Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 14
Epigenetic Histone Modifications in the Pathogenesis of Diabetic Kidney Disease
Received 23 October 2020
Accepted for publication 22 December 2020
Published 22 January 2021 Volume 2021:14 Pages 329—344
DOI https://doi.org/10.2147/DMSO.S288500
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Heng-Cheng Lu, Wen-Ni Dai, Li-Yu He
Department of Nephrology, The Second Xiangya Hospital, Central South University, Hunan Key Laboratory of Kidney Disease and Blood Purification, Changsha, Hunan, People’s Republic of China
Correspondence: Li-Yu He
Department of Nephrology, The Second Xiangya Hospital, Central South University, Hunan Key Laboratory of Kidney Disease and Blood Purification, 139 Renmin Road, Changsha, Hunan, People’s Republic of China
Tel +8673185292064
Fax +8673185295843
Email [email protected]
Abstract: Diabetic kidney disease (DKD), as the main complication of diabetes mellitus, is the primary cause of the end-stage renal disease (ESRD) and the most common chronic kidney disease. Overall, 30– 40% of patients with type 1 and type 2 diabetes eventually develop DKD. Although some diabetes patients have intensified glycemic control, they still develop diabetic kidney disease. Current treatment methods can alleviate but do not markedly halt disease development, resulting in renal failure and severe complications, even contributing to elevated morbidity and mortality rates. DKD is a disease with interactions of genes and the environment. Emerging evidence indicates that DKD-associated key genes are also regulated by the epigenetic mechanism. Recently, increasing researches involving cells and experimental animals demonstrated that histone post-translational modifications can mediate gene expression, which correlated with diabetic kidney disease. Novel therapeutic strategies for epigenetic events could be beneficial for the early detection and treatment of DKD to prevent it from developing into end-stage renal disease (ESRD). In this review, we discuss prior findings in the field of histone modifications in DKD, especially histone acetylation and histone methylation. We then focus on recent developments in histone acetylation and methylation involved in the pathogenesis of DKD.
Keywords: histone, epigenetics, kidney disease, diabetes, acetylation, methylation
Introduction
Diabetic nephropathy, now commonly recognized as diabetic kidney disease (DKD), occurs in diabetic patients, especially those with poor long-term glycemic control,1 causing high morbidity and risk of death. As lifestyles and diets have changed in recent years, the global incidence of diabetes mellitus (DM) has increased annually, as reported by the International Diabetes Federation, which estimates that the incidence of diabetes mellitus (DM) will increase to 700 million in 2045.2 Consequently, diabetic kidney disease will increase social financial costs and threaten human health. DKD is defined by changes in the structure and function of the kidneys. The primary renal structural changes in DKD are mesangial matrix expansion, extracellular matrix accumulation, glomerular and tubular basement membrane thickening, and podocyte injury, eventually contributing to glomerulosclerosis and tubulointerstitial fibrosis.3,4 Clinically, microalbuminuria refers to the daily amount of urinary albumin between 30 mg-300 mg, also known as incipient nephropathy.5 Persistent microalbuminuria (>300mg/day) is called macroalbuminuria or overt nephropathy, followed by renal injury, ultimately leading to ESRD.3–5 The pathogenesis of DKD is multifactorial and involves many mechanisms such as oxidative stress, metabolic disturbance, activation of the renin-angiotensin-aldosterone system (RAAS), production of inflammatory factors, profibrotic transforming growth factor-β1 (TGF-β1), and genetic susceptibility, among which genetic predisposition explains some diabetic patients who do not properly control their glycemia but do not develop DKD.4,6 While some patients with diabetes still develop DKD even if they maintain good glycemic control,7 growing evidence demonstrates the relationship between epigenetic mechanisms and gene expression associated with diabetic complications,8 stressing the role of epigenetic modifications and metabolic memory in diabetic complications. Current diabetic kidney disease treatment strategies include blood pressure control, glycemic control, weight reduction, and intensive lifestyle interventions.9 The most effective drugs for DKD treatment are the combined use of RAAS blockers and Sodium-glucose cotransporter 2 (SGL2) inhibitors.10,11 However, the implementation of these measures seems not likely to improve longer-term outcomes, for example, the reversal and reduction of DKD progression in many patients. The increasing rates of DKD suggest a further understanding of underlying mechanisms to find better novel therapies for the clinical management of DKD.12–14
Over the past decade, epigenetics has expanded rapidly in many fields and participates in the pathophysiology of DKD. Furthermore, environmental stimuli and inflammation are considered to mediate epigenetic mechanisms.15 Environmental factors, such as metabolites, contribute to the development of diabetes and DKD, which could regulate epigenetic status. That is to say, epigenetic modifications may play a critical role in DKD.12,16,17 In recent years, histone post-translational modifications (PTMs) have been reported to involve regulating DKD-related gene expression, such as connective tissue growth factor (CTGF), collagen-α1[Ι], and plasminogen activator inhibitor-1 (PAI-1).18,19 However, our knowledge of the magnitude of the relationship for histone modifications at individual gene sites with diabetic kidney disease and the underlying mechanisms is limited. In this review, we summarize current findings of histone modifications and DNA methylation in DKD, mainly highlighting the role of histone acetylation and methylation in DKD.
Re-Recognition of the Pathogenesis of DKD
Traditional Pathogenesis
The term “diabetic nephropathy” was replaced by “diabetic kidney disease” (DKD) by the Kidney Disease Outcomes Quality Initiative (KDOQI), in line with the chronic kidney disease (CKD) classification.20 Traditionally, DKD is characterized by diffuse or nodular glomerulosclerosis. DKD has traditionally been recognized as a change in hemodynamics and metabolic disturbance. These changes can activate the expression of metabolic product, cytokines, chemokines, and immune system,21,22 as well as dysregulating signaling pathways, such as the oxidative stress23–26 and profibrotic pathways.27,28 Furthermore, endothelial cell apoptosis exposed to sustained hyperglycemia could lead to kidney injury. These circulating renal factors have a cross-talk relationship to accelerate kidney damage and cause irreversible injury. DKD not only destroys the kidney itself but damages other organs, resulting in more serious diabetic comorbidities.
Novel Potential Pathogenesis
Microenvironmental changes and multidimensional pathology in DKD have updated conventional knowledge. Several different factors can enhance the pathogenic pathway initiated and maintained by hyperglycemia in the kidney. These include haemodynamic factors, including impaired autoregulation, hyperperfusion and hypoperfusion, as well as metabolic factors, including excess fatty acids, carbonyl and oxidative stress, and activation of the renin–angiotensin–aldosterone system (RAAS).29 These factors by themselves do not cause DKD but promote and enhance common pathogenic mechanisms in the presence of diabetes, including increased levels of growth factors, vasoactive hormones, cytokines and chemokines in the kidney. For example, vascular susceptibility to oxidative stress could be increased by endothelial dysfunction under high glucose condition, then endothelial dysfunction and subsequent microvascular rarefaction could also reduce blood flow, ultimately leading to hypoxia.4 The pathological manifestation of DKD is the accumulation of extracellular matrix in the glomeruli and tubules and the increased deposition of collagen, fibronectin, and laminin in the mesangial matrix, glomerular basement membrane, and tubulointerstitium.30
The conventional view is that the relationship between diabetes and mitochondrial dysfunction and complications are related to the harmful effects of hyperglycemia; however, further recognition of the stimulation of mitochondrial biogenesis and mitochondrial electron transport chain activity is more conducive to the treatment of DKD.23 Advanced technology in metabolomics, bioinformatics, and systems biology tools open a new window to find the mechanism of DKD. Similarly, considerable evidence shows that macrophage accumulation is a feature of kidney injury and it can produce pro-inflammatory factors, reactive oxygen species (ROS), and metalloproteinases, which lead to renal damage.31,32 Many studies have demonstrated that macrophages are closely associated with the decrease in the glomerular filtration rate (GFR) and histological changes.33,34 However, the mechanism of M2 macrophages how to promote renal repair and reduce the progression of DKD remains controversial, which is worthy of further study. Meanwhile, autophagy involved in the pathogenesis of diabetic kidney disease is also well described in the literature,35 which highlights the regulation of autophagy in diabetic kidney disease. Although long after attainment of glycemic control, the incidence of diabetic complications can still be induced by the exposure of target cells in memory to hyperglycemia.36–38 The Diabetes Control and Complications Trial (DCCT) indicated that patients with type 1 diabetes in the intensive glycemic control group had a lower rate and severity of complications compared with those in the conventional group,39,40 in the follow-up observational Epidemiology of Diabetes Intervention and Complications (EDIC) study, patients in the initial intensive treatment group sustained to maintain a low risk of complications relative to those on conventional therapy.40 The continuous effects of high glucose on metabolic memory are a major obstacle to the effective management of diabetic complications. Substantial evidences support the role of epigenetic mechanisms in metabolic memory, recommending to target these mechanisms which may provide a new therapy to treat the diabetic complications.
Epigenetic Modification
Epigenetic Regulation in Kidney Disease
Epigenetic modifications regulate gene expression, not the change in DNA sequences.41,42 They can occur in response to environmental stimuli, including diet, metabolic disorders, exercise, oxidative stress, inflammation, and drugs.41 These changes can be passed down to offspring, but can potentially be reversed. Epigenetic modifications include DNA methylation, histone modifications, and non-coding RNAs. At present, more than 100 kinds of modifications have been described, including methylation, acetylation, phosphorylation, sumoylation, ubiquitylation, citrullination, biotinylation, crotonylation, and ADP ribosylation.
