Back to Journals » Clinical Epidemiology » Volume 11
Epidemiology of systemic sclerosis and systemic sclerosis-associated interstitial lung disease
Authors Bergamasco A, Hartmann N, Wallace L, Verpillat P
Received 19 October 2018
Accepted for publication 21 February 2019
Published 18 April 2019 Volume 2019:11 Pages 257—273
DOI https://doi.org/10.2147/CLEP.S191418
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Henrik Toft Sørensen
Aurore Bergamasco,1 Nadine Hartmann,2 Laura Wallace,3 Patrice Verpillat4
1YolaRx Consultants, Paris, France; 2Boehringer Ingelheim International GmbH, Ingelheim am Rhein, Germany; 3Boehringer Ingelheim Pharmaceuticals, Inc., Ridgefield, CO, USA; 4Boehringer Ingelheim Pharma GmbH & Co. KG, Ingelheim am Rhein, Germany
Background: Interstitial lung disease (ILD) is one of the leading causes of mortality in patients with systemic sclerosis (SSc). To further understand this patient population, we present the first systematic review on the epidemiology of SSc and SSc-associated ILD (SSc-ILD).
Methods: Bibliographic databases and web sources were searched for studies including patients with SSc and SSc-ILD in Europe and North America (United States and Canada). The systematic review was limited to publications in English, German, French, Spanish, Italian, and Portuguese, published between January 1, 2000 and February 29, 2016. For all publications included in the review, the methodologic quality was assessed. For each dimension and region, data availability in terms of quantity and consistency of reported findings was evaluated.
Results: Fifty publications reporting epidemiologic data (prevalence, incidence, demographic profile, and survival and mortality) were included; 39 included patients with SSc and 16 included patients with SSc-ILD. The reported prevalence of SSc was 7.2–33.9 and 13.5–44.3 per 100,000 individuals in Europe and North America, respectively. Annual incidence estimates were 0.6–2.3 and 1.4–5.6 per 100,000 individuals in Europe and North America, respectively. Associated ILD was present in ∼35% of the patients in Europe and ∼52% of the patients in North America. In Europe, a study estimated the prevalence and annual incidence of SSc-ILD at 1.7–4.2 and 0.1–0.4 per 100,000 individuals, respectively. In both Europe and North America, SSc-ILD was diagnosed at a slightly older age than SSc, with both presentations of the disease affecting 2–3 times more women than men. Ten-year survival in patients with SSc was reported at 65–73% in Europe and 54–82% in North America, with cardiorespiratory manifestations (including ILD) associated with poor prognosis.
Conclusion: This systematic review confirms that SSc and SSc-ILD are rare, with geographic variation in prevalence and incidence.
Keywords: epidemiology, interstitial lung disease, systemic sclerosis
Plain language summary
Systemic sclerosis (SSc) is an uncommon condition that results in hard, thickened areas of skin and additional problems with internal organs and blood vessels. Interstitial lung disease (ILD) affects the tissue and space around the air sacs of the lungs and is one of the leading causes of death in patients with SSc. As such, it is important to understand how this condition manifests to better recognize and treat patients. We identified 50 papers published between January 2000 and February 2016 that assessed how common SSc and SSc-associated ILD (SSc-ILD) are and/or provided information on the affected patient populations as well as survival/mortality rates in these patients. We found that there was wide variation in the reported frequency of SSc and SSc-ILD across Europe and North America. Both SSc and SSc-ILD most commonly affected women, and patients with SSc-ILD were diagnosed at a slightly older age. Manifestations of the disease affecting the heart and lungs, including ILD, were found to be associated with poorer prognosis in patients with SSc. Few reports on patients with SSc-ILD were identified in our review, so additional studies would be helpful to add to the currently available literature and enable a deeper understanding of the impact of SSc and SSc-ILD.
Introduction
Systemic sclerosis (SSc) is a rare, heterogeneous autoimmune disease characterized by sclerodermatous skin changes, vasculopathy, and involvement of internal organs, including the lungs.1 The etiology of SSc is largely unknown but is thought to involve both genetic and environmental factors.2 The primary event in SSc pathogenesis is thought to be injury to endothelial cells; this is followed by aberrant vascular and immune responses that lead to the excessive deposition and accumulation of extracellular matrix. The resultant progressive tissue remodeling can destroy tissue architecture and cause loss of organ function.3
Early studies consider the American College of Rheumatology (ACR) 1980 criteria to classify SSc. These require finding proximal involvement of the skin by sclerodermatous changes or two or more of the following minor criteria: sclerodactyly; digital pitting scars on the fingertips or loss of substance of the distal finger pad; or bilateral basilar pulmonary fibrosis.4 However, as these criteria have low sensitivity, particularly in cases of early or mild SSc, and do not consider the assessment of serum antibodies or vasculopathy,2 there have been subsequent attempts to improve on their limitations. The classification by LeRoy et al (19885) and the updated version by LeRoy and Medsger (20016) were used to distinguish the various cutaneous forms of the disease, depending on the extent of skin involvement. In patients with limited cutaneous SSc (lcSSc), skin sclerosis is distal and limited to the elbows, knees, and clavicles, whereas the diffuse cutaneous form of the disease (dcSSc) affects the whole body.7 The classification by LeRoy and Medsger also proposes a limited SSc subset with healthy skin.6 In 2013, a new 9-point classification system, endorsed by the ACR/European League Against Rheumatism (EULAR), was validated. This enabled the identification of early disease, thereby leading to earlier management, which may slow disease progression.8
Organ involvement occurs early in the course of the disease.9 The occurrence of associated complications is related to the severity and progressiveness of disease, with approximately 25% of the patients developing significant pulmonary involvement within 3 years of SSc diagnosis.10 Interstitial lung disease (ILD) constitutes one of the most common types of direct pulmonary involvement in SSc patients, characterized by diffuse parenchymal infiltrative processes that may lead to fibrosis. ILD is most frequently observed in those with dcSSc9,11,12 and is a major contributor to the morbidity and mortality associated with SSc.1 There is currently no consensus on how ILD in SSc should be diagnosed, resulting in discrepancies in the assessment and reported frequency of occurrence of this complication. Diagnosis of SSc-associated ILD may be performed using several auxiliary diagnostic tools such as pulmonary function tests and chest high-resolution computed tomography (HRCT), with the latter generally accepted as the “gold standard” for evaluating the extent of lung involvement.1,13–16 It should be noted that there is no systematic use of biopsies or histology to aid diagnosis due to the presence of an ILD-associated disease.
The aim of therapeutic treatment for SSc and SSc-associated ILD (SSc-ILD) is to improve quality of life by minimizing specific organ involvement and subsequent life-threatening diseases. In 2009, the EULAR and EULAR Scleroderma Trials and Research (EUSTAR) groups published evidence-based recommendations for the treatment of SSc.17 The treatment of lung disease is not yet fully defined; however, immunosuppressive therapies are often used with the aim of reducing pulmonary interstitial inflammation.18 Based on the results of the Scleroderma Lung Study II19 and several other low-quality studies20 that suggest that mycophenolate alone could control active ILD with a tolerable safety profile, most experts are transitioning from cyclophosphamide to this immunosuppressive therapy for long-term disease control. An update to the EULAR guidelines was published in 2016 to include new treatment options for SSc-related organ complications. It also provides direction for future clinical research in SSc.21
It is important to gain further understanding of the SSc and SSc-ILD patient populations to better inform the diagnosis and treatment of these patients. In this paper, we present the results of a systematic review on the epidemiology of SSc and SSc-ILD, including data on prevalence, incidence, demographic profile, and survival and mortality.
Materials and methods
Search strategy
Literature searches were conducted using Medline and Embase electronic bibliographical databases. Search strategies combined thesaurus terms (MeSH and Emtree terms for Medline and Embase, respectively) and free-text keywords; full search strategies are provided in
In addition, using similar combinations of the previously defined keywords, pragmatic searches of web sources were conducted using Google and Google Scholar. Websites of relevant scientific societies, medical associations, and conference proceedings were also reviewed (
Study selection process
Results from the literature searches were uploaded into EPPI-Reviewer 4 (Social Science Research Unit, UCL Institute of Education, University of London, UK). Duplicate publications were identified and excluded using an automated procedure followed by manual quality control. The titles and abstracts of all remaining publications were independently screened by two reviewers, with any conflicts resolved by a third reviewer, according to eligibility criteria. Inclusion criteria included: studies conducted in humans; studies on SSc or SSc-ILD; studies reporting epidemiologic data; studies conducted in, or reporting data from, European countries or North America (United States and Canada) (data for the rest of the world were not considered); and studies published in English, German, French, Spanish, Italian, or Portuguese. Exclusion criteria included: case reports, case series, and opinion pieces. Following screening, the eligibility of all identified publications was confirmed via an in-depth review of the full-text articles, if available.
