Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 12
Epidermolytic hyperkeratosis: clinical update
Authors Peter Rout D ![]() , Nair A
, Nair A ![]() , Gupta A
, Gupta A ![]() , Kumar P
, Kumar P ![]()
Received 15 October 2018
Accepted for publication 1 April 2019
Published 8 May 2019 Volume 2019:12 Pages 333—344
DOI https://doi.org/10.2147/CCID.S166849
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jeffrey Weinberg
Denice Peter Rout,* Anushka Nair,* Anand Gupta, Piyush Kumar
Amity Institute of Biotechnology, Amity University Mumbai, Navi Mumbai, India
*These authors contributed equally to this work
Abstract: Epidermolytic hyperkeratosis (EHK), earlier termed as bullous congenital ichthyosiform erythroderma is a skin disorder characterized as an autosomal dominant and rare disorder which has been observed to affect 1 in over 200,000 infants as a consequence of a significant mutation in the genes responsible for the keratin proteins, mostly keratin 1 and 10. The features present at birth include erythema and blistering. In adults, the hallmarks include hyperkeratosis, erosions, and blisters. The major symptoms including xerosis, pruritus, and painful fissuring lead not only to cosmetic problems but also stress, inferiority complex and other psychological conditions. While clinical inspection followed by confirmatory tests including histopathology and electron microscopic assessment is used for diagnosis, treatment modalities can be further improved for better diagnosis. This article reviews subtypes of ichthyosis, with a focus on EHK, genetics behind the disease, recently reported mutations, the existing diagnostics and treatments for the same and potential of new modalities in diagnosis/treatment.
Keywords: epidermolytic hyperkeratosis, ichthyosis, skin, skin disorder
Introduction
Epidermolytic hyperkeratosis (EHK), earlier termed as bullous congenital ichthyosiformerythroderma (BCIE),1 is a skin disorder characterized as an autosomal dominant and rare disorder which has been observed to affect 1 in over 200,000 infants as a consequence of a significant mutation in the genes responsible for the keratin proteins.2,3 This disorder is histopathologically perceived as a heritable keratinization skin disorder which is typically identified by blistering of the skin, clumping of the tonofilament (keratin bundles), and cytolysis in the terminally differentiating epidermal cells.4–8 Newborns are at a high risk of developing electrolytic diseases which may be fatal, but as the age progresses the blistering and redness reduces and are, over time, replaced by a progressive hyperkeratosis in the stratum corneum; the uppermost layer of skin starts to thicken and shows blistering.9 For the renewal of the normal human epidermis, keratinocytes are on a constant move from the proliferating basal strata to the terminally well differentiated squames that form the stratum corneum,10 but in the individuals infected by EHK, this process is hampered due to which the regular epidermal function of being a barrier is ineffective.11 Insights from cutaneous lesions suggest that the cleavage of the tissue happens intraepidermally with the separation of the mutated suprabasal keratinocytes along with focal hyperkeratosis.12 The examination of epidermolytic hyperkeratosis skin using electron microscopy has revealed the formation of clumps in the suprabasal epidermal cells of keratin intermediate filaments (KIF)13,14 along with the presence of keratin 1 and keratin 10.15,16 Such observations suggested that EHK is linked to keratin 1 and keratin 10. This pair exist as multiple heterodimers and form the KIF in the upper epidermal layer, and a mutation in them leads to EHK.17 The expression of the keratin proteins is specific to epithelial cells depending on the cell type and the state of differentiation.11
The keratin protein’s general structure is formed by a conserved rod domain along with distinct four alpha-helical structures which are separated by linker sequences that are nonhelical and also by globular and nonhelical sequences which are of varied compositions and size.18,19 On chromosome 17, the small, acidic type I keratins are encoded (keratins 9 to 19), while on chromosome 12, the more basic and large type II keratins are encoded (keratins 1 to 8).11,20–23 Each of the two keratin types contributes one protein to form the heterodimer subunit which is a coiled structure that then forms the tonofilaments.11,24,25 Initially, the basal epidermal cells are observed to be expressing only keratins 5 and 14, but as the cells begin to terminally differentiate and start the migration towards the spinous layer, the expression of keratin proteins 5 and 14 is reduced and instead the expression of keratin 1 and keratin 10 is promoted.26–29 In the suprabasal spinous and granular cells, clumping of the tonofilaments along with a collapsed keratin filament network surrounds the nuclear region and is the hallmark of EHK.5,30–32 Such a collapse generally suggests an error in the differentiation specific keratin,16,33 although the other probable causes for EHK have been identified as deformed filaggrin which is an associated protein for keratin filament,31 an envelope precursor, involucrin34 or an abnormality in the metabolism of lysosome.35 It has been observed that most families that have EHK are predisposed to have linkage near the gene locus of chromosome 12 for type II keratins.36,37 While several mutations are already known, discovery of new mutations including c.475T>C, p.Ser159Pro), and c.562A>C in the recent years suggest further explorations to understand genetics and heredity of EHK in order to identify newer methods of detection and treatment.
This article reviews subtypes of ichthyosis, with a focus on epidermolytic hyperkeratosis, the genetics behind the disease, recently discovered mutations, the existing diagnostics and treatments for the same and potential of new modalities in diagnosis/treatment.
Subtypes of ichthyosis
Ichthyosis is a set of disorders of cornification which are identified clinically by significant scaling patterns and by hyperkeratosis which is a histopathological characterization. The clinical subtypes of this disorder are usually based on the mode of inheritance and the observed clinical and pathological data.38 The word “Ichthy” originates from the Greek word for “fish”. As the skin becomes thick and appears like fish scales, the disorder has been named as “Ichthyosis”. This disorder is either acquired or inherited and around 28 recognized types of ichthyosis and related skin ailments exist.39 Ichthyosis is usually categorized as; true ichthyosis, epidermolytic hyperkeratosis and ichthyosiform states. Each of these types further is divided into several subtypes. Primarily, the autosomal recessive, autosomal dominant and X-linked recessive types fall under the true ichthyosis type.40 Some of the major types of inherited ichthyosis that are recognized are: ichthyosis vulgaris, Harlequin ichthyosis, epidermolytic hyperkeratosis, congenital ichthyosiform erythroderma, X-linked ichthyosis, localized ichthyosis and lamellar ichthyosis. Of the various types, ichthyosis vulgaris is the most common type of ichthyosis which occurs in every 1 out of 250 people.41 The X-linked ichthyosis is specific to only males and its occurrence is approximately around 1 in every 6,000 births.42 Harlequin ichthyosis is one of the rare but extremely severe kind of inherited ichthyosis wherein an affected child looks like it is wearing a harlequin costume43 The classical lamellar ichthyosis also called the autosomal recessive congenital ichthyosis is one of the severe forms which effects 1 in every 300,000 births and is associated with recessive genes.44 Ichthyosiform dermatoses group represent a huge clinical spectrum of disorders with lack of properly defined pathogenetic mechanism responsible for these conditions.45 Acquired ichthyosis is a condition similar to ichthyosis vulgaris both clinically and histologically and it usually occurs during adulthood in association with other diseases or lymphoma.46–52
A rare form of ichthyosis with clinical manifestations at birth is epidermolytic hyperkeratosis (EHK) which is an autosomal dominant disease of unknown etiology which has been recorded to affect approximately every 1 in 30,000 human beings. The disease is characterized primarily with hyperkeratotic scaliness along with severe blistering as a subsequent effect of cytolysis in the suprabasal cells, and also hyperproliferation in the basal cells. As per the histological approach, the epidermis of patients suffering from EHK exhibits a thickened, granular layer and stratum corneum, with irregularly shaped and enlarged cells.53 On the ulterior structure, only the suprabasal layers are affected, wherein three major aberrancies are observed namely: clumping of tonofilaments, keratohyalin granules and nuclei of irregular size and shape, and degeneration of the cell. Figure 1 shows clinical features of three keratin-related conditions associated with EHK.
