Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 21
Epigenetic Mechanisms Linking Chronic Obstructive Pulmonary Disease and Atrial Fibrillation: A Multi-Omics Mendelian Randomization Study
Authors Zhang M, Li T, Tan L, Yu J, Yao Q, Chen L ![]()
Received 27 August 2025
Accepted for publication 2 December 2025
Published 8 January 2026 Volume 2026:21 563463
DOI https://doi.org/10.2147/COPD.S563463
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jill Ohar
MiaoAn Zhang,1 TianFeng Li,1 LingLin Tan,1 Jinhua Yu,1 Qiying Yao,2 Liang Chen1
1Department of Cardiology, The Second Affiliated Hospital of Dalian Medical University, Dalian, People’s Republic of China; 2Department of Physiology, Dalian Medical University, Dalian, People’s Republic of China
Correspondence: Qiying Yao, Email [email protected]; Liang Chen, Email [email protected]
Background and Objective: Chronic obstructive pulmonary disease (COPD) and atrial fibrillation (AF) frequently co-occur, yet the underlying molecular mechanisms remain unclear. We assessed whether genetic liability to COPD increases AF risk and mapped epigenetic mediators using multi-omics data.
Methods: We integrated genome-wide association data for COPD and AF with epigenome-wide DNA methylation profiles and gene expression datasets. Summary-data-based Mendelian randomization served as the primary approach to link disease-associated variants with methylation, gene expression, and AF, complemented by two-sample Mendelian randomization, colocalization, and checks for pleiotropy. We prioritized COPD-related CpG sites and genes, examined functional context using protein–protein interaction networks and pathway enrichment, and nominated candidate therapeutics with drug-prediction algorithms followed by molecular docking. Analyses were conducted primarily in cohorts of European ancestry.
Results: Genetic liability to COPD was associated with a higher risk of AF (odds ratio [OR] = 1.156, 95% confidence interval [CI] 1.025– 1.304; p = 0.018). We identified 109 COPD-related CpG sites associated with AF risk. Cross-omics triangulation highlighted seven candidate genes, with FES showing consistent evidence across genomic, epigenetic, and transcriptomic layers. Functional analyses indicated enrichment in immune signaling and signal transduction pathways. Docking analyses suggested favorable binding between predicted drugs and their targets.
Conclusion: These findings support a pathway by which COPD may influence AF risk through epigenetic regulation and prioritize CpG sites, genes, and potentially druggable targets for further study. Because the underlying datasets are predominantly from individuals of European ancestry, replication in more diverse populations is needed to assess generalizability.
Keywords: chronic obstructive pulmonary disease, atrial fibrillation, summary-data-based mendelian randomization, multi-omics, methylation, gene expression, protein, drug target
Introduction
Chronic obstructive pulmonary disease (COPD) and atrial fibrillation (AF) represent two predominant chronic conditions afflicting middle-aged and elderly populations, imposing formidable health and economic burdens on healthcare systems worldwide.1,2 This comorbidity has direct implications for patient outcomes and management, including lower success of rhythm-control strategies, higher bleeding risk with anticoagulation, and increased cardiovascular and all-cause mortality.3,4 These conditions frequently coexist, with epidemiological studies indicating that approximately 13% of AF patients manifest concurrent COPD.5 The incidence of AF among COPD patients exceeds that observed in the general population by 1.23-fold,6 with this association maintaining statistical significance across Asian populations.7 Furthermore, concurrent COPD substantially elevates recurrence rates and adverse event incidence following catheter ablation in AF patients.3 Concurrently, AF patients with comorbid COPD demonstrate significantly heightened risks of hemorrhagic complications, cardiovascular mortality, and all-cause mortality.4 Consequently, investigating COPD-AF comorbidity mechanisms represents a critically important and urgently needed research priority.
Mechanistically, three complementary rationales have been proposed to explain COPD-related AF susceptibility: (i) biological pathways, whereby COPD-associated inflammatory responses and oxidative stress may precipitate structural remodeling8 and electrical conduction alterations,9 thereby amplifying AF susceptibility; (ii) genetic convergence, with hub genes linking COPD and AF, encompassing RPS3, FABP5, and EPHA4,10 alongside common biomarkers—such as CDK8—which may contribute to an adverse cycle via activation of NF-κB-mediated immune-inflammatory cascades;11 and (iii) epigenetic regulation, wherein DNA methylation, as a pivotal modification, may mediate inter-disease associations and provide a tractable layer for mechanistic dissection.12
In recent years, epigenome-wide association studies (EWAS) have furnished novel perspectives for elucidating complex disease molecular mechanisms, with DNA methylation as a pivotal epigenetic modification potentially exerting key roles in inter-disease associations.12 However, previous investigations predominantly remained constrained to association analysis levels, lacking comprehensive exploration of causal relationships. Mendelian randomization analysis predicated on methylation quantitative trait loci (mQTL) can leverage EWAS-identified disease-related CpG sites as exposures, inferring causal associations with health outcomes through genetic instrumental variables, thereby providing robust analytical tools for investigating epigenetic-mediated mechanisms between diseases.13 Simultaneously, integrated analysis of multi-omics data has emerged as a pivotal strategy for revealing complex disease regulatory networks. Cross-validation through DNA methylation, gene expression, and protein abundance levels can furnish more reliable biological evidence.14
Although existing epidemiological, basic research, and observational studies demonstrate associations between COPD and AF, underlying molecular mechanisms remain incompletely elucidated. Therefore, this investigation utilized large-scale population EWAS, quantitative trait loci (QTL), and genome-wide association study (GWAS) datasets, within a summary-data-based Mendelian randomization framework complemented by two-sample Mendelian randomization and colocalization, and employed multi-omics integration strategies to systematically explore causal linkages between COPD and AF across DNA methylation, gene expression, and protein abundance levels. We endeavored to screen candidate genes potentially mediating this association based on integrated omics evidence, aiming to provide novel insights for comprehensive understanding of COPD-AF comorbidity molecular mechanisms and developing innovative prevention and therapeutic strategies.
Materials and Methods
Study Design and Data Sources
Research Framework
This investigation constituted a comprehensive secondary analysis employing publicly accessible summary-level EWAS, GWAS, and QTL datasets. The analytical framework integrated epigenetic and multi-omics methodologies, utilizing genetic instrumental variables to mitigate confounding influences and combining multi-omics data to identify potential epigenetic pathways mediating COPD to AF associations (Figure 1). The research workflow encompassed: (1) assessment of genetic correlations between COPD and AF; (2) exploration of preliminary genetically predicted causal associations; (3) epigenetic mechanism analysis (core research focus); (4) multi-level integrative validation; (5) functional annotation and therapeutic target prediction.
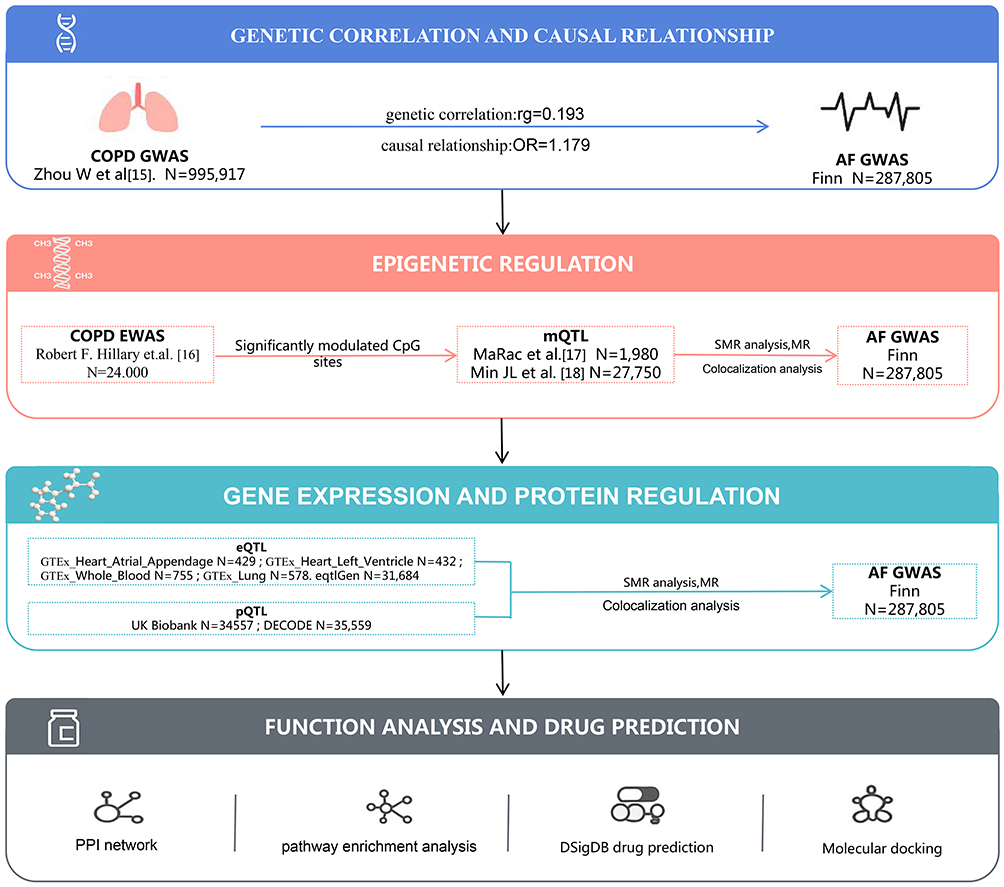 |
Figure 1 The overall design of this study. Abbreviations: COPD, chronic obstructive pulmonary disease; AF, atrial fibrillation; mQTL, methylation quantitative trait loci; eQTL, expression quantitative trait loci; pQTL, protein quantitative trait loci; SMR, summary-data-based Mendelian randomization; PPI, protein–protein interaction. |
Data Sources
All EWAS, GWAS, and QTL datasets comprised individuals of European ancestry; dataset-level ancestries are summarized in Table 1 and Supplementary Table S1.
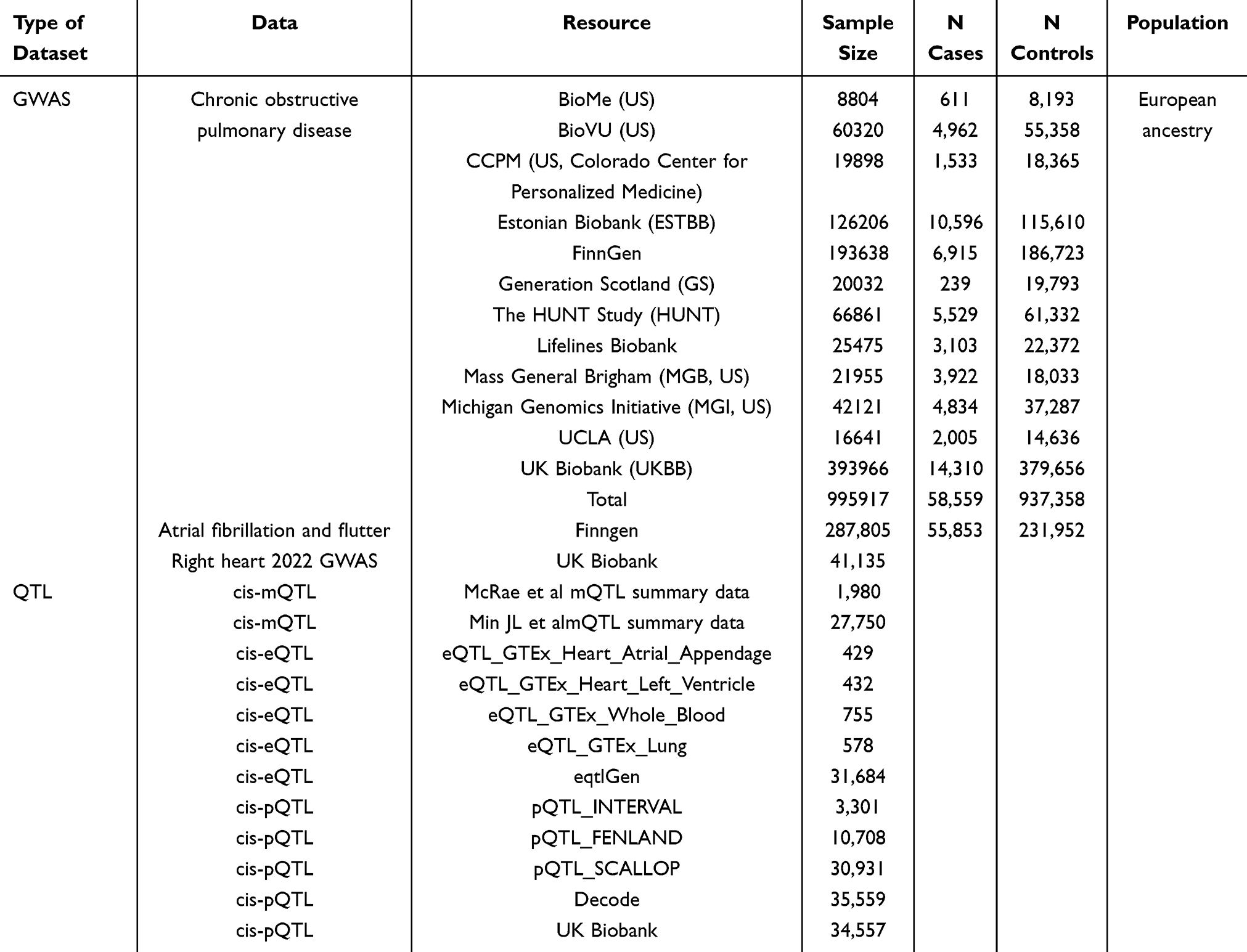 |
Table 1 Summary of Datasets, Sample Sizes, and Ancestry |
i) GWAS Data: COPD GWAS data were procured from Zhou W et al’s comprehensive investigation,15 encompassing 12 cohorts including FinnGen and UK Biobank repositories. The FinnGen cohort utilized ICD-10 codes J43 and J44, ICD-9 codes 492 and 4912, and ICD-8 codes 492 and 49104 for COPD diagnosis. Lifelines Biobank employed spirometry-based diagnostic criteria (FEV1/FVC × 100 < 70). HUNT, MGI, UCLA Precision Health Biobank, and UK Biobank employed phecode-based diagnoses, comprising 58,559 cases and 937,358 controls. AF GWAS data originated from FinnGen R11, utilizing ICD-10 code I48 definitions, encompassing 55,853 cases and 231,952 controls. Right heart-related phenotypic data were derived from UK Biobank cardiac MRI measurements following Pirruccello et al’s methodology, encompassing 41,135 subjects with measurements including pulmonary artery-to-aortic diameter ratio, pulmonary artery-to-aortic diastolic diameter ratio, right atrial maximum area, right atrial minimum area, right atrial area change fraction (RAFAC), right ventricular end-diastolic volume (RVEDV), right ventricular end-systolic volume (RVESV), right ventricular stroke volume (RVSV), right ventricular ejection fraction (RVEF), and pulmonary artery diameter.
ii) Epigenetic Data: COPD-EWAS summary statistics were obtained from published literature sources.16 CpG sites with FDR < 0.05 were defined as “COPD-associated” DNA methylation sites, yielding 155,862 sites for identifying COPD-related DNA methylation CpG sites. Methylation QTL (mQTL) data were sourced from: (1) McRae et al’s investigation (n = 1,980 Europeans);17 (2) Min et al’s (GoDMC) study (n = 27,750 Europeans).18
iii) Gene Expression Data: (1) Expression QTL (eQTL) from the eQTLGen consortium (n = 31,684 Europeans); (2) tissue-specific data from the Genotype-Tissue Expression Consortium (GTEx V8), including atrial appendage tissue (n = 429 Europeans), left ventricular tissue (n = 432 Europeans), whole blood tissue (n = 755 Europeans), and lung tissue (n = 578 Europeans), representing cis-eQTL data exclusively.
iv) Protein Expression Data: (1) Protein QTL (pQTL) from Sun et al (n = 3,301); (2) pQTL from Pietzner et al (n = 10,708 Finns); (3) pQTL from Folkersen et al (n = 30,931); (4) pQTL from Ferkingstad et al (n = 35,559 Icelanders); (5) pQTL from Benjamin B. Sun et al (n = 34,557 Europeans). For MR analysis pQTL data selection: we selected UKB and deCODE database pQTL data, utilizing UKB as the primary analysis when proteins were available, with deCODE data serving as sensitivity validation; proteins exclusively available in deCODE were designated as secondary studies for sensitivity analysis. All original GWAS/QTL analyses were adjusted for age, sex, and the first 10 genomic principal components in their respective studies; summary statistics employed in this investigation were directly retrieved without additional adjustments.