Quality assessment
For all publications included in the review, a qualitative assessment of methodologic quality and individual risk of bias was performed considering criteria from the Newcastle–Ottawa scale (NOS;
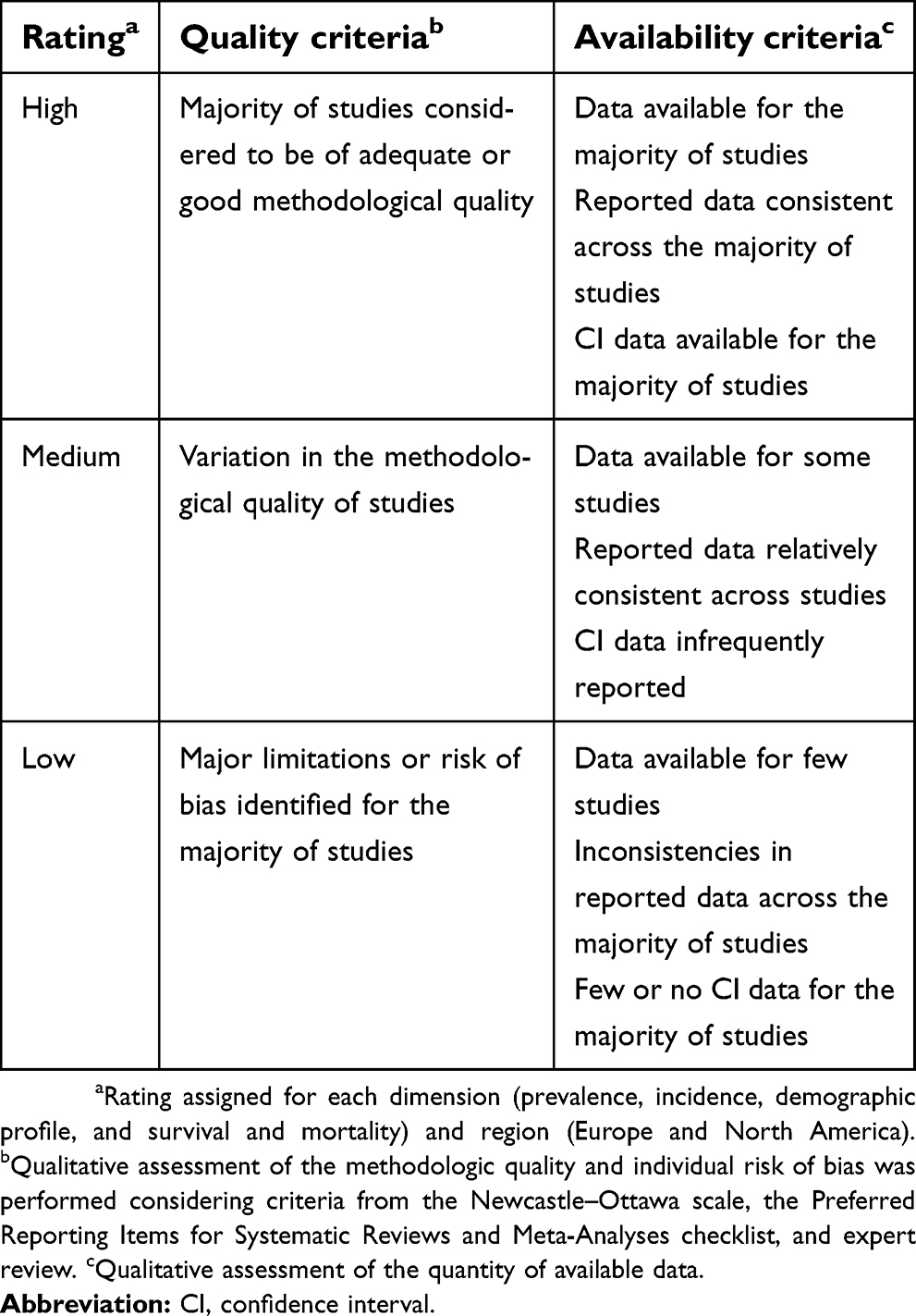 | Table 1 Quality assessment criteria |
Results
In total, 1,637 publications were identified through literature searches conducted in Medline and Embase. After the removal of 249 duplicates and the exclusion of 1,229 publications following screening, 159 publications were assessed for eligibility (based on in-depth review of the full-text article, where available), of which 11 did not meet eligibility criteria, resulting in 148 publications undergoing quality assessment. Web-based searches and manual review of reference lists yielded one additional reference, providing a total of 149 sources from which data were extracted (Figure 1).
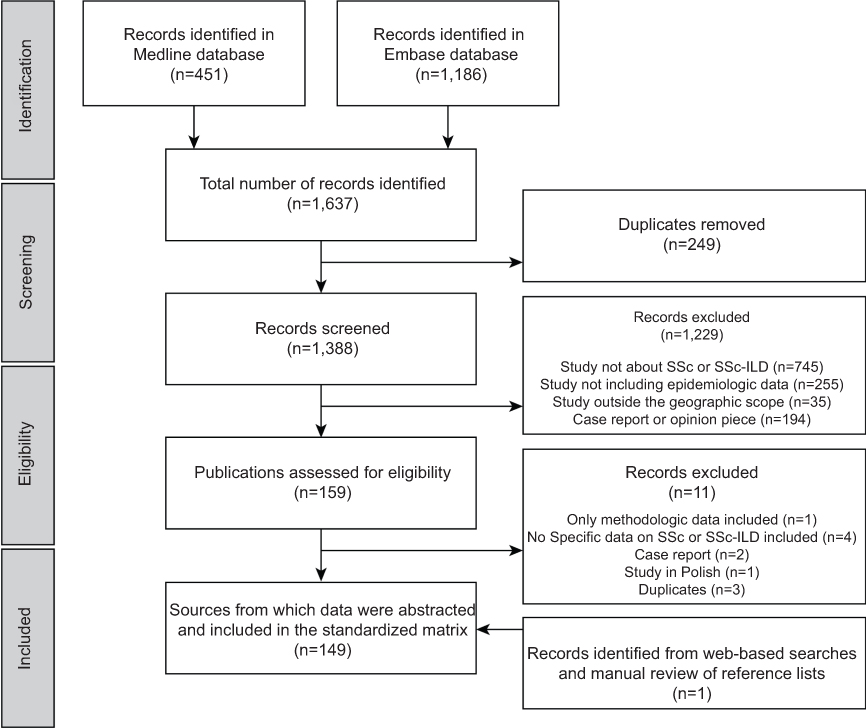 | Figure 1 Flow diagram of publications identified for inclusion.Abbreviations: ILD, interstitial lung disease; SSc, systemic sclerosis. |
For the purposes of this paper, the presented results are restricted to 50 publications that included data on prevalence, incidence, demographic profile, and survival and mortality of the populations of interest, and an overview of all included publications is presented in
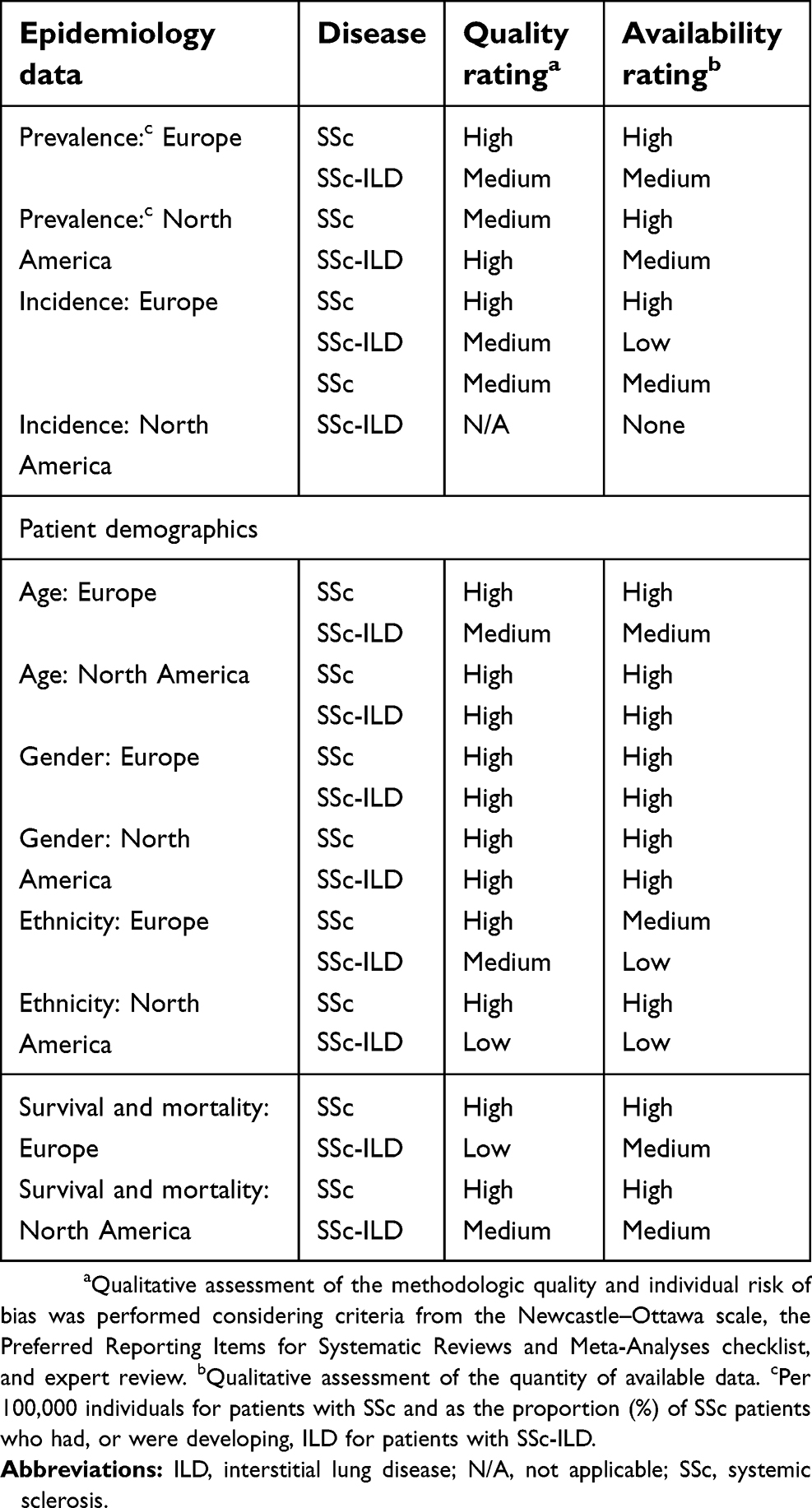 | Table 2 Overview of quality assessment results |
Prevalence
A total of 18 studies were identified that assessed the prevalence of SSc across Europe and North America (Figure 2). No dedicated studies reported prevalence data for SSc-ILD; however, seven studies were identified that reported the proportion of SSc patients who had, or were developing, ILD.