 | Figure 1 Clinical features of three keratin diseases. (A) Blisters on the soles of a patient with the milder Weber–Cockayne form of epidermolysis bullosa simplex. (B) Widespread epidermolytic hyperkeratosis in a bullous congenital ichthyosiform erythroderma patient. (C) Site-restricted epidermolytic hyperkeratosis of the palms in a mother and child with epidermolytic palmoplantar keratoderma.Note: Reproduced from McLean WI. Genetic disorders of palm skin and nail. J Anat. 2003;202(1):133–141 with permission from John Wiley and Sons.54 |
Pathology of epidermolytic hyperkeratosis
The most important proteins required for the structural development of the epidermis are the keratin proteins.55 By studying the keratin gene sequences, understanding the switching of the keratin gene expression which occurs as epidermal cells continue to terminally differentiate and analyzing how keratins aggregate into 10 nm filaments has led to the basic understanding of the genetic basis of epidermolytic hyperkeratosis. At the junction between the environmental conditions and the human body, the epidermis plays a protective role.56 It carries out this role by forming a massive cytoskeletal structure formed by 10 nm keratin filaments. Basal epidermal cells portray a complex network of relatively spread out keratin filaments, formed of the type I keratin K14 and type II keratin K5.57,58 Once the basal cells cease their division and commence their journey to the surface of the skin, they switch from expressing the K14 and K5 genes to K10 (type I) and K1 (type II).26,59 Simultaneously, K1/K10 filaments of the suprabasal cells clump together to form tonofibrillar bundles which are thicker than the tonofilament bundles K5/K14 of the basal cells, leading to the enhanced ability of the keratins to survive through the damaging phases when the production of flattened squames occurs, which leaves 85% of the keratinocytes which are terminally differentiated as keratin. Keratins are part of the superfamily of the intermediate filament (IF) proteins, which can self assemble in vitro into a 10 nm nuclear or cytoskeletal matrix fibers.60,61 The classical trait of the cytoskeletal IF polypeptides is the 310 amino acid α-helical coil which is centrally located and termed as the rod comprising heptad repetitions of hydrophobic moieties that allow them to wind around another IF protein along a seal which is also hydrophobic. This rod is further divided into four different segments which are known as helix 1A, 1B, 2A and 2B and is also connected by three nonhelical, short linker segments. The rod regions of keratins type I and type II form unstaggered, parallel, heterodimers, of which two then align in antiparallel directions to form structurally stable heterodimers.61 Over 5,000 heterotetramers then tend to form a network of a complex hierarchy of end to end and lateral associations to form a single 10-nm-IF of length 10–20 µm.62,63
EHK is histopathologically characterized by (1) clumping of tonofilaments and aggregation of perinuclear shells of tonofilaments within the suprabasal cells; (2) a basal epidermal layer which is normal but sometimes hyperproliferative; (3) a granular, thickened layer and stratum corneum and (4) degeneration of cells in suprabasal layers, starting in the lower spinous area and aggrandizing while terminal differentiation of the cells occur.55 Ichthyosis hystrix and palmoplantar keratoderma both have characteristics that imply that they are milder versions of EHK. A basic understanding at the genetic level of EHK roots from the clear similarity between the clumping of the tonofilaments in the suprabasal cells, that occurs in the skin for a person suffering from EHK, with the occurrence in the basal cells of the skin of a person suffering from Epidermolysis Bullosa Simplex (EBS).13 EHK clumps associate with only those antibodies which are specific for K10 and K1.15,16 It was predicted from the initial transgenic mouse based studies that EHK is a genetic disorder for which K1 and K10 are responsible, considering the mirror image parallels among the ultrastructural and morphological traits of EHK and EBS, along with the understanding that as the epidermal cells begin their terminal differentiation, they transcend into the expression of K1 and K10 genes.64 Pathobiological and biochemical features of human disorder EHK were portrayed by the transgenic mice that were expressing a reduced human K10 gene.53 In other studies, the linkages of keratin gene clumps on specific chromosomes 12 and 17 of the EHK affected members and palmoplantar keratoderma (PPK) affected members were studied, which further strengthened the idea that these diseases of the suprabasal layers are keratin-based disorders.10,65–70 Full proof evidence resulted from the direct sequencing of the two genes namely K1 and K10 isolated directly from the DNA of the patient. Point mutations in K1 or K10 genes were observed in three independent studies of separate cases of human EHK initially.11,37,53 Now, several other significant point substitutions have been reported.17,71–74
Inheritance pattern of EHK and new classifications of EHK
As per the results of the First Ichthyosis Consensus Conference in Soreze, 2009, it has been established that ichthyosis falls under the broad category of Mendelian disorders of cornification (MEDOC).75,76 Inherited ichthyosis is considered as a disease group within the larger group of MEDOC which is characterized by mutations caused in the genes responsible for the skin barrier formation. At present, based on the molecular genetics of the disorder, the inherited ichthyosis is further classified as nonsyndromic ichthyoses and ichthyosis syndromes.77 Of the nonsyndromic ichthyosis, keratinopathic ichthyosis is a proposed umbrella term under which EHK, annular epidermolytic ichthyosis (AEI), superficial epidermolytic ichthyosis (SEI), etc are covered. The keratinopathic ichthyosis are all characterized by mutations in the KRT1, KRT2 and KRT10 genes of the keratin family. The inheritance pattern of EHK has been studied extensively and it is predominantly understood to be an autosomal dominant inheritance with several cases also being identified as a result of spontaneous mutations. Family history of the disease is studied through pedigree analysis of the specific keratin genes namely KRT1, KRT2 and KRT10. Techniques such as immunohistochemistry, immunocytochemistry, light microscopy, electron microscopy, etc are used to screen the tissue and cellular level expression of the genes and help to determine and diagnose the inheritance pattern.
Mutations and genotype-phenotype correlations
It has been observed that EHK is caused predominantly due to mutations in the KRT1 and KRT10 genes. Several studies report that point mutations in the conserved regions are responsible for the development of this disease. Chipev et al identified a point mutation in the H1 sub-domain of the intermediate keratin 1 filament wherein a leucine to proline mutation occurs.37 This mutation was observed using electron microscopy and quantitative competition assay and it was concluded that the mutation resulted in seriously affected structure and organization of keratin filaments. Syder et al studied the correlation between disease severity and the extent of mutation for severe EHK incidences.73 They revealed through their studies that a mutation of arginine to histidine in the amino end of the α-helical rod domain of KRT10 and the tyrosine to cysteine mutation in the carboxy-domain of KRT1 hold significance over the severity of EHK. Virtanen et al . studied an interesting mutation in which a complex insertion/deletion led to the first ever reported case of deletion of an entire exon that is exon 6 of KRT1 which resulted in the removal of 42 amino acids from the 2B helix of the protein resulting in a severe form of EHK.78 It has been observed that the genotype-phenotype correlations in patients suffering from EHK are very complex. It is a disorder in which the genotype-phenotype relationships are dependent on the mutations in the keratin gene and on the position of the mutation within the gene. The correlation is also understood based on level of mutated allele expression and the functional significance of the amino acid substitution. Clinical studies along with in vivo studies on transgenic mice have been shown to support the theory of mutation in keratin genes resulting in varied phenotype.79 Research also states that there are mutations in the keratin genes wherein the genetic background is responsible in the modulation of the phenotype. Understanding the molecular mechanisms of these mutations can lead the path to gene therapy and gene replacement technologies in order to alleviate these syndromes.