Additionally, we queried UK Biobank cardiac MRI phenotypes for left atrial structural measures (eg, left atrial diameter/volume). These measures were not available as standardized fields in the imaging-derived dataset used here; therefore, left atrial diameter–based analyses were not performed. We also screened UK Biobank and public GWAS resources for pulmonary hypertension, but available datasets had insufficient case numbers to support adequately powered two-sample MR with our instruments; consequently, pulmonary hypertension was not analyzed as an outcome.
Genetic Correlation Analysis
Linkage disequilibrium score regression (LDSC) analysis was executed using the R package “ldscr” (version 4.3.1) to estimate genetic correlation (rg) between COPD and AF based on GWAS summary statistics. LDSC analysis employed the 1000 Genomes Project European population as the reference LD panel, calculating standardized LD scores. P < 0.005 was considered statistically significant genetic correlation, with P values between 0.005 and 0.05 indicating suggestive associations.19
Two-Sample Mendelian Randomization Analysis
Two-sample Mendelian randomization (MR) analyses were conducted using the R package TwoSampleMR (v0.5.7). MR rests on three core assumptions: (i) relevance—the genetic instruments (IVs) are associated with the exposure; (ii) independence—the IVs are independent of confounders of the exposure–outcome relationship; and (iii) exclusion restriction—the IVs influence the outcome only via the exposure (ie, no horizontal pleiotropy). Because horizontal pleiotropy can bias causal estimates, we applied multiple complementary MR estimators to ensure robustness to horizontal pleiotropy and outliers. Specifically, we implemented MR-Egger,20 Weighted Median,21 Inverse-Variance Weighted (IVW),22 Simple Mode,21 and Weighted Mode.23 IVW provides the most precise estimates under no directional pleiotropy; the Weighted Median yields a consistent estimate if ≥50% of the total weight comes from valid instruments; MR-Egger allows for directional pleiotropy via a non-zero intercept under the InSIDE assumption; and mode-based estimators assume that the largest cluster of similar ratio estimates is formed by valid instruments. For mQTL, eQTL, and pQTL analyses, IVW was used when multiple SNP instruments were available, and the Wald ratio was used when only a single SNP instrument was present.
Instrument selection criteria were: (1) genome-wide significant association with the exposure (p < 5×10−8); (2) LD independence (r2 < 0.001 within a 10,000 kb window) with LD clumping and screening performed using PLINK v1.90b7 (64-bit);24 (3) F-statistic > 10 to limit weak instrument bias, with F = (N − 2) × R2/(1 − R2);25 (4) exclusion of SNPs directly associated with AF at genome-wide significance (p < 5×10−8); and (5) harmonization and SNP handling followed TwoSampleMR v0.5.7 defaults (action=2): effect alleles were aligned across datasets; strand issues were corrected by complementing where applicable; palindromic A/T or C/G SNPs with intermediate allele frequency were excluded; and SNPs with unresolved allele conflicts were removed. Outcome variants were additionally filtered to 0.01 < EAF < 0.99. In this analysis, no selected instruments were missing in the outcome dataset, and no LD proxy replacement was applied.
Significant cis-mQTLs were defined as variants located within <1 Mb of CpG sites with P < 1×10−8. For pQTLs, deCODE cis-pQTL data included SNPs within ±100 kb (excluding the MHC region), and UK Biobank pQTL data likewise used ±100 kb windows with MHC region variants removed. Because many CpG-site MR analyses relied on single cis-SNP instruments, these results are primarily hypothesis-generating and should be interpreted cautiously.
We controlled multiple testing using the Benjamini–Hochberg procedure (FDR < 0.05). Heterogeneity across instruments was evaluated using Cochran’s Q (p < 0.05 indicating heterogeneity); random-effects IVW was applied when heterogeneity was present, otherwise fixed-effects IVW was used. Directional (unbalanced) pleiotropy was assessed with the MR-Egger intercept (p < 0.05 indicating pleiotropy), and MR-PRESSO was used to detect and correct outlier SNP effects. Steiger directionality tests were conducted to confirm the exposure→outcome direction and reduce the possibility of reverse causation. Leave-one-out analyses were performed to evaluate the influence of individual instruments on overall estimates. We considered an MR association robust when estimates were directionally consistent across multiple estimators, the MR-Egger intercept did not indicate pleiotropy, no residual MR-PRESSO outliers remained, and fixed- and random-effects IVW results were comparable.
Summary-Data-Based Mendelian Randomization Analysis (SMR)
SMR methodology was employed to initially assess statistical associations between CpG sites and AF at the DNA methylation level utilizing mQTL data. Subsequently, for significantly associated CpG sites, eQTL and pQTL data were utilized to explore associations with AF at gene and protein expression levels. For each QTL type, the most statistically significant cis-acting variant (top cis-variant) served as the instrumental variable. The Benjamini–Hochberg method was applied for multiple testing correction, with FDR < 0.05 considered statistically significant. All significant SMR results (FDR < 0.05) underwent heterogeneity testing (HEIDI) (P_HEIDI > 0.05) to exclude potential linkage disequilibrium (LD) effects. SMR and HEIDI analyses were completed using the online SMR portal (https://yanglab.westlake.edu.cn/smr-portal/, accessed on [2024–11-10]).
Colocalization Analysis
Colocalization analysis was performed for significant sites (p_SMR_FDR < 0.05, p_HEIDI > 0.05) in SMR analyses of mQTL, eQTL, and pQTL with AF utilizing the R package “coloc” (version 5.2.3). Colocalization analysis employed a Bayesian statistical framework to evaluate posterior probabilities of two phenotypes sharing identical causal variants. The analysis considered five hypotheses: (1) PPH0: no association at the locus; (2) PPH1: association exclusively with COPD; (3) PPH2: association exclusively with AF; (4) PPH3: distinct causal variants for the two phenotypes; (5) PPH4: shared causal variant for both phenotypes.26 Default prior parameters (p1 = 1×10−4, p2 = 1×10−4, p12 = 1×10−5) were utilized, executing within ±1 Mb range around CpG sites. Posterior probability PPH4 > 0.70 was considered robust statistical evidence supporting shared causal variants, based on Claudia Giambartolomei et al’s research.27 The “locuscomparer” package (version 1.0.0) generated regional association comparison plots for intuitive visualization of shared signals between phenotypes.
Protein–Protein Interaction (PPI) and Functional Enrichment Analysis
Protein–protein interaction networks estimated correlations among COPD- and AF-related proteins (p_SMR_FDR < 0.05) utilizing the Search Tool for Retrieval of Interacting Genes (STRING, https://string-db.org/)28 and GeneMANIA (https://genemania.org/).29
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses evaluated relationships between potential enriched pathways and proteins. R language and related packages including “GO.db” (v3.18.0), “clusterProfiler” (v4.10.1), “AnnotationHub” (v3.10.1), “org.Hs.eg.db” (v3.18.0), “org.Mm.eg.db” (v3.18.0), “DOSE” (v3.28.2), “ReactomePA” (v1.46.0), “limma” (v3.58.1), “circlize” (v0.4.16), “RColorBrewer” (v1.1–3), “ComplexHeatmap” (v2.18.0), and “ggplot2” (v3.5.1) were employed for enrichment analysis and result visualization.
Drug Prediction and Molecular Docking
To translate the statistical signals into therapeutic hypotheses, we integrated prioritized targets from the MR/eQTL/pQTL and expression analyses into a two-step in-silico workflow: (i) drug prediction to retrieve compounds with known mechanisms acting on the prioritized genes, and (ii) structure-based molecular docking to assess the physical plausibility of drug–target interactions. These in-silico analyses are hypothesis-generating, intended to rank candidates for experimental validation.
Drug Prediction
The Drug–Gene Interaction Database (DSigDB) was utilized to evaluate potential protein–drug interactions, predicting candidate pharmaceuticals targeting genes indicated at gene expression and protein expression levels. DSigDB comprises a comprehensive repository containing 22,527 genomic signatures and 17,389 distinct compounds, encompassing 19,531 gene sets connecting drugs and compounds with their target genes.30 Screening criteria included: (1) drug-gene pairs with established mechanisms of action. For each gene, potential drug numbers, action types (inhibitors, agonists, etc.), and clinical application status were systematically recorded.
Molecular Docking
Molecular docking was subsequently utilized to evaluate predicted interactions between candidate drugs and their targets. This approach facilitates prioritization of drug targets through identifying protein characteristics and protein–drug interaction patterns, providing foundations for future experimental validation.
Component structures were obtained from the PubChem database (https://pubchem.ncbi.nlm.nih.gov/), preserved in SDF format, imported into Chem3D 19.0 software, and energy-minimized using the MM2 module to obtain the lowest-energy dominant conformation, preserved in mol2 format and converted to pdbqt files using AutodockTools 1.5.7. Three-dimensional structures of core target proteins were obtained from the Uniprot database (https://www.uniprot.org/), preserved as PDB structure files, and processed using AutodockTools 1.5.7 to incorporate hydrogen atoms, defining them as receptors and preserving as pdbqt files. The vina plugin was employed for ligand-receptor docking, with visualization analysis performed using PyMOL 3.2 software following docking completion.
Statistical Analysis
All statistical analyses were completed using R (version 4.3.1). Multiple p-value calculations employed the Benjamini–Hochberg method for false discovery rate control, with FDR significance threshold established at 0.05, explicitly reporting effect estimates and confidence intervals.31
Reporting Standards
This investigation adhered to STROBE-MR guidelines, with the STROBE-MR checklist provided in Supplementary Table S49.
The language in this manuscript was polished using Claude sonnet 4 solely for grammatical refinement and clarity enhancement, with all scientific content and interpretations remaining entirely our own.
Results
Genetic Correlation Between COPD and AF
LDSC genetic correlation analysis established significant genetic correlation between COPD and AF (rg = 0.193, p = 1.04×10−9), notwithstanding relatively modest heritability estimates for both conditions (COPD: h2 = 0.0161, p < 1×10−50; AF: h2 = 0.0722, p < 1×10−40) (Supplementary Table S2).
Genetic Association and Mendelian Randomization Analysis Between COPD and AF
Two-sample Mendelian randomization (MR) analysis demonstrated that COPD significantly elevated AF risk (OR = 1.156, 95% CI: 1.025–1.304, p = 0.018) (Supplementary Table S3). Sensitivity analyses corroborated this potential causal association (Supplementary Table S4-S5). Following MR-PRESSO exclusion of instrumental variables exhibiting pleiotropy (Supplementary Table S6), COPD remained significantly associated with AF risk (OR = 1.179, 95% CI: 1.073–1.296, p = 0.00065) (Supplementary Table S7 and Supplementary Figure 1), with no horizontal pleiotropy detected (MR Egger intercept test p = 0.696) (Supplementary Figures 2–3) Furthermore, leave-one-out analysis validated result robustness (Supplementary Figure 4) (Supplementary Tables S8–S9). Steiger directionality test precluded reverse causation (p = 3.69×10−19) (Supplementary Table S10).
Epigenetic Analysis of Molecular Mechanisms Underlying COPD’s Impact on AF
Identification of COPD-Associated DNA Methylation Sites
Through comprehensive epigenome-wide association study (EWAS), we identified 155,862 CpG sites demonstrating significant associations with COPD (FDR < 0.05) (Supplementary Table S11), ultimately extracting mQTL corresponding to 2,605 CpG sites from McRae mQTL data.
Causal Association Between COPD-Related Methylation Sites and AF
Utilizing stepwise SMR methodology, we established linkage between 109 CpG sites and atrial fibrillation (AF) through genetic instrumental variables (p_SMR_FDR < 0.05), with all sites successfully passing HEIDI testing (p_HEIDI > 0.05), representing single instrumental variable-based analyses. This furnished statistical evidence for potential causal associations whereby methylation status of these 109 sites through genetic regulation might influence AF risk (p_SMR_FDR < 0.05, p_HEIDI > 0.05) (Figure 2A–C and Supplementary Table S12). Among these, 53 CpG sites exhibited positive correlation between methylation levels and AF risk, encompassing ATF5 (cg04315771) and RPS2 (cg02871659); 56 sites demonstrated negative correlation, such as FKBP2 (cg16977872) and VSTM4 (cg07260003). Forty-eight CpG sites demonstrated consistent β-value directions between COPD EWAS data and mQTL-SMR results, whereas 61 CpG sites exhibited opposite directions (Figures 3A–C, 4A–C and Supplementary Tables S13-S14).