In this report, we briefly introduce the epigenetic regulations that have been studied in kidney disease, especially DKD, and then introduce research and the latest developments in histone modification of acetylation and methylation involved in the pathogenesis of DKD. Non-coding RNA has been extensively reviewed and is beyond the scope of this study.43
DNA Methylation
DNA methylation, recognized as a “silencing” marker, involves the addition of a methyl group to the 5-position of cytosines and primarily occurs on 5ʹ-cytosines of CpG dinucleotides and to a lesser extent in non-CG contexts.44,45 The function of DNA methylation is highly dependent on the location of the CpG in the genome.46 In general, methylation of DNA at gene promoter areas can repress transcription and gene expression, whereas at gene bodies it can activate transcription and modulate alternative splicing.12
Histone Modifications
Histones are highly conserved, alkaline, positively charged proteins, that contain core histones (H2A, H2B, H3, and H4) and linker histones (H1 and H5).45,47 Similar structures in the four core histones are characterized by a conserved central motif domain and an unstructured amino-terminal tail. The unit structure components of chromatin, called the nucleosome, consists of DNA wrapped around an octamer of histone proteins, which contains an H3-H4 tetramer and two H2A-H2B dimers.48 The post-translational modification (PTM) of histones is the main mechanism to regulate chromatin structure, commonly occurring on the amino acid residues lysine, arginine, serine, tyrosine, and threonine and ultimately influencing transcriptional activity.49 The enzymes that mediate histone modification including acetyltransferases, methyltransferases are called epigenetic writers; deacetylases and demethylases are called epigenetic erasers; and proteins recognizing acetylated proteins at promoters and enhancers are called epigenetic readers (for example, bromodomain-containing protein 4).50–52
Post-translational modifications (PTMs) of histone proteins include lysine acetylation (KAc), lysine methylation (Kme), arginine methylation, serine ubiquitylation, and threonine phosphorylation. To date, lysine (K), arginine (R) in both methylation and acetylation are the most widely researched, and phosphorylation, ubiquitination, and sumoylation have also been described.
Histone Acetylation
Histone acetylation involves histone acetyltransferases (HATs) transferring an addition of an acetyl group to the lysine of core histones. Generally, histone acetylation is enriched at promoters and enhancers of actively transcribed genes but decreased in the suppressed genomic region. As acetyl can reduce the negative charge of DNA and make chromatin more easily accessible to transcription factors (TFs) and their coactivators,53 acetylation of lysine residual histones is usually associated with transcriptionally active genes. Three main HATs families include Gcn5-related N-acetyltransferases (GNATs: GCN5 and PCAF), MYST (MOZ and Ybf2/Sas3), Sas2, Tip60, and p300/CREB-binding protein (CBP). Conversely, histone deacetylation is catalyzed by histone deacetylases (HDACs), and histone deacetylation can regulate transcriptional repression by allowing chromatin compaction. Acetyl groups in lysine residues of histones and non-histone proteins can be removed by HDACs.54 Four classes of HDAC have been identified, Class I (HDACs 1, 2, 3, and 8), Class II (HDACs 4, 5, 6, 7, 9, and 10), Class III (SIRT1-7), and Class IV HDAC (HDAC11), which shares similar conserved residues with Classes I and II HDACs. Class I HDACs are expressed ubiquitously, mainly localizing in the nucleus, and demonstrate high enzymatic activity, whereas Class II HDACs show different expression patterns with tissue-specific roles and localize in the nucleus and cytoplasm.63 Classes, I, II, and IV are dependent on Zn2+, whereas Class III HDACs require NAD+ as a co-factor rather than Zn2+ for their enzymatic functions.55 HDAC inhibitors are ineffective against Class III (SIRT1-7). In general, H3K9ac, H3K14ac, H3K18ac, H3K23ac, and H3K27ac are enriched at promoters of transcriptional genes and contribute to gene expression. The association of histone acetylation in DKD progression has been verified in human renal tissues.56–58 H3K9 acetylation (H3K9ac) levels were significantly increased in renal biopsies from patients with DKD.56 CHIP assays obtained from the glomeruli of diabetic mice compared with normal conditions in vivo exhibited increased induction of Pai1 (as pro-fibrotic genes) and p21 were related to the enrichment of H3K9ac and H3K14ac at the two gene transcriptional promoters.59 Overexpression of H3K9/14Ac levels was reported at the CTGF, PAI-1, and FN-1 promoters in kidneys of diabetic mice, which were associated with p300/CBP-mediated histone acetylation.60 The antidiabetic agent metformin, is widely used as a first-line treatment for patients with type 2 diabetes mellitus, and it has been reported metformin improves glucose metabolism by stimulating CBP phosphorylation, then triggers the dissociation of the CREB-CBP-TORC2 transcription complex, leading to reducing gluconeogenic enzyme gene expression.61 A recent study showed that STZ-induced diabetic mice kidney could reduce acetylation of nephrin, while reduction of nephrin acetylation may be alleviated by MicroRNA-29a, thus protecting against hyperglycemia-induced podocyte dysfunction.62 In another study, high glucose-induced hyperacetylation of the redox-regulating protein p66Shc promoter in podocytes with diabetic kidney disease increased protein p66Shc expression. Protease-activated protein C (aPC) reversed hyperacetylation of the p66Shc promoter and decreased mitochondrial ROS formation in podocytes.63 Taken together, these studies indicate an important role of histone acetylation in kidney diseases.
Histone Methylation
Histone methylation mainly occurs on the amino acid residues of lysine and arginine. Unlike histone acetylation, histone methylation functions as an information marker to store rather than change the charge of histones to disturb its contact with DNA.64 Histone methylation has three different forms, mono-, di-, or trimethyl, for lysine or arginine residues, which increases the complexity of PTMs. Therefore, either gene activation or repression in histone methylation is determined by the extent of methylation as well as different residues modified.65 Generally, H3K4me1/2/3, H3K36me2/3, and H3K79me1/2 are related to transcriptionally active gene regions, whereas H3K9me2/3, H4K20me3, and H3K27me3 are associated with repressive gene regions. H3K4me2 andH3K4me3 are fully enriched at transcriptional promoters, leading to gene expression. In contrast, H3K9me3 and H3K27me3 are enriched at inactive or slicing gene promoters, which could inhibit gene expression.65,66 Lysine methylation (Kme) is catalyzed by histone methyltransferases (HMTs). However, histone lysine demethylases remove methyl groups from histones, resulting in the demethylation of histones. The first histone demethylase was lysine methylase 1 (LSD1), which could specifically remove the methylation of H3K4 and H3K9.67,68 Many lysine demethylases were identified and renamed lysine demethylases (KDMs) due to their different specificity to various histone lysine residues and non-histone proteins.69–71 Given the high selectivity of these enzymes to targeted histone residues, HMTs are classified as two types: arginine methyltransferases (PRMTs) and lysine methyltransferases (KMTs). Lysine methyltransferases (KMTs) include two families based on the catalytic sequence, one is the SET domain-containing KMTs (Su(var)3–9, enhancer of zeste and trithorax), and the other is a non-SET domain, for example, DOT1L. Histone methylation is dynamic, reversible modification that is regulated by HMTs and KDMs.17 Interestingly, HMTs and KDMs possess substrates specific for lysine residues. For example, methylation of H3K36 is specifically mediated by SET2, and the demethylation of trimethyl H3K36 andH3K9 is regulated by JHDM3/JMJD (trimethyl demethylases), whereas H3K36me2 and H3K36me1 are only demethylated by JHDM1A instead of JHDM3/JMJD. Similarly, methylation of H3K27 and H3K79 is mediated by EZH2 and DOT1, respectively.64 Histone methylation has been considered one of the most stable PTMs and considerable evidence has demonstrated that histone methylation plays a key role in contributing to the pathogenesis of DKD in preclinical in vivo and in vitro models. Because HMTs are involved in histone methylation, some of the targeted HMTs, small-molecule modulators, have been used to test the therapeutic effects in experimental kidney disease. Interestingly, in type 1 diabetic rat kidney, a decreased level of H3K9me2 on the Col1a1 gene promoter was observed, accompanied by decreased expression of SUV39H1 (a histone methylase, specifically catalyzed H3K9me2/3), which is involved in the development of diabetic renal fibrosis. In diabetic conditions, decreased expression of SUV39H1 HMTs was also demonstrated by Villeneuve et al72 and Chen et al.73 Additionally, decreased histone H3K9me3 levels at the promoters of some pro-inflammatory or pro-fibrotic genes also contributed to the development of DKD.58,74,75 Lin et al found HG decreased histone H3K9me3 levels at the promoters of the fibronectin and p21WAF1 genes in mesangial cells while accelerating HG-induced cell hypertrophy, which was attenuated by Suv39h1 overexpression.76 A recent study of DKD patients showed the overexpression of SUV39H1 and H3K9me3, with reduced renal inflammation and apoptosis, suggesting that SUV39H1 may be a protective target for the treatment of DKD.77 Further research should be conducted to test whether the overexpression chromatin marker, such as H3K9me3, could be used as a marker indicating the progression of DKD. In two rodents models of type 1 diabetes, OVE26 mice and streptozotocin rats, the levels of H3K4m2, a histone methylation activating mark, are increased, while the levels of H3K27m3, a repressive mark, are reduced in key genes, such as Mcp-1, vimentin and the fibrosis marker Fsp150, suggesting differential kidney gene expression in DKD is associated with aberrant histone methylation.
Histone Crotonylation
Histone crotonylation consists of the transfer of crotonyl groups to lysine residues of histones, that similar to acetylation, confers histones with negative charge.51,52 Lysine crotonylation (Kcr) has been considered as the conserved histone post-translational modification in the kidney.51 Nonetheless, the genomic pattern of histone crotonylation differs from histone acetylation.51 Histone crotonylation can activate or repress gene transcription in a gene- and/or environment-dependent manner.51 Recently, a potential role of histone Kcr was described in acute kidney injury (AKI).78 Most recently, the protective part of HDAC inhibitors in kidney diseases may be related to their role in crotonylation regulation, which could promote some nephroprotective genes, such as PGC1α and Sirt-3.79 This opens the door to explore therapeutic strategies based on the modulation of histone crotonylation.
Histone Ubiquitination
Ubiquitin is a small, highly conserved 76 amino acid protein that can be covalently linked to lysine residues on histone and non-histone target proteins;80 And it targets the proteins for degradation through the ubiquitin proteasome system (UPS). Ubiquitination involves a multistep process mediated by an enzymatic cascade with ubiquitin ligases, including E1 activating enzymes, E2 conjugating enzymes, and E3 ubiquitin ligases. Studies found ubiquitination is widely involved in the occurrence of DN.81 The key step in Nuclear factor-kappa B (NF-κB) activation is through ubiquitination of IκB and NF-κB dissociation, which plays a vital role in the expression of inflammatory cytokines related to DN. As previously reported, ubiquitination is involved in the progression of DN through activating NF-κB, TGF-β by degrading the related signal proteins. Most recently, a study in vitro and vivo suggested the tripartite motif-containing (TRIM13, a well-defined E3 ubiquitin ligase) promoted ubiquitination and degradation of C/EBP homologous protein (CHOP, associated with renal injury), which attenuated DN-induced collagen synthesis and restored renal function.82 This finding provides new insights into the application of histone ubiquitin in the treatment of diabetic nephropathy. Based on existing literature and studies, additional research is required to expose the hidden targets of histone ubiquitination to prevent DN.