Recent updates in EHK-associated mutations
Chen et al reported a case of heterozygous missense mutation (exon 1 of KRT10) with a T to Ctransition (c.475T>C) leading to proline at amino acid position 159 (p.Ser159Pro). The cases showed severe forms of EHK.80 A novel mutation in KRT1 (c.562A>C; p.N188H) was reported by Eskin-Schwartz et al. While histology showed cell-cell dissociation in epidermis with foci of acantholysis, epidermolytic changes were not observed. Reduced expression of several desmosomal proteins including desmoglein 1, desmoplakin, plakoglobin, E-cadherin as well as keratin 10 were revealed by immunostaining, suggesting limitations in diagnosis of EHK by histology alone.81 Some other mutations that could be interesting to explore are KDSR (3-ketodihydrosphingosine reductase),82 PNPLA1,83 SDR9C7,84 and ELOVL1.85 Terrinoni et al (2018) have shown additional roles of KRT1 and KRT10 associated mutations in development of a rare manifestation of epidermolytic ichthyosis—ichthyosis hystrix of Curth-Macklin.86 While co-occurrence of other diseases is not common, recently, coincidental latent tuberculosis and erythema annulare centrifugum was reported in a 12-year-old female.87 Such associations need further explorations.
Diagnosis of the disease
The diagnosis of patients with EHK is generally made on clinical grounds; nonetheless, until now just a single clinical scoring framework that incorporates light dim scaling, keratosis pilaris and expanded palmoplantar markings has been proposed for the finding of EHK. In addition to cosmetic problems, EHK patients normally suffer from a progression of medicinal issues that are specifically or indirectly identified with pathophysiological parameters such as cellular collapse, blistering, and impaired barrier function. The compensatory hyperproliferation of these physiological entities leads to hyperkeratosis.
Clinical inspection and dermoscopy
A dermatologist can assess the condition and use a dermoscope for better visualization. Hyperkeratosis can vary from mild to severe and is typically more prominent over joints and in flexural sites. The scale is classically described as corrugated or cardboard-like. Some patients also experience joint contractures. Hair, nails, and teeth are normal and ectropion is generally absent. Superficial bacterial infections are common and are often associated with a characteristic odor of the skin. Some of the basic symptoms that are in primary focus for the prognosis includes xerosis, pruritus, painful fissuring of the thickened skin, erythema, anhidrosis, ectropion, and decreased range of motion at joints and are mentioned in Table 1.
 | Table 1 Common symptoms and pathophysiological parameters for epidermolytic hyperkeratosis |
Biomarker and electron microscopy
Immunohistochemistry (IHC) can be of assistance in the conclusion of EHK and ARCI, if antibodies are accessible against the suspected applicant protein and the patient’s mutation(s) causes a deletion of particularly anomalous protein articulation in the epidermis.88 Electron microscopy patterns in the upper epidermis were one of the diagnostic methods for commenting upon the presence of a condition of EHK or its types in the past.89 In any case, electron microscopy is expensive, tedious and requires unique neurotic skill not generally accessible. Electron microscopy investigation is nevertheless, extremely valuable in describing the cell organelles in granular cells, and unusual lipid structures in the epidermis utilizing ruthenium tetroxide postobsession, which perfectly images the lipid envelopes and lipid bilayers in the stratum corneum.90–92
Histopathological
The histological findings using hematoxylin and eosin cannot be uniquely associated with EHK. Typical findings include marked hyperkeratosis, a thick granular layer, coarse keratohyaline granules, and vacuolar degeneration of the upper epidermis. Occasionally, deeper granular cells become dense, enlarged, and irregularly shaped, and these masses appear to be keratohyaline granules. In addition to this, dyskeratosis is also found . in varying concentrations.93 Patients whose pathologic slides demonstrate continuous involvement of the entire horizontal epidermis with these distinctive findings are more likely to have generalized disease; those with focal involvement revealing skip areas of normal epidermis are more likely to have a mosaic form of epidermolytic ichthyosis.39
Genetic testing
Dideoxy sequencing (Sanger sequencing method) was considered to be the inimitable standard for the diagnosis of EHK by which the coding some portion of the suspected EHK causing quality was dissected, exon by exon. In recent times, advances in massive parallel sequencing have been produced and applied for substantial scale hereditary diagnostics, particularly in genetically heterogeneous maladies. However, the next generation sequencing cannot match up with the clinical analysis of the different forms of ichthyosis, but the multigene-panel sequencing that comprises all suspected candidate genes within one analysis is the gold standard diagnostic approach.94,95
Fetal screening for EHK
Various approaches are employed for screening an affected fetus, namely: (1) amniocentesis for chromosome analysis, biochemical analysis and for cultured fibroblasts; (2) for fetal skin biopsy and for visual inspection, fetoscopy; (3) maternal serum screening.96–99 Fetoscopy along with skin biopsy and amniocentesis are the most commonly used diagnostic procedures.99,100 The epidermal development starts in the initial weeks of life, wherein it comprises of a monolayered sheet of cells and eventually by the end of the second trimester, it develops into a stratified squamous epithelium.101 For the onset of keratinization, there is a regional variation, that is it occurs by 24 weeks throughout the skin whereas it occurs in about 15 weeks in the hair follicles.102 Before 24 weeks’ gestation, occurrence of precocious hyperkeratosis and keratinization observed via fetal skin biopsy can be diagnosed as epidermolytic hyperkeratosis.97 Irrespective of these advanced techniques being present, the broad applications of diagnostic screening are being curbed due to several limitations such as limited sensitivity of analysis of a mutation or genetic heterogeneity.103
Several modern methods based on optical and spectroscopic techniques such as Raman spectroscopy and optical coherence tomography have shown potential in in vivo identification of diseases such as cancers, including skin cancers and other skin disorders.104–111 These modalities can be explored for EHK and other skin conditions as well. A recent study by Lima et al has shown classification of nonmelanoma skin cancer from actinic keratosis (AK) and normal skin112 suggesting potential of the technique in exploring other skin conditions including EHK.
Strategies for treatment of EHK
A systematic review of therapies, clinical trials of drugs and treatment strategies have been discussed in the following section corroborated from different sources.
One of the primary aims in EHK therapy is to evacuate scales and to diminish uncomfortable dryness of the skin (xerosis) without causing excessively irritation. To achieve this, the following viewpoints must be taken into account before endorsing a treatment:
- The age and sex of the patient (children have a slender skin and a higher skin surface zone or body: weight proportion, in this manner expanding the hazard for systemic toxicity; pregnant women ought not to be presented to possibly teratogenic compounds).
- The type and seriousness of the disease (thick scales require keratolytic agents, xerosis requires just emollients, fissures, crevices and erosions may block the utilization of keratolytics and require antimicrobial treatment).
- The degree and location of the skin lesions (entire body application increases the risk for systemic toxicity; face and flexural locales normally require less potent treatment and are more in danger of skin irritation).113
Although there is no definitive treatment yet for EHK, several treatment strategies are based on the use of emollients, topical keratolytics, and oral retinoids.
Hydration, lubrication, keratolysis and antimicrobials
The first line of therapy includes the use of wide assortments of emollients and topical keratolytics that aim to expedite desquamation and enhance the appearance of the skin. These include urea, lactic acid, glycolic acid, glycerol, paraffin, propylene glycol, ammonium lactate, salicylic acid, tazarotene, N-acetyl-cysteine and a diversity of fatty creams.114
The mechanism of action of common additives in the emollients can be stated as compounds such as NaCl, urea, glycerol particularly act on hydrating the skin. Similarly, petrolatum and other lipids work on the lubrication and accompaniments such as α-hydroxy acids,21urea (>5%), propylene glycol, salicylic acid, N-acetylcysteinamide act on keratolysing the epidermal layer. Similarly, retinoids and calcipotriol act as modulators of differentiation antimicrobials.115 A summary is provided in Table 2. Aqua-glycolic lotion is a commercially available form of α-hydroxy acid. Lactic acid is available in a readymade prescription form of 12% ammonium lactate lotion (Lac-Hydrin) or as compounded by a prescription wherein 5–10% is suitable in a vehicle solvent.