 |
Figure 2 Epigenome-wide analysis of COPD-associated DNA methylation sites in relation to AF risk. (A) Manhattan plot of SMR analysis between COPD-associated mQTLs andAF, marking the genes with significant CpG sites (FDR < 0.05). (B) Volcano plot depicting effect sizes (β) and significance levels of SMR results for COPD-associated methylation sites and AF risk. (C) The forest plot shows the top methylation sites that exhibited significant associations when conducting an SMR analysis of COPD methylation and AF. In the plot, small triangular symbols represent odds ratios (OR), and straight lines represent 95% confidence intervals (CI). FDR = false discovery rate. |
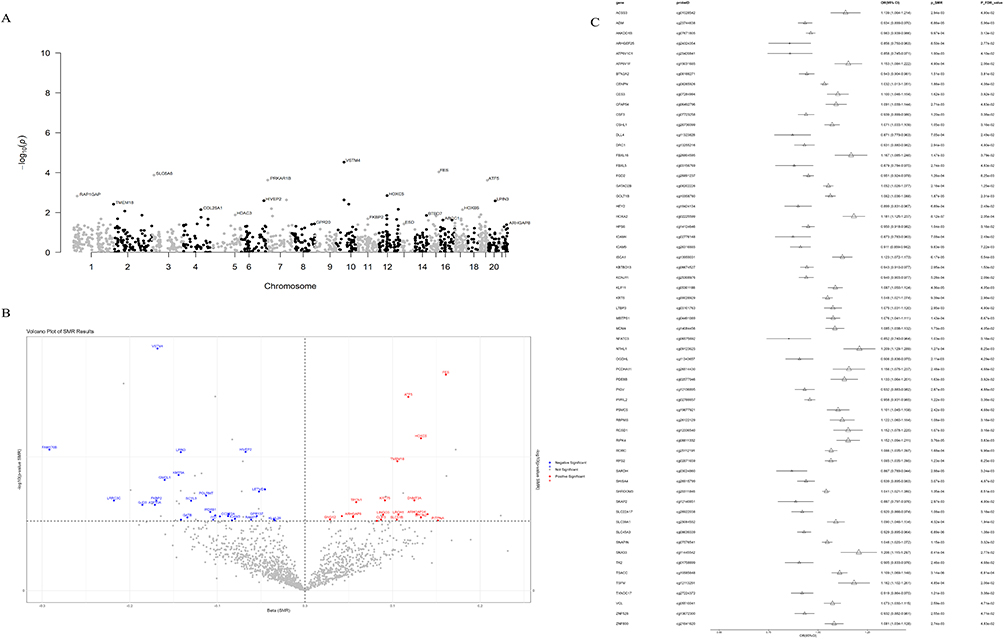 |
Figure 3 Integrative analysis of genes with consistent effect directions between COPD EWAS data and mQTL-SMR results. (A) Manhattan plot displaying SMR analysis results of COPD-associated mQTLs with consistent effect directions in relation to AF risk, marking the genes with significant CpG sites (FDR < 0.05). (B) The volcano plot depicts the effect size (β) and significance level of the SMR results for the methylated sites with consistent β values related to COPD and the AF risk. (C) The forest plot shows the main methylated sites that demonstrated significant association when performing SMR analysis on the CpG sites and AF that were directionally consistent with the β values related to COPD. In the plot, small triangular symbols represent odds ratios (OR), and straight lines represent 95% confidence intervals (CI). Abbreviations: FDR, false discovery rate. |
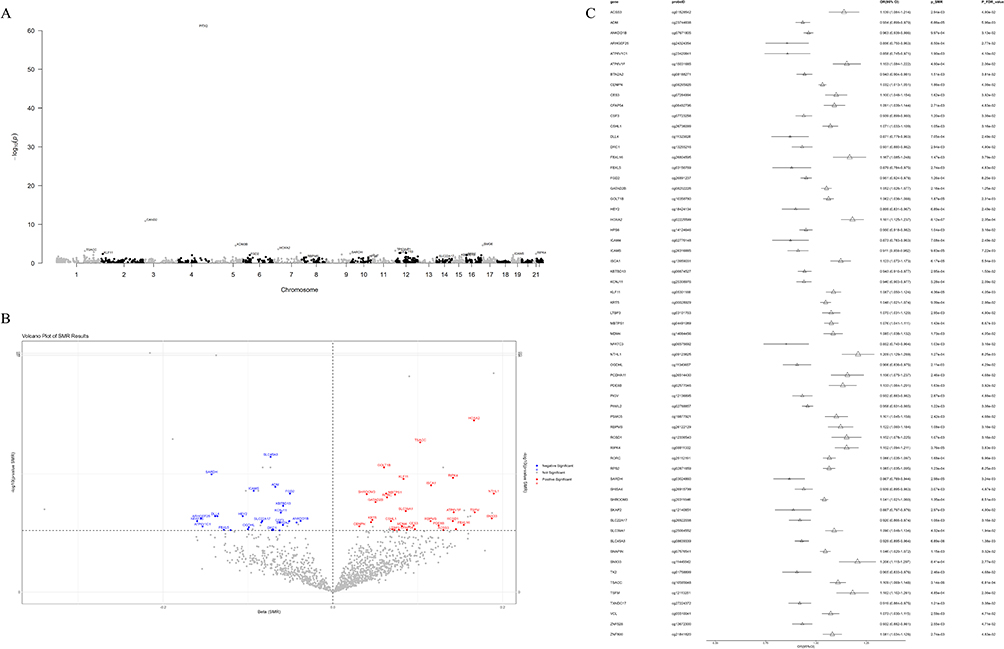 |
Figure 4 Analysis of genes with opposite effect directions between COPD EWAS data and mQTL-SMR results. (A) Manhattan plot displaying SMR analysis results of COPD-associated mQTLs with opposite effect directions in relation to AF risk, marking the genes with significant CpG sites (FDR < 0.05). (B) The volcano plot depicts the effect size (β) and significance level of the SMR results for the36793methylated sites with opposite β values related to COPD and the AF risk. (C) The forest plot shows the main methylated sites that demonstrated significant association when performing SMR analysis on the CpG sites and AF that were directionally opposite with the β values related to COPD. In the plot, small triangular symbols represent odds ratios (OR), and straight lines represent 95% confidence intervals (CI). Abbreviations: FDR, false discovery rate. |
To validate SMR results, we conducted two-sample MR analysis on 101 of the 109 positive CpG sites amenable to MR analysis (Supplementary Table S15). Among 46 CpG sites with consistent β-value directions between COPD EWAS data and mQTL-SMR results, MR analysis indicated 24 CpG sites demonstrated significant positive or negative associations with AF risk (p_FDR < 0.05), with 11 CpG sites exhibiting significant positive associations (beta > 0) and 13 sites demonstrating significant negative associations (beta < 0) (Supplementary Table S16). Among 55 CpG sites with inconsistent β-value directions between COPD EWAS data and mQTL-SMR results, MR analysis indicated 31 CpG sites demonstrated significant positive or negative associations with AF risk (p_FDR < 0.05), with 18 sites exhibiting significant positive associations (p_FDR < 0.05, beta > 0) and 13 sites demonstrating significant negative associations (p_FDR < 0.05, beta < 0) (Supplementary Table S17). Steiger directionality tests excluded reverse causation possibilities (Supplementary Tables S18–S20).
Colocalization Validation of Methylation-AF Associations
Subsequent colocalization analysis utilizing GoDMC data revealed 25 CpG sites (including cg04510874 in the FES gene region) with PP.H4 > 0.7, suggesting their function as shared causal variant sites linking COPD with AF (Figure 5A–D, Supplementary Figure 5A–5S and Supplementary Table S21), representing novel molecular-level discoveries of association mechanisms. Among CpG sites with consistent β-value directions between COPD EWAS data and mQTL-SMR results, 14 sites demonstrated PP.H4 > 0.7, with 9 sites exhibiting high colocalization probability (PP.H4 > 0.9), encompassing FES (cg04510874), FKBP2 (cg16977872), ATF5 (cg04315771), VSTM4 (cg07260003), and LPIN3 (cg24845165) (Figure 6A–D and Supplementary Table S22). Among CpG sites with inconsistent β-value directions between COPD EWAS data and mQTL-SMR results, 11 sites demonstrated PP.H4 > 0.7 (Figure 7A–D and Supplementary Table S23).
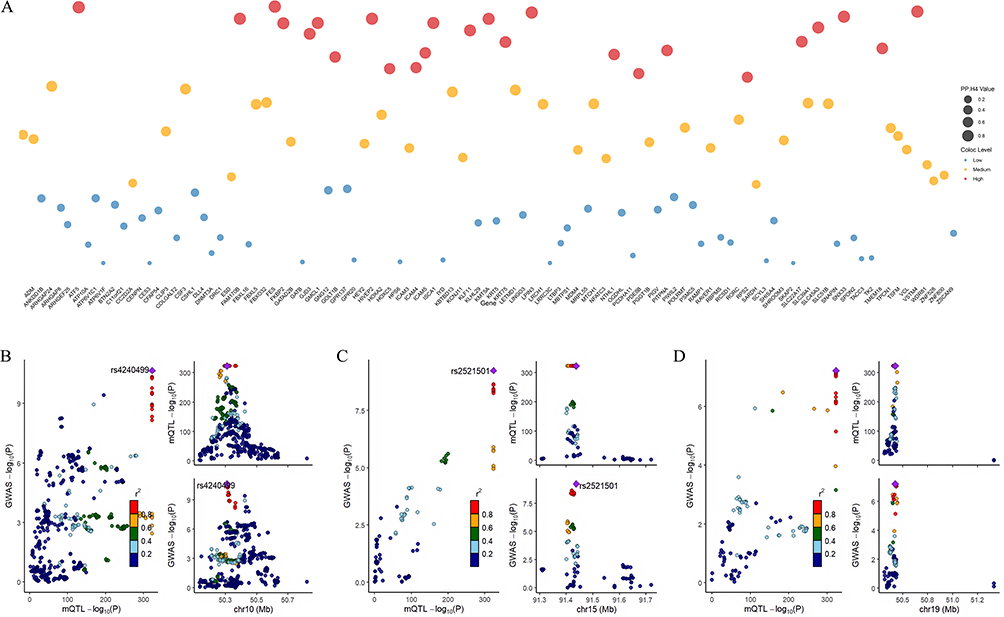 |
Figure 5 Co-localization analysis of COPD-associated DNA methylation sites in relation to AF risk. (A) Colocalization analysis results for significant SMR findings, with posterior probability (PP.H4) values indicating shared causal variants between methylation sites and AF. (B-D) Regional association plots for the three CpG sites with highest colocalization evidence 35771 (PP.H4 > 0.9), associated with ATF5, FES, and VSTM4 genes, demonstrating shared genetic signals between methylation quantitative trait loci and AF risk. |
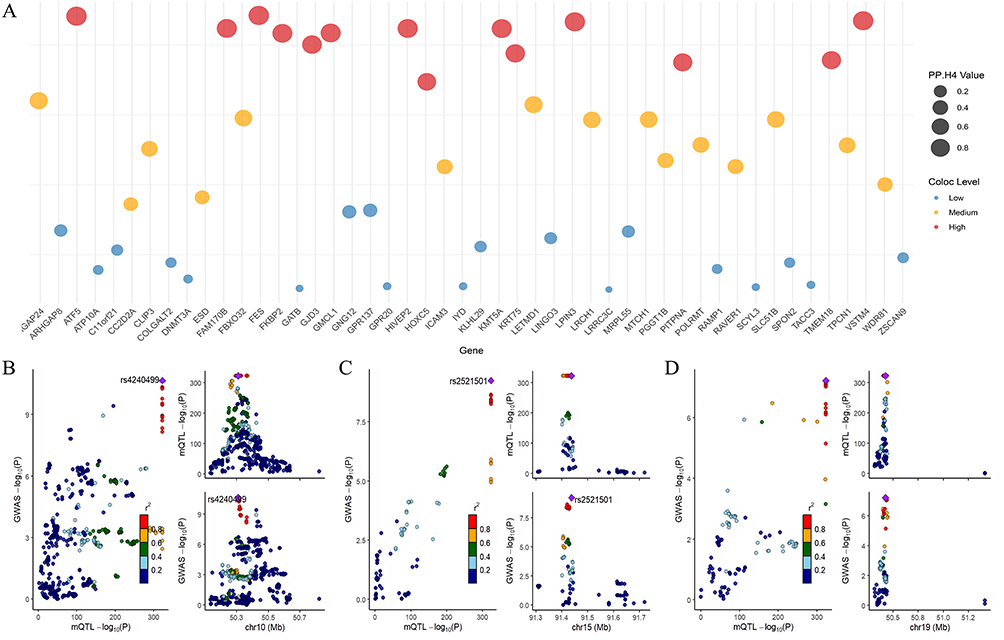 |
Figure 6 Co-localization analysis of genes with consistent effect directions between COPD EWAS data and mQTL-SMR results. (A) Colocalization analysis results for methylation sites with consistent β-direction, with higher PP.H4 values indicating stronger evidence for shared causal variants. (B-D) Regional association plots for ATF5, FES, and VSTM4, the three genes demonstrating the highest colocalization evidence (PP.H4 > 0.9), suggesting their roles as key mediators in the COPD-AF causal cascade. |
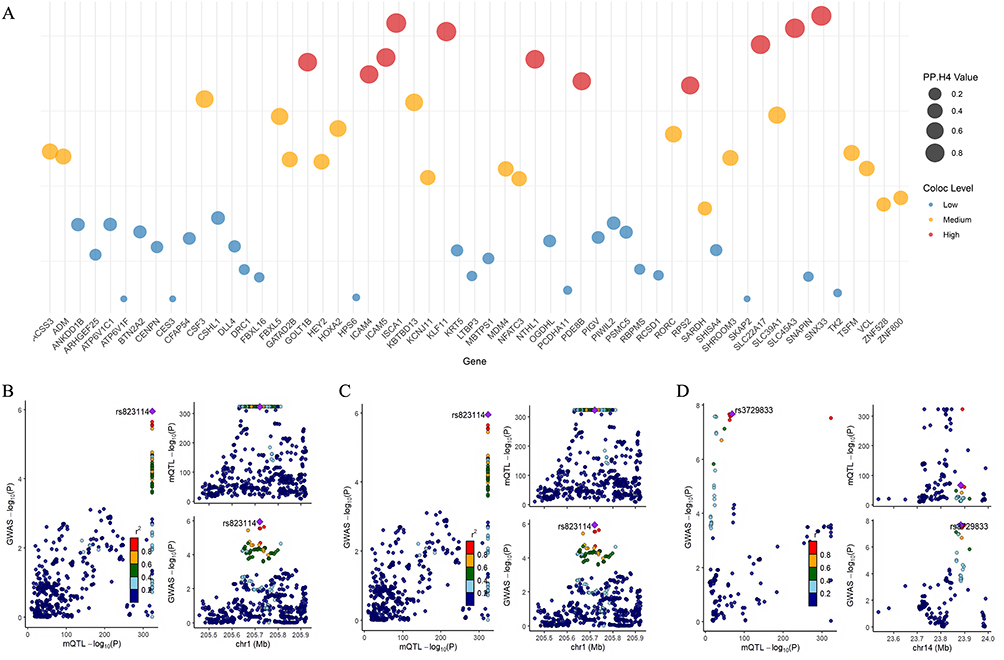 |
Figure 7 Co-localization analysis of genes with opposite effect directions between COPD EWAS data and mQTL-SMR results. (A) Colocalization analysis results for methylation sites with opposite β-direction, with higher PP.H4 values indicating stronger evidence for shared causal variants. (B–D) Regional association plots for SLC45A3, KLF11, and SLC22A17, the three genes with opposite effect directions showing the highest colocalization evidence, suggesting their involvement in masking effect mechanisms between COPD and AF. |
Investigation of the EWAS Atlas website (https://ngdc.cncb.ac.cn/ewas/atlas/index) among the 25 colocalization-positive CpG sites revealed that RPS2 (cg02871659), KLF11 (cg05301188), KMT5A (cg05677062), FKBP2 (cg16977872), and PITPNA (cg20426671) were associated with smoking exposure, whereas remaining CpG sites exhibited no significant smoking associations. All CpG sites were unrelated to harmful particulate matter exposure (PM2.5, PM10).