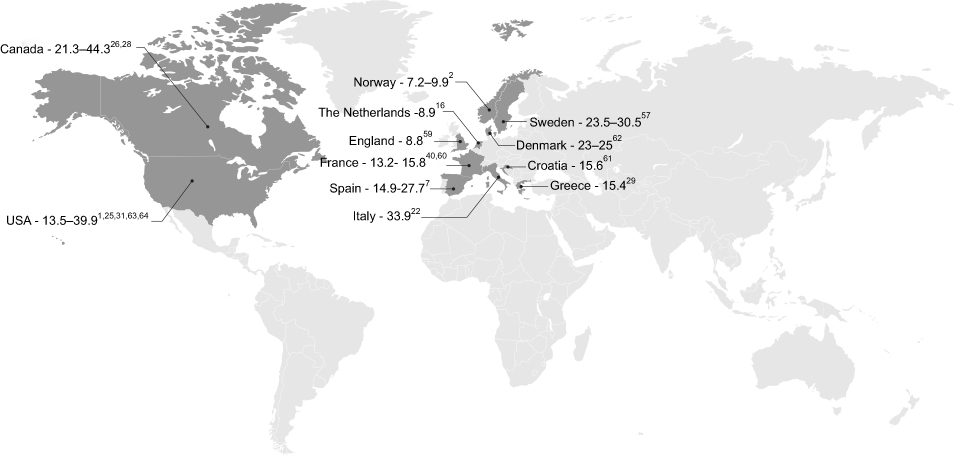 | Figure 2 Prevalence of SSc across Europe and North America (per 100,000 individuals).1,2,7,16,22,25,26,28,29,31,40,57,59–64 |
Europe
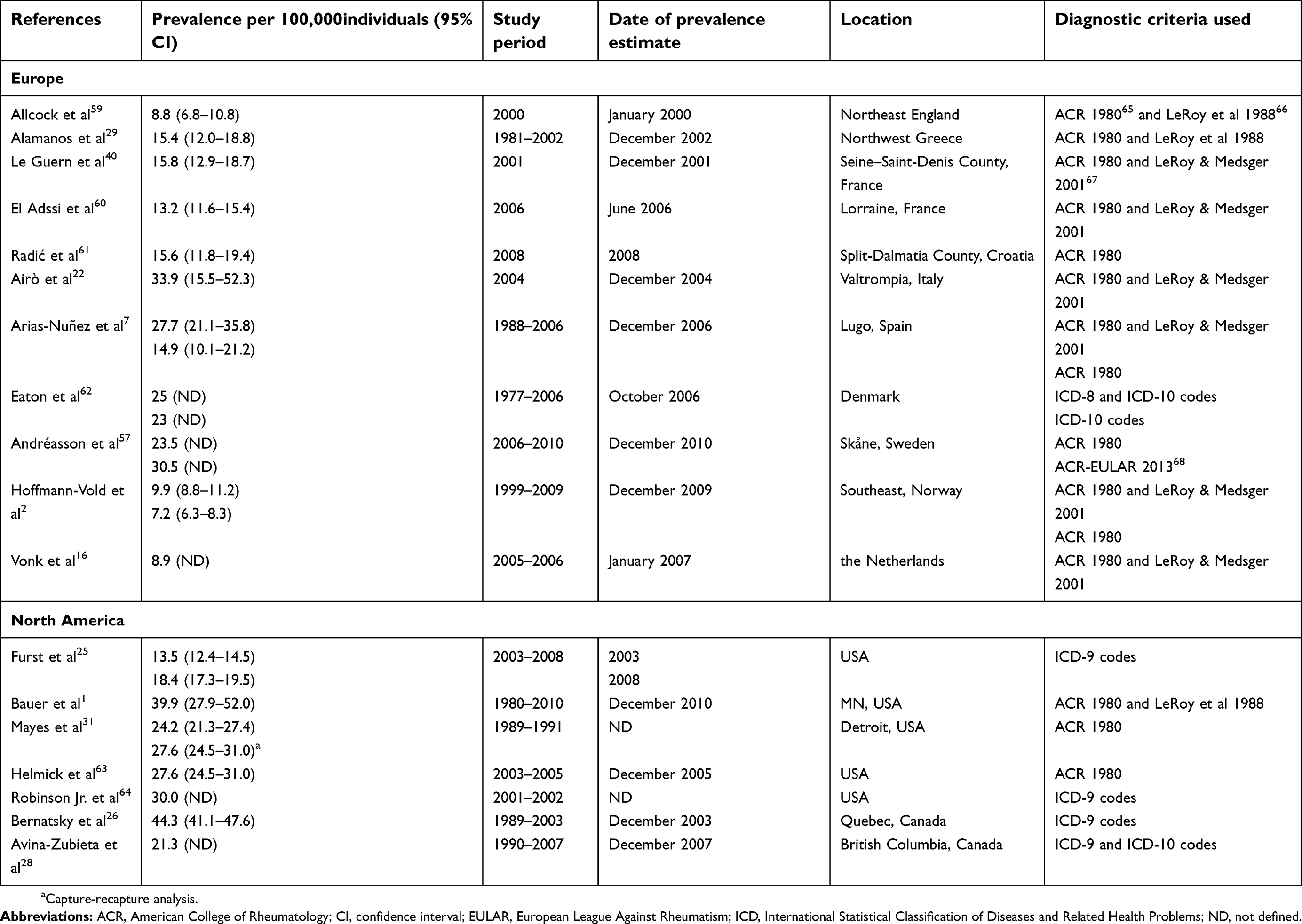
In Europe, 11 studies have been conducted to evaluate the prevalence of SSc (Table 3). Reported estimates vary widely, ranging from 7.2 per 100,000 individuals in Norway in 20092 to 33.9 per 100,000 individuals in Italy in 2004.22 Differences between genders have been reported, with reported prevalence estimates of 4.2 (95% confidence interval [CI]: 3.2–5.4) per 100,000 men, and 15.6 (95% CI: 13.6–17.8) per 100,000 women in Norway in 2009.2
 | Table 3 Prevalence of systemic sclerosis |
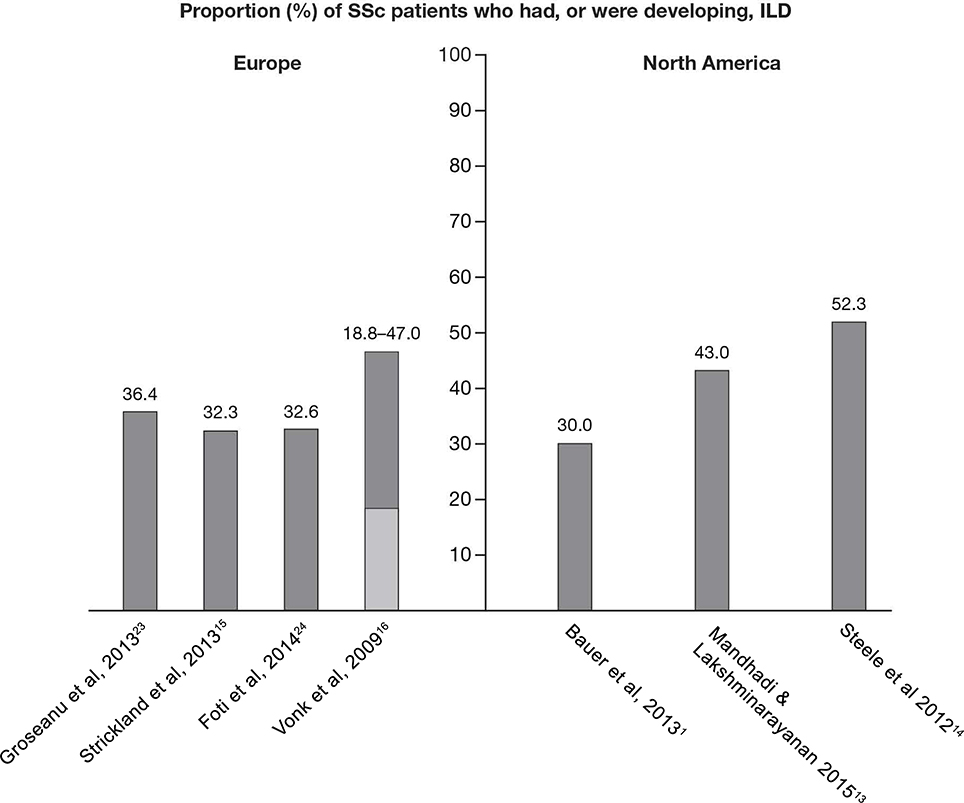
No prevalence data specific to SSc-ILD were identified in the systematic review. Four publications did, however, describe the proportion of SSc patients who had, or were developing, ILD (Figure 3), of which one also reported the prevalence of SSc, thereby enabling an estimate of SSc-ILD prevalence to be derived. Over the period of 2010–2012, in a cohort study of 44 SSc patients conducted in Romania, ILD was reported to affect 36.4% of the patients (method of diagnosis not reported).23 Similar findings were reported from a study including 204 SSc patients from the Royal National Hospital for Rheumatic Diseases connective tissue disease database in the UK between 1999 and 2000. It was estimated that 32.3% of the SSc patients also had ILD, diagnosed using HRCT or lung biopsy.15 These results were also supported by findings from the Rheumatology Unit at Catania Hospital, Italy, where 32.6% of the 44 SSc patients diagnosed between 2006 and 2013 had co-morbid ILD (method of diagnosis not reported).24 Based on these data, the proportion of ILD among SSc patients was estimated at ~35% in Europe. However, the reported proportion of ILD among SSc patients in the Netherlands varied from 18.8% (diagnosed using total lung capacity <70% predicted) to 47.0% (diagnosed using HRCT score >3), when assessed using data from the Pulmonary Hypertension Screening, a Multidisciplinary Approach in Scleroderma (POEMAS) registry combined with nationwide questionnaires collected between 2005 and 2007.16 Based on the reported prevalence of SSc in this study, the derived prevalence of SSc-ILD was estimated as 1.7–4.2 per 100,000 individuals.16
 | Figure 3 Proportion (%) of SSc patients who had, or were developing, ILD. Where a range is presented, the light grey bar represents the lower limit and the dark grey bar represents the upper limit.Abbreviations: ILD, interstitial lung disease; SSc, systemic sclerosis. |
North America
Seven studies were identified that evaluated the overall prevalence of SSc in North America (Table 3). The reported prevalence of SSc in North America was slightly higher compared to Europe, ranging from 13.5 per 100,000 individuals in the United States in 200325 to 44.3 per 100,000 individuals in Quebec, Canada, in 2003.26 As with European estimates, differences between genders were also reported in three Canadian database studies. Prevalence rates of 13.3 (95% CI: 11.1–16.1) per 100,000 men, and 74.4 (95% CI: 69.3–79.7) per 100,000 women were reported in Quebec in 2003;26 however, slightly lower estimates were reported in both Alberta (9.8 [95% CI: 7.2–13.6] per 100,000 men and 57.7 [95% CI: 51.3–65.3] per 100,000 women27), and British Columbia (6.4 per 100,000 men and 35.6 per 100,000 women28) in 2007.
As with Europe, no specific prevalence data were identified for SSc-ILD during the systematic review. Consequently, the proportion of SSc patients with, or developing, ILD was derived using reported data from three studies (Figure 3). A historical, population-based cohort study (1980–2010) conducted in Olmsted County, MN, USA, identified 64 patients with SSc, of whom 30% developed ILD, as diagnosed by HRCT.1 Higher proportions were, however, reported in other published studies. A medical chart review of 128 SSc patients from a single center between 2004 and 2014 reported 43.0% also had ILD, as diagnosed by HRCT.13 In addition, in a cohort of 1,168 SSc patients, identified through the Canadian Scleroderma Research Group registry from 2004 to 2010, the proportion of patients with SSc-ILD was 52.3%.14 This latter estimate, covering a 6-year period, represents the most compelling figure for the proportion of ILD among the SSc patient population in North America due to the use of a multiple imputation method for patients without HRCT data to correct for verification bias.