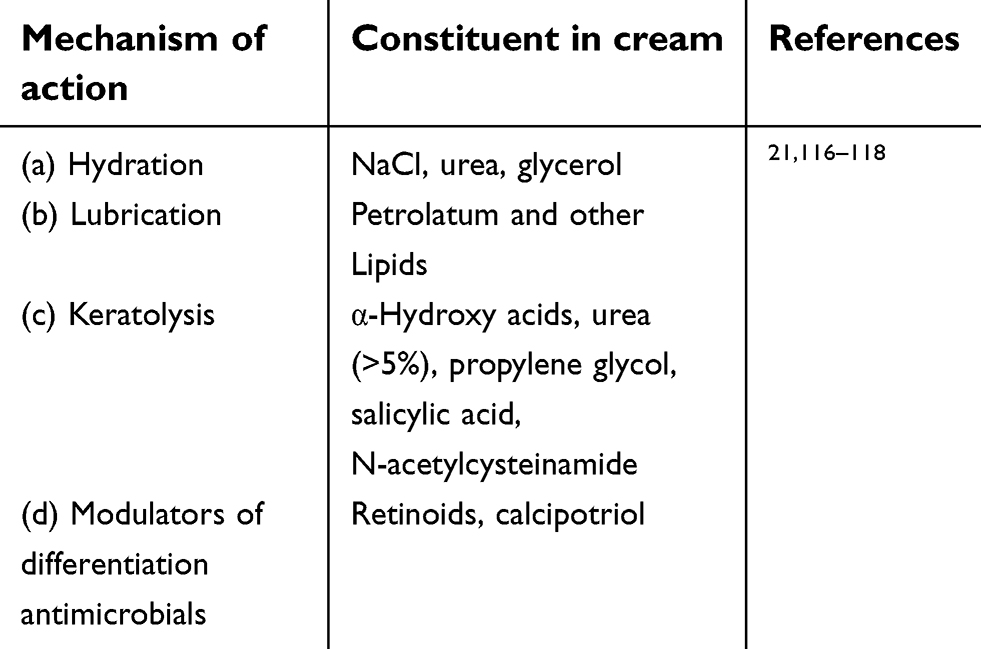 | Table 2 Mechanism of action of common additives in ointments |
Therapies for mild EHK
The most discerning method for treating ichthyosis vulgaris and X-linked recessive ichthyosis (XRI) would be to make up for absent or surplus components in the horny layer scilicet, inadequate breakdown products of filaggrin and intemperate cholesterol sulfate, respectively. Anyway, some achievement has been accounted for utilizing cholesterol-containing creams to offset the elevated cholesterol sulfate levels in XRI skin.119 Emollients comprising a physiological blend of ceramides and other skin lipids have been shown to reestablish the skin barrier in different conditions.120
Therapies for relentless EHK
By utilizing a combination of at least two keratolytic specialists and lotions in the same lipophilic cream base it is frequently conceivable to accomplish added or even synergistic impacts in lamellar ichthyosis without the need to use chafing concentrations of either agent alone.121,122
The therapy for EHK is quite assertive as hyperkeratosis must be decreased to limit the distorting and putrid scales, which harbor numerous microorganisms. An excessively strong keratolytic treatment will aggravate the condition by upsetting the epidermal obstruction further, expanding the danger of excruciating blisters and skin disintegrations inclined to disease. Applications of a few drugs like tazarotene,123 N-acetylcysteine,124,125 liarozole and calcipotriol126-128 have found some success as these medications likely act through decreasing epidermal hyperproliferation and affecting keratinocyte differentiation and hence, corneocyte function.
Effects of topical therapies
Aside from ephemeral stinging, pruritus or skin inflammation while applying the ointments, the early local side effects of topical treatment are generally negligible, and any long-haul lethal impact can ordinarily be limited if the topical cures are utilized effectively. Topical treatment requires significant investment of time and must be administered regularly, as per instructions.
The determination of a reasonable cream base (hydrophilic or lipophilic, nonocclusive or semi-occlusive) is in this manner of significance not only for ideal pharmacological impacts of the dynamic ingredients but also for their pharmacokinetics and transcutaneous penetration.129
Oral retinoids
Retinoids have a keratolytic impact that encourages the slivering of scales from the surface and counteracts unreasonable hyperkeratosis, prompting a more typical density and enhanced functioning of the horny layer.115 While the range of viability and toxicity of various retinoids are comparative and congruent, they are not indistinguishable. The aromatic retinoids generally have a more prominent impact on volar skin prompting advantage in the treatment of palmoplantar hyperkeratosis.130–132 One of the side effects of consumption of retinoids is the increased case of skin fragility.133 When the skin is prone to blistering in the case of EHK, the consumption of retinoids enhances the condition at a more intense level. It has been discovered that retinoid treatment, given topically as tretinoin, tazarotene or adapalene, and foundationally as acitretin, is more viable in patients with mutations in KRT10 in contrast with those with changes in KRTl. Most likely the former patients can more easily endure the unavoidable down-direction of KRT2 by retinoids.134 KRT2 and KRT1 give off an impression of being commonly replaceable in the heterodimerization of keratin fibers, clarifying why the presence of the former protein may decrease the negative effect of KRTlmutations. On the other hand, a down-direction of KRT2 is the in all probability explanation to why ichthyosis bullosa of Siemens (IBS), a shallow variation of EHK because of predominant KRT2 mutations,135 reacts so well to low doses of retinoids.136 An equivalent option to engineered retinoid treatment is to control the endogenous level of all-trans retinoic acid (tretinoin) by hindering its cellular catabolism in the skin with retinoic acid metabolism blocking agents (RAMBAs).137 Despite the fact that the danger of teratogenicity should still be considered amid RAMBA treatment, other retinoidal reactions give off an impression of being insignificant and there is no extended impact of the medication after cessation of treatment.
Isotretinoin (Accutane) treatment
Isotretinoin, a retinoid, is commercially available as Accutane (Roche, Switzerland). Due to known adverse effects, isotretinoin should be prescribed to patients with serious instances of ichthyosis when conventional treatment, including systemic antibiotics are not much effective. Further, among females, isotretinoin is indicated only for non-pregnant cases as it can cause severe birth defects, with abnormalities affecting face, eyes, ears, skull, central nervous system, cardiovascular framework, and thymus and parathyroid organs; teratogenicity is well recognized as a serious potential adverse effect. Fifty percent of pregnancies spontaneously abort, and of the remainder about half of the infants are born with cardiovascular or skeletal deformities.138 Isotretinoin acts by influencing cell-cycle progression, cellular differentiation, cell survival and apoptosis.139–141 It results in a significant reduction in sebum production, influences comedogenesis, lowers the effect of ichthyosis, has anti-inflammatory properties,141 and shows diminishing hyperkeratinization. However, accurate mechanism of action of isotretinoin as well as its influence on metabolics of keratinocytes can be further investigated.142
Conclusion
Addressing EHK is extremely important for a patient. Not only are the genetics and physiology of the disease important but also the psychological and social impact on the patient needs to be assessed. Preventive measures for EHK as such do not exist since EHK is a form of a genetic condition but specific medications for EHK such as α-hydroxyl acids or topical antibiotics have been used. α-Hydroxy acids are a group of naturally occurring, simple, hygroscopic acids which aid in the hydration of skin.143 Several compounds and formulations, including aqua-glycolic lotion, when used twice a day are more effective in reducing the severity of EHK than petrolatum-based creams.144 EHK is usually accompanied with a secondary infection which mostly is malodorous. To reduce odor as well as for a reduction in the incidences of such infections, topical antibiotics and sodium bicarbonate salts can be used.143 In order to distinguish EHK from the other subtypes of ichthyosis, prenatal diagnosis can be done for better prognosis. These specialized screening tests are generally labor-intensive, very expensive and the biggest drawback of them all being that they are performed in relatively few specific laboratories.