Multi-Omics Evidence Validation of Important Pathways
Building on pivotal genes discovered at the methylation level, we integrated transcriptomic and proteomic data to triangulate putative mediators linking COPD to AF. We retain detailed eQTL-SMR results in Tables 1–3 in the main text for transparency and readability, with extended statistics in Supplementary Tables S24–S35.
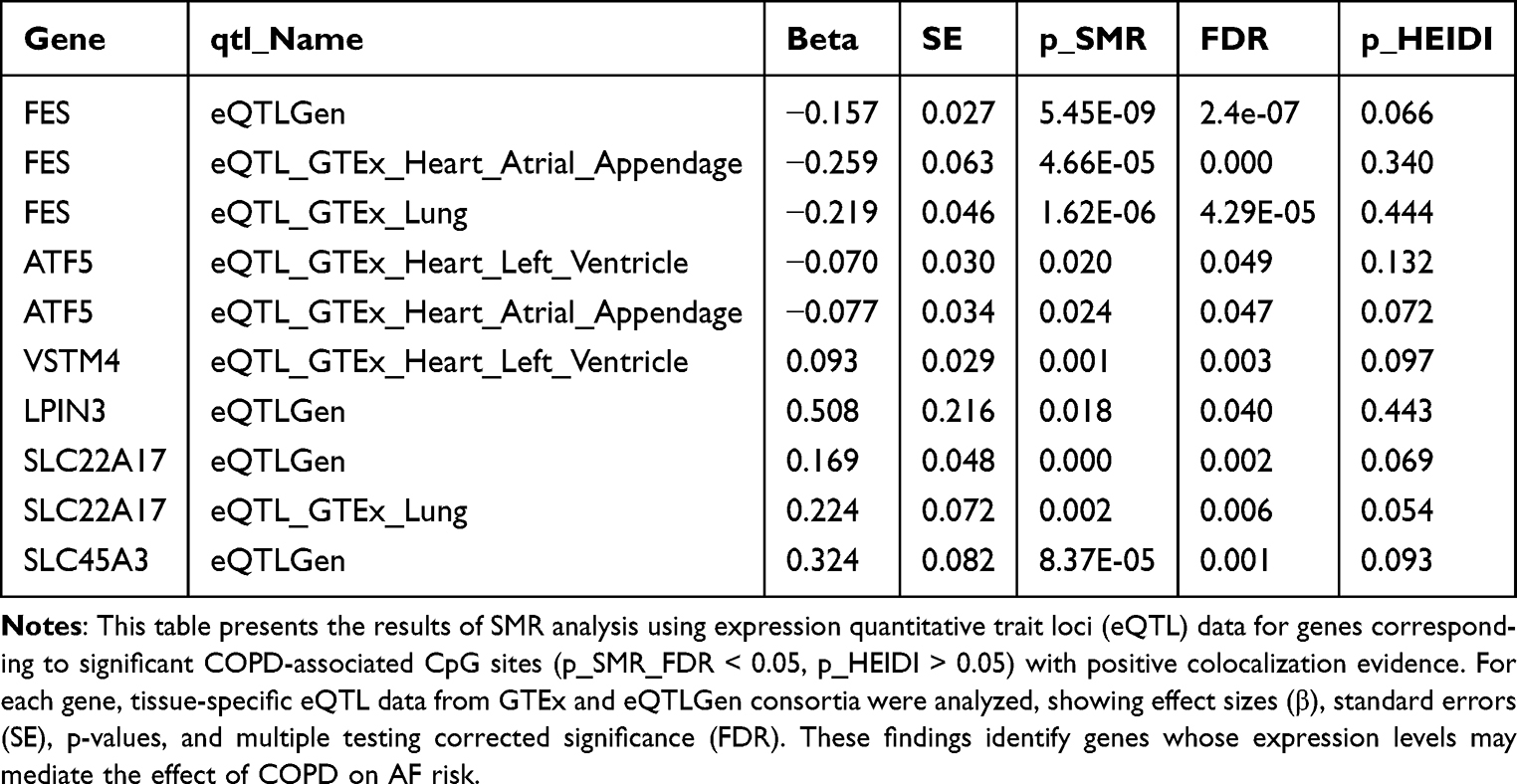 |
Table 2 SMR Analysis of Gene Expression Associations with AF for Significant COPD-Related Methylation Sites |
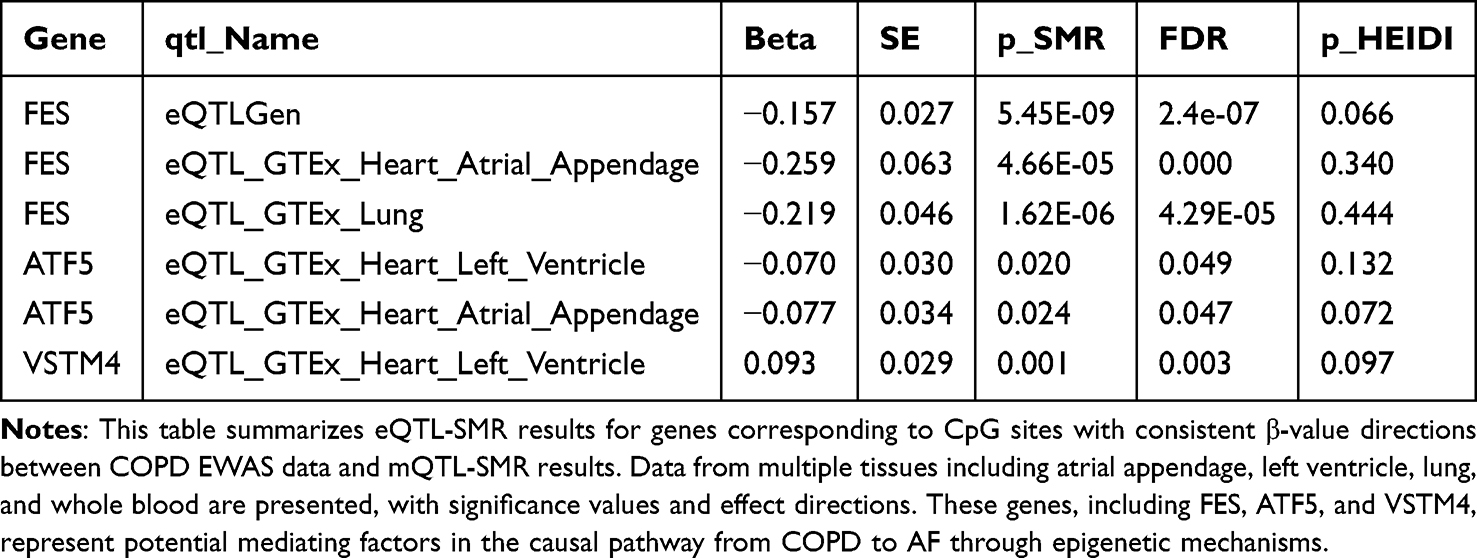 |
Table 3 SMR Analysis of Gene Expression Associations with AF for Methylation Sites with Consistent Effect Directions |
Gene Expression Level Validation
For genes significant at the methylation level, we performed eQTL-SMR analysis across GTEx and eQTLGen datasets (Table 1 and Supplementary Table S24). Table 1 summarizes representative gene–tissue associations with AF, using FDR control for p_SMR and HEIDI p > 0.05 as consistency with colocalization. At a glance, FES showed inverse associations with AF across eQTLGen and GTEx (atrial appendage, lung); ATF5 showed inverse associations in atrial appendage and left ventricle; VSTM4 showed a positive association in left ventricle; LPIN3, SLC22A17, and SLC45A3 showed positive associations in eQTLGen and/or GTEx lung (Table 2).
To align methylation and expression directions, we stratified genes by whether CpG effects in COPD EWAS and mQTL-SMR pointed in the same (consistent) or opposite (inconsistent) direction. Among genes with consistent directions (Table 3 and Supplementary Table S25), six genes showed expression levels associated with AF risk in at least one tissue. Integrating methylation and expression directions, four genes maintained significant associations, and two were validated by MR. Specifically, in atrial appendage, ATF5 (OR: 0.926, 95% CI: 0.854–0.998) and FES (OR: 0.772, 95% CI: 0.611–0.933) exhibited negative correlations with AF. In left ventricle, ATF5 (OR: 0.932, 95% CI: 0.869–0.996) was negatively correlated with AF, while VSTM4 (OR: 1.098, 95% CI: 1.047–1.149) was positively correlated (Supplementary Tables S26–S27). Colocalization supported shared signals for FES across GTEx atrial appendage, lung, whole blood, and eQTLGen, and for ATF5 in GTEx left ventricle and lung (PP.H4 > 0.9; Supplementary Table S28).
Among genes with opposite methylation–expression directions (Table 4 and Supplementary Table S29), four genes showed expression levels associated with AF in at least one tissue. After integrating methylation and expression directions, two genes maintained significant associations, both showing negative results in MR (Supplementary Tables S30–S31). Colocalization analysis indicated robust evidence for SLC45A3 with eQTLGen (PP.H4 > 0.9; Supplementary Table S32).
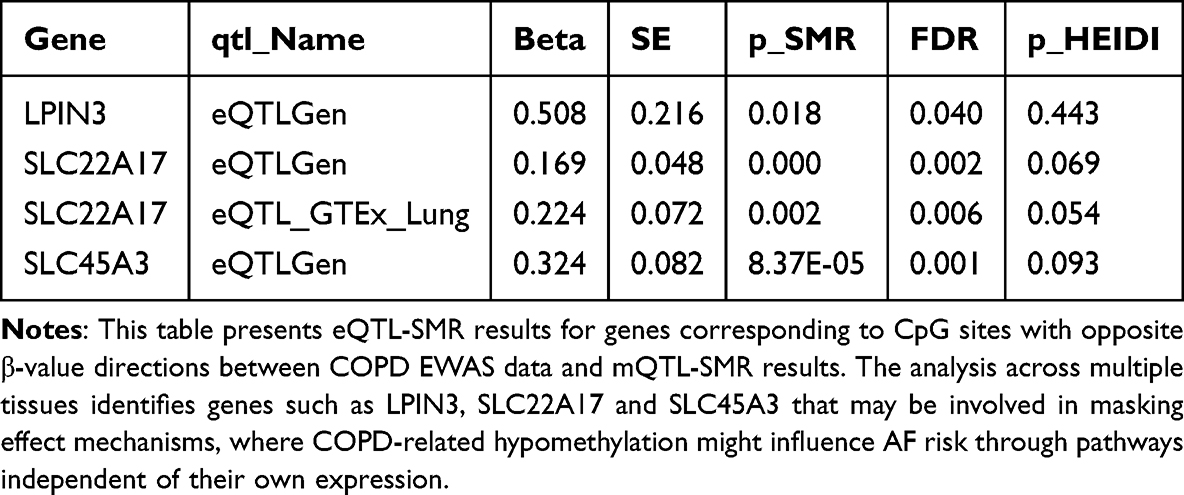 |
Table 4 SMR Analysis of Gene Expression Associations with AF for Methylation Sites with Opposite Effect Directions |
Protein Level Validation
At the protein level, ICAM4 abundance was positively associated with AF (pQTL-SMR; MR replicated in deCODE), whereas FES protein levels showed an inverse association with AF in UK Biobank-based MR (Supplementary Tables S33–S35). These protein-level findings converge with the expression-level directions for FES and suggest an orthogonal protein-based signal for ICAM4.
Multi-Level Evidence Integration and Functional Stratification
Integrating evidence across methylation, gene expression, and protein levels, we classified candidates into three functional tiers (Supplementary Tables S24–S35):
Tier 1 (highest): FES—consistent signals across methylation (positive direction), expression (negative), and protein (negative), with colocalization and MR support.
Tier 2 (moderate): ATF5, VSTM4, LPIN3—supported by methylation plus ≥1 additional omics layer or MR consistency in at least one tissue/dataset.
Tier 3 (suggestive/masking): ICAM4, SLC22A17, SLC45A3—signals present but limited to specific omics layers and/or with opposite directions between methylation and expression, suggesting masking or tissue/context dependencies.
Integrative Summary of Candidate Genes (with Clinical Significance)
Table 5 summarizes cross-omics directions, colocalization, MR replication, and tier assignments. Effect sizes correspond to modest odds-ratio shifts typical for complex traits (approximately OR 0.77–1.10 for key signals per SD/log-odds scale), implying small individual-level risk changes but potential population relevance via pathway-level modulation.
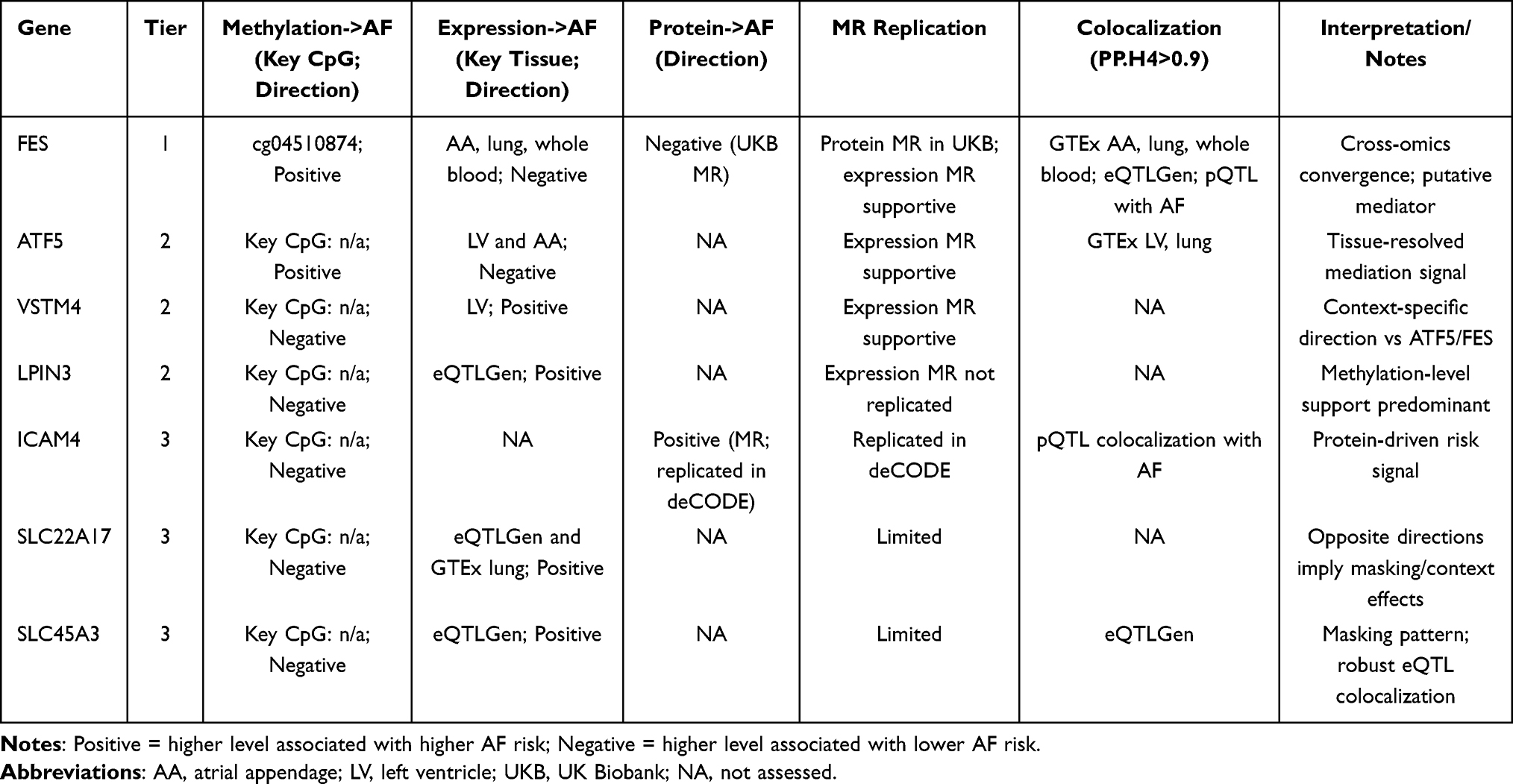 |
Table 5 Integrative Summary of Candidate Genes Across Methylation, Expression, and Protein Layers |
Right Heart Phenotype Mediation Analysis
To explore intermediate links in the COPD–methylation–AF pathway, we performed a two-step MR using right-heart imaging phenotypes as candidate mediators (Figure 8 and Supplementary Table S36-S43). First, cg18054172 (HOXC5) methylation was inversely associated with the pulmonary artery/aortic (PA/Ao) diameter ratio (beta = -0.073, p = 1.93×10−4, 95% CI: -0.112 to -0.035), remaining significant after multiple testing (BH-adjusted q = 0.0487). Second, higher genetically proxied PA/Ao ratio was associated with lower AF risk beta = -0.214, SE = 0.0798, p = 0.0072, 95% CI: -0.371 to -0.058), with a consistent weighted median estimate (beta = -0.179, p = 0.046) and a directionally similar but non-significant MR-Egger slope (beta = -0.422, p = 0.36). The combination of these two negative paths implies a positive indirect effect of HOXC5 methylation on AF risk via the PA/Ao ratio. Consistent with the upstream association of COPD with higher HOXC5 methylation (beta = 0.019), the overall COPD→methylation→PA/Ao→AF chain points to a positive mediated effect on AF risk. We note that this inverse PA/Ao→AF direction differs from some observational reports; because the PA/Ao ratio integrates both pulmonary artery and aortic diameters, the MR signal may be influenced by denominator (aortic) variation, and thus should be interpreted cautiously pending component-specific analyses.