Histone Phosphorylation
Nephrin, a critical podocyte membrane component, has been shown to activate phosphotyrosine signaling pathways in human podocytes, then reduce cell death induced by apoptotic stimuli. High glucose and diabetes result in upregulation of SH2 domain-containing phosphatase 1 (SHP-1) in podocytes, thereby contributing to nephrin dephosphorylation and podocyte apoptosis.83 Additionally, an increased level of SHP-1 was also found in diabetic mice, causing decreased nephrin phosphorylation, which may lead to diabetic nephropathy. Another enzyme, Nicotinamide adenine dinucleotide phosphate oxidase (NOX) is the source of reactive oxygen species in hyperglycaemia; Especially when phosphorylation of the cytosolic components of NOX, the development of oxidative stress worsens the kidney in a series of stages.84 Accordingly, investigating phosphorylation targets may benefit patients with diabetic kidney disease.
Histone Acetylation and Methylation Participate in the Regulation of Diabetic Kidney Disease
Some studies investigated in peripheral blood cells have included histone modifications in type 2 diabetics.85,86 Additionally, histone modification variations have been shown in human monocytes cultured under high glucose at a genome-wide level.87 However, plasma levels may not reflect their status in tissues and the cell nucleus.88 Therefore, further epigenome-wide studies in tissues from T2D patients are needed, especially in single-cell analyses. In some cases, histone acetylation and methylation are in a similar pattern and it is difficult to discern the specific contribution of each histone modification to gene expression differences.57 Although the overall profile of histone methylation in DKD has not been fully described, there is still information on individual modifications and genes.
Diabetic kidney disease is one of the major complications caused by persistent hyperglycemia.85,89 Inflammation and fibrosis are the two main factors implicated in the development of DKD. In this section, we focus on the roles of histone acetylation and methylation in the regulation of inflammation and fibrosis in DKD. Generally, ROS is considered to activate nuclear factor-kappaB (NF-κB), resulting in a series of inflammation responses.90 NF-kB, a transcription factor, is involved in diabetic complications. Bierhaus et al91 confirmed that hyperglycemia induces activation of NF-kB, then activates its downstream target molecules such as adhesion molecules (monocyte chemoattractant protein-1 [MCP-1]),92 also known as chemokines CCL2, which participate in the pathogenesis of DKD. Pro-inflammatory cytokines and adhesion molecules are also activated by NF-kB. ROS-mediated inflammatory signaling regulated by lysine methyltransferase SETD7 was observed in an experiment conducted by He et al.93 Previous work has demonstrated that SET9 promotes ECM deposition in fibrosis.94,95 SET9 is shown to be recruited to the α-SMA gene, and SET9 inhibition to treat CKD.96 In diabetic mice, high-glucose conditions increased the expression of Set7 and NF-κB; both were related with elevated ROS production.97 These findings suggest that histone modifications mediated by ROS are involved in the inflammatory reaction of DKD. Likewise, high blood glucose levels (>15 mM) of stimuli such as TGF-β have been implicated in the pathogenesis of DKD due to the adverse influence in renal cells.98–100 A body of evidence has shown that TGF-β-mediated histone modifications are correlated with the development of DKD.101−104
Histone Acetylation Involves Renal Fibrosis of DKD
Many studies have demonstrated that under high glucose and TGF-β1-induced conditions, profibrotic cytokines associated with diabetic nephropathy can be regulated by histone acetylation. Significant induction of PAI-1 and p21 mRNA in TGF-β1 treatment of RMCs was associated with elevated H3K9/14Ac levels and overexpression of CREB-binding protein (CBP) or p300 at PAI-1 and p21 promoters. Meanwhile, high-glucose treatment increased H3K9/14Ac at TGF-β1-inducible genes PAI-1 and p21 (the key players in DN) in rat renal mesangial cells. Furthermore, increased expression of PAI-1 and p21 in glomeruli from diabetic mice was also associated with elevated levels of promoter H3K9/14Ac, demonstrating abnormal histone acetylation in gene regulation both in vivo and vitro relevance to DN. A previous experiment in human renal proximal tubular epithelial cells (RPTEC) showed that epithelial-to-mesenchymal transition (EMT) induced by TGF-β1 could be suppressed by TSA, an HDAC inhibitor.103
CHIP assays from glomeruli of diabetic mice also showed that increased expression of PAI-1 and p21 was related to the enrichment of H3K9/14Ac at their gene promoters.59 Conversely, co-transfection experiments confirmed that the overexpression of HDAC1 and HDAC5 could suppress TGF-β-induced gene expression (PAI-1 and p21). These studies suggest a key role of histone acetylation in the pathogenesis of gene expression and provide further therapeutic targets for DKD. In another study with a type 1 diabetes mouse model, significantly increased levels of connective tissue growth factor (CTGF), plasminogen activator inhibitor (PAI-1), and fibronectin (FN-1) in the kidney were related to increased HAT activity and enrichment of H3K9/14Ac and HAT p300/CBP at the CTGF, PAI-1, and FN-1 gene,60 suggesting a relationship between histone acetylation and renal fibrosis, which may provide a precise mechanism of glomerulosclerosis and interstitial fibrosis to prevent the development of DKD. It is well established that DKD is characterized by the accumulation of extracellular matrix (ECM) proteins including collagen, laminin, and fibronectin. Some evidence shows that the transcription of ECM proteins is regulated by epigenetic histone modifications. Fibroblasts incubated with TGF-β revealed that elevated histone acetyltransferase activity of p300 and histone H4 acetylation accelerated COL1A2 expression.105 Interestingly, research in mice with diabetic kidney disease showed that high glucose induces the expression of myocardin-related transcription factor A (MRTF-A), which could activate collagen transcription. Further analysis revealed that MRTF-A recruited p300 and WD repeat-containing protein 5 (WDR5), an important component of histone H3K4 methyltransferase, to the collagen promoters, eventually leading to its gene expression.106 MRTF-A silencing makes acetylated histone H3K18/K27 and trimethylated histone H3K4 disappear and diminishes diabetic tubulointerstitial fibrosis.107 This study indicates that MRTF-A-associated histone modifications might provide a novel mechanism against DN-associated renal fibrosis. In another study, mice with streptozotocin-induced diabetic kidney disease showed low expression of the histone deacetylase SIRT1, and increased albuminuria. However, overexpression of SIRT1 in renal tubular could induce hypermethylation of the Cldn1 gene (the tight junction protein) and then prevented albuminuria.108 On the other hand, proximal tubule-specific deletion of Sirt1 in mice showed increased albuminuria related with reduced Cldn1 methylation, increased histone acetylation, and upregulation of Claudin-1.108 Thus, it can alleviate renal fibrosis with reduced albuminuria. These studies show the protective effects of SIRT1 in DKD and can further as a therapeutic target for DKD with in-depth evaluation. In rat DKD, the HDAC inhibitors trichostatin A (TSA) and valproic acid (VPA) were protective. TSA blocked extracellular matrix accumulation by TGF-β1-induced. And it is thought to increase E-cadherin expression through HDAC inhibition, leading to increased acetylation of E-cadherin promoter, but it is unclear the mechanism on TGF-β1 expression.102 HDAC 2/4/5/9 have been shown to be upregulated in kidney biopsy tissue obtained from patients with diabetes. More specifically, the mRNA level of HDAC2/4/5 was negatively correlated with eGFR in patients with DKD. Noh et al102 also reported that markedly increased HDAC-2 activity was proved in kidneys of diabetic rats and rat tubular epithelial cells. New observations demonstrate diabetes and TGF-β1 could activate HDAC-2 in the kidneys, eventually involving the accumulation of ECM and EMT, and EMT has been reported to cause podocyte loss.109–111 Podocytes stimulated by harmful factors such as high glucose, advanced glycation end products, and transforming growth factor-β showed high expression of HDAC4. However, renal injury was alleviated by silencing the HDAC4 gene.112 After in-depth studies, Wang et al reported that inhibition of autophagy and increasing renal inflammation were associated with the effects of HDAC4112. In vivo gene silencing of HDAC4 ameliorated renal injury in STZ-induced diabetic rats, as evidenced by reduced albuminuria, ameliorated podocyte injury and mesangial expansion.113 Hence, HDAC4 plays a critical role in regulating the pathogenesis of DKD as an epigenetic mediator. Cultured murine proximal tubular cells treated with TGF-β1 also showed protective effects through treatment with PCI34051 (a highly selective inhibitor of HDAC8) or HDAC8 siRNA, suppressing EMT.114 All of the above indicates that HDAC changes influence the progression of DKD, which may promote renal fibrosis and EMT. In view of individual HDAC isoforms playing different roles, additional studies are needed to clarify the relationship between renal fibrosis and histone acetylation and deacetylation, further giving a therapeutic target for individual patients with DKD.