Molecular biology research and genetic advancements in ichthyosis have significantly improved the conventional nosology as well as prognosis of the different conditions. Gene therapy approaches can also be explored, however, safe and practical gene therapy techniques to either treat or prevent EHK still need to be developed.103 Future scope and advancements can be broadly categorized as: (1) genetic engineering mediated correction of disorders, (2) improved retinoid therapy to develop better oral medicines, (3) an enhanced understanding of fetal keratinization to aid in precise and an earlier prenatal diagnosis, (4) a developed mechanism for detection of carrier states as well as novel noninvasive methods based on optical and spectroscopic techniques for early detection.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Oji V, Tadini G, Akiyama M, et al. Revised nomenclature and classification of inherited ichthyoses: results of the first ichthyosis consensus conference in soreze 2009. J Am Acad Dermatol. 2010;63(4):607–641. doi:10.1016/j.jaad.2009.11.020
2. Müller FB, Huber M, Kinaciyan T, et al. A human keratin 10 knockout causes recessive epidermolytic hyperkeratosis. Hum Mol Genet. 2006;15(7):1133–1141. doi:10.1093/hmg/ddl028
3. Goldsmith LAJPMG. The ichthyosis. Prog Med Genet. 1976;1:185–240.
4. Esterly NBJP. The ichthyosiform dermatoses. Pediatrics. 1968;42(6):990–1004.
5. Hirone TJM. Electron microscopic studies of ichthyosis and congenital ichthyosiform erythroderma. J Electron Microsc (Tokyo). 1969;18(1):63–72.
6. Ishibashi Y, Klingmüller G. Erythrodermia ichthyosiformis congenita bullosa Brocq. Über die sogenannte granulöse Degeneration. Arch Dermatol Res. 1968;233(1):11–32.
7. Klaus S, Weinstein GD, Frost P. Localized epidermolytic hyperkeratosis: a form of keratoderma of the palms and soles. Arch Dermatol. 1970;101(3):272–275.
8. Schnyder U. Inherited ichthyoses. Arch Dermatol. 1970;102(3):240–252.
9. Gutierrez JA, Hannoush ZC, Vargas LG, et al. A novel nonsense mutation in keratin 10 causes a familial case of recessive epidermolytic ichthyosis. Mol Genet Genomic Med. 2013;1(2):108–112. doi:10.1002/mgg3.6
10. Bonifas JM, Bare JW, Chen MA, et al. Linkage of the epidermolytic hyperkeratosis phenotype and the region of the type II keratin gene cluster on chromosome 12. J Invest Dermatol. 1992;99(5):524–527.
11. Rothnagel J, Dominey A, Dempsey L, et al. Mutations in the rod domains of keratins 1 and 10 in epidermolytic hyperkeratosis. Science. 1992;257(5073):1128–1130.
12. Ackerman ABJAD. Histopathologic concept of epidermolytic hyperkeratosis. Arch Dermatol. 1970;102(3):253–259.
13. Anton-Lamprecht I. Genetically induced abnormalities of epidermal differentiation and ultrastructure in ichthyoses and epidermolyses: pathogenesis, heterogeneity, fetal manifestation, and prenatal diagnosis. J Invest Dermatol. 1983;81:S149–S156.
14. Williams ML, Elias P. Genetically transmitted, generalized disorders of cornification: the ichthyoses. Dermatol Clin. 1987;5(1):155–178.
15. Cheng J, Syder AJ, Yu Q-C, Letal A, Paller AS, Fuchs EJC. The genetic basis of epidermolytic hyperkeratosis: a disorder of differentiation-specific epidermal keratin genes. Cell. 1992;70(5):811–819.
16. Ishida-Yamamoto A, McGrath JA, Judge MR, Leigh IM, Lane EB, Eady R. Selective involvement of keratins K1 and K10 in the cytoskeletal abnormality of epidermolytic hyperkeratosis (bullous congenital ichthyosiform erythroderma). J Invest Dermatol. 1992;99:1.
17. Chipev CC, Yang J-M, DiGiovanna JJ, et al. Preferential sites in keratin 10 that are mutated in epidermolytic hyperkeratosis. Am J Hum Genet. 1994;54(2):179.
18. Nishikawa N, Tanizawa Y, Tanaka S, Horiguchi Y, Matsuno H, Asakura T. pH dependence of the coiled-coil structure of keratin intermediate filament in human hair by 13 C NMR spectroscopy and the mechanism of its disruption. Polym J. 1998;30(2):125.
19. Steinert PM, Roop D. Molecular and cellular biology of intermediate filaments. Annu Rev Biochem. 1988;57(1):593–625. doi:10.1146/annurev.bi.57.070188.003113
20. Bader B, Jahn L, Franke W. Low level expression of cytokeratins 8, 18 and 19 in vascular smooth muscle cells of human umbilical cord and in cultured cells derived therefrom, with an analysis of the chromosomal locus containing the cytokeratin 19 gene. Eur J Cell Biol. 1988;47(2):300–319.
21. Lessin SR, Huebner K, Isobe M, Croce CM, Steinert P. Chromosomal mapping of human keratin genes: evidence of non-linkage. J Invest Dermatol. 1988;91(6):572–578.
22. Popescu N, Bowden P, DiPaolo J. Two type II keratin genes are localized on human chromosome 12. Hum Genet. 1989;82(2):109–112.
23. Rosenberg M, Fuchs E, Le Beau M, Eddy R, Shows TJC, Research G. Three epidermal and one simple epithelial type II keratin genes map to human chromosome 12. Cytogenet Cell Genet. 1991;57(1):33–38. doi:10.1159/000133109
24. Coulombe PA, Fuchs E. Elucidating the early stages of keratin filament assembly. J Cell Biol. 1990;111(1):153–169.
25. Steinert P. The two-chain coiled-coil molecule of native epidermal keratin intermediate filaments is a type I-type II heterodimer. J Biol Chem. 1990;265(15):8766–8774.
26. Fuchs E, Green HJC. Changes in keratin gene expression during terminal differentiation of the keratinocyte. Cell. 1980;19(4):1033–1042.
27. Roop DR, Hawley-Nelson P, Cheng CK, Yuspa S. Keratin gene expression in mouse epidermis and cultured epidermal cells. Proc Natl Acad Sci U S A. 1983;80(3):716–720.
28. Schweizer J, Kinjo M, Fürstenberger G, Winter HJC. Sequential expression of mRNA-encoded keratin sets in neonatal mouse epidermis: basal cells with properties of terminally differentiating cells. Cell. 1984;37(1):159–170.
29. Woodcock-Mitchell J, Eichner R, Nelson WG, Sun -T-T. Immunolocalization of keratin polypeptides in human epidermis using monoclonal antibodies. J Cell Biol. 1982;95(2):580–588.
30. Anton-Lamprecht I, Schnyder U. Ultrastructure of inborn errors of keratinization. Arch Dermatol Forsch. 1974;250(3):207–227.
31. Holbrook KA, Dale BA, Sybert VP, Sagebiel R. Epidermolytic hyperkeratosis: ultrastructure and biochemistry of skin and amniotic fluid cells from two affected fetuses and a newborn infant. J Invest Dermatol. 1983;80(4).
32. Wilgram GF, Caulfield J. An electron microscopic study of epidermolytic hyperkeratosis: with a special note on the keratinosome as the fourth structural factor in the formation of the horny layer. Arch Dermatol. 1966;94(2):127–143.
33. Ogawa H, Hattori M, Ishibashi Y. Abnormal fibrous protein isolated from the stratum corneum of a patient with bullous congenital ichthyosiform erythroderma (BCIE). Arch Dermatol Res. 1979;266(2):109–116.
34. Kanitakis J, Misery L, Nicolas J, et al. Disseminated superficial porokeratosis in a patient with AIDS. Br J Dermatol. 1994;131(2):284–289.
35. Rothnagel JJCOCB. Keratin 5 has also been implicated as the genetic defect in another family affected with EBS (24). Cellular Organelles, Volume 2 (Principles of Medical Biology). 1992;4:94.
36. Bonifas JJJID. Epidermolytic hyperkeratosis: linkage to keratin gene regions on chromosomes 12q and 17q in two families. J Invest Dermatol. 1992;98(573).
37. Chipev CC, Korge BP, Markova N, et al. A leucine→ proline mutation in the H1 subdomain of keratin 1 causes epidermolytic hyperkeratosis. Cell. 1992;70(5):821–828.
38. Obu H, Adimora G, Obumneme-Anyim I, Ndu I, Asinobi I. Collodion baby: A report of 4 cases. Niger J Paediatr. 2013;40(3):307–310.