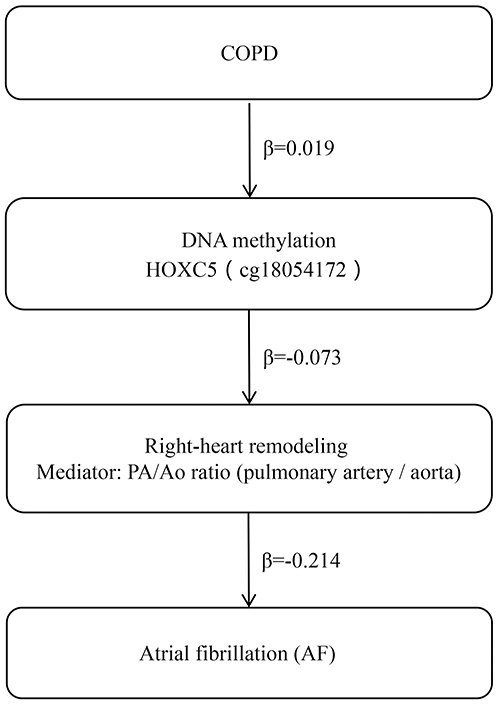 |
Figure 8 Two-step MR schematic linking COPD, HOXC5 methylation (cg18054172), the PA/Ao ratio, and AF. Vertical schematic illustrating the hypothesized chain from COPD to AF via HOXC5 methylation and the pulmonary artery/aortic (PA/Ao) diameter ratio. Links are labeled only with effect direction (Positive/Negative); no effect sizes are shown, and AF effects are on the log-odds scale. In our data, Step 1 (methylation to PA/Ao) is Negative and Step 2 (PA/Ao to AF) is Negative; the product-of-coefficients implies a positive indirect effect of HOXC5 methylation on AF via PA/Ao. Two-sample, two-step MR was used with mQTL instruments for cg18054172, imaging GWAS for PA/Ao, and AF GWAS; inverse-variance weighting provided primary estimates with sensitivity analyses as noted. |
PPI and Enrichment Analysis
Protein–protein interaction analysis of genes corresponding to CpG sites with consistent β-value directions between COPD EWAS data and mQTL-SMR results revealed functional interconnectivity including co-expression patterns (Figure 9A). STRING network analysis identified no meaningful functional connections (Supplementary Figure 6). GO enrichment analysis revealed that these genes were predominantly enriched in immune response mechanisms, signal transduction processes, metabolic pathways, developmental differentiation, cellular structural organization, and transport-related biological processes (Figure 9B–E and Supplementary Table S44). KEGG enrichment identified significant signaling pathways including lysine degradation and hormonal signaling pathway regulation (Figure 9F–H and Supplementary Table S45).
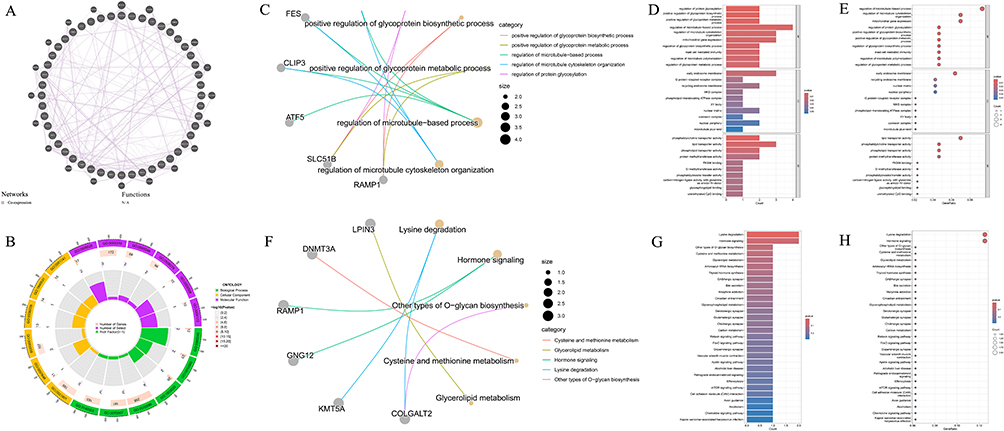 |
Figure 9 Functional analysis of genes with consistent effect directions between COPD EWAS and mQTL-SMR results. (A) Protein–protein interaction network of genes with consistent β-value directions, demonstrating functional connections and co-expression patterns.The purple line in the graph: Networks: Co-expression. (B) Circular representation of GO enrichment analysis for mediating effect genes, highlighting immune response, signal transduction, and metabolic pathway involvement. (C) Gene functional association network based on GO analysis, illustrating interconnections between biological processes. (D) Bar plot of significantly enriched GO terms for mediating effect genes. (E) Bubble plot of GO enrichment results, with larger bubbles indicating more genes associated with each term. (F) Gene functional association network based on KEGG pathway analysis, revealing connections between signaling cascades. (G) Bar plot of significantly enriched KEGG pathways, including lysine degradation and hormonal signaling regulation. (H) Bubble plot of KEGG pathway enrichment, with significance37815level indicated by color intensity. |
Protein–protein interaction analysis of genes corresponding to CpG sites with opposite β-value directions between COPD EWAS data and mQTL-SMR results revealed functional interconnectivity patterns (Figure 10A). STRING network analysis identified 63 nodes and 12 functional connections (Supplementary Figure 7), revealing potential collaborative participation in specific biological processes. GO functional enrichment analysis demonstrated these genes were primarily associated with cardiac and vascular development-related pathways, including atrial development, vascular smooth muscle cell differentiation, Notch signaling pathway in cardiac development, and atrial septal development. Additionally, enrichment in steroid and hormone metabolism regulation processes was observed (Figure 10B–E and Supplementary Table S46). KEGG pathway analysis identified significantly enriched signaling pathways including Notch signaling pathway (Figure 10F–H and Supplementary Table S47).
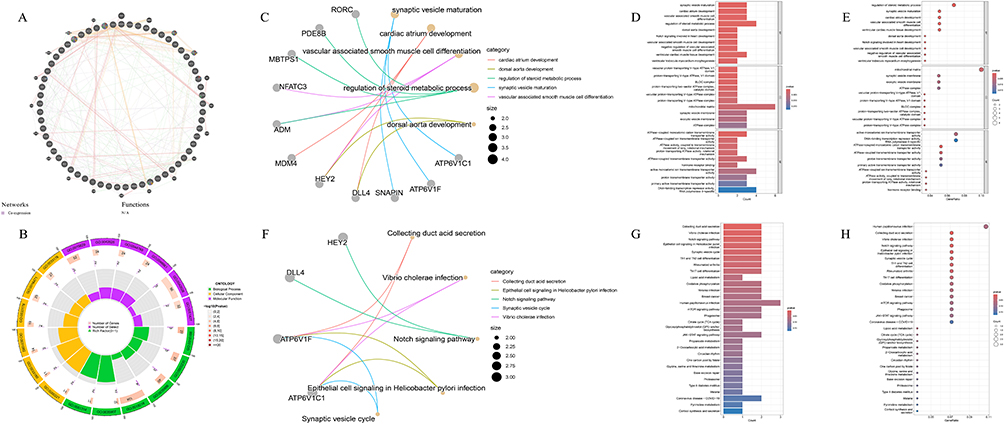 |
Figure 10 Functional analysis of genes with opposite effect directions between COPD EWAS and mQTL-SMR results. (A) Protein–protein interaction network of genes with opposite β-value directions, demonstrating functional connections and co-expression patterns. Networks (presented as lines in the diagram): Purple: Co-expression; Orange: Predicted; Red: Physical Interactions; Green: Genetic Interactions; Yellow-green: Shared protein domains.Functions (presented by the color inside the circle in the diagram):Red: Iron ion transport; Blue: Transition metal ion transport; Orange: ATPase-coupled transmembrane transporter activity; Purple: Primary active transmembrane transporter activity; Green: Active ion transmembrane transporter activity; Light purple: Phagosome maturation; Turquoise: Intracellular pH reduction. (B) Circular representation of GO enrichment analysis for masking effect genes, highlighting cardiac development and vascular-related pathways. (C) Gene functional association network based on GO analysis, illustrating interconnections between development-related processes. (D) Bar plot of significantly enriched GO terms, showing predominance of cardiac and vascular development pathways. (E) Bubble plot of GO enrichment results for masking effect genes. (F) Gene functional association network based on KEGG pathway analysis, emphasizing Notch signaling pathway connections. (G) Bar plot of significantly enriched KEGG pathways, including Notch signaling and mTOR pathways. (H) Bubble plot of KEGG pathway enrichment for masking effect genes, suggesting involvement in cardiac development regulation. |
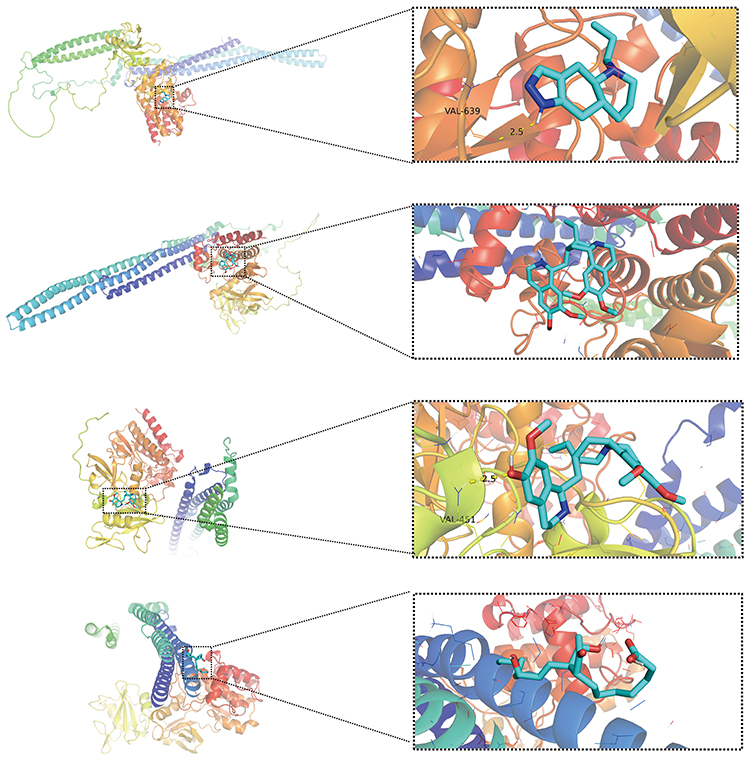 |
Figure 11 Molecular docking analysis of candidate drugs with target proteins identified through multi-omics integration. Visualization of molecular docking results for FES protein with four candidate compounds showing high binding affinity: FES-Quinpirole, FES-Emetine, FES-Cephaeline, and FES-Alprostadil. Each panel displays the three-dimensional protein structure with the compound in its predicted binding pose, highlighting key interaction sites. |
These functional analysis results suggest COPD might influence AF risk through epigenetic regulation of cardiac development and angiogenesis-related genes, particularly through Notch and mTOR signaling pathway mediation.
Candidate Drug Prediction and Molecular Docking
Based on multi-omics integration-identified 4 moderate-to-high evidence mediating genes, DSigDB database predicted 222 potential therapeutic compounds. FES and ATF5 demonstrated significant associations with the most candidate drugs, including Emetine, Cephaeline, Alprostadil, Quinpirole, Gossypol, Irinotecan, Deptropine, Daunorubicin, Ajmaline, Minocycline, and Chlorhexidine (Supplementary Table S48).
AutoDock molecular docking analysis identified 11 drug–protein interactions displaying high binding affinity (Figure 11, Table 6 and Supplementary Figure 8A-8H). ATF5 with Gossypol [PMID: 3503] exhibited the lowest binding energy (-7.1 kcal/mol), demonstrating the most thermodynamically stable binding conformation (Table 6). Molecular docking results indicated this compound formed 2 hydrogen bonds with ATF5 active sites, suggesting potential for effective ATF5 functional regulation. These findings provide potential targets for developing epigenetic regulatory drugs targeting AF risk in COPD patients, particularly through FES and ATF5-mediated epigenetic modifications.