Histone Acetylation Involves Renal Inflammation in DKD
In the Diabetes Control and Complication Trial (DCCT), the progress of microvascular results in the long-term Epidemiology of Diabetes Intervention and Complication (EDIC) studies showed that the hyperacetylation promoter (P < 0.05) in the top 38 cases contained more than 15 genes related to the NF-κB inflammatory pathway and rich in genes related to diabetic complications.114 Preliminary work in endothelial cells showed that the sustained expression of p65 was associated with enrichment in Set7 and H3K4me1 on the p65 gene promoter, although cultured in transient hyperglycemia, and changes will always exist, even if returning to normoglycemia.71,115 The study highlighted that short-term hyperglycemia could have long-lasting effects on gene expression through epigenetic histone modification. Evans et al116 reported that stress-activated protein kinases stress pathways such as the NF-κB, p38 MAPK, and kinases resulted in late diabetic complications. The first study of human blood monocytes showed that, under diabetic conditions, increased levels in acetylation of histones H3K9/14Ac and H4K5, 8, and 12Ac at the promoters of inflammatory genes such as TNF-α and COX-2 led to gene transcription.117 Another study in advanced diabetic kidney disease mice after unilateral nephrectomy showed significantly increased global renal histone H3K9 and H3K23 acetylation, whereas CCL2 antagonist not only reversed histone acetylation abnormalities but also alleviated the progression of diabetic kidney disease.57 These studies demonstrated that histone acetylation under diabetic conditions is involved in the pathogenesis of DKD, which was associated with the continuous expression of the inflammatory gene. HDAC1 is downregulated both in Akita mice and in rat glomerular mesangial cells exposed to high glucose, resulting in overexpression of inflammatory gene through histone hyperacetylation.118 In another study, elevation of RNA polymerase II recruitment and H3K4me2 was found but decreased repressive H3K27m3 markers at the MCP-1 gene were observed in an OVE26 mice model of T1DM instead of in rat models, which eventually contributed to the increased expression of MCP-1 in a mouse model.119 The difference between rats and mice suggests that individual differences in epigenetics also need to be taken into account when translating into human DKD.
Histone Methylation Involves Renal Fibrosis of DKD
Under normal and high-glucose conditions, histone methylation is associated with TGF-β-1-mediated ECM gene expression, such as Colla1, plasminogen activator inhibitor-1 (PAI-1), and connective tissue growth factor (CTGF).120–123
TGF-β1 plays an important role that drives collagen myofibroblasts in injured kidneys and their signaling is also a key mediator in the expression of fibrotic and ECM genes involved in the pathogenesis of diabetic kidney diseases. In models of TGF-β1-induced renal fibrosis, hypermethylation of Rasal1 promoter was induced by TGF-β1, which increased fibroblast activation, and fibrosis.108 In another study of 18 in RMCs stimulated by TGF-β1 showed not only the increasing recruitment of H3K4, HMT, and SET7/9 at ECM gene promoters but also in the expression of SET7/9. However, knockdown of SET7/9 could decrease global H3K4me1 but not H3K4me2 or H3K4me3 levels, indicating SET7/9-mediated H3K4me1 could play an important role in ECM gene expression. Involvement of histone methyltransferase (HMT) SET7/9 in p21 gene expression related to cellular hypertrophy, which results in the pathogenesis of DN, has also been demonstrated both in glomeruli of STZ-induced rats and HG-induced RMCs.124 These studies suggest that SET7/9 could participate in renal fibrosis by regulating methylation of H3 lysine 4 at fibrotic gene promoters. Then, SET7/9 may be a potential target for fibrotic gene disorders, which could provide potential therapeutic targets for DKD.
H3K9me3 levels in vascular smooth muscle cells (VSMCs) and endothelial cells in diabetic db/db mice were lower than those in a control db/db group.125,126 They were consistent with the result of a study of RMCs stimulated by TGF-β1, which were related to HG-induced upregulation of these fibrotic genes. It was found that in patients with diabetic kidney disease, H3K9me3 overexpression in renal tubules has been verified as a protective role by decreasing renal inflammation and apoptosis. Accordingly, more details should be researched in DKD with the methylation of H3K9 for new insights to delay the progression of DKD. Loss of H3K27me3, EZH2, and heightened UTX (also known as KDM6A, a histone demethylase) were detected in the human podocytes in glomeruli of DKD, which increased podocyte dedifferentiation and aggravated glomerular injury by regulating Jagged-1 overexpression.127 Increased expression of H3K27me3 demethylases accompanied by decreased levels of Ezh2 protein and H3K27me3 were observed in rodent models with diabetic kidney disease. In the same experiment, RMCs stimulated by TGF-β showed that the reduction of H3K27me3 at the CTGF, Serpine1, and CCl2 gene promoters upregulated profibrotic and inflammatory gene expression.101 Moreover, in streptozotocin-diabetic rats and in podocytes cultured under a high glucose with the inhibition of EZH reduced H3K27me marks at the Pax6 promoter, and then promoting PAX6 expression and aggravating podocyte injury, oxidative stress and proteinuria.128 However, another report showed that high-glucose stimulation promoted EMT and significantly up-regulated EZH2 expression in renal tubular epithelial cells, which may participate in the development of DN.129 The existence of the above biases may be related to the balance of histone methylation in epigenetic processes. SUV39H1, another histone methyltransferase as repressive mark H3K9m3, has been observed downregulated in kidneys from streptozotocin mice and in mesangial cells under high glucose. Interestingly, overexpression of SUV39H1 decreased extracellular matrix production in mesangial cells, reducing the renal fibrosis.76,130
Histone Methylation Involves Renal Inflammation in DKD
In experimental diabetic nephropathy, histone methylation was associated with severe glomerulosclerosis, albuminuria and glomerular filtration rate reduction and CCL2 antagonist prevented the histopathological damage, indicating a role for CCL2 or inflammation in epigenetic regulation.57
In vitro, endothelial cells cultured in transient hyperglycemia showed that an increase in p65 expression was correlated with elevated levels of H3K4me1 and SET7 at the p65 promoter, but there was no change in H3K4me2/3.115,126 In the same study, EI-Osta et al115 indicated that sustained increase in monocyte chemoattractant protein 1 (MCP-1) and vascular cell adhesion molecule 1 (VCAM-1) were induced by activation of p65, both involved in the pathological process of DKD. HMT SET7 (the H3K4 methyltransferase) promotes the expression of inflammatory genes such as TNF-alpha in monocytes, which is a downstream inflammatory factor regulated by NF-κB.131 These findings revealed the key role of HMT involved in mediating the expression of inflammatory genes related to DKD. Diminished H3K27me3 and increased expression of UTX were observed in glomerular podocytes from humans with glomerulosclerosis or DKD, whereas inhibition of UTX alleviated the established glomerular injury in db/db mice; UTX was recently found overexpressed in DKD patients and an elevated level of UTX and reduced H3K27me2/3 were also observed in db/db mice. Further studies to reveal the role of UTX in DKD showed that the transcriptional activation of inflammatory genes is mediated by UTX through removing H3K27me3 from these gene promoters.131 These results suggest the pathogenesis of DKD is associated with histone methylation and inflammation in the process could be regulated by histone methylation. Although TGF-β is considered the key cytokine in contributing to fibrosis,132 the suppressed expression of matrix metalloproteinase 9 (MMP9) could also alleviate pathological conditions of early diabetic kidney disease,133 such as mesangial expansion, proteinuria, and podocyte foot effacement in the early stage of diabetic kidney disease. The decreased expression of lncRNA growth arrest-specific transcript5 (GAS5) and matrix metalloproteinase 9 (MMP9), a key inflammatory protein associated with DKD pathogenesis, was shown in a rat model with diabetic kidney disease. ChIP assays demonstrated that dramatic enrichment of EZH2 (a methyltransferase that induces histone H3 lysine 27 trimethylation) after overexpression of GAS5 could also elevate H3K27me3 as well as EZH2 to the promoter region of MMP9, leading to downregulated MMP9 expression and attenuating early diabetic kidney injury.134 The experimental data provide a promising target for DKD in the GAS5/MMP9 regulatory mechanism. EZH2 treatment can also impede the progression of DN. However, elevated recruitment of RNA polymerase II and H3K4me2 but decreased repressive H3K27m3 markers at MCP-1 genes were observed in a mouse model of T1DM instead of in male Sprague-Dawley rat models, which eventually contributed to increased expression of Monocyte chemoattractant protein-1 (MCP-1) in a mouse model.119 The difference between rats and mice suggests that individual differences in epigenetics also need to be taken into account when translating into human DKD.
Histone Methylation and Acetylation Interactions in DKD
A single histone modification does not always function in isolation. There is a complex interplay of indistinct histone modification. As previously mentioned, increased expression of pro-fibrotic genes (Colla1, PAI-1, and CTGF) induced by TGFβ1 and high glucose were enriched with active histone methylation markers, H3K4me, and histone acetylation markers H3K9/14ac at their promoters, hence their co-modifications may lead to more transcription of pro-fibrotic genes. Similarly, H3K18/K27ac and H3K4me3 recruited to the collagen promoters participate jointly in renal fibrosis of DKD.107 In other DKD experimental mice, increased histone acetylation (H3K9 and H3K23) and histone methylation (H3K4 dimethylation) were associated with progressive glomerulosclerosis,57 so these co-expressed chromatin markers can provide a useful signal to indicate advanced diabetic nephropathy, which could support clinicians to provide individualized treatment for patients. Considerable evidence has suggested that injured kidneys increased the expression of DOT1L and H3K79 dimethylation, particularly in renal tubular epithelial cells and myofibroblasts, eventually aggravating renal fibrosis, even developing end-stage renal disease. However, emerging evidence by Zhang et al135 showed that Dot1l has an antifibrotic effect. Using several approaches in groups of mice, Dot1a-HDAC2 complex regulated H3K79me2 and H3 acetylation at the endothelin 1 (Edn1) promoter, ensuring the balance of endothelin transcription. This could represent a new mechanism between Dot1a and HDAC2 in modulating kidney fibrosis.
Translating Histone Modifications Findings in DKD
Some epigenetic drugs focus on cancer, neuronal diseases, hematological diseases and inflammatory disease,136,137 such as HDAC inhibitors.138 HDAC inhibitors were used for the modulation of insulin signaling and β-cell functioning,139,140 as it could release the glucose transporter 4, GLUT4,141 and then transfers the glucose from the outside cell to the inside of the cell, avoiding producing a series of harmful factors to the kidney. The class III HDAC protein, SIRT1, plays a protective role in the upregulation of the antioxidant gene in glomerular mesangial cells.142 In diabetic OVE26 mice, administration of the SIRT1 agonist BF175 attenuated podocyte injury.143 However, HDAC inhibitors were generally non-specific; Hence, it may be valuable to develop more selective inhibitors or activators of HDACs. Meanwhile, considering safety and specific populations that may benefit from epigenetic interventions, there is not yet enough information. Accordingly, more clinical trial stages should be target epigenetic modifications in DKD. The most tough and unresolved but critical issue is to define the tissue-specific relative contributions of epigenetic writers and erasers; It has been shown HDAC3 deletion from the macrophage is vasculo-protective,144 while deletion of the HDAC3 from endothelial cells aggravates the macrovascular disease.145 Recently, whereas HDAC9 is a protective target for Medial artery calcification (MAC) in CKD patients,146 overexpression of the same enzyme in diabetic nephropathy exacerbates podocyte injury.147 In the future, Genome editing via CRISPR–Cas9 and other methods148–151 will be a strong method to modify epigenetic changes, benefiting from its locus-specific epigenetic modification, eventually improve the efficacy of pharmacological therapy.
Summary and Future Perspectives
The pathogenesis of DKD is complicated with interactions between injury factors, growth factors/cytokines, and metabolic products. The epigenetic mechanism can integrate these connections to mediate the development of DKD. With accumulating investigations in both animals and renal cells, as well as the data from clinical diabetes patients, as previously described, we can conclude that “metabolic memory” exists in DKD. Histone modifications and DNA methylation participate in DKD-associated gene expression, including fibrotic and inflammatory genes (Figure 1). The epigenetic mechanism provides an insight to thoroughly understand the DKD mechanism. Cellular heterogeneity and individual differences were found in histone modifications, so the cell-type-specific gene expression affected by histone modifications makes it difficult to identify the stage of DKD progression in clinical patients, even using epigenome-wide association studies (EWAS). Overall, HDAC inhibitors may protect against fibrosis in diabetic kidney disease. However, this field remains challenging, since the enzymes have a broad substrate specificity and deacetylate many proteins that are not related to epigenetic regulation.136 Although these data indicate that histone acetylation and methylation may play a key role in altered gene expression during DKD, it is more necessary to specifically target individual enzymes function in vivo. The detailed analysis of DKD between histone modifications requires more in-depth research. For example, high glucose may induce more transcriptional factors that regulate DKD-associated genes with epigenetic histone modifications. Understanding the regulation of fibrosis by TGF-β can help identify more potential antifibrotic targets, which could prevent or halt renal fibrosis in DKD. Exploring the precise pathological mechanisms in epigenetic histones and novel biomarkers is necessary to translate these preclinical findings into treatment strategies for DKD patients. Also, when single-cell epigenetic techniques are developed and together with currently available single-cell transcriptomics data, it can pinpoint the effect of certain histone post-translational modifications to a specific cell type or a specific molecule.
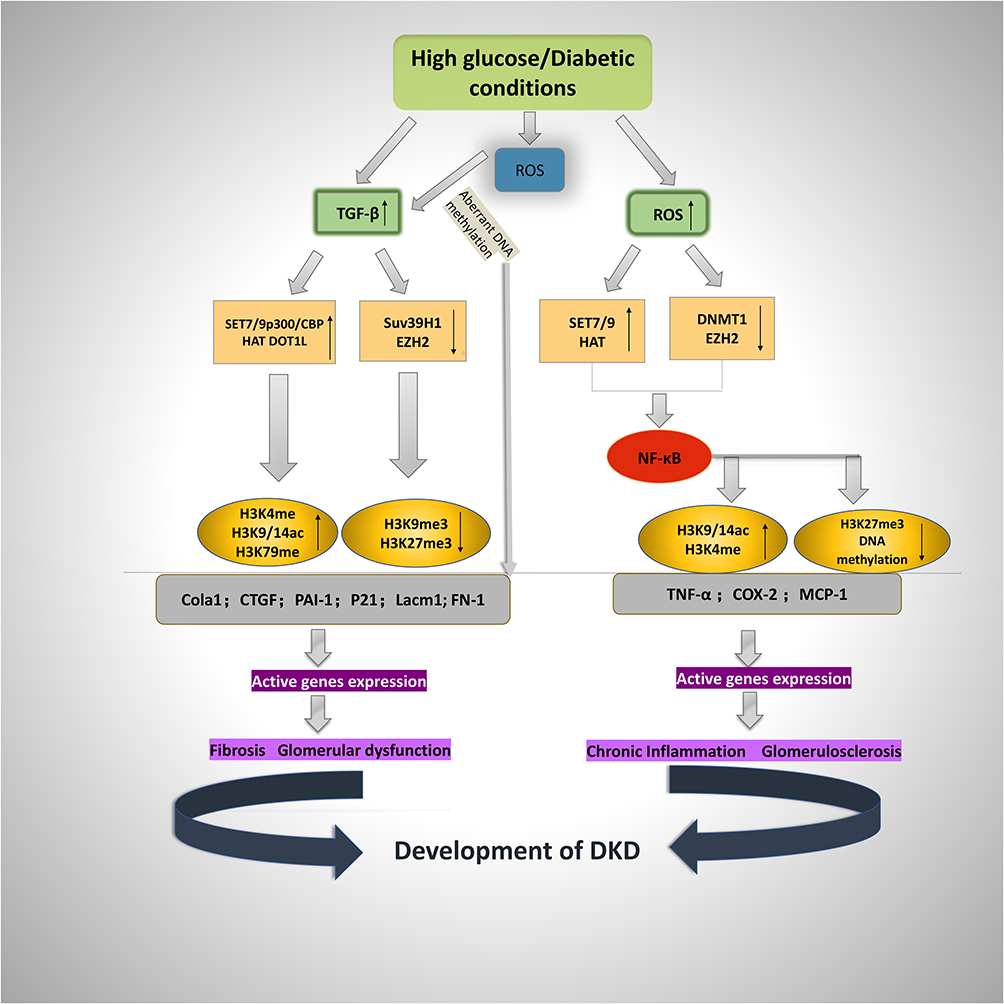 |
Figure 1 Schematic representation of histone modifications in diabetic-induced fibrotic and inflammatory gene expression. High-glucose conditions cause the expression of ECM-associated genes Col1a1, CTGF, PAI-1, FN-1, Lacm1, and P21 as well as inflammatory genes TNF-α, COX-2, and MCP-1, leading to fibrosis and glomerulosclerosis in the pathogenesis of DKD. The gene expression is based on the increased active chromatin markers (H3K4me, H3K9/14ac, and H3K79me) and decreased repressive markers (H3K9me3 and H3K27me3) on the promoters of fibrotic and inflammatory genes. Under diabetic conditions (high glucose), TGF-β antibodies, some specific HDACs, TSA, and antioxidants could have renoprotective effects. |
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This study was funded by research grants (81870500 and 81770692) from the National Natural Science Foundation of China. It was also supported by the Hunan Provincial Clinical Medical Technology Innovation Guide Project (S2020SFYLJS0334).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gross JL, de Azevedo MJ, Silveiro SP, Canani LH, Caramori ML, Zelmanovitz T. Diabetic nephropathy: diagnosis, prevention, and treatment. Diabetes Care. 2005;28(1):164–176. doi:10.2337/diacare.28.1.164
2. Saeedi P, Petersohn I, Salpea P, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the International Diabetes Federation Diabetes Atlas, 9(th) edition. Diabetes Res Clin Pract. 2019;157:107843. doi:10.1016/j.diabres.2019.107843
3. Anders HJ, Huber TB, Isermann B, Schiffer M. CKD in diabetes: diabetic kidney disease versus nondiabetic kidney disease. Nat Rev Nephrol. 2018;14(6):361–377. doi:10.1038/s41581-018-0001-y
4. Thomas MC, Brownlee M, Susztak K, et al. Diabetic kidney disease. Nat Rev Dis Primers. 2015;1:15018. doi:10.1038/nrdp.2015.18
5. Mogensen CE, Christensen CK, Vittinghus E. The stages in diabetic renal disease. With emphasis on the stage of incipient diabetic nephropathy. Diabetes. 1983;32(Suppl 2):64–78. doi:10.2337/diab.32.2.s64
6. McKnight AJ, McKay GJ, Maxwell AP. Genetic and epigenetic risk factors for diabetic kidney disease. Adv Chronic Kidney Dis. 2014;21(3):287–296. doi:10.1053/j.ackd.2014.03.010
7. Thomas MC. Glycemic exposure, glycemic control, and metabolic karma in diabetic complications. Adv Chronic Kidney Dis. 2014;21(3):311–317. doi:10.1053/j.ackd.2014.03.004
8. Zhang L, Chen B, Tang L. Metabolic memory: mechanisms and implications for diabetic retinopathy. Diabetes Res Clin Pract. 2012;96(3):286–293. doi:10.1016/j.diabres.2011.12.006
9. Doshi SM, Friedman AN. Diagnosis and management of Type 2 diabetic kidney disease. Clin J Am Soc Nephrol. 2017;12(8):1366–1373. doi:10.2215/cjn.11111016
10. Zou H, Zhou B, Xu G. SGLT2 inhibitors: a novel choice for the combination therapy in diabetic kidney disease. Cardiovascular Diabetology. 2017;16(1):65. doi:10.1186/s12933-017-0547-1
11. Feng Y, Huang R, Kavanagh J, et al. Efficacy and safety of dual blockade of the renin-angiotensin-aldosterone system in diabetic kidney disease: a meta-analysis. Am J Cardiovasc Drugs. 2019;19(3):259–286. doi:10.1007/s40256-018-00321-5
12. Kato M, Natarajan R. Diabetic nephropathy–emerging epigenetic mechanisms. Nat Rev Nephrol. 2014;10(9):517–530. doi:10.1038/nrneph.2014.116
13. Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-β: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12(6):325–338. doi:10.1038/nrneph.2016.48
14. Reidy K, Kang HM, Hostetter T, Susztak K. Molecular mechanisms of diabetic kidney disease. J Clin Invest. 2014;124(6):2333–2340. doi:10.1172/jci72271
15. Horsburgh S, Robson-Ansley P, Adams R, Smith C. Exercise and inflammation-related epigenetic modifications: focus on DNA methylation. Exerc Immunol Rev. 2015;21:26–41.