39. Ross R, DiGiovanna JJ, Capaldi L, Argenyi Z, Fleckman P, Robinson-Bostom L. Histopathologic characterization of epidermolytic hyperkeratosis: a systematic review of histology from the national registry for ichthyosis and related skin disorders. J Am Acad Dermatol. 2008;59(1):86–90. doi:10.1016/j.jaad.2008.02.031
40. Güzel AI, Tokmak A, Kara AS, AJBJoM Y, Research M. Harlequin Ichthyosis: case Report of a Rare Type of Ichthyosis. BMJ Case Rep. 2015;5(4):557.
41. Vidyadhar TVVJP. A review on different types of ichthyosis and management. PharmaTutor 2015;3(1):25–31.
42. Küster WJDA. Ichthyosen: vorschläge für eine verbesserte Therapie. Dtsch Ärztebl 2006;103(24):A1684–A1689.
43. Rand RE, Baden H. The ichthyoses—a review. J Am Acad Dermatol. 1983;8(3):285–305.
44. Huber M, Rettler I, Bernasconi K, et al. Mutations of keratinocyte transglutaminase in lamellar ichthyosis. Science. 1995;267(5197):525–528.
45. Frost P, Weinstein GD, Bothwell JW, Wildnauer R. Ichthyosiform dermatoses: III. Studies of transepidermal water loss. Arch Dermatol. 1968;98(3):230–233.
46. COOPER MF W, SHUSTER SJBJoD P, SHUSTER SJBJoD P, Shuster S. Acquired ichthyosis and impaired dermal lipogenesis in Hodgkin‘s disease. Br J Dermatol. 1980;102(6):689–693.
47. Glazebrook A, Tomaszewski W. Syphilology. Ichthyosiform atrophy of the skin in hodgkin‘s disease: report of a case, with reference to vitamin A metabolism. Archives of Dermatology and Syphilology. 1944;50(2):85–89.
48. McCann S, Barry D, Temperley I, Weir D. Ichthyosis and marrow involvement in malignant histiocytosis of the intestine. Br J Haematol. 1981;48(2):281–285.
49. Ronchese F, Gates D. Ichthyosiform atrophy of the skin in Hodgkins disease. N Engl J Med. 1956;255(6):287–289. doi:10.1056/NEJM195608092550608
50. Sneddon IJBMJ. Acquired ichthyosis in Hodgkin‘s disease. J Chronic Dis. 1955;1(4916):763.
51. Stevanović D. Hodgkin‘s disease of the skin: acquired ichthyosis preceding tumoral and ulcerating lesions for seven years. Arch Dermatol. 1960;82(1):96–99.
52. Welsh J, Epstein EJJ. Acquired ichthyosis in Hodgkin‘s disease: report of a case. J Am Med Assoc. 1952;148:1221–1223.
53. Fuchs E, Esteves RA, Coulombe PA. Transgenic mice expressing a mutant keratin 10 gene reveal the likely genetic basis for epidermolytic hyperkeratosis. Proc Natl Acad Sci U S A. 1992;89(15):6906–6910.
54. McLean WI. Genetic disorders of palm skin and nail. J Anat. 2003;202(1):133–141.
55. Fuchs E, Coulombe P, Cheng J, Burks AW. Genetic bases of epidermolysis bullosa simplex and epidermolytic hyperkeratosis. Int Arch Allergy Immunol. 1994;103. doi:10.1159/000236606
56. Wolf FT, Frredberg IN, Austen KF. Dermatology in General Medicine.
57. Moll R, Franke WW, Schiller DL, Geiger B, Krepler RJC. The catalog of human cytokeratins. Patterns of Expression in Normal Epithelia, Tumors and Cultured Cells. 1982;31(1):11–24.
58. Nelson WG, Sun -T-T. The 50-and 58-kdalton keratin classes as molecular markers for stratified squamous epithelia: cell culture studies. J Cell Biol. 1983;97(1):244–251.
59. Roop DR, Huitfeldt H, Kilkenny A, Yuspa SHJD. Regulated expression of differentiation-associated keratins in cultured epidermal cells detected by monospecific antibodies to unique peptides of mouse epidermal keratins. Differentiation. 1987;35(2):143–150.
60. Conway JF, Parry D. Intermediate filament structure: 3. Analysis of sequence homologies. J Biol Macromol. 1988;10(2):79–98.
61. Fuchs E, Weber K. Intermediate filaments: structure, dynamics, function and disease. Annu Rev Biochem. 1994;63(1):345–382. doi:10.1146/annurev.bi.63.070194.002021
62. Geisler N, Schünemann J, Weber K. Chemical cross‐linking indicates a staggered and antiparallel protofilament of desmin intermediate filaments and characterizes one higher‐level complex between protofilaments. Eur J Biochem. 1992;206(3):841–852.
63. Steinert PM, Marekov LN, Fraser RB, Parry D. Keratin intermediate filament structure: crosslinking studies yield quantitative information on molecular dimensions and mechanism of assembly. J Mol Biol. 1993;230(2):436–452. doi:10.1006/jmbi.1993.1161
64. Vassar R, Coulombe PA, Degenstein L, Albers K, Fuchs EJC. Mutant keratin expression in transgenic mice causes marked abnormalities resembling a human genetic skin disease. Cell. 1991;64(2):365–380.
65. Compton JG, DiGiovanna JJ, Santucci SK, et al. Linkage of epidermolytic hyperkeratosis to the type II keratin gene cluster on chromosome 12q. Nat Genet. 1992;1(4):301.
66. Kimonis V, Yang J-M, Doyle SZ, Bale SJ, Compton JG, DiGiovanna J. A mutation in the V1 end domain of keratin 1 in non-epidermolytic palmar-plantar keratoderma. J Invest Dermatol. 1994;103(6):764–769.
67. Pulkkinen L, Christiano AM, Knowlton RG, Uitto J. Epidermolytic hyperkeratosis (bullous congenital ichthyosiform erythroderma). Genetic linkage to chromosome 12q in the region of the type II keratin gene cluster. J Clin Invest. 1993;91(1):357–361.
68. Reis A, Hennies H-C, Langbein L, et al. Keratin 9 gene mutations in epidermolytic palmoplantar keratoderma (EPPK). Nat Genet. 1994;6(2):174.
69. Rogaev EI, Rogaeva EA, Ginter EK, et al. Identification of the genetic locus for keratosis palmaris et plantaris on chromosome 17 near the RARA and keratin type I genes. Nat Genet. 1993;5(2):158.
70. Torchard D, Blanchet-Bardon C, Serova O, et al. Epidermolytic palmoplantar keratoderma cosegregates with a keratin 9 mutation in a pedigree with breast and ovarian cancer. Nat Genet. 1994;6(1):106.
71. Huber M, Scaletta C, Benathan M, et al. Abnormal keratin 1 and 10 cytoskeleton in cultured keratinocytes from epidermolytic hyperkeratosis caused by keratin 10 mutations. J Invest Dermatol. 1994;102(5):691–694.
72. McLean WI, Eady RA, Dopping-Hepenstal PJ, et al. Mutations in the rod 1A domain of keratins 1 and 10 in Bullous Congenital Ichthyosiform Erythoderma (BCIE). J Invest Dermatol. 1994;102(1):24–30.
73. Syder AJ, Yu Q-C, Paller AS, Giudice G, Pearson R, Fuchs E. Genetic mutations in the K1 and K10 genes of patients with epidermolytic hyperkeratosis. Correlation between location and disease severity. J Clin Invest. 1994;93(4):1533–1542. doi:10.1172/JCI117132
74. Yang J-M, Chipev CC, DiGiovanna JJ, et al. Mutations in the H1 and 1A domains in the keratin 1 gene in epidermolytic hyperkeratosis. J Invest Dermatol. 1994;102(1):17–23.
75. Williams ML, Elias PM. Ichthyosis. Genetic heterogeneity, genodermatoses, and genetic counseling.. Arch Dermatol. 1986;122(5):529–531.