Incidence
In total, nine studies reported data on the incidence of SSc in Europe and North America. As with prevalence, no studies specifically assessed the incidence of SSc-ILD. Only one European study reported the proportion of SSc patients who had, or were developing, ILD, therefore enabling the incidence of SSc-ILD to be estimated.
Europe
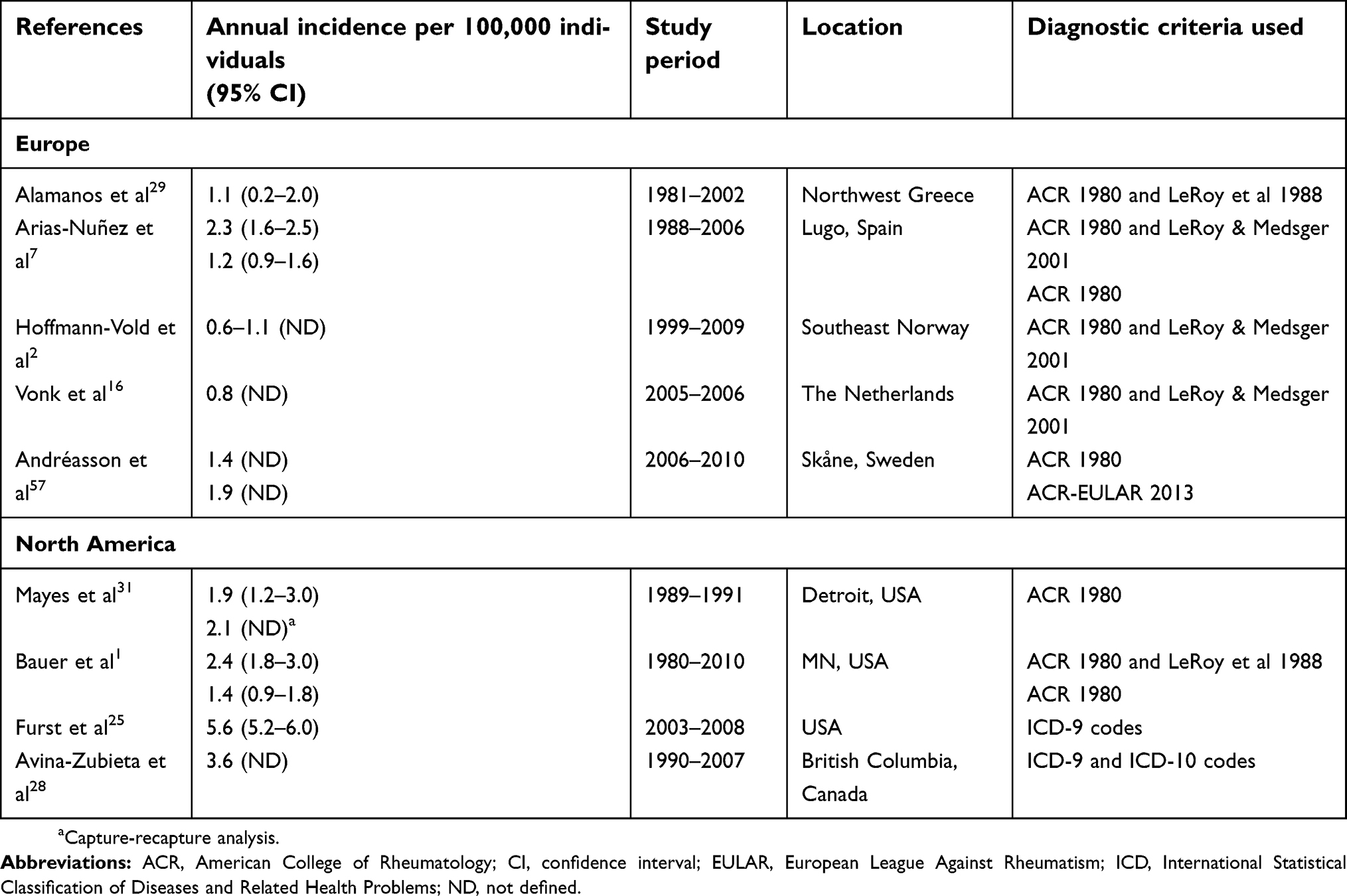
In Europe, five studies have been conducted to evaluate the overall incidence of SSc (Table 4), with annual incidence estimates ranging from 0.6 per 100,000 individuals in Norway from 1999–20092 to 2.3 per 100,000 individuals in Spain from 1988–2006.7 The overall annual incidence rate per 100,000 individuals for the period between 1988 and 1992 (1.4 [95% CI: 0.6–2.3]) was lower than those for the periods 1993–1996 (2.7 [1.4–3.7]), 1997–2002 (3.9 [2.8–5.8]), and 2003–2006 (2.5 [1.4–4.1]). Note that the confidence intervals for the periods 1993–1996 and 2003–2006 overlap with that of the period 1988–1992. The increased incidence rates over the course of the study were attributed to a progressive increase in SSc diagnosis in women between 1993–2002.7 Considering gender and age differences, using the ACR 1980 criteria, the annual-adjusted incidence rate was reported to be higher in women (1.8 [95% CI: 1.2–2.5] per 100,000 compared to 0.7 [95% CI: 0.3–1.2] per 100,000 men) and to significantly increase in individuals aged >45 years (3.1 [95% CI: 2.0–4.1] per 100,000 individuals aged 45–64 years and 3.0 [95% CI: 2.1–4.1] per 100,000 individuals aged >65 years) compared with individuals aged <45 years (0.7 [95% CI: 0.15–1.41] per 100,000) in a long-term cohort study conducted in Spain from 1988 to 2006.7 Similar findings were reported in a Greek cohort study covering the period of 1981–2002, in which annual incidence was reported to be 0.2 and 1.9 per 100,000 men and women, respectively, and was highest in men aged >65 years (0.7 per 100,000) and women aged 45–64 years (3.9 per 100,000).29 Conversely, an annual incidence of 0.3 (95% CI: 0.1–0.5) per million children (aged <16 years) was reported using data reported by members of specialist medical associations in the UK and Ireland over the period of 2005–2007.30
 | Table 4 Annual incidence of systemic sclerosis |
Similar to prevalence, no specific incidence data were identified for SSc-ILD during the systematic review. Combining data from the POEMAS registry and nationwide questionnaires collected in the Netherlands in 2006, the incidence of SSc-ILD in Europe was then calculated using the reported incidence of SSc and the proportion of SSc patients with ILD. Thus, estimated annual incidence ranged from 0.1 to 0.4 per 100,000 individuals, when defined using total lung capacity <70% predicted or HRCT score >3, respectively.16
North America
Four studies have been conducted in North America, with results suggesting the incidence of SSc is similar to that in Europe (Table 4). However, estimates were observed to vary according to the data source used, with studies based on health care databases providing higher annual estimates (3.6–5.6 per 100,000 individuals25,28) compared to medical chart review (1.4–2.4 per 100,000 individuals1,31). A slight increase in incidence rate over time was observed in a historical population-based cohort study conducted through a review of electronic medical records of SSc patients in Olmsted County, MN, USA, from 1980 to 2010. Higher annual incidence rates were reported for the period of 2000–2010 compared to 1980–1989: 2.7 (95% CI: 1.7–3.7) versus 1.7 (95% CI: 0.7–2.7) per 100,000 individuals, respectively, using the ACR 1980 and/or LeRoy et al 1988 criteria; 1.7 (95% CI: 1.0–2.5) versus 1.1 (95% CI: 0.4–1.9) per 100,000 individuals, respectively, considering the ACR 1980 criteria only.1
No incidence data specific to SSc-ILD in North America were identified through the systematic review. In addition, in this region, available data were insufficient to derive estimates of the incidence of this condition.