16. Keating ST, van Diepen JA, Riksen NP, El-Osta A. Epigenetics in diabetic nephropathy, immunity and metabolism. Diabetologia. 2018;61(1):6–20. doi:10.1007/s00125-017-4490-1
17. Susztak K. Understanding the epigenetic syntax for the genetic alphabet in the kidney. J Am Soc Nephrol. 2014;25(1):10–17. doi:10.1681/asn.2013050461
18. Sun G, Reddy MA, Yuan H, Lanting L, Kato M, Natarajan R. Epigenetic histone methylation modulates fibrotic gene expression. J Am Soc Nephrol. 2010;21(12):2069–2080. doi:10.1681/asn.2010060633
19. Kato M, Dang V, Wang M, et al. TGF-β induces acetylation of chromatin and of Ets-1 to alleviate repression of miR-192 in diabetic nephropathy. Sci Signal. 2013;6(278):ra43. doi:10.1126/scisignal.2003389
20. KDOQI clinical practice guidelines and clinical practice recommendations for diabetes and chronic kidney disease. Am J Kidney Dis. 2007;49(2Suppl 2):S12–154. doi:10.1053/j.ajkd.2006.12.005
21. Flyvbjerg A. The role of the complement system in diabetic nephropathy. Nat Rev Nephrol. 2017;13(5):311–318. doi:10.1038/nrneph.2017.31
22. Li L, Chen L, Zang J, et al. C3a and C5a receptor antagonists ameliorate endothelial-myofibroblast transition via the Wnt/β-catenin signaling pathway in diabetic kidney disease. Metabolism. 2015;64(5):597–610. doi:10.1016/j.metabol.2015.01.014
23. Lin YC, Chang YH, Yang SY, Wu KD, Chu TS. Update of pathophysiology and management of diabetic kidney disease. J Formos Med Assoc. 2018;117(8):662–675. doi:10.1016/j.jfma.2018.02.007
24. Sharma D, Bhattacharya P, Kalia K, Tiwari V. Diabetic nephropathy: new insights into established therapeutic paradigms and novel molecular targets. Diabetes Res Clin Pract. 2017;128:91–108. doi:10.1016/j.diabres.2017.04.010
25. Warren AM, Knudsen ST, Cooper ME. Diabetic nephropathy: an insight into molecular mechanisms and emerging therapies. Expert Opin Ther Targets. 2019;23(7):579–591. doi:10.1080/14728222.2019.1624721
26. Liu YN, Zhou J, Li T, et al. Sulodexide protects renal tubular epithelial cells from oxidative stress-induced injury via upregulating klotho expression at an early stage of diabetic kidney disease. J Diabetes Res. 2017;2017:4989847. doi:10.1155/2017/4989847
27. Li SY, Huang PH, Tarng DC, et al. Four-and-a-Half LIM domains Protein 2 is a coactivator of Wnt signaling in diabetic kidney disease. J Am Soc Nephrol. 2015;26(12):3072–3084. doi:10.1681/asn.2014100989
28. Deshpande SD, Putta S, Wang M, et al. Transforming growth factor-β-induced cross talk between p53 and a microRNA in the pathogenesis of diabetic nephropathy. Diabetes. 2013;62(9):3151–3162. doi:10.2337/db13-0305
29. Cooper ME. Pathogenesis, prevention, and treatment of diabetic nephropathy. Lancet. 1998;352(9123):213–219. doi:10.1016/s0140-6736(98)01346-4
30. Ban CR, Twigg SM. Fibrosis in diabetes complications: pathogenic mechanisms and circulating and urinary markers. Vascular Health and Risk Management. 2008;4(3):575–596. doi:10.2147/vhrm.s1991
31. Tian S, Chen SY. Macrophage polarization in kidney diseases. Macrophage (Houst). 2015;2:1. doi:10.14800/macrophage.679
32. Matoba K, Takeda Y, Nagai Y, Kawanami D, Utsunomiya K, Nishimura R. Unraveling the role of inflammation in the pathogenesis of diabetic kidney disease. Int J Mol Sci. 2019;20:14. doi:10.3390/ijms20143393
33. Klessens CQF, Zandbergen M, Wolterbeek R, et al. Macrophages in diabetic nephropathy in patients with type 2 diabetes. Nephrol Dial Transplant. 2017;32(8):1322–1329. doi:10.1093/ndt/gfw260
34. Huang J, Xiao Y, Xu A, Zhou Z. Neutrophils in type 1 diabetes. J Diabetes Investig. Sep 2016;7(5):652-63. doi:10.1111/jdi.12469
35. Yang D, Livingston MJ, Liu Z, et al. Autophagy in diabetic kidney disease: regulation, pathological role and therapeutic potential. Cell Mol Life Sci. 2018;75(4):669–688. doi:10.1007/s00018-017-2639-1
36. Reddy MA, Zhang E, Natarajan R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia. 2015;58(3):443–455. doi:10.1007/s00125-014-3462-y
37. Reddy MA, Natarajan R. Epigenetic mechanisms in diabetic vascular complications. Cardiovasc Res. 2011;90(3):421–429. doi:10.1093/cvr/cvr024
38. Villeneuve LM, Natarajan R. The role of epigenetics in the pathology of diabetic complications. Am J Physiol Renal Physiol. 2010;299(1):F14–25. doi:10.1152/ajprenal.00200.2010
39. Effect of intensive therapy on the microvascular complications of type 1 diabetes mellitus. JAMA. 2002;287(19):2563–2569. doi:10.1001/jama.287.19.2563
40. Nathan DM. The diabetes control and complications trial/epidemiology of diabetes interventions and complications study at 30 years: overview. Diabetes Care. 2014;37(1):9–16. doi:10.2337/dc13-2112
41. Wing MR, Ramezani A, Gill HS, Devaney JM, Raj DS. Epigenetics of progression of chronic kidney disease: fact or fantasy? Semin Nephrol. 2013;33(4):363–374. doi:10.1016/j.semnephrol.2013.05.008
42. Beckerman P, Ko YA, Susztak K. Epigenetics: a new way to look at kidney diseases. Nephrol Dial Transplant. 2014;29(10):1821–1827. doi:10.1093/ndt/gfu026
43. Kota SK, Kota SB. Noncoding RNA and epigenetic gene regulation in renal diseases. Drug Discov Today. 2017;22(7):1112–1122. doi:10.1016/j.drudis.2017.04.020
44. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–492. doi:10.1038/nrg3230
45. Lister R, Pelizzola M, Dowen RH, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315–322. doi:10.1038/nature08514
46. Zeisberg EM, Zeisberg M. The role of promoter hypermethylation in fibroblast activation and fibrogenesis. J Pathol. 2013;229(2):264–273. doi:10.1002/path.4120
47. Guo C, Dong G, Liang X, Dong Z. Epigenetic regulation in AKI and kidney repair: mechanisms and therapeutic implications. Nat Rev Nephrol. 2019;15(4):220–239. doi:10.1038/s41581-018-0103-6
48. Chen R, Kang R, Fan XG, Tang D. Release and activity of histone in diseases. Cell Death Dis. 2014;5(8):e1370. doi:10.1038/cddis.2014.337
49. Jezek M, Green EM. Histone modifications and the maintenance of telomere integrity. Cells. 2019;8(2). doi:10.3390/cells8020199
50. Martinez-Moreno JM, Fontecha-Barriuso M, Martin-Sanchez D, et al. Epigenetic modifiers as potential therapeutic targets in diabetic kidney disease. Int J Mol Sci. 2020;21(11). doi:10.3390/ijms21114113
51. Tan M, Luo H, Lee S, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146(6):1016–1028. doi:10.1016/j.cell.2011.08.008
52. Fontecha-Barriuso M, Martin-Sanchez D, Ruiz-Andres O, et al. Targeting epigenetic DNA and histone modifications to treat kidney disease. Nephrol Dial Transplant. 2018;33(11):1875–1886. doi:10.1093/ndt/gfy009
53. Xu L, Natara R, Chen Z. Epigenetic risk profile of diabetic kidney disease in high-risk populations. Curr Diab Rep. 2019;19(3):9. doi:10.1007/s11892-019-1129-2
54. Tang J, Zhuang S. Histone acetylation and DNA methylation in ischemia/reperfusion injury. Clin Sci (Lond). 2019;133(4):597–609. doi:10.1042/cs20180465
55. Pang M, Zhuang S. Histone deacetylase: a potential therapeutic target for fibrotic disorders. J Pharmacol Exp Ther. 2010;335(2):266–272. doi:10.1124/jpet.110.168385
56. Liu M, Liang K, Zhen J, et al. Sirt6 deficiency exacerbates podocyte injury and proteinuria through targeting Notch signaling. Nat Commun. 2017;8(1):413. doi:10.1038/s41467-017-00498-4
57. Sayyed SG, Gaikwad AB, Lichtnekert J, et al. Progressive glomerulosclerosis in type 2 diabetes is associated with renal histone H3K9 and H3K23 acetylation, H3K4 dimethylation and phosphorylation at serine 10. Nephrol Dial Transplant. 2010;25(6):1811–1817. doi:10.1093/ndt/gfp730
58. De Marinis Y, Cai M, Bompada P, et al. Epigenetic regulation of the thioredoxin-interacting protein (TXNIP) gene by hyperglycemia in kidney. Kidney Int. 2016;89(2):342–353. doi:10.1016/j.kint.2015.12.018
59. Yuan H, Reddy MA, Sun G, et al. Involvement of p300/CBP and epigenetic histone acetylation in TGF-β1-mediated gene transcription in mesangial cells. Am J Physiol Renal Physiol. 2013;304(5):F601–13. doi:10.1152/ajprenal.00523.2012
60. Wang Y, Wang Y, Luo M, et al. Novel curcumin analog C66 prevents diabetic nephropathy via JNK pathway with the involvement of p300/CBP-mediated histone acetylation. Biochim Biophys Acta. 2015;1852(1):34–46. doi:10.1016/j.bbadis.2014.11.006
61. He L, Sabet A, Djedjos S, et al. Metformin and insulin suppress hepatic gluconeogenesis through phosphorylation of CREB binding protein. Cell. 2009;137(4):635–646. doi:10.1016/j.cell.2009.03.016
62. Lin CL, Lee PH, Hsu YC, et al. MicroRNA-29a promotion of nephrin acetylation ameliorates hyperglycemia-induced podocyte dysfunction. J Am Soc Nephrol. 2014;25(8):1698–1709. doi:10.1681/asn.2013050527