76. Traupe H. The Ichthyoses: A Guide to Clinical Diagnosis, Genetic Counseling, and Therapy. Berlin, Heidelberg: Springer Science & Business Media; 2012.
77. Yoneda K. Inherited ichthyosis: syndromic forms. J Dermatol. 2016;43(3):252–263. doi:10.1111/1346-8138.13284
78. Virtanen M, Vahlquist A, Smith SK, Vahlquist A, Bowden PE. Splice site and deletion mutations in keratin (KRT1 and KRT10) genes: unusual phenotypic alterations in Scandinavian patients with epidermolytic hyperkeratosis. J Invest Dermatol. 2003;121(5):1013–1020. doi:10.1046/j.1523-1747.2003.12534.x
79. Bickenbach JR, Longley MA, Bundman DS, et al. A transgenic mouse model that recapitulates the clinical features of both neonatal and adult forms of the skin disease epidermolytic hyperkeratosis. Differentiation. 1996;61(2):129–139. doi:10.1046/j.1432-0436.1996.6120129.x
80. Chen PJ, Li CX, Wen J, et al. S159P mutation of keratin 10 gene causes severe form of epidermolytic hyperkeratosis. J Eur Acad Dermatol Venereol. 2016;30(10):e102–e104. doi:10.1111/jdv.13345
81. Eskin-Schwartz M, Drozhdina M, Sarig O, et al. 381 Epidermolytic ichthyosis sine epidermolysis. J Invest Dermatol. 2016;136(5):S67.
82. Boyden LM, Vincent NG, Zhou J, et al. Mutations in KDSR cause recessive progressive symmetric erythrokeratoderma. Am J Human Genet. 2017;100(6):978–984. doi:10.1016/j.ajhg.2017.05.003
83. Ohno Y, Kamiyama N, Nakamichi S, Kihara A. PNPLA1 is a transacylase essential for the generation of the skin barrier lipid ω-O-acylceramide. Nat Commun. 2017;8:14610. doi:10.1038/ncomms14610
84. Takeichi T, Nomura T, Takama H, et al. Deficient stratum corneum intercellular lipid in a Japanese patient with lamellar ichthyosis with a homozygous deletion mutation in SDR 9C7. Br J Dermatol. 2017;177(3):e62–e64. doi:10.1111/bjd.15315
85. Kutkowska-Kaźmierczak A, Rydzanicz M, Chlebowski A, et al. Dominant ELOVL1 mutation causes neurological disorder with ichthyotic keratoderma, spasticity, hypomyelination and dysmorphic features. J Med Genet. 2018;55(6):408–414. doi:10.1136/jmedgenet-2017-105172
86. Terrinoni A, Didona B, Caporali S, et al. Role of the keratin 1 and keratin 10 tails in the pathogenesis of ichthyosis hystrix of Curth Macklin. PLoS One. 2018;13(4):e0195792. doi:10.1371/journal.pone.0195792
87. Hirt P, Price A, Alwunais K, Schachner L. An interesting case of coincidental epidermolytic hyperkeratosis and erythema annulare centrifugum in the setting of latent tuberculosis in a 12‐year‐old female. Int J Dermatol. 2019. doi:10.1111/ijd.14339
88. Diociaiuti A, Fortugno P, El Hachem M, et al. Early immunopathological diagnosis of ichthyosis with confetti in two sporadic cases with new mutations in keratin 10. Acta Derm Venereol. 2014;94(5):579–582. doi:10.2340/00015555-1796
89. Anton-Lamprecht I, Brenner RJ, Williams LW, Burks AW. Ultrastructural identification of basic abnormalities as clues to genetic disorders of the epidermis. Int Arch Allergy Immunol. 1994;103. doi:10.1159/000236606
90. Elias PM. Ichthyoses: Clinical, Biochemical, Pathogenic and Diagnostic Assessment. Basel, Switzerland: Karger AG; 2010.
91. Lin TK, Crumrine D, Ackerman LD, et al. Cellular changes that accompany shedding of human corneocytes. J Invest Dermatol. 2012;132(10):2430–2439. doi:10.1038/jid.2012.173
92. Paller AS, van Steensel MAM, Rodriguez-Martín M, et al. PATHOGENESIS-BASED THERAPY REVERSES CUTANEOUS ABNORMALITIES IN AN INHERITED DISORDER OF DISTAL CHOLESTEROL METABOLISM. J Invest Dermatol. 2011;131(11):2242–2248. doi:10.1038/jid.2011.189
93. Bergman R, Khamaysi Z, Sprecher E. A unique pattern of dyskeratosis characterizes epidermolytic hyperkeratosis and epidermolytic palmoplantar keratoderma. Am J Dermatopathol. 2008;30(2):101–105. doi:10.1097/DAD.0b013e3181614898
94. Eckl KM, de Juanes S, Kurtenbach J, et al. Molecular analysis of 250 patients with autosomal recessive congenital ichthyosis: evidence for mutation hotspots in ALOXE3 and allelic heterogeneity in ALOX12B. J Invest Dermatol. 2009;129(6):1421–1428. doi:10.1038/jid.2008.409
95. Fischer J. Autosomal recessive congenital ichthyosis. J Invest Dermatol. 2009;129(6):1319–1321. doi:10.1038/jid.2009.57
96. Arnold M-L, Anton-Lamprecht I. Prenatal diagnosis of epidermal disorders. In: Prenatal Diagnosis of Heritable Skin Diseases. Vol. 16. Gedde-Dahl Jr T, Wuepper KD, editors. Switzerland: Karger Publishers; 1987:120–128.
97. Baden H. Keratinizing disorders. Genetic Disorders of the Skin. Alper JC, editor. St Louis: Mosby Yearbook; 1991:170–194.
98. Sybert VP, Holbrook K. Prenatal diagnosis and screening. Dermatoligic clinics. 1987;5(1):17–41.
99. Williams M. The ichthyoses—pathogenesis and prenatal diagnosis: a review of recent advances. Pediatr Dermatol. 1983;1(1):1–24.
100. Elias S. Use of fetoscopy for the prenatal diagnosis of hereditary skin disorders. In: Prenatal Diagnosis of Heritable Skin Diseases. Vol. 16. Basel, Switzerland; Karger Publishers; 1987:1–13.
101. Holbrook KA, Wolff K. The structure and development of skin. In: Fitzpatrick TB, Eisen AZ, Wolff K, et al, editors. Dermatology in general medicine. 4th ed. New York: McGraw-Hill; 1993:159–171.
102. Perry TB, Holbrook KA, Hoff MS, Hamilton EF, Senikas V, Fisher C. Prenatal diagnosis of congenital non‐bullous ichthyosiform erythroderma (lamellar ichthyosis). Prenat Diagn. 1987;7(3):145–155.
103. Richard G. Molecular genetics of the ichthyoses. Am J Med Genet. 2004year>;131C:32–44. doi:10.1002/ajmg.c.30032
104. Prashanth Panta C-W-L, Kumar P, Tuan-Shu H, et al. Optical coherence tomography: emerging in vivo optical biopsy technique for oral cancer. In: Oral Cancer Detection. Springer Nature; 2018.
105. Kumar P, Cm K. Optical Techniques: Investigations in Oral Cancers.
106. Bhattacharjee T, Kumar P, Fillipe L Automated pre-processing and multivariate vibrational spectra analysis software for rapid results in clinical settings.
107. Kumar P. Raman spectroscopy as a promising noninvasive tool in brain cancer detection. J Innov Opt Health Sci. 2017;10(05):1730012. doi:10.1142/S1793545817300129
108. Kumar P, Bhattacharjee T, Pandey M, Hole AR, Ingle A, Murali Krishna C. Raman spectroscopy in experimental oral carcinogenesis: investigation of abnormal changes in control tissues. J Raman Spectrosc. 2016;47:11. doi:10.1002/jrs.4977
109. Kumar P, Bhattacharjee T, Ingle A, Maru G, Krishna CM. Raman spectroscopy of experimental oral carcinogenesis: study on sequential cancer progression in hamster buccal pouch model. Technol Cancer Res Treat. 2016;15(5):NP60–72. doi:10.1177/1533034615598622
110. Dalal K, Elanchezhiyan D, Maran VB, et al. Optical, spectroscopic, and Doppler evaluation of “normal” and “abnormal” reflexology areas in lumbar vertebral pathology: a case study. Case Rep Med. 2012;2012:904729. doi:10.1155/2012/904729
111. Sahu A, Yélamos O, Iftimia N, et al. Evaluation of a combined reflectance confocal microscopy–optical coherence tomography device for detection and depth assessment of basal cell carcinoma. JAMA Dermatol. 2018. doi:10.1001/jamadermatol.2018.2446
112. Lima AMF, Daniel CR, Navarro RS, et al. Discrimination of non-melanoma skin cancer and keratosis from normal skin tissue in vivo and ex vivo by Raman spectroscopy. Vib Spectrosc. 2019;100:131–141. doi:10.1016/j.vibspec.2018.11.009
113. Vahlquist A, Ganemo A, Virtanen M. Congenital ichthyosis: an overview of current and emerging therapies. Acta Derm Venereol. 2008;88(1):4–14. doi:10.2340/00015555-0415
114. Hernandez-Martin A, Aranegui B, Martin-Santiago A, Garcia-Doval I. A systematic review of clinical trials of treatments for the congenital ichthyoses, excluding ichthyosis vulgaris. J Am Acad Dermatol. 2013;69(4):544–549.e548. doi:10.1016/j.jaad.2013.05.017
115. Digiovanna JJ, Mauro T, Milstone LM, Schmuth M, Toro JR. Systemic retinoids in the management of ichthyoses and related skin types. Dermatol Ther. 2013;26(1):26–38. doi:10.1111/j.1529-8019.2012.01527.x
116. Tomita Y, Akiyama M, Shimizu H. Stratum corneum hydration and flexibility are useful parameters to indicate clinical severity of congenital ichthyosis. Exp Dermatol. 2005;14(8):619–624. doi:10.1111/j.0906-6705.2005.00341.x
117. Vahlquist A, Gånemo A, Virtanen M. Congenital ichthyosis: an overview of current and emerging therapies. Acta Derm Venereol. 2008;88(1):4–14. doi:10.2340/00015555-0415
118. Long MC. Ichthyosis with confetti: a rare diagnosis and treatment plan. BMJ Case Rep. 2014;2014:bcr2014204509. doi:10.1136/bcr-2014-204509
119. Lykkesfeldt G, Hyer H. TOPICAL CHOLESTEROL TREATMENT OF RECESSIVE X-LINKED ICHTHYOSIS. Lancet. 1983;322(8363):1337–1338. doi:10.1016/S0140-6736(83)91093-0
120. Chamlin SL, Kao J, Frieden IJ, et al. Ceramide-dominant barrier repair lipids alleviate childhood atopic dermatitis: changes in barrier function provide a sensitive indicator of disease activity. J Am Acad Dermatol. 2002;47(2):198–208.
121. Gånemo A, Virtanen M, Vahlquist A. Improved topical treatment of lamellar ichthyosis: a double-blind study of four different cream formulations. Br J Dermatol. 1999;141(6):1027–1032.
122. Kiistala R, Lauharanta J, Kanerva L. Transepidermal water loss and sweat gland response in lamellar ichthyosis before and during treatment with etretinate: report of three cases. Acta Derm Venereol. 1982;62(3):268–270.
123. Stege H, Hofmann B, Ruzicka T, Lehmann P. Topical application of tazarotene in the treatment of nonerythrodermic lamellar ichthyosis. Arch Dermatol. 1998;134(5):640–641.
124. Redondo P, Bauzá A. Topical N-acetylcysteine for lamellar ichthyosis. Lancet. 1999;354(9193):1880. doi:10.1016/S0140-6736(99)04245-2
125. Sarici SU, Sahin M, Yurdakök M. Topical N-acetylcysteine treatment in neonatal ichthyosis. Turk J Pediatr. 2003;45(3):245–247.
126. Bogenrieder T, Landthaler M, Stolz W. Bullous Congenital Ichthyosiform Erythroderma: Safe and Effective Topical Treatment with Calcipotriol Ointment in a Child. Vol. 83. 2003.
127. Kerkhof PCM. Biological activity of vitamin D analogues in the skin, with special reference to antipsoriatic mechanisms. Br J Dermatol. 1995;132(5):675–682.
128. Lucker GPH, Kerkhof PCM, Mr DÏJK, Steijlen PM. Effect of topical calcipotriol on congenital ichthyoses. Br J Dermatol. 1994;131(4):546–550.
129. Loden M, Maibach HI. Dry Skin and Moisturizers: Chemistry and Function. Taylor & Francis; 1999.
130. Melnik B, Glück S, Jungblut RM, Goerz G. Retrospective radiographic study of skeletal changes after long-term etretinate therapy. Br J Dermatol. 1987;116(2):207–212.
131. Roos TC, Jugert FK, Merk HF, Bickers DR. Retinoid Metabolism in the Skin. Pharmacol Rev. 1998;50(2):315–333.
132. Steijlen PM, Dooren-Greebe RJ, Kerkhof PCM. Acitretin in the treatment of lamellar ichthyosis. Br J Dermatol. 1994;130(2):211–214.
133. Katsambas AD, Lotti TM, Dessinioti C, D‘Erme AM. European Handbook of Dermatological Treatments. Springer Berlin Heidelberg; 2015.
134. Virtanen M, Gedde-Dahl T, Mörk JN, Leigh I, Bowden PE, Vahlquist A. Phenotypic/genotypic correlations in patients with epidermolytic hyperkeratosis and the effects of retinoid therapy on keratin expression. Acta Derm Venereol. 2001;81:163-170.
135. McLean WH, Morley SM, Lane EB, et al. Ichthyosis Bullosa of Siemens–A Disease Involving Keratin 2e. J Invest Dermatol. 1994;103(3):277–281.
136. Steijlen PM, Doorkn-Greebe RJ, Happle R, Kerkhof PCM. Ichthyosis bullosa of Siemens responds well to low-dosage oral retinoids. Br J Dermatol. 1991;125(5):469–471.
137. Stoppie P, Borgers M, Borghgraef P, et al. R115866 inhibits all-trans -retinoic acid metabolism and exerts retinoidal effects in rodents. J Pharmacol Exp Ther. 2000;293(1):304–312.
138. Stern RS. When a uniquely effective drug is teratogenic. Mass Medical Soc. 1989.
139. Fogh K, Voorhees JJ, Astrom A. Expression, purification, and binding properties of human cellular retinoic acid-binding protein type I and type II. Arch Biochem Biophys. 1993;300(2):751–755. doi:10.1006/abbi.1993.1104
140. Allenby G, Bocquel M-T, Saunders M, et al. Retinoic acid receptors and retinoid X receptors: interactions with endogenous retinoic acids. Proc National Acad Sci. 1993;90(1):30–34. doi:10.1073/pnas.90.1.30
141. Tsukada M, Schröder M, Orfanos CE, et al. 13-cis retinoic acid exerts its specific activity on human sebocytes through selective intracellular isomerization to all-trans retinoic acid and binding to retinoid acid receptors. J Investig Dermatol. 2000;115(2):321–327. doi:10.1046/j.1523-1747.2000.00066.x
142. Dalziel K, Barton S, Marks R. The effects of isotretinoin on follicular and sebaceous gland differentiation. Br J Dermatol. 1987;117(3):317–323.
143. Shwayder T, Ott F. All about ichthyosis. Pediatr Clin North Am. 1991;38(4):835–857.
144. Buxman M, Hickman J, Ragsdale W, Stretcher G, Krochmal L, Wehr R. Therapeutic activity of lactate 12% lotion in the treatment of ichthyosis: active versus vehicle and active versus a petrolatum cream. J Am Acad Dermatol. 1986;15(6):1253–1258.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.