. Front Immunol. 2019;10:100. doi:10.3389/fimmu.2019.00100
90. Abd-Elmawla MA, Hassan M, Elsabagh YA, et al. Deregulation of long noncoding RNAs ANCR, TINCR, HOTTIP and SPRY4-IT1 in plasma of systemic sclerosis patients: SPRY4-IT1 as a novel biomarker of scleroderma and its subtypes. Cytokine. 2020;133:155124. doi:10.1016/j.cyto.2020.155124
91. Takata M, Pachera E, Frank-Bertoncelj M, et al. OTUD6B-AS1 might be a novel regulator of apoptosis in systemic sclerosis. Front Immunol. 2019;10:1100. doi:10.3389/fimmu.2019.01100
92. Pachera E, Assassi S, Salazar GA, et al. Long noncoding RNA H19X is a key mediator of TGF-beta-driven fibrosis. J Clin Invest. 2020;130(9):4888–4905. doi:10.1172/JCI135439
93. Muntyanu A, Le M, Ridha Z, et al. Novel role of long non-coding RNAs in autoimmune cutaneous disease. J Cell Commun Signal. 2021. doi:10.1007/s12079-021-00639-x
94. Wang Z, Jinnin M, Nakamura K, et al. Long non-coding RNA TSIX is upregulated in scleroderma dermal fibroblasts and controls collagen mRNA stabilization. Exp Dermatol. 2016;25(2):131–136. doi:10.1111/exd.12900
95. Wasson CW, Ross RL, Wells R, et al. Long non-coding RNA HOTAIR induces GLI2 expression through Notch signalling in systemic sclerosis dermal fibroblasts. Arthritis Res Ther. 2020;22(1):286. doi:10.1186/s13075-020-02376-9
96. Dolcino M, Tinazzi E, Puccetti A, et al. In systemic sclerosis, a unique long non coding RNA regulates genes and pathways involved in the three main features of the disease (vasculopathy, fibrosis and autoimmunity) and in carcinogenesis. J Clin Med. 2019;8(3):320. doi:10.3390/jcm8030320
97. Servaas NH, Mariotti B, van der Kroef M, et al. Characterization of long non-coding RNAs in systemic sclerosis monocytes: a potential role for PSMB8-AS1 in altered cytokine secretion. Int J Mol Sci. 2021;22(9):4365. doi:10.3390/ijms22094365
98. Wermuth PJ, Piera-Velazquez S, Jimenez SA. Exosomes isolated from serum of systemic sclerosis patients display alterations in their content of profibrotic and antifibrotic microRNA and induce a profibrotic phenotype in cultured normal dermal fibroblasts. Clin Exp Rheumatol. 2017;35 Suppl 106(4):21–30.
99. Fang S, Xu C, Zhang Y, et al. Umbilical cord-derived mesenchymal stem cell-derived exosomal MicroRNAs suppress myofibroblast differentiation by inhibiting the transforming growth factor-beta/SMAD2 pathway during wound healing. Stem Cells Transl Med. 2016;5(10):1425–1439. doi:10.5966/sctm.2015-0367
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.