63. Bock F, Shahzad K, Wang H, et al. Activated protein C ameliorates diabetic nephropathy by epigenetically inhibiting the redox enzyme p66Shc. Proc Natl Acad Sci U S A. 2013;110(2):648–653. doi:10.1073/pnas.1218667110
64. Klose RJ, Zhang Y. Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Biol. 2007;8(4):307–318. doi:10.1038/nrm2143
65. Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi:10.1016/j.cell.2007.02.005
66. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi:10.1038/nature11247
67. Shi Y, Lan F, Matson C, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119(7):941–953. doi:10.1016/j.cell.2004.12.012
68. Metzger E, Wissmann M, Yin N, et al. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437(7057):436–439. doi:10.1038/nature04020
69. Shi Y, Whetstine JR. Dynamic regulation of histone lysine methylation by demethylases. Mol Cell. 2007;25(1):1–14. doi:10.1016/j.molcel.2006.12.010
70. Agger K, Cloos PA, Christensen J, et al. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature. 2007;449(7163):731–734. doi:10.1038/nature06145
71. Kato M, Natara R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat Rev Nephrol. 2019;15(6):327–345. doi:10.1038/s41581-019-0135-6
72. Villeneuve LM, Kato M, Reddy MA, Wang M, Lanting L, Natara R. Enhanced levels of microRNA-125b in vascular smooth muscle cells of diabetic db/db mice lead to increased inflammatory gene expression by targeting the histone methyltransferase Suv39h1. Diabetes. 2010;59(11):2904–2915. doi:10.2337/db10-0208
73. Chen J, Guo Y, Zeng W, et al. ER stress triggers MCP-1 expression through SET7/9-induced histone methylation in the kidneys of db/db mice. Am J Physiol Renal Physiol. 2014;306(8):F916–25. doi:10.1152/ajprenal.00697.2012
74. Cai M, Bompada P, Atac D, Laakso M, Groop L, De Marinis Y. Epigenetic regulation of glucose-stimulated osteopontin (OPN) expression in diabetic kidney. Biochem Biophys Res Commun. 2016;469(1):108–113. doi:10.1016/j.bbrc.2015.11.079
75. Reddy MA, Sumanth P, Lanting L, et al. Losartan reverses permissive epigenetic changes in renal glomeruli of diabetic db/db mice. Kidney Int. 2014;85(2):362–373. doi:10.1038/ki.2013.387
76. Lin SH, Ho WT, Wang YT, Chuang CT, Chuang LY, Guh JY. Histone methyltransferase Suv39h1 attenuates high glucose-induced fibronectin and p21(WAF1) in mesangial cells. Int J Biochem Cell Biol. 2016;78:96–105. doi:10.1016/j.biocel.2016.06.021
77. Wang J, Yan W, Peng X, et al. Functional role of SUV39H1 in human renal tubular epithelial cells under high-glucose ambiance. Inflammation. 2018;41(1):1–10. doi:10.1007/s10753-017-0657-7
78. Ruiz-Andres O, Sanchez-Niño MD, Cannata-Ortiz P, et al. Histone lysine crotonylation during acute kidney injury in mice. Dis Model Mech. 2016;9(6):633–645. doi:10.1242/dmm.024455
79. Martinez-Moreno JM, Fontecha-Barriuso M, Martín-Sánchez D, et al. The contribution of histone crotonylation to tissue health and disease: focus on kidney health. Front Pharmacol. 2020;11:393. doi:10.3389/fphar.2020.00393
80. Aquila L, Atanassov BS. Regulation of histone ubiquitination in response to DNA double strand breaks. Cells. 2020;9:7. doi:10.3390/cells9071699
81. Aghdam SY, Sheibani N. The ubiquitin-proteasome system and microvascular complications of diabetes. J Ophthalmic Vis Res. 2013;8(3):244–256.
82. Li Y, Ren D, Shen Y, Zheng X, Xu G. Altered DNA methylation of TRIM13 in diabetic nephropathy suppresses mesangial collagen synthesis by promoting ubiquitination of CHOP. EBioMedicine. Jan. 2020;51:102582. doi:10.1016/j.ebiom.2019.11.043
83. Denhez B, Lizotte F, Guimond MO, Jones N, Takano T, Geraldes P. Increased SHP-1 protein expression by high glucose levels reduces nephrin phosphorylation in podocytes. J Biol Chem. 2015;290(1):350–358. doi:10.1074/jbc.M114.612721
84. Laddha AP, Kulkarni YA. NADPH oxidase: A membrane-bound enzyme and its inhibitors in diabetic complications. Eur J Pharmacol. 2020;881:173206. doi:10.1016/j.ejphar.2020.173206
85. Peng J, Li X, Zhang D, et al. Hyperglycemia, p53, and mitochondrial pathway of apoptosis are involved in the susceptibility of diabetic models to ischemic acute kidney injury. Kidney Int. Jan 2015;87(1):137-50. doi:10.1038/ki.2014.226
86. Paneni F, Costantino S, Battista R, et al. Adverse epigenetic signatures by histone methyltransferase Set7 contribute to vascular dysfunction in patients with type 2 diabetes mellitus. Circ Cardiovasc Genet. 2015;8(1):150–158. doi:10.1161/circgenetics.114.000671
87. Miao F, Wu X, Zhang L, Yuan YC, Riggs AD, Natarajan R. Genome-wide analysis of histone lysine methylation variations caused by diabetic conditions in human monocytes. J Biol Chem. 2007;282(18):13854–13863. doi:10.1074/jbc.M609446200
88. Duncan TM, Reed MC, Nijhout HF. The relationship between intracellular and plasma levels of folate and metabolites in the methionine cycle: a model. Mol Nutr Food Res. 2013;57(4):628–636. doi:10.1002/mnfr.201200125
89. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK Prospective Diabetes Study (UKPDS) Group. Lancet. 1998;352(9131):854–865.
90. Marui N, Offermann MK, Swerlick R, et al. Vascular cell adhesion molecule-1 (VCAM-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cells. J Clin Invest. 1993;92(4):1866–1874. doi:10.1172/jci116778
91. Bierhaus A, Schiekofer S, Schwaninger M, et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes. 2001;50(12):2792–2808. doi:10.2337/diabetes.50.12.2792
92. Ueda A, Ishigatsubo Y, Okubo T, Yoshimura T. Transcriptional regulation of the human monocyte chemoattractant protein-1 gene. Cooperation of two NF-kappaB sites and NF-kappaB/Rel subunit specificity. J Biol Chem. 1997;272(49):31092–31099. doi:10.1074/jbc.272.49.31092
93. He S, Owen DR, Jelinsky SA, Lin LL. Lysine Methyltransferase SETD7 (SET7/9) Regulates ROS Signaling through mitochondria and NFE2L2/ARE pathway. Sci Rep. Oct. 2015;5:14368. doi:10.1038/srep14368
94. Elkouris M, Kontaki H, Stavropoulos A, et al. SET9-mediated regulation of TGF-β signaling links protein methylation to pulmonary fibrosis. Cell Rep. 2016;15(12):2733–2744. doi:10.1016/j.celrep.2016.05.051
95. Sasaki K, Doi S, Nakashima A, et al. Inhibition of SET domain-containing lysine methyltransferase 7/9 ameliorates renal fibrosis. J Am Soc Nephrol. 2016;27(1):203–215. doi:10.1681/asn.2014090850
96. Shuttleworth VG, Gaughan L, Nawafa L, et al. The methyltransferase SET9 regulates TGFB1 activation of renal fibroblasts via interaction with SMAD3. J Cell Sci. Jan. 2018;131:1. doi:10.1242/jcs.207761
97. Chokpaisarn J, Urao N, Voravuthikunchai SP, Koh TJ. Quercus infectoria inhibits Set7/NF-κB inflammatory pathway in macrophages exposed to a diabetic environment. Cytokine. Jun. 2017;94:29–36. doi:10.1016/j.cyto.2017.04.005
98. Sharma K, Ziyadeh FN. Hyperglycemia and diabetic kidney disease. The case for transforming growth factor-beta as a key mediator. Diabetes. 1995;44(10):1139–1146. doi:10.2337/diab.44.10.1139
99. Kato M, Putta S, Wang M, et al. TGF-beta activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat Cell Biol. 2009;11(7):881–889. doi:10.1038/ncb1897
100. Kato M, Yuan H, Xu Z-G, et al. Role of the Akt/FoxO3a pathway in TGF-β1–mediated mesangial cell dysfunction: a novel mechanism related to diabetic kidney disease. Journal of the American Society of Nephrology. 2006;17(12):3325–3335. doi:10.1681/asn.2006070754
101. Jia Y, Reddy MA, Das S, et al. Dysregulation of histone H3 lysine 27 trimethylation in transforming growth factor-β1–induced gene expression in mesangial cells and diabetic kidney. J Biol Chem. 2019;294(34):12695–12707. doi:10.1074/jbc.RA119.007575
102. Noh H, Oh EY, Seo JY, et al. Histone deacetylase-2 is a key regulator of diabetes- and transforming growth factor-β1-induced renal injury. American Journal of Physiology-Renal Physiology. 2009;297(3):F729–39. doi:10.1152/ajprenal.00086.2009
103. Yoshikawa M, Hishikawa K, Marumo T, Fujita T. Inhibition of histone deacetylase activity suppresses epithelial-to-mesenchymal transition induced by TGF-β1 in human renal epithelial cells. J Am Soc Nephrol. 2007;18(1):58–65. doi:10.1681/asn.2005111187
104. Guo Q, Li X, Han H, et al. Histone Lysine Methylation in TGF-β1 Mediated p21 gene expression in rat mesangial cells. Biomed Res Int. 2016;2016:6927234. doi:10.1155/2016/6927234
105. Ghosh AK, Bhattacharyya S, Lafyatis R, et al. p300 is elevated in systemic sclerosis and its expression is positively regulated by TGF-β: epigenetic feed-forward amplification of fibrosis. J Invest Dermatol. 2013;133(5):1302–1310. doi:10.1038/jid.2012.479
106. Shao J, Xu H, Wu X, Xu Y. Epigenetic activation of CTGF transcription by high glucose in renal tubular epithelial cells is mediated by myocardin-related transcription factor A. Cell Tissue Res. 2020;379(3):549–559. doi:10.1007/s00441-019-03124-5