Demographic profile
Age: Europe
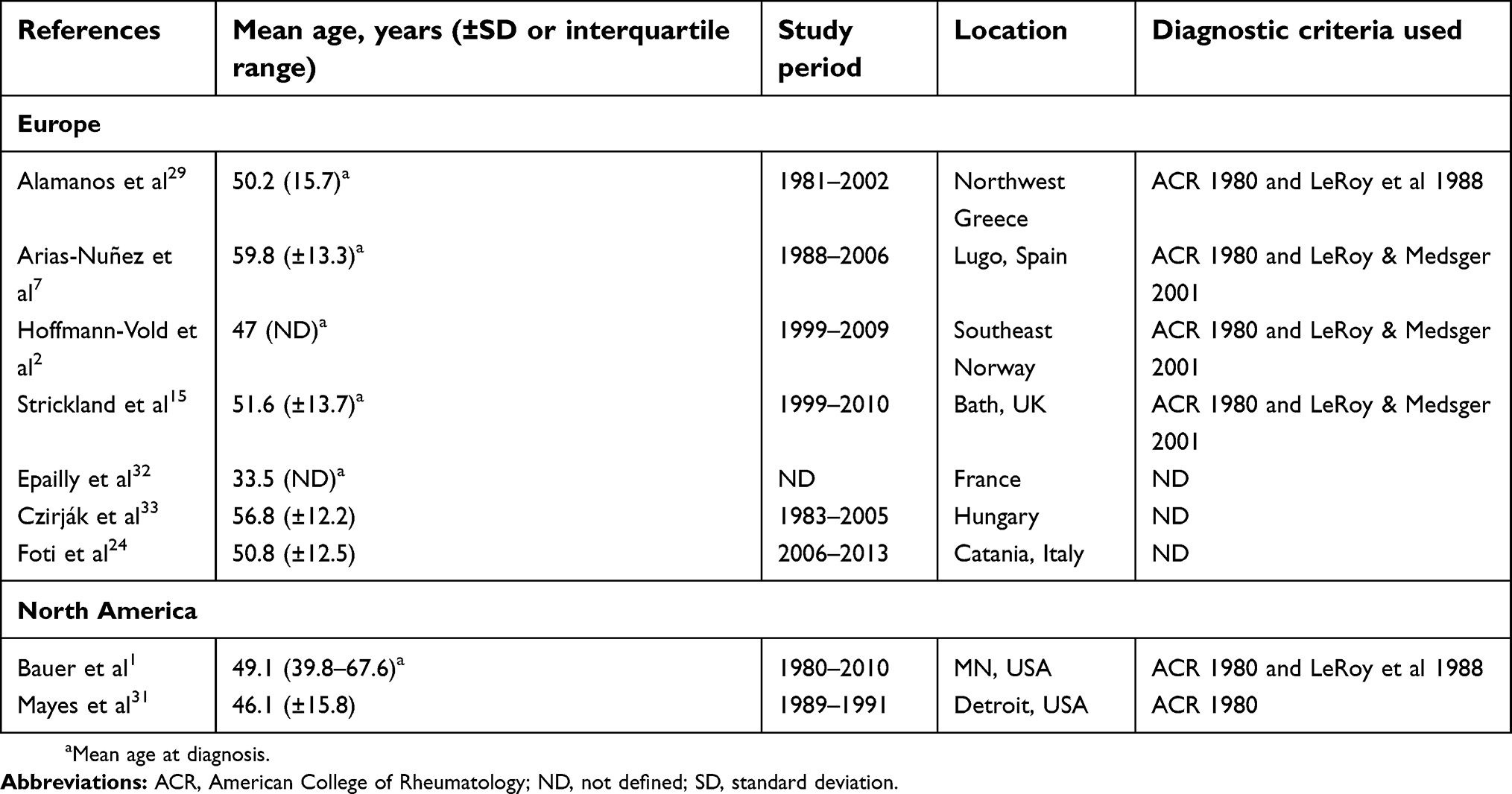
Seven studies were identified that assessed age in relation to SSc in Europe (Table 5). Diagnosis of SSc was reported to occur at an age of 33.5–59.8 years.2,7,15,24,29,32,33 Gender differences in age at diagnosis were suggested by cases of SSc identified through a systematic recording system in Greece during 1981–2002, where women were reported to have a lower mean age at diagnosis compared to men (49.2±15.7 and 58.9±13.5 years, respectively).29 Regarding prevalent cases of SSc, a cohort study conducted in southeast Norway reported a mean age of 56.7±13.5 years for women and 56.1±13.2 years for men in December 2009,2 which is comparable to the age of the overall population reported in two other studies (50.8±12.524 and 56.8±12.2 years33).