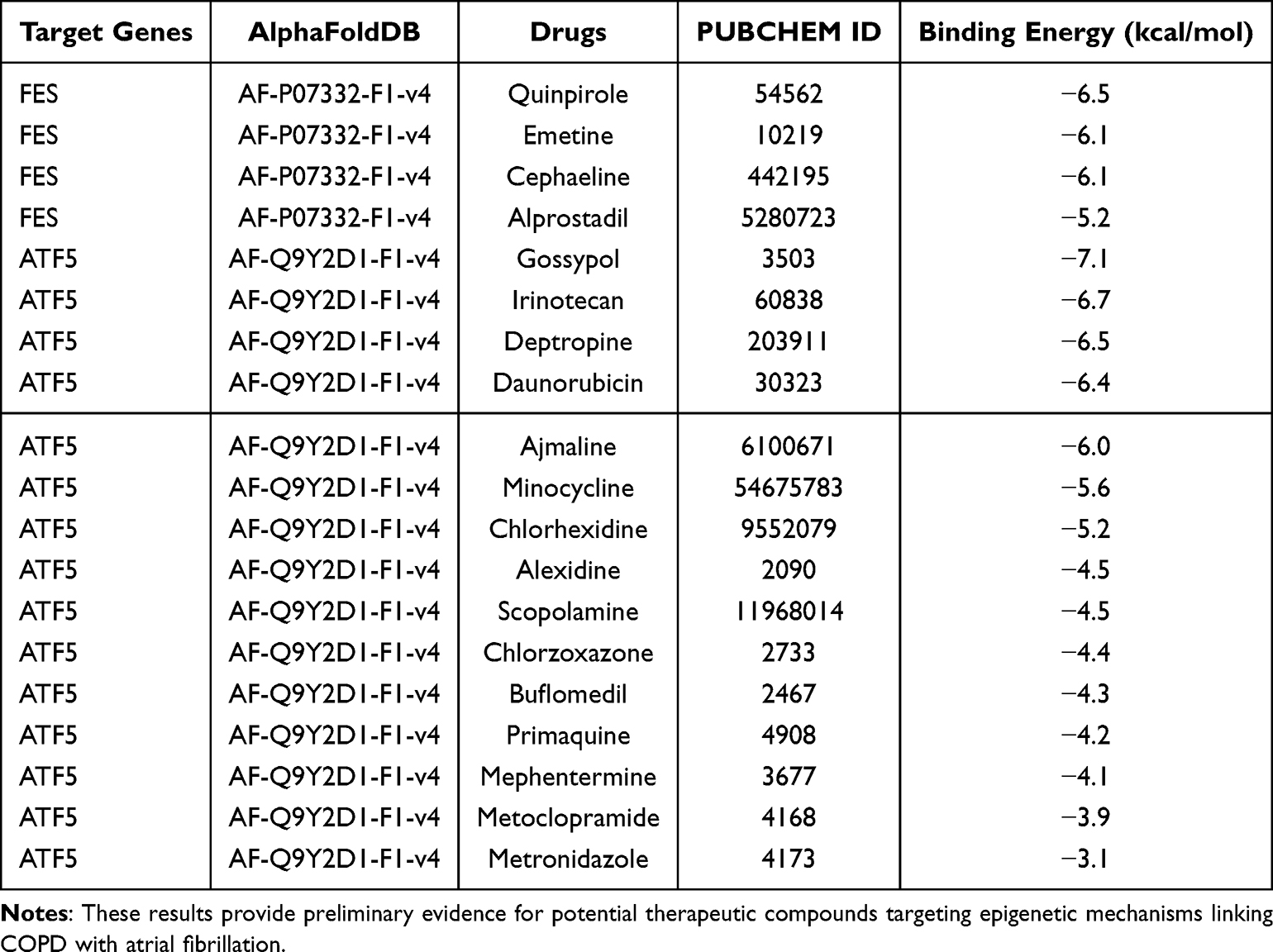 |
Table 6 Comparative Analysis of 38837 Binding Energy Scores for All Predicted Drug–Protein Interactions, with Lower Scores Indicating More Thermodynamically Stable Binding Conformations |
Discussion
To our knowledge, this is the first study to systematically integrate EWAS with multi-layer QTLs (mQTL, eQTL, pQTL) and causal inference tools (SMR, two-sample MR, HEIDI, and colocalization) to investigate the COPD–AF comorbidity and to prioritize putative mediators by cross-omics convergence. Through SMR, we identified 7 candidate genes with multi-omics evidence, encompassing 4 mediating effect genes and 3 masking effect genes. Among these, FES emerged as the most evidence-supported pivotal mediating gene, validated across multiple levels and demonstrating consistent alterations across DNA methylation, gene expression, and protein levels, providing preliminary human genetic support for its potential as a therapeutic target. Additionally, 7 genes including ATF5, ICAM4, and SLC22A17 function at minimally two regulatory levels, representing genes associated with incident AF in COPD. Compared to FES, these candidate targets should be considered preliminary findings necessitating validation through additional independent investigations. In contrast to prior COPD–AF studies that were predominantly observational or single-omics, our framework requires cross-layer consistency and colocalization (with protein-level replication where available), exemplified by FES convergence, ICAM4 protein replication, and a two-step MR illustrating a right-heart mediation chain (HOXC5 methylation → PA/Ao → AF).
Multiple COPD-related mechanisms may predispose to AF, including blood-gas disturbances (hypoxemia, hypercapnia), sympathetic activation, pulmonary hypertension with right-heart remodeling,32 chronic inflammation/oxidative stress (eg, Nox2) leading to atrial fibrosis,8,33 electrophysiological remodeling (prolonged atrial electromechanical delay, increased P-wave dispersion),9 and medication effects such as prolonged β2-agonist use.34 These observations support a close COPD–AF link and are consistent with our discovery of significant genetic correlation and unidirectional causal associations.
Our MR analysis demonstrated that COPD significantly elevates atrial fibrillation risk, with Steiger directionality testing further corroborating this causal association. Previous studies suggested potential shared genetic susceptibility between COPD and AF,35 consistent with our findings, suggesting that incident AF in COPD patients may involve underlying genetic mechanisms, providing crucial clues for subsequent epigenetic research. However, research findings regarding COPD-AF causal relationships are not entirely consistent. Guangzan Yu et al’s two-sample Mendelian randomization analysis demonstrated no significant causal relationship between COPD and AF (p = 0.991), whereas reverse Mendelian randomization results suggested causal relationships between AF and COPD (p = 0.018).36
Discrepancies in these research findings can be analyzed from both sample characteristics and methodological dimensions. Our investigation integrated multi-cohort data encompassing 58,559 COPD cases, whereas Yu et al utilized smaller-scale European population data with 13,530 COPD cases, representing approximately 4.3-fold increase in sample size and providing substantially higher statistical power. Methodological rigor differences are equally noteworthy, as our investigation employed more stringent methodological strategies, encompassing five complementary MR methods for cross-validation, MR-PRESSO pleiotropy control, and Steiger directionality testing excluding reverse causation. Despite our study’s distinct advantages in sample size and methodology, potential causal associations between COPD and AF may still require corroboration and validation from independent investigations.
Epigenetic studies suggest that DNA methylation in COPD can modulate oxidative stress and inflammation, contributing to atrial fibrosis and electrical remodeling; aberrant methylation has been reported in ion-channel (eg, KCNQ1) and fibrosis genes (eg, TGF-β) in blood and lung tissues.37,38 However, previously published EWAS combined with MR either ignored the sign of CpG β in EWAS data or selected only positive β values,39,40 introducing potential logical or selection biases. Our investigation systematically compared consistency between COPD EWAS data CpG site β values and SMR result β value positivity/negativity, discovering complex regulatory mechanisms of DNA methylation in COPD-induced atrial fibrillation. Through SMR, we identified 109 sites whose methylation status through genetic regulation might influence AF risk. Among CpG sites with consistent β-value directions between COPD EWAS data and mQTL-SMR results, 48 sites were identified, with 20 CpG sites exhibiting positive correlation between methylation levels and AF risk and 28 demonstrating negative correlation. Among CpG sites with opposite β-value directions between COPD EWAS data and mQTL-SMR results, 61 sites were identified, with 33 exhibiting positive correlation and 28 demonstrating negative correlation. However, the 109 CpG sites identified as potentially causally related (putative causal) to AF through SMR screening were predominantly limited by single cis-SNP instrumental variables, requiring further validation through multi-SNP IV or independent datasets.
Long-term smoking and particulate exposure are major risk factors for COPD and can reshape DNA methylation and activate inflammatory/oxidative pathways.37,41–43 Therefore, we systematically validated associations between colocalization-positive CpG sites and environmental factors. Results demonstrated that among 25 candidate sites, only 5 were smoking-related (20%), with none related to harmful particulate matter, indicating that most epigenetic signals reflect disease-specific mechanisms rather than environmental exposure effects, ensuring result robustness. Subsequently combining multi-omics evidence, we ultimately identified 7 important genes functioning in AF risk during COPD development: FES, ATF5, VSTM4, LPIN3, ICAM4, SLC22A17, and SLC45A3, with FES, ATF5, VSTM4, and LPIN3 serving as mediating genes, and ICAM4, SLC22A17, and SLC45A3 serving as masking effect genes.
FES, ATF5, VSTM4, and LPIN3 demonstrated β-direction consistency, suggesting these genes may participate in mediating COPD-induced atrial fibrillation mechanisms. Among these, FES exhibited consistency across multi-omics levels (methylation, gene expression, protein), potentially representing the most critical gene in COPD-AF comorbidity. We discovered that hypomethylation of the FES promoter region site cg01943198 associated with COPD risk, through enhancing FES expression (blood eQTL) and protein levels (plasma pQTL), ultimately resulting in elevated AF risk. This complete causal cascade from DNA methylation to gene expression to protein levels to disease risk provides important molecular-level insights for understanding how COPD might influence AF through epigenetic mechanisms. FES (Feline Sarcoma Oncogene), as a non-receptor tyrosine kinase,44 has been previously reported in inflammatory responses and cytoskeletal remodeling,45 with preliminary exploration of its specific roles in cardiovascular systems. For example, previous investigations suggested FES may reduce CAD (Coronary Artery Disease) incidence risk through protection against atherosclerosis,46 suggesting FES may participate in common regulatory pathways in CAD and AF development. However, FES’s important mediating role in COPD-AF comorbidity has not been previously elucidated. COPD-induced chronic inflammation may promote atrial remodeling through activating such kinases, requiring experimental validation. Additionally, ATF5, VSTM4, and LPIN3 also serve as mediating genes. Previous research suggested that ATF5 may regulate macrophage polarization, precipitating airway inflammation and subsequently leading to COPD progression and acute exacerbations. Additionally, ATF5 may mitigate mitochondrial dysfunction and oxidative stress damage in pathological cardiac hypertrophy through activating UPRmt (Mitochondrial Unfolded Protein Response) pathways, participating in atrial structural remodeling and thereby reducing AF risk.47 COPD precipitates hypoxia, which may affect myocardial stress through the ATF5-SIRT1-UPRmt axis.48 VSTM4 assumes important roles in cancer development49 but demonstrates no direct associations with COPD or atrial fibrillation. Additionally, LPIN3 relates to lipid metabolism and inflammatory responses,50 potentially participating in atrial fibrillation through affecting myocardial energy metabolism or inflammatory responses. In previous investigations, these three genes exhibited no direct associations with COPD or AF, potentially reflecting epigenetic mechanism complexity in disease development, requiring further experimental research for validation. Furthermore, at protein expression levels, ATF5, VSTM4, and LPIN3 demonstrated no associations with AF.
In the medium term, peripheral-blood methylation assays targeting FES and ATF5 (eg, targeted bisulfite amplicon sequencing or droplet digital PCR) could be assembled into a concise risk panel for COPD populations (Figure 12). A prespecified workflow would (i) compute a biomarker score from FES/ATF5 methylation (and expression where available) alongside clinical covariates (age, sex, smoking status, FEV1, hypoxemia, β2-agonist use), (ii) triage high-score patients to intensified AF surveillance (eg, 7–14-day patch ECG monitoring and echocardiographic evaluation), and (iii) evaluate incremental discrimination and clinical net benefit (AUC, NRI, and decision-curve analysis) over standard predictors. For therapy, the convergent multi-omics signals nominate FES- and ATF5-linked pathways for mechanistic follow-up and pharmacologic exploration; directional modulation should be guided by tissue-specific functional studies, with safety profiling prioritized given kinase and mitochondrial-stress biology. These translational steps do not change the current standard of care but define a path to prospective validation.
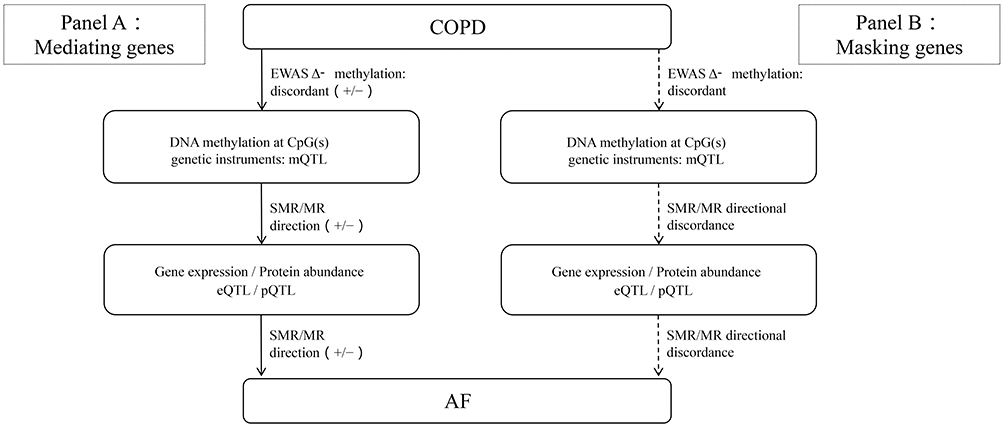 |
Figure 12 Conceptual flowchart distinguishing mediating vs masking genes along the COPD to methylation to expression/protein to AF pathway. Two parallel panels share a common start node (COPD) and end node (AF; log-odds scale), with intermediate layers: DNA methylation at CpG(s) (genetic instruments: mQTL) and gene expression/protein abundance (eQTL/pQTL). Arrows are labeled only with effect direction (+/−): the COPD to methylation link reflects the delta methylation direction from EWAS; the methylation to expression/protein and expression/protein to AF links reflect SMR/colocalization directions. Panel A (Mediating genes): all links are directionally coherent and the implied net COPD to AF effect matches the overall MR direction (examples: FES, ATF5, VSTM4, LPIN3). Panel B (Masking genes): at least one link is directionally discordant (shown as dashed and annotated “discordant”), indicating that COPD-related methylation changes do not propagate to AF risk via the observed expression/protein direction (examples: ICAM4, SLC22A17, SLC45A3; ICAM4 shows protein-level signal without expression-level concordance). |
For genes such as SLC22A17, SLC45A3, and ICAM4 displaying masking effects, where COPD-related hypomethylation elevates AF risk through mechanisms independent of their own expression, we hypothesize several possible biological mechanisms (Figure 12). From regulatory network perspectives, CpG sites discovered in our investigation may represent regulatory hubs simultaneously negatively regulating ICAM4 and other genes while positively regulating another (unmeasured) pivotal gene promoting AF risk. Another possible explanation involves dynamic regulatory characteristics during disease progression, such as compensatory responses under chronic inflammatory stimulation leading to seemingly contradictory statistical associations. Unlike previous investigations primarily focusing on tumor microenvironment-related mechanisms,51 our investigation first revealed roles of SLC22A17 and SLC45A3 genes in AF development in COPD. ICAM4 gene may mediate cell adhesion and migration through integrin binding, precipitating inflammatory responses and thereby elevating AF risk in COPD patients,52 but direct evidence remains insufficient. Exploring main biological mechanisms of these genes in AF occurrence following COPD requires further experimental validation. These genes can undergo further cellular and animal experimental validation exploring biological mechanisms and may be incorporated into polygenic risk assessment models for further randomized controlled or cohort studies investigating effects of gene methylation interventions on AF incidence.
Through chronic hypoxia, inflammation, and vascular remodeling, COPD can lead to pulmonary hypertension and progressive right-heart structural changes.32,53 AF commonly involves pulmonary vein–left atrial triggers alongside ion-channel dysfunction, calcium-handling abnormalities, structural remodeling, and autonomic dysregulation;54,55 consequently, COPD-related right-heart remodeling may increase AF risk. To further elucidate right heart structural alterations’ roles in the COPD-methylation-AF causal cascade, we utilized mQTL data and right heart system GWAS data for MR analysis. To further probe right-heart structural pathways within the COPD–methylation–AF cascade, we conducted a two-step MR linking HOXC5 (cg18054172) methylation to the pulmonary artery/aortic (PA/Ao) diameter ratio and then to AF. Higher genetically proxied HOXC5 methylation was associated with a lower PA/Ao ratio, and in turn, a higher PA/Ao ratio was associated with a lower AF risk. The product of these two negative paths implies a positive indirect (mediated) effect of HOXC5 methylation on AF via a reduction in PA/Ao. Accordingly, contrary to our earlier phrasing, the data do not support an increase in PA/Ao as a mechanism; rather, they are consistent with HOXC5-related methylation changes contributing to AF risk through processes that lower PA/Ao. Because PA/Ao is a ratio surrogate of cardiopulmonary remodeling and the first step used a limited number of instruments, these findings should be interpreted cautiously and validated with direct right-heart measures (eg, absolute PA diameter, right-ventricular indices, pulmonary pressures) in independent cohorts.
Notably, our investigation’s epigenetic and functional annotation analyses were primarily based on peripheral blood and left heart system data. COPD-induced pulmonary hypertension and pulmonary heart disease primarily affect right heart systems, particularly right ventricle and pulmonary arteries. Although we validated main findings across multiple tissues, tissue-specific differences may influence clinical interpretation of results. Expression patterns and functions of important genes such as FES in right heart systems require further research. Our findings should be considered important associative clues, while their precise roles in COPD-related right heart lesions require validation through targeted right heart system experimental research. Future investigations should focus on expression alterations and functional significance of these candidate genes in pulmonary hypertension and right ventricular remodeling processes.
Genes with consistent β-direction mapped to immune/secretory pathways and lipid metabolism, aligning with shared inflammatory biology of COPD and AF.56–59 Direct mechanistic links between lipid metabolism and the COPD–AF axis remain to be established and will require targeted experimentation.
For opposite-direction genes, enrichment highlighted cardiac development, Notch signaling, angiogenesis, and immune/secretory pathways. Notch signaling has been implicated in COPD airway remodeling and in cardiac development/conduction and AF.60–65 mTOR signaling connects airway inflammation with atrial remodeling relevant to AF.66–68 Angiogenesis-related methylation (eg, VEGF) may influence cardiac remodeling and AF susceptibility.69 These pathway-level findings are hypothesis-generating and need experimental confirmation.
For 4 moderate-to-high evidence mediating genes identified in COPD-induced AF risk, we conducted exploratory candidate drug prediction, with results suggesting FES and ATF5 as the most promising potential therapeutic targets, yielding 11 high-affinity candidate compounds. This “in-silico” list provides reference for subsequent in vitro/in vivo screening. Research demonstrated FES’s protective effects against coronary heart disease and myocardial infarction, with therapeutic potential in controlling blood glucose, blood pressure, and lipid levels, emphasizing the importance of FES-targeted drug development.70 ATF5 is primarily utilized in tumor-related treatments,71 with no discovered applications in respiratory and cardiovascular systems. Molecular docking results in our investigation suggested high-affinity drug-protein binding between FES and Emetine, Cephaeline, Alprostadil, Quinpirole; and between ATF5 and Gossypol, Irinotecan, Deptropine, Daunorubicin, Ajmaline, Minocycline, Chlorhexidine. However, these in-silico predicted drugs exclusively suggest potential affinity, requiring cellular-animal experiments and preclinical pharmacological systematic assessment before determining their true efficacy and safety for COPD-AF risk. Among these, Alprostadil can vasodilate and improve cardiovascular microcirculation, providing cardioprotective effects.72 Additionally, animal experiments demonstrated Alprostadil’s protective effects against acute respiratory distress syndrome.73 Although no evidence demonstrates direct relationships with COPD and AF, this still provides candidate molecular targets for subsequent functional experiments and early pharmacological screening. Ajmaline represents a sodium channel blocker utilized for Brugada syndrome diagnosis,74 with unclear therapeutic effects on AF and no respiratory system disease applications. However, Ajmaline prolongs QT intervals and may even precipitate torsades de pointes ventricular tachycardia,75,76 highlighting the enormous challenges and necessity of translating in-silico predictions to clinical applications, requiring thorough cardiac toxicity assessment before any clinical application. Additionally, no corresponding therapeutic drugs were identified for targets including VSTM4, LPIN3, ICAM4, SLC22A17, and SLC45A3, requiring further research. To operationalize translation, we propose a pragmatic pathway: analytical validation of FES/ATF5 methylation assays, independent case–control replication, and a prospective COPD cohort with AF endpoints to test predefined biomarker cut-points and decision thresholds. Integration into clinical decision algorithms for ambulatory ECG deployment should be assessed using AUC/NRI and decision-curve analyses, prior to any interventional trial.
Our investigation’s main innovations and advantages encompass: 1. Methodological innovation: First integration of EWAS data with multi-level QTL data, applying epigenetic SMR and MR analytical strategies, surpassing traditional association studies to explore DNA methylation’s mediating roles in COPD-induced AF from causal inference perspectives. 2. Discovery novelty and systematicity: Through multi-omics evidence integration and cross-validation (methylation, gene expression, protein abundance), combined with colocalization analysis, we first discovered a cohort of candidate genes (particularly FES) mediating COPD-AF associations with high confidence, providing more comprehensive and reliable molecular evidence. 3. Potential translational value: Candidate drug prediction and molecular docking based on newly discovered molecular targets provide preliminary insights for developing novel strategies targeting AF prevention or treatment in COPD patients. Additionally, combining epigenetic data may facilitate individual-level COPD patient management, individualizing more appropriate regimens to reduce concurrent AF risk.
Despite providing valuable scientific discoveries, our investigation possesses inevitable limitations requiring full consideration during result interpretation. Regarding data sources and population representativeness, research data primarily originated from European ancestry populations with single methylation data sources, potentially limiting result generalizability. Due to allele frequency and linkage disequilibrium pattern differences across populations, genetic variations demonstrate population specificity, making validation of these findings in multi-ethnic populations particularly important. Additionally, since analyses relied on cross-sectional EWAS data, temporal relationships between methylation as COPD cause, consequence, or common manifestation cannot be clearly determined. Prospective cohort studies will facilitate further elucidation of temporal relationships among these factors.
We also acknowledge potential residual confounding at the phenotype level by smoking behavior and treatment differences. Detailed smoking phenotypes (eg, cumulative exposure/pack-years, intensity trajectories, time since cessation, secondhand exposure) and medication regimens (eg, beta2-agonists, anticholinergics, inhaled corticosteroids, theophylline, systemic steroids) were not harmonized across cohorts and could influence both COPD severity and AF detection. Although genetic designs mitigate measured confounding and our smoking-annotation of colocalized CpGs suggested limited direct overlap (5/25 sites), phenotype-level bias from smoking and treatment differences cannot be fully excluded.
Several related risk factors could not be assessed in the present work. Left atrial diameter (or standardized left atrial indices) was not available in the UK Biobank cardiac MRI phenotype set accessed for this study, precluding LAD-based analyses. While we explored pulmonary hypertension as a clinically relevant outcome, available GWAS had too few cases to enable reliably powered two-sample MR; we therefore did not proceed with formal analysis to avoid unstable estimates. Future updates to cardiac MRI phenotypes and larger pulmonary hypertension GWAS will allow us to revisit these questions.
Methodologically, SMR analysis primarily relied on single cis-QTL instrumental variables. Despite rigorous HEIDI testing, unknown linkage disequilibrium interference cannot be theoretically completely excluded. Additionally, limited instrumental variable numbers for some CpG sites may possess insufficient statistical power. Despite FDR correction, false-positive results may still exist. Regarding tissue specificity, our investigation’s QTL data primarily originated from peripheral blood, lung, and left heart systems, while COPD-related pulmonary heart disease more involves right heart systems. This mismatch may limit clinical discovery interpretation.
In translational research, molecular docking predictions provided theoretical foundations for drug–target interactions but their accuracy is influenced by protein structure quality, making it difficult to fully reveal biological system complexity. Therefore, key CpG site and gene regulatory relationships require cellular and animal experimental validation. Simultaneously, in vitro and in vivo studies should be conducted to evaluate candidate drug pharmacokinetics, efficacy, and safety, ensuring statistical discoveries can effectively translate to clinical applications.
Cross-omics convergence supports FES/ATF5 methylation (±expression) with ICAM4 protein as a compact blood panel to triage COPD patients for intensified AF surveillance and to refine prognostic models. Next steps are assay validation, independent replication, and prospective testing with decision-curve evaluation; integration with clinical covariates and right-heart indices (eg, PA/Ao) is essential. FES kinase and ATF5 stress pathways provide tractable targets for preclinical modulation prior to any interventional studies.
Conclusion
In conclusion, using an integrated multi-omics causal framework, we provide genetic evidence supporting a causal link between COPD and AF, with part of this effect mediated by DNA methylation. Cross-omics convergence prioritizes FES and ATF5 as key mediating genes. These insights nominate a compact peripheral-blood panel based on FES/ATF5 methylation (and, where available, expression) to refine AF risk stratification and guide intensified surveillance in COPD, while highlighting tractable pathways for prevention pending prospective validation.
Abbreviations
COPD, Chronic Obstructive Pulmonary Disease; AF, Atrial Fibrillation, EWAS, epigenome-wide association studies; GWAS, genome-wide association studies; mQTL, methylation quantitative trait loci; eQTL, expression quantitative trait loc; pQTL, protein quantitative trait loci; SMR, Summary-data-based Mendelian Randomization; HEIDI, HEIDI Testing; MR, Mendelian Randomization; IVW, Inverse-Variance Weighted; SNP, single nucleotide polymorphism; IV, Instrumental Variables; OR, Odds Ratio; CI, Confidence Interval; FDR, False Discovery Rate; β, Beta Coefficient; SE, Standard Error; LDSC, LD Score Regression; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; PPI, Protein–Protein Interaction Network; UKB, UK Biobank; GTEx, Genotype-Tissue Expression Consortium; DSigDB, Drug–Gene Interaction Database; RVEF, Right Ventricular Ejection Fraction.
Data Sharing Statement
All data related to our study are presented in the article or uploaded as supplementary information. Reasonable requests for further data queries could be addressed to the corresponding author.
Ethics Approval and Consent to Participate
This investigation utilized publicly accessible summary-level data. All original studies generating these summary data received approval from their respective Institutional Review Boards (IRBs) or ethics committees. Participants in original studies provided informed consent.
My study is exempt from ethical approval based on national legislation guidelines. The relevant article is Article 32 of the Measures for Ethical Review of Life Science and Medical Research Involving Human Subjects (China, promulgated on February 18, 2023), which states that the following types of research involving human data or biological samples, which do not harm the human body and do not involve sensitive personal information or commercial interests, may be exempt from ethical review to reduce unnecessary burdens on researchers and promote the development of life science and medical research involving humans:
1. Research using lawfully obtained public data or data generated by observing public behavior without interference;
2. Research using anonymized information data.
Acknowledgments
All authors are grateful to all those who participated in this study and thank the International Genetics Consortium for UK Biobank database, FinnGen database, and quantitative trait loci sources for providing publicly available statistics.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
No funding was received for this article.
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Chen S, Kuhn M, Prettner K, et al. The global economic burden of chronic obstructive pulmonary disease for 204 countries and territories in 2020-50: a health-augmented macroeconomic modelling study. Lancet Glob Health. 2023;11(8):e1183–26. doi:10.1016/S2214-109X(23)00217-6
2. Chugh SS, Havmoeller R, Narayanan K, et al. Worldwide epidemiology of atrial fibrillation: a Global Burden of Disease 2010 Study. Circulation. 2014;129(8):837–847. doi:10.1161/CIRCULATIONAHA.113.005119
3. Proietti M, Laroche C, Drozd M, et al. Impact of chronic obstructive pulmonary disease on prognosis in atrial fibrillation: a report from the EURObservational Research Programme Pilot Survey on Atrial Fibrillation (EORP-AF) general registry. Am Heart J. 2016;181:83–91. doi:10.1016/j.ahj.2016.08.011
4. Romiti GF, Corica B, Pipitone E, et al. Prevalence, management and impact of chronic obstructive pulmonary disease in atrial fibrillation: a systematic review and meta-analysis of 4,200,000 patients. Eur Heart J. 2021;42(35):3541–3554. doi:10.1093/eurheartj/ehab453
5. Warming PE, Garcia R, Hansen CJ, et al. Atrial fibrillation and chronic obstructive pulmonary disease: diagnostic sequence and mortality risk. Eur Heart J Qual Care Clin Outcomes. 2023;9(2):128–134. doi:10.1093/ehjqcco/qcac059
6. Liao KM, Kuo LT, Lu HY. Increased risk of peripheral arterial occlusive diseases in patients with chronic obstructive pulmonary disease: a nationwide study in Taiwan. Int J Chron Obstruct Pulmon Dis. 2019;14:1455–1464. doi:10.2147/COPD.S202029
7. Bucci T, Romiti GF, Shantsila A, et al. Risk of death and cardiovascular events in asian patients with atrial fibrillation and chronic obstructive pulmonary disease: a report from the prospective APHRS registry. J Am Heart Assoc. 2024;13(7):e032785. doi:10.1161/JAHA.123.032785
8. Pastori D, Andreozzi P, Carnevale R, et al. Does the coexistence of chronic obstructive pulmonary disease and atrial fibrillation affect Nox2 activity and urinary isoprostanes excretion? Antioxid Redox Signal. 2019;31(11):786–790. doi:10.1089/ars.2019.7811
9. Cimci M, Borekci S, Kilickiran Avci B, et al. Assessment of atrial electromechanical delay and P wave dispersion in patients with chronic obstructive pulmonary disease. Turk Kardiyol Dern Ars. 2020;48(3):263–269. doi:10.5543/tkda.2019.58665
10. Xiong S, Liu Q, Zhou S, et al. Identification of key genes and regulatory networks involved in the Comorbidity of atrial fibrillation and chronic obstructive pulmonary disease. Heliyon. 2023;9(11):e22430. doi:10.1016/j.heliyon.2023.e22430
11. Sun Z, Lin J, Zhang T, et al. Combining bioinformatics and machine learning to identify common mechanisms and biomarkers of chronic obstructive pulmonary disease and atrial fibrillation. Front Cardiovasc Med. 2023;10:1121102. doi:10.3389/fcvm.2023.1121102
12. Liu X, Shi J, Jiao Y, et al. Integrated multi-omics with machine learning to uncover the intricacies of kidney disease. Brief Bioinform. 2024;25(5). doi:10.1093/bib/bbae364
13. Singh M, Dolan CV, Lapato DM, et al. Unidirectional and bidirectional causation between smoking and blood DNA methylation: evidence from twin-based Mendelian randomisation. Eur J Epidemiol. 2025;40(1):55–69. doi:10.1007/s10654-024-01187-5
14. Akiyama M. Multi-omics study for interpretation of genome-wide association study. J Hum Genet. 2021;66(1):3–10. doi:10.1038/s10038-020-00842-5
15. Zhou W, Kanai M, Wu K-H-H, et al. Global biobank meta-analysis initiative: powering genetic discovery across human disease. Cell Genom. 2022;2(10):100192. doi:10.1016/j.xgen.2022.100192
16. Hillary RF, McCartney DL, Bernabeu E, et al. Blood-based epigenome-wide analyses on the prevalence and incidence of nineteen common disease states. medRxiv. 2023:
17. Wu Y, Zeng J, Zhang F, et al. Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits. Nat Commun. 2018;9(1):918. doi:10.1038/s41467-018-03371-0
18. Min JL, Hemani G, Hannon E, et al. Genomic and phenotypic insights from an atlas of genetic effects on DNA methylation. Nat Genet. 2021;53(9):1311–1321. doi:10.1038/s41588-021-00923-x
19. Srivastava AK, Williams SM, Zhang G. Heritability estimation approaches utilizing genome-wide data. Curr Protoc. 2023;3(4):e734. doi:10.1002/cpz1.734
20. Bowden J, Smith GD, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–525. doi:10.1093/ije/dyv080
21. Bowden J, Davey Smith G, Haycock PC, et al. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304–314. doi:10.1002/gepi.21965
22. Slob EAW, Burgess S. A comparison of robust Mendelian randomization methods using summary data. Genet Epidemiol. 2020;44(4):313–329. doi:10.1002/gepi.22295
23. Hartwig FP, Smith GD, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol. 2017;46(6):1985–1998. doi:10.1093/ije/dyx102
24. Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi:10.1086/519795
25. Burgess S, Thompson SG, Collaboration CCG. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol. 2011;40(3):755–764. doi:10.1093/ije/dyr036
26. Rasooly D, Peloso GM, Giambartolomei C. Bayesian genetic colocalization test of two traits using coloc. Curr Protoc. 2022;2(12):e627. doi:10.1002/cpz1.627
27. Giambartolomei C, Vukcevic D, Schadt EE, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10(5):e1004383. doi:10.1371/journal.pgen.1004383
28. Szklarczyk D, Kirsch R, Koutrouli M, et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023;51(D1):D638–D646. doi:10.1093/nar/gkac1000
29. Warde-Farley D, Donaldson SL, Comes O, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38(Web Server issue):W214–20. doi:10.1093/nar/gkq537
30. Yoo M, Shin J, Kim J, et al. DSigDB: drug signatures database for gene set analysis. Bioinformatics. 2015;31(18):3069–3071. doi:10.1093/bioinformatics/btv313
31. Glickman ME, Rao SR, Schultz MR. False discovery rate control is a recommended alternative to Bonferroni-type adjustments in health studies. J Clin Epidemiol. 2014;67(8):850–857. doi:10.1016/j.jclinepi.2014.03.012
32. Zakynthinos E, Daniil Z, Papanikolaou J, et al. Pulmonary hypertension in COPD: pathophysiology and therapeutic targets. Curr Drug Targets. 2011;12(4):501–513. doi:10.2174/138945011794751483
33. Graul EL, Nordon C, Rhodes K, et al. Factors associated with non-fatal heart failure and atrial fibrillation or flutter within the first 30 days post COPD exacerbation: a nested case-control study. BMC Pulm Med. 2024;24(1):221. doi:10.1186/s12890-024-03035-4
34. Liang H, Tan J, Xu W, et al. The impact of beta-blockers and renin-angiotensin-aldosterone system inhibitors on the prognosis of atrial fibrillation patients with chronic obstructive pulmonary disease: a nation-wide registry study. Int J Chron Obstruct Pulmon Dis. 2025;20:699–708. doi:10.2147/COPD.S511117
35. Volchkova EA, Zateishchikov DA. Genetic features of factors of inflammation and fibrosis possible common pathway of formation of predisposition to atrial fibrillation and chronic obstructive pulmonary disease. Kardiologiia. 2015;55(12):81–89. doi:10.18565/cardio.2015.12.81-89
36. Yu G, Liu L, Ma Q, et al. Bidirectional causal association between chronic obstructive pulmonary disease and cardiovascular diseases: a mendelian randomization study. Int J Chron Obstruct Pulmon Dis. 2024;19:2109–2122. doi:10.2147/COPD.S475481
37. Sarker RSJ, Conlon TM, Morrone C, et al. CARM1 regulates senescence during airway epithelial cell injury in COPD pathogenesis. Am J Physiol Lung Cell Mol Physiol. 2019;317(5):L602–L614. doi:10.1152/ajplung.00441.2018
38. Morrow JD, Cho MH, Hersh CP, et al. DNA methylation profiling in human lung tissue identifies genes associated with COPD. Epigenetics. 2016;11(10):730–739. doi:10.1080/15592294.2016.1226451
39. Zheng Z, Chen H, Chen Y, et al. Causal association between epilepsy and its DNA methylation profile and atrial fibrillation. Heart Rhythm. 2024;22(6):1588–1597. doi:10.1016/j.hrthm.2024.09.008
40. Zhan ZQ, Li J-X, Chen Y-X, et al. The effects of air and transportation noise pollution-related altered blood gene expression, DNA methylation, and protein abundance levels on gastrointestinal diseases risk. Sci Total Environ. 2024;951:175633. doi:10.1016/j.scitotenv.2024.175633
41. Chung C, Park SY, Huh J-Y, et al. Fine particulate matter aggravates smoking induced lung injury via NLRP3/caspase-1 pathway in COPD. J Inflamm. 2024;21(1):13. doi:10.1186/s12950-024-00384-z
42. Delavar MA, Jahani MA, Sepidarkish M, et al. Relationship between fine particulate matter (PM(2.5)) concentration and risk of hospitalization due to chronic obstructive pulmonary disease: a systematic review and meta-analysis. BMC Public Health. 2023;23(1):2229. doi:10.1186/s12889-023-17093-6
43. Chen Q, Nwozor KO, van den Berge M, et al. From differential DNA methylation in COPD to mitochondria: regulation of AHRR expression affects airway epithelial response to cigarette smoke. Cells. 2022;11(21):3423. doi:10.3390/cells11213423
44. Kim J, Ogata Y, Ali H, et al. The Fes tyrosine kinase: a signal transducer that regulates myeloid-specific gene expression through transcriptional activation. Blood Cells Mol Dis. 2004;32(2):302–308. doi:10.1016/j.bcmd.2003.12.004
45. Parsons SA, Mewburn JD, Truesdell P, et al. The Fps/Fes kinase regulates leucocyte recruitment and extravasation during inflammation. Immunology. 2007;122(4):542–550. doi:10.1111/j.1365-2567.2007.02670.x
46. Karamanavi E, McVey DG, van der Laan SW, et al. The FES gene at the 15q26 coronary-artery-disease locus inhibits atherosclerosis. Circ Res. 2022;131(12):1004–1017. doi:10.1161/CIRCRESAHA.122.321146
47. Shi L, Tan Y, Zheng W, et al. CTRP3 alleviates mitochondrial dysfunction and oxidative stress injury in pathological cardiac hypertrophy by activating UPRmt via the SIRT1/ATF5 axis. Cell Death Discov. 2024;10(1):53. doi:10.1038/s41420-024-01813-x
48. Chang X, Zhou S, Huang Y, et al. Zishen Huoxue decoction (ZSHX) alleviates ischemic myocardial injury (MI) via Sirt5-beta-tubulin mediated synergistic mechanism of “mitophagy-unfolded protein response” and mitophagy. Chin J Nat Med. 2025;23(3):311–321. doi:10.1016/S1875-5364(25)60838-7
49. Ren Y, Feng L, Tan Z, et al. Constructing a novel prognostic model for triple-negative breast cancer based on genes associated with vasculogenic mimicry. Aging. 2024;16(9):8086–8109. doi:10.18632/aging.205806
50. Zhu H, Zhong Z, Zhao G, et al. Serum soluble asialoglycoprotein receptor 1: a potential predictor marker linked to type 2 diabetes mellitus, demonstrating positive correlation with high sensitive c-reactive protein. Diabetes Metab Syndr Obes. 2025;18:663–675. doi:10.2147/DMSO.S511208
51. Zhang X, Fang H, Wu W, et al. LPIN3 promotes colorectal cancer growth by dampening intratumoral CD8(+) T cell effector function. Cancer Immunol Immunother. 2025;74(4):135. doi:10.1007/s00262-025-03989-2
52. Spring FA, Parsons SF, Ortlepp S, et al. Intercellular adhesion molecule-4 binds alpha(4)beta(1) and alpha(V)-family integrins through novel integrin-binding mechanisms. Blood. 2001;98(2):458–466. doi:10.1182/blood.V98.2.458
53. Ferrer MI. Cor pulmonale (pulmonary heart disease): present-day status. Am Heart J. 1975;89(5):657–664. doi:10.1016/0002-8703(75)90513-X
54. Nattel S. Basic electrophysiology of the pulmonary veins and their role in atrial fibrillation: precipitators, perpetuators, and perplexers. J Cardiovasc Electrophysiol. 2003;14(12):1372–1375. doi:10.1046/j.1540-8167.2003.03445.x
55. Heijman J, Voigt N, Nattel S, et al. Cellular and molecular electrophysiology of atrial fibrillation initiation, maintenance, and progression. Circ Res. 2014;114(9):1483–1499. doi:10.1161/CIRCRESAHA.114.302226
56. Kotlyarov S, Lyubavin A. Early detection of atrial fibrillation in chronic obstructive pulmonary disease patients. Medicina. 2024;60(3):352. doi:10.3390/medicina60030352
57. Wen X, Wu X, Deng Z, et al. The nonlinear relationship between high-density lipoprotein and changes in pulmonary structure function and pulmonary function in COPD patients in China. Int J Chron Obstruct Pulmon Dis. 2024;19:1801–1812. doi:10.2147/COPD.S467976
58. Wu R, Gong H. The association between non-high-density lipoprotein cholesterol to high-density lipoprotein cholesterol ratio and chronic obstructive pulmonary disease: the mediating role of dietary inflammatory index. Front Nutr. 2024;11:1427586. doi:10.3389/fnut.2024.1427586
59. Su Y, Ning Y, Jiang Z, et al. Blood metabolome mediates the effect of the plasma lipidome on the risk of atrial fibrillation: a mendelian randomization study. Clin Cardiol. 2025;48(3):e70112. doi:10.1002/clc.70112
60. Sulague RM, Whitham T, Danganan LML, et al. The left atrial appendage and atrial fibrillation-a contemporary review. J Clin Med. 2023;12(21):6909. doi:10.3390/jcm12216909
61. Artavanis-Tsakonas S, Muskavitch MA. Notch: the past, the present, and the future. Curr Top Dev Biol. 2010;92:1–29.
62. Di Stefano A, Gnemmi I, Rosani U, et al. Upregulation of notch signaling and cell-differentiation inhibitory transcription factors in stable chronic obstructive pulmonary disease patients. Int J Mol Sci. 2024;25(6):3287. doi:10.3390/ijms25063287
63. de la Pompa JL, Epstein JA. Coordinating tissue interactions: notch signaling in cardiac development and disease. Dev Cell. 2012;22(2):244–254. doi:10.1016/j.devcel.2012.01.014
64. Quillard T, Charreau B. Impact of notch signaling on inflammatory responses in cardiovascular disorders. Int J Mol Sci. 2013;14(4):6863–6888. doi:10.3390/ijms14046863
65. Lipovsky CE, Jimenez J, Guo Q, et al. Chamber-specific transcriptional responses in atrial fibrillation. JCI Insight. 2020;5(18). doi:10.1172/jci.insight.135319
66. Dong L, Wang Y, Chen H, et al. MTOR suppresses cigarette smoke-induced airway inflammation and MMP12 expression in macrophage in chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2024;19:269–279. doi:10.2147/COPD.S426333
67. Ebana Y, Sun Y, Yang X, et al. Pathway analysis with genome-wide association study (GWAS) data detected the association of atrial fibrillation with the mTOR signaling pathway. Int J Cardiol Heart Vasc. 2019;24:100383. doi:10.1016/j.ijcha.2019.100383
68. Jin C, Zhong Z, Gao L, et al. Focus on the role of inflammation as a bridge between ferroptosis and atrial fibrillation: a narrative review and novel perspective. Rev Cardiovasc Med. 2024;25(4):110. doi:10.31083/j.rcm2504110
69. Han MR, Jeong JH, Kim YG, et al. Epigenetic regulation on left atrial function and disease recurrence after catheter ablation in atrial fibrillation. Clin Clin Epigenet. 2024;16(1):183. doi:10.1186/s13148-024-01794-9
70. Sun Z, Yun Z, Lin J, et al. Comprehensive mendelian randomization analysis of plasma proteomics to identify new therapeutic targets for the treatment of coronary heart disease and myocardial infarction. J Transl Med. 2024;22(1):404. doi:10.1186/s12967-024-05178-8
71. Zhang X, Fan Y, Tan K. A bird’s eye view of mitochondrial unfolded protein response in cancer: mechanisms, progression and further applications. Cell Death Dis. 2024;15(9):667. doi:10.1038/s41419-024-07049-y
72. Qin Z, Kong B, Zheng J, et al. Alprostadil injection attenuates coronary microembolization-induced myocardial injury through GSK-3beta/Nrf2/HO-1 signaling-mediated apoptosis inhibition. Drug Des Devel Ther. 2020;14:4407–4422. doi:10.2147/DDDT.S272877
73. Yan X, Li Y, Choi YH, et al. Protective effect and mechanism of alprostadil in acute respiratory distress syndrome induced by oleic acid in rats. Med Sci Monit. 2018;24:7186–7198. doi:10.12659/MSM.909678
74. Monasky MM, Micaglio E, D’Imperio S, et al. The mechanism of ajmaline and thus brugada syndrome: not only the sodium channel! Front Cardiovasc Med. 2021;8:782596. doi:10.3389/fcvm.2021.782596
75. Thapanasuta M, Chokesuwattanaskul R, Leelapatana P, Rungpradubvong V, Prechawat S. T-Peak to T-End interval for prediction of positive response to ajmaline challenge test in suspected brugada syndrome patients. Med Sci. 2022;10(4):69.
76. Haverkamp W, Mönnig G, Kirchhof P, et al. Torsade de pointes induced by ajmaline. Z Kardiol. 2001;90(8):586–590. doi:10.1007/s003920170128
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.