107. Xu H, Wu X, Qin H, et al. Myocardin-related transcription factor A epigenetically regulates renal fibrosis in diabetic nephropathy. J Am Soc Nephrol. 2015;26(7):1648–1660. doi:10.1681/asn.2014070678
108. Hasegawa K, Wakino S, Simic P, et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat Med. 2013;19(11):1496–1504. doi:10.1038/nm.3363
109. Jinde K, Nikolic-Paterson DJ, Huang XR, et al. Tubular phenotypic change in progressive tubulointerstitial fibrosis in human glomerulonephritis. Am J Kidney Dis. 2001;38(4):761–769. doi:10.1053/ajkd.2001.27693
110. Oldfield MD, Bach LA, Forbes JM, et al. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J Clin Invest. 2001;108(12):1853–1863. doi:10.1172/jci11951
111. Yamaguchi Y, Iwano M, Suzuki D, et al. Epithelial-mesenchymal transition as a potential explanation for podocyte depletion in diabetic nephropathy. Am J Kidney Dis. 2009;54(4):653–664. doi:10.1053/j.ajkd.2009.05.009
112. Wang X, Liu J, Zhen J, et al. Histone deacetylase 4 selectively contributes to podocyte injury in diabetic nephropathy. Kidney Int. 2014;86(4):712–725. doi:10.1038/ki.2014.111
113. Zhang Y, Zou J, Tolbert E, Zhao TC, Bayliss G, Zhuang S. Identification of histone deacetylase 8 as a novel therapeutic target for renal fibrosis. FASEB J. 2020;34(6):7295–7310. doi:10.1096/fj.201903254R
114. Miao F, Chen Z, Genuth S, et al. Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes. 2014;63(5):1748–1762. doi:10.2337/db13-1251
115. El-Osta A, Brasacchio D, Yao D, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. 2008;205(10):2409–2417. doi:10.1084/jem.20081188
116. Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev. 2002;23(5):599–622. doi:10.1210/er.2001-0039
117. Miao F, Gonzalo IG, Lanting L, Natarajan R. In vivo chromatin remodeling events leading to inflammatory gene transcription under diabetic conditions. J Biol Chem. 2004;279(17):18091–18097. doi:10.1074/jbc.M311786200
118. Chen H, Li J, Jiao L, et al. Apelin inhibits the development of diabetic nephropathy by regulating histone acetylation in Akita mouse. J Physiol. 2014;592(3):505–521. doi:10.1113/jphysiol.2013.266411
119. Komers R, Mar D, Denisenko O, Xu B, Oyama TT, Bomsztyk K. Epigenetic changes in renal genes dysregulated in mouse and rat models of type 1 diabetes. Lab Invest. 2013;93(5):543–552. doi:10.1038/labinvest.2013.47
120. Brosius FC
121. Baricos WH, Reed JC, Cortez SL. Extracellular matrix degradation by cultured mesangial cells: mediators and modulators. Exp Biol Med (Maywood). 2003;228(9):1018–1022. doi:10.1177/153537020322800905
122. Sakharova OV, Taal MW, Brenner BM. Pathogenesis of diabetic nephropathy: focus on transforming growth factor-beta and connective tissue growth factor. Curr Opin Nephrol Hypertens. 2001;10(6):727–738. doi:10.1097/00041552-200111000-00001
123. Guha M, Xu ZG, Tung D, Lanting L, Natarajan R. Specific down-regulation of connective tissue growth factor attenuates progression of nephropathy in mouse models of type 1 and type 2 diabetes. FASEB J. 2007;21(12):3355–3368. doi:10.1096/fj.06-6713com
124. Li X, Li C, Li X, et al. Involvement of histone lysine methylation in p21 gene expression in rat kidney in vivo and rat mesangial cells in vitro under diabetic conditions. J Diabetes Res. 2016;2016:3853242. doi:10.1155/2016/3853242
125. Villeneuve LM, Reddy MA, Lanting LL, Wang M, Meng L, Natarajan R. Epigenetic histone H3 lysine 9 methylation in metabolic memory and inflammatory phenotype of vascular smooth muscle cells in diabetes. Proc Natl Acad Sci U S A. 2008;105(26):9047–9052. doi:10.1073/pnas.0803623105
126. Brasacchio D, Okabe J, Tikellis C, et al. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes. 2009;58(5):1229–1236. doi:10.2337/db08-1666
127. Majumder S, Thieme K, Batchu SN, et al. Shifts in podocyte histone H3K27me3 regulate mouse and human glomerular disease. J Clin Invest. 2018;128(1):483–499. doi:10.1172/jci95946
128. Siddiqi FS, Majumder S, Thai K, et al. The histone methyltransferase enzyme enhancer of zeste homolog 2 protects against podocyte oxidative stress and renal injury in diabetes. J Am Soc Nephrol. 2016;27(7):2021–2034. doi:10.1681/asn.2014090898
129. Ruan Y, Zhang Y, Zhao J, et al. [EZH2 promotes development and progression of diabetic nephropathy]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2020;36(3):212–219.Chinese.
130. Goru SK, Kadakol A, Pandey A, Malek V, Sharma N, Gaikwad AB. Histone H2AK119 and H2BK120 mono-ubiquitination modulate SET7/9 and SUV39H1 in type 1 diabetes-induced renal fibrosis. Biochem J. 2016;473(21):3937–3949. doi:10.1042/bcj20160595
131. Li Y, Reddy MA, Miao F, et al. Role of the histone H3 lysine 4 methyltransferase, SET7/9, in the regulation of NF-kappaB-dependent inflammatory genes. Relevance to diabetes and inflammation. J Biol Chem. 2008;283(39):26771–26781. doi:10.1074/jbc.M802800200
132. Rao P, Pang M, Qiao X, et al. Promotion of β-catenin/Foxo1 signaling ameliorates renal interstitial fibrosis. Lab Invest. 2019;99(11):1689–1701. doi:10.1038/s41374-019-0276-z
133. Zhong Y, Zhang X, Cai X, Wang K, Chen Y, Deng Y. Puerarin attenuated early diabetic kidney injury through down-regulation of matrix metalloproteinase 9 in streptozotocin-induced diabetic rats. PLoS One. 2014;9(1):e85690. doi:10.1371/journal.pone.0085690
134. Zhang L, Zhao S, Zhu Y. Long noncoding RNA growth arrest-specific transcript 5 alleviates renal fibrosis in diabetic nephropathy by downregulating matrix metalloproteinase 9 through recruitment of enhancer of zeste homolog 2. FASEB J. 2020;34(2):2703–2714. doi:10.1096/fj.201901380RR
135. Zhang L, Chen L, Gao C, et al. Loss of histone H3 K79 methyltransferase Dot1l facilitates kidney fibrosis by upregulating endothelin 1 through histone deacetylase 2. J Am Soc Nephrol. 2020;31(2):337–349. doi:10.1681/asn.2019070739
136. Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11(5):384–400. doi:10.1038/nrd3674
137. Morgado-Pascual JL, Rayego-Mateos S, Tejedor L, Suarez-Alvarez B, Ruiz-Ortega M. Bromodomain and extraterminal proteins as novel epigenetic targets for renal diseases. Front Pharmacol. 2019;10:1315. doi:10.3389/fphar.2019.01315
138. Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17(10):630–641. doi:10.1038/nrg.2016.93
139. Sharma S, Taliyan R. Histone deacetylase inhibitors: future therapeutics for insulin resistance and type 2 diabetes. Pharmacol Res. 2016;113(PtA):320–326. doi:10.1016/j.phrs.2016.09.009
140. Christensen DP, Dahllöf M, Lundh M, et al. Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus. Mol Med. 2011;17(5–6):378–390. doi:10.2119/molmed.2011.00021
141. Singh R, Chandel S, Dey D, et al. Epigenetic modification and therapeutic targets of diabetes mellitus. Biosci Rep. 2020;40:9. doi:10.1042/bsr20202160
142. Huang K, Huang J, Xie X, et al. Sirt1 resists advanced glycation end products-induced expressions of fibronectin and TGF-β1 by activating the Nrf2/ARE pathway in glomerular mesangial cells. Free Radic Biol Med. Dec. 2013;65:528–540. doi:10.1016/j.freeradbiomed.2013.07.029
143. Hong Q, Zhang L, Das B, et al. Increased podocyte Sirtuin-1 function attenuates diabetic kidney injury. Kidney Int. 2018;93(6):1330–1343. doi:10.1016/j.kint.2017.12.008
144. Hoeksema MA, Gijbels MJ, Van den Bossche J, et al. Targeting macrophage Histone deacetylase 3 stabilizes atherosclerotic lesions. EMBO Mol Med. 2014;6(9):1124–1132. doi:10.15252/emmm.201404170
145. Zampetaki A, Zeng L, Margariti A, et al. Histone deacetylase 3 is critical in endothelial survival and atherosclerosis development in response to disturbed flow. Circulation. 2010;121(1):132–142. doi:10.1161/circulationaha.109.890491
146. He P, Yu H, Jiang L, et al. Hdac9 inhibits medial artery calcification through down-regulation of Osterix. Vascul Pharmacol. 2020;132:106775. doi:10.1016/j.vph.2020.106775
147. Liu F, Zong M, Wen X, et al. Silencing of Histone Deacetylase 9 expression in podocytes attenuates kidney injury in diabetic nephropathy. Sci Rep. 2016;6:33676. doi:10.1038/srep33676
148. Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346(6213):1258096. doi:10.1126/science.1258096
149. Dominguez AA, Lim WA, Qi LS. Beyond editing: repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat Rev Mol Cell Biol. 2016;17(1):5–15. doi:10.1038/nrm.2015.2
150. Liao HK, Hatanaka F, Araoka T, et al. In vivo target gene activation via CRISPR/Cas9-mediated trans-epigenetic modulation. Cell. 2017;171(7):1495–1507.e15. doi:10.1016/j.cell.2017.10.025
151. Komor AC, Badran AH, Liu DR. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. 2017;169(3):559. doi:10.1016/j.cell.2017.04.005
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.