 | Table 5 Mean age of patients with systemic sclerosis |
Two European studies reported age-related data for patients with SSc-ILD. For hospitalized SSc patients with clinically significant ILD in the UK, a mean (±standard deviation [SD]) age at initial presentation of 46 (±11) years was reported over the period of 1985–2001.34 More recently, using data from The Health Improvement Network (THIN) general practice database from 2000 to 2009, a mean age at diagnosis of SSc-ILD of 61.8 (±11.1) years was reported.35
Age: North America
Two studies conducted in North America were identified, which reported a mean age at SSc diagnosis of 46.1 years31 and 49.1 years1 in the overall population (Table 5). Inconsistent data on gender differences have been reported in two studies, with a mean (±SD) age at SSc diagnosis of 49.7 (±1.2) years in women and 41.3 (±2.8) years in men reported following medical chart review from a single center in Ontario, Canada,36 while no difference in age at diagnosis between women (46.0±15.8 years) and men (46.7±16.9 years) was reported in a cross-sectional US study.31 Additionally, in the US cross-sectional study, mean age at diagnosis was reported to be lower in black/African-American patients (41.0±14.6 years) compared to white patients (48.1±15.9 years).31
Mean age at diagnosis was found to be slightly higher in patients with SSc-ILD in three identified studies. A US cohort study from 1997 to 2013, including 156 SSc-ILD patients, reported a mean age at diagnosis of 54.5 (±13.2) years,37 consistent with results from two longitudinal US cohort studies assessing 225 SSc-ILD patients (mean age: 54 years).38 Similarly, the median age of SSc patients with severe ILD, identified as patients with forced vital capacity (FVC) <60% from the Pulmonary Hypertension Assessment and Recognition of Outcomes in Scleroderma (PHAROS) registry in the United States and Canada, was estimated to be 52.5 years.39
Gender: Europe
Results from eight studies across Europe suggested that SSc predominantly affects women (Table 6), with reported ratios ranging from 3.8:1 (female:male) in Norway2 to 11.5:1 in France.40
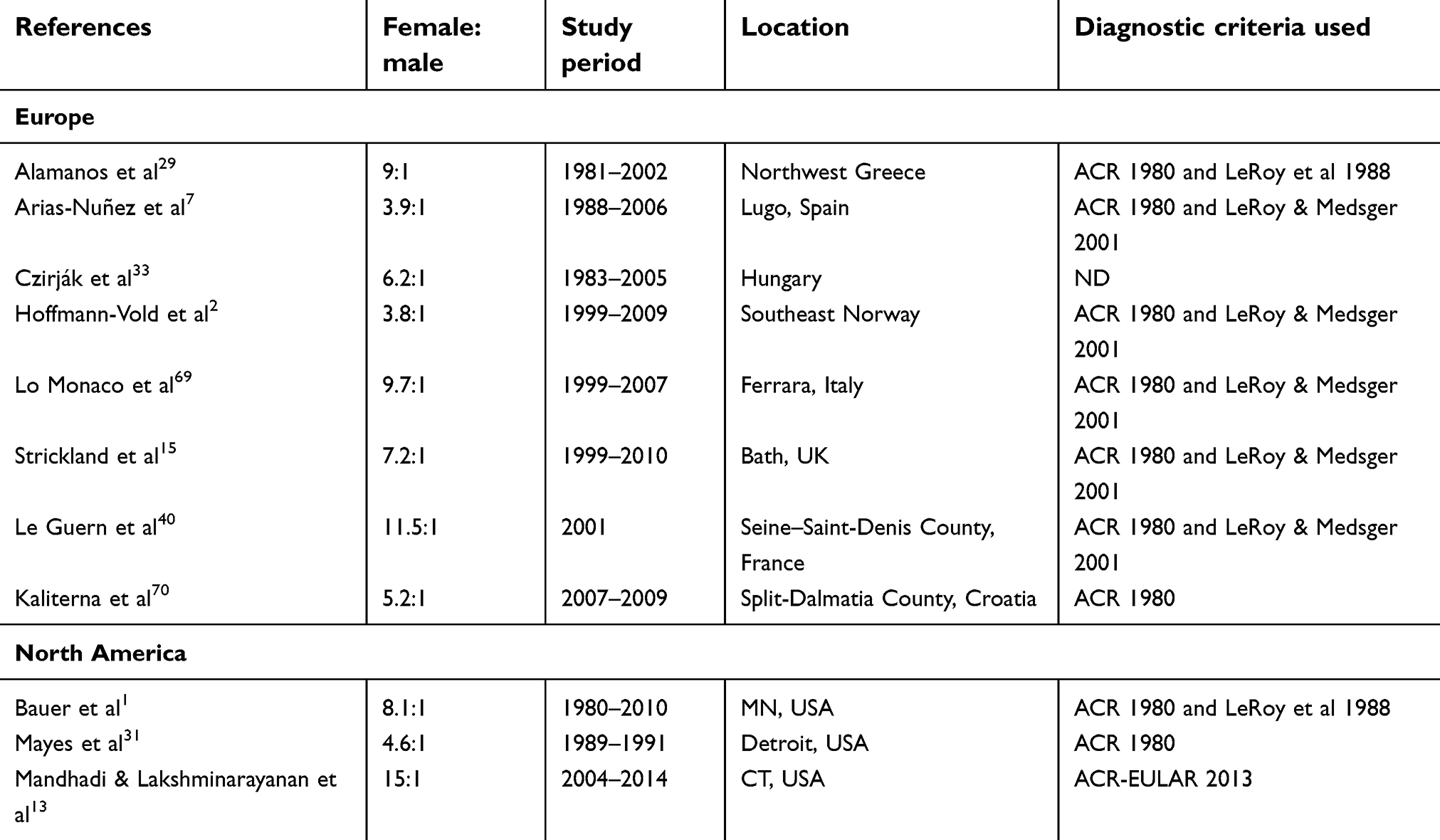 | Table 6 Gender of patients with systemic sclerosis |
A similar distribution between genders was observed in patients with SSc-ILD, with women representing 82% and 75% of the cases in two UK cohort studies: one conducted over the period of 1985–200134 and one using THIN data from 2000 to 2009,35 respectively.
Gender: North America
Similarly, a female predominance of SSc was reported in North America, with three studies reporting a greater proportion of women with the condition compared to men (Table 6), with female:male ratios ranging from 4.6:1 to 15:1.13,31
Female gender was also found to predominate in five studies assessing SSc-ILD patients in North America, with women representing 78% of a cohort of SSc patients with severe ILD from the PHAROS registry39 and accounting for 89% of SSc-ILD patients who had undergone HRCT in a cross-sectional study conducted in Canada.41 Additional publications from the United States37,38 and Canada14 reported similar results (69%, 84%, and 86% female, respectively).
Ethnicity: Europe
In Europe, two studies considered ethnicity in relation to SSc. One study conducted within a French multi-ethnic county during 2001 reported higher SSc prevalence rates in individuals aged >15 years with a non-European ethnic background, compared to Europeans (21.1 per 100,000 vs 14.0 per 100,000, respectively).40 Conversely, data from the connective tissue disease database at the University Hospital of Leicester in the UK identified 14% of the SSc patients as being South Asian, 58% as being Caucasian, and 3% as being black/African-American.42 It should be considered that the reported ethnicity may be a function of the location of the study, so geography may have an influence on the observed results.
No publications reporting specific data on the influence of ethnicity in SSc-ILD were identified. In an analysis of data from the University Hospital of Leicester connective tissue disease database in the UK, SSc-ILD was observed in 60% of the South Asian patients with SSc (n=10), and in 29.8% of the Caucasian patients with SSc (n=58). However, these data were gathered from a small (N=70) patient population, have not been fully published, and should be interpreted with caution.42
Ethnicity: North America
In North America, two studies considered ethnicity in relation to SSc. Prevalence of SSc was found to be similar in First Nation compared to non-First Nation women (64.6 [95% CI: 43.4–94.0] and 57.2 [95% CI: 50.4–65.3] per 100,000).27 A US cross-sectional study from 1989 to 1991 reported a higher prevalence among black/African-American patients (31.5 [95% CI: 28.2–35.2] per 100,000) compared to white patients (22.5 [95% CI: 19.7–25.6] per 100,000), which may be related to the higher frequency of dcSSc observed in black/African-American patients (60.3% vs 26.6% in non-black/African-American patients).31
Regarding the influence of ethnicity on SSc-ILD, only one publication was identified. The majority of SSc patients with severe ILD identified through the PHAROS registry were found to be Caucasian (63%; 24% black/African-American; 11% Hispanic).39
Survival and mortality
Europe
In Europe, 10 publications were identified that reported data on survival and mortality in patients with SSc. Patients with SSc were reported to have 5- and 10-year survival rates of 83–84% and 65–73%, respectively.7,29,33 The reported standardized mortality ratio (SMR) ranged from 1.3 (95% CI: 1.0–1.8) in the UK between 1999 and 201015 to 2.0 in both Greece between 1981 and 2002 (95% CI: 1.2–2.8)29 and Norway between 1999 and 2009 (95% CI: 1.4–2.6).43 Better prognosis was observed in the UK in patients with lcSSc compared to those with dcSSc over the period of 1999–2010 (SMR: 1.7 [95% CI: 0.8–3.0] and 1.3 [95% CI: 0.9–1.7], respectively).15 In addition, 5- and 10-year survival in Hungarian SSc patients over the period of 1983–2005 were reported to be 90.5% and 81.8% versus 67.0% and 48.6% for lcSSc and dcSSc, respectively.33 Cardiorespiratory manifestations were reported to represent the leading cause of death in SSc patients, accounting for 65–67% of all deaths.7,33,44 These findings were supported by results from a European multicenter cohort study (2002–2007) in which the main causes of death included pulmonary disease (35%), cardiac disease (29%), and renal failure (3%);45 a study based on data from the EUSTAR database (2004–2008) in which 35% of the SSc-related deaths were attributed to pulmonary fibrosis, 26% to pulmonary arterial hypertension (PAH), and 26% to cardiac causes;46 a Spanish cohort study (1986–2011) in which the major causes of death were reported as cardiac involvement (23.5%), severe infections (23.5%), pulmonary manifestations (20.6%), and renal involvement (5.8%);47 and a Greek study (1981–2002) using death certificates on which cardiopulmonary insufficiency (58.3%), coronary disease (8.3%), and renal failure (5.6%) were among the leading causes of death.29 Prognostic factors reported to be associated with increased mortality and poorer survival included: male sex (odds ratio [OR] 1.9);45 older age (hazard ratio [HR] 1.3 per 10 years;46 HR 1.1);48 increased disease duration (HR 1.1 per year);48 anti-Ro antibodies (HR 3.9);47 proteinuria (HR 3.3;46 OR 2.345); increased erythrocyte sedimentation rate (OR 1.9);45 higher Rodnan skin score (HR 1.2 per 10 points;46 HR 1.1 per point48); PAH (HR 7.3;48 HR 5.6;47 HR 2.046), ILD (HR 6.8);47 reduced diffusing capacity of the lung (OR 1.9;45 HR 1.2 per 10% decrease46); and dyspnea (HR 1.6).46
Regarding SSc-ILD, three publications reporting European survival and mortality data were identified. In the UK, results from a cohort study of 40 patients with SSc-ILD over the period 2000–2009 reported a mean survival duration of 8.8 years and mortality rate of 70.9 per 1000 person-years (95% CI: 36.9–136.3).35 In a second UK cohort of 36 SSc-ILD patients treated with intravenous cyclophosphamide (1999–2009) at the Royal Derby, King's Mill and Nottingham University Hospitals, overall survival was 76.1% at 5 years (62.9% for SSc-ILD presenting with respiratory symptoms and 91.5% for SSc-ILD diagnosed due to decline in screening pulmonary function tests), with a reported mean survival of 7.7 years (95% CI: 6.3–9.2) (4.7 [95% CI:3.3–6.1] and 8.9 [95% CI:7.6–10.2] years for those with respiratory symptoms and pulmonary function decline, respectively).49 Moreover, in Spain between 2005 and 2013, reported mean survival for patients who underwent a lung transplant (n=9 with six cases due to ILD) was 26 months (range: 7–52 months).50
North America
Eight publications reporting survival and mortality data in SSc patients in North America were identified during the systematic review. Data suggest that survival duration after SSc diagnosis has improved over time, with an increase in the reported 10-year survival rate from 62.6% over the period of 1989–199131 to 82% over the period of 1994–2004.36 Similarly, based on data from the Pittsburgh Scleroderma Databank, an improvement in 10-year survival was observed (from 54% over the period of 1972–1976, to 66% over the period of 1987–1991).51 In addition, median survival time after SSc diagnosis was reported to be ~11 years over the period of 1989–1991 in Detroit31 and 22.9 years over the period of 1980–2010 in Minnesota.1 Based on analysis of the mortality database of the National Center of Health Statistics (1999–2002), the age-adjusted death rate associated with SSc was reported as 4.7 per million in the overall population, with rates of 2.1 and 6.8 per million reported for men and women, respectively. Furthermore, mortality rates were observed to consistently increase with age in both sexes, with a peak of 8.0 cases per million in men aged 65–74 years, and 33.3 cases per million in women aged 75–84 years.52 An SMR of 4.7 (95% CI: 3.6–5.7) among incident cases, and 3.7 (95% CI: 3.2–4.2) among prevalent cases was reported in a large cohort study in Canada (2005–2012).53 Two separate studies have reported overall US in-hospital mortality rates for SSc patients as being 6.3% (95% CI: 5.8–6.7) between 2002 and 200354 and 7.1% in 1995.55 Over time, the frequency of deaths due to renal crisis in SSc patients in the United States has decreased considerably, from 42% in 1972 to only 6% in 1996, with the proportion of patients who died of pulmonary fibrosis having increased from 6% to 33%.51 In a cohort study in Canada (1994–2004), primary causes of death included ILD (30.3%), renal complications (27.2%), cardiac complications (27.2%), and PAH (15.2%).36 Similarly, a cross-sectional US analysis over 2002 and 2003 reported SSc (13.9%), respiratory failure (8.3%), respiratory infection (7.8%), and congestive heart failure (5.8%) as the most common principal diagnoses for patients who died in the hospital.54 As in Europe, poor prognostic factors for survival included male sex (female sex OR 0.754), PAH (HR 4.81), and ILD/pulmonary fibrosis (HR 2.91 2.7;55 OR 2.654), in addition to smoking (HR 1.71), chronic kidney disease (HR 2.81), and congestive heart failure (OR 1.7;55 OR 1.4;54 HR 1.21).
Three publications conducted in North America reported survival and mortality data for patients with SSc-ILD. Data from the Stanford ILD database in the United States over the period of 2002–2009 reported survival rates of 100%, 90%, and 77%, for 1, 3, and 5 years, respectively.56 Results from a US regression analysis in 2015 highlighted that increased age (HR 1.07), male sex (female:male HR 0.38), decreased diffusing capacity for carbon monoxide percent predicted (HR 0.94), and long-term oxygen therapy (HR 5.25) were significant predictors of mortality in patients with SSc-ILD.38 Reduced pulmonary function measurements were also identified as important independent predictors of 1-year mortality (normal pulmonary function:reduced pulmonary function HR 0.4–0.6) based on data from a US specialized SSc-ILD clinic (1997–2013).37
Discussion
Based on the results from our systematic review, data on the epidemiology of SSc and SSc-ILD are scarce. Regarding SSc, wide variation in prevalence was observed, with slightly higher estimates reported in North America (13.5–44.3 per 100,000 individuals) compared to Europe (7.2–33.9 per 100,000 individuals), which may be a true reflection of epidemiological variation or an artifact of clinical data analyses, as further discussed. Estimated annual incidence of SSc was less than 10 per 100,000 individuals in both Europe (0.6–2.3 per 100,000 individuals) and North America (1.4–5.6 per 100,000 individuals), with an increase in reported incidence observed over time. Diagnosis of SSc was reported to occur at the age of 33.5–59.8 years in Europe and 46.1–49.1 years in North America, and to be observed more frequently in women (ratio of 3.8–11.5:1 female:male in Europe and 4.6–15:1 in North America). The consistently higher prevalence and incidence rates among women suggest a clinically important difference in the occurrence of SSc between genders. Within Europe, SSc was observed more frequently in non-Europeans when compared to Europeans and in Caucasians when compared to South Asians. In North America, a higher prevalence of SSc was reported in black/African-American patients. Ten-year survival in patients with SSc was reported at 65–73% in Europe and 54–82% in North America, with cardiorespiratory manifestations, including ILD and PAH, reported as poor prognostic factors (HR range 1.2–6.8).
Few publications (n=16) reported findings specific to the SSc-ILD population. Data on prevalence and incidence were derived based on the proportion of SSc patients who also reported ILD; however, it should be considered that these estimates may not be fully representative of the overall SSc-ILD patient population. ILD was estimated to affect ~35% of the SSc patients in Europe and ~52% in North America; however, the method of ILD assessment may potentially contribute to differences in the observed frequencies. For example, when diagnosed via HRCT, ILD was estimated to affect 32.3–47.0% of the SSc patients in Europe; whereas only 18.8% of the patients were reported to be affected when diagnosed based on reduced lung function.16 Based on the available data, SSc-ILD was calculated to have an estimated prevalence and annual incidence of 1.7–4.2 per 100,000 individuals and 0.1–0.4 per 100,000 individuals in Europe, respectively. Incidence estimates could not be obtained for North America due to lack of published data; however, based on reported data for SSc, it may be expected that SSc-ILD incidence rates would be similar to those observed in Europe. In both Europe and North America, a slightly higher mean age at diagnosis was observed in patients with SSc-ILD compared to those with SSc (46–61.8 years and 52.5–54.5 years, respectively). Similar gender ratios were observed in patients with SSc and SSc-ILD, with a female predominance in both Europe (75–82%) and North America (≥69%). Data from North America suggest that SSc-ILD and associated complications are observed most frequently in Caucasian patients; however, these data must be interpreted with caution due to the paucity of available information. Limited data on survival and mortality were available in patients with SSc-ILD; however, the presence of respiratory symptoms and reduced pulmonary function was reported to be associated with poorer prognosis.
It should be considered that differences in the underlying patient populations, data sources, methodology, definitions, and diagnostic criteria existed between studies (
This systematic review also has some limitations. Although the majority of data were considered “medium” or “high” for both availability and quality of evidence in our review (Table 2), the number of identified publications that assessed each epidemiologic category was relatively small (n=21, 10, 23, and 24 for prevalence, incidence, demographic profile, and survival and mortality, respectively). The identified studies included a range of sample sizes, with some including as few as seven patients. In addition, a quarter of the identified publications were in abstract format only (n=16), meaning limited methodologic and results data were included. Furthermore, publications were limited to those in English, German, French, Spanish, Italian, or Portuguese, and studies outside Europe or North America were not included, which may have restricted the available evidence.
This first systematic review on the epidemiology of SSc and SSc-ILD provides a useful overview. Additional evidence in relation to prevalence, incidence, demographic profile, and survival and mortality would be beneficial to gain a better understanding of the burden of SSc and SSc-ILD.
Conclusions
This systematic review confirms our assumptions that SSc and SSc-ILD are rare, with wide-ranging estimates of SSc prevalence and incidence reported across Europe and North America. Both SSc and SSc-ILD were reported to predominantly affect women, with cardiorespiratory manifestations associated with poorer prognosis. Additional observational studies would be beneficial to provide more accurate estimates of prevalence and incidence, as well as demographic and survival/mortality data. In particular, more epidemiologic information on patients with SSc-ILD would enable a deeper understanding of the burden and impact of this rare disease.
Abbreviations list
ACR, American College of Rheumatology; CI, confidence interval; dcSSc, diffuse cutaneous systemic sclerosis; EULAR, European League Against Rheumatism; EUSTAR, EULAR Scleroderma Trials and Research; HRCT, high-resolution computed tomography; ILD, interstitial lung disease; NOS, Newcastle–Ottawa scale; OR, odds ratio; PHAROS, Pulmonary Hypertension Assessment and Recognition of Outcomes in Scleroderma; POEMAS, Pulmonary Hypertension Screening, a Multidisciplinary Approach in Scleroderma; PRISMA, Preferred Reporting Items for Systematic reviews and Meta-Analyses checklist; SD, standard deviation; SRC, scleroderma renal crisis; SSc, systemic sclerosis; SSc-ILD, systemic sclerosis-associated interstitial lung disease; THIN, The Health Improvement Network.
Acknowledgments
The authors received no compensation related to the development of the manuscript. Medical writing assistance, in the form of the preparation and revision of the manuscript, was supported financially by Boehringer Ingelheim International GmbH and provided by Lianne Young, BSc (Hons), ISMPP CMPP™, of Complete HealthVizion, and Ben Daniels, BSc (Hons), BA, of AMICULUM Limited under the authors’ conceptual direction and based on feedback from the authors. Patrice Verpillat’s current affiliation is Merck KGaA, Darmstadt, Germany.
Author contributions
The authors meet the criteria for authorship as recommended by the International Committee of Medical Journal Editors. All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work. AB and PV were involved in the planning and conduct of the systematic review, interpretation of data, and development of the manuscript. NH and LW were involved in the interpretation of data and development of the manuscript.
Disclosure
AB is an employee of YolaRx Consultants, which received funding from Boehringer Ingelheim for the conduct of this systematic literature review. NH and LW are employees of Boehringer Ingelheim. PV is a former employee of Boehringer Ingelheim. The authors report no further conflicts of interest in this work.
References
1. Avina-Zubieta JA, Sayre EC, Bernatsky S, et al. Adult Prevalence of Systemic Autoimmune Rheumatic Diseases (SARDs) in British Columbia, Canada [abstract]. Arthritis Rheum. 2011;63(Suppl 10):S719
2. Hoffmann-Vold A-M, Midtvedt Ø, Molberg Ø, Garen T, Gran JT. Prevalence of systemic sclerosis in South-East Norway. Rheumatology (Oxford). 2012;51(9):1600–1605. doi:10.1093/rheumatology/ker154
3. Ho YY, Lagares D, Tager AM, Kapoor M. Fibrosis—a lethal component of systemic sclerosis. Nat Rev Rheumatol. 2014;10(7):390–402. doi:10.1038/nrrheum.2014.53
4.
5. LeRoy EC, Black C, Fleischmajer R, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988;15(2):202–205.
6. LeRoy EC, Medsger TA
7. Arias-Nuñez MC, Llorca J, Vazquez-Rodriguez TR, et al. Systemic sclerosis in northwestern Spain: a 19-year epidemiologic study. Medicine (Baltimore). 2008;87(5):272–280. doi:10.1097/MD.0b013e318189372f
8. Romanowska-Próchnicka K, Walczyk M, Olesinska M. Recognizing systemic sclerosis: comparative analysis of various sets of classification criteria. Reumatologia. 2016;54(6):296–305. doi:10.5114/reum.2016.64906
9. Jaeger VK, Wirz EG, Allanore Y, et al. Incidences and risk factors of organ manifestations in the early course of systemic sclerosis: a longitudinal EUSTAR study. PLoS One. 2016;11(10):e0163894. doi:10.1371/journal.pone.0163894
10. McNearney TA, Reveille JD, Fischbach M, et al. Pulmonary involvement in systemic sclerosis: associations with genetic, serologic, sociodemographic, and behavioral factors. Arthritis Rheum. 2007;57(2):318–326. doi:10.1002/art.22532
11. Siméon-Aznar CP, Fonollosa-Plá V, Tolosa-Vilella C, et al. Registry of the Spanish Network for Systemic Sclerosis: clinical pattern according to cutaneous subsets and immunological status. Semin Arthritis Rheum. 2012;41(6):789–800. doi:10.1016/j.semarthrit.2011.10.004
12. Nihtyanova SI, Schreiber BE, Ong VH, et al. Prediction of pulmonary complications and long-term survival in systemic sclerosis. Arthritis Rheumatol. 2014;66(6):1625–1635. doi:10.1002/art.38390
13. Mandhadi R, Lakshminarayanan S. Pulmonary involvement in systemic sclerosis at initial presentation: a single center experience. Arthritis Rheumatol. 2015;67(Suppl 10):S3575–3576.
14. Steele R, Hudson M, Lo E, Baron M. Group obotCSR. Clinical decision rule to predict the presence of interstitial lung disease in systemic sclerosis. Arthritis Care Res (Hoboken). 2012;64(4):519–524. doi:10.1002/acr.21583
15. Strickland G, Pauling J, Cavill C, Shaddick G, McHugh N. Mortality in systemic sclerosis—a single centre study from the UK. Clin Rheumatol. 2013;32(10):1533–1539. doi:10.1007/s10067-013-2289-0
16. Vonk MC, Broers B, Heijdra YF, et al. Systemic sclerosis and its pulmonary complications in The Netherlands: an epidemiological study. Ann Rheum Dis. 2009;68(6):961–965. doi:10.1136/ard.2008.091710
17. Kowal-Bielecka O, Landewé R, Avouac J, et al. EULAR recommendations for the treatment of systemic sclerosis: a report from the EULAR Scleroderma Trials and Research group (EUSTAR). Ann Rheum Dis. 2009;68(5):620–628. doi:10.1136/ard.2008.096677
18. Adler S, Huscher D, Siegert E, et al. Systemic sclerosis associated interstitial lung disease - individualized immunosuppressive therapy and course of lung function: results of the EUSTAR group. Arthritis Res Ther. 2018;20(1):17. doi:10.1186/s13075-018-1517-z
19. Silver KC, Silver RM. Management of systemic-sclerosis-associated interstitial lung disease. Rheum Dis Clin North Am. 2015;41(3):439–457. doi:10.1016/j.rdc.2015.04.006
20. Tashkin DP, Roth MD, Clements PJ, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med. 2016;4(9):708–719. doi:10.1016/S2213-2600(16)30152-7
21. Kowal-Bielecka O, Fransen J, Avouac J, et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis. 2017;76(8):1327–1339. doi:10.1136/annrheumdis-2016-209909
22. Airò P, Tabaglio E, Frassi M, Scarsi M, Danieli E, Rossi M. Prevalence of systemic sclerosis in Valtrompia in northern Italy. A collaborative study of rheumatologists and general practitioners. Clin Exp Rheumatol. 2007;25(6):878–880.
23. Groseanu L, Saulescu I, Balanescu A, et al. Vitamin D status and clinical significance in a group of scleroderma patients. Ann Rheum Dis. 2013;72(Suppl 3):A647. doi:10.1136/annrheumdis-2013-eular.1922
24. Foti R, Converso G, Benenati A, et al. Iloprost as cyclic six-day per month. Long term efficacy in scleroderma patients. Clin Exp Rheumatol. 2014;32(2 Suppl 81):S113.
25. Furst DE, Fernandes AW, Iorga SR, Greth W, Bancroft T. Epidemiology of systemic sclerosis in a large US managed care population. J Rheumatol. 2012;39(4):784–786. doi:10.3899/jrheum.111106
26. Bernatsky S, Joseph L, Pineau CA, Belisle P, Hudson M, Clarke AE. Scleroderma prevalence: demographic variations in a population-based sample. Arthritis Rheum. 2009;61(3):400–404. doi:10.1002/art.v61:3
27. Barnabe C, Joseph L, Edworthy S, et al. Scleroderma prevalence in Alberta: a population-based assessment [abstract]. J Rheumatol. 2011;38(6):1151. doi:10.3899/jrheum.100724
28. Fischer A, Molitor JA, Hummers LK, et al. Severe Interstitial Lung Disease within the PHAROS Registry: Baseline Characteristics and Clinical Features [abstract]. Arthritis Rheum. 2011;63(Suppl 10):S269–S270.
29. Alamanos Y, Tsifetaki N, Voulgari PV, et al. Epidemiology of systemic sclerosis in northwest Greece 1981 to 2002. Semin Arthritis Rheum. 2005;34(5):714–720. doi:10.1016/j.semarthrit.2004.08.004
30. Herrick AL, Ennis H, Bhushan M, Silman AJ, Baildam EM. Incidence of childhood linear scleroderma and systemic sclerosis in the UK and Ireland. Arthritis Care Res (Hoboken). 2010;62(2):213–218. doi:10.1002/acr.20070
31. Mayes MD, Lacey JV
32. Epailly E, Ikic A, Emmanuel C, et al. Heart transplantation in 8 patients with systemic sclerosis: results of a French national registry [abstract]. Transpl Int. 2015;28(Suppl S4):267.
33. Czirják L, Kumánovics G, Varjú C, et al. Survival and causes of death in 366 Hungarian patients with systemic sclerosis. Ann Rheum Dis. 2008;67(1):59–63. doi:10.1136/ard.2006.066340
34. Bouros D, Wells AU, Nicholson AG, et al. Histopathologic subsets of fibrosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am J Respir Crit Care Med. 2002;165(12):1581–1586. doi:10.1164/ajrccm.165.8.2106104
35. Navaratnam V, Ali N, Smith CJP, McKeever T, Fogarty A, Hubbard RB. Does the presence of connective tissue disease modify survival in patients with pulmonary fibrosis?. Respir Med. 2011;105(12):1925–1930. doi:10.1016/j.rmed.2011.03.020
36. Al-Dhaher FF, Pope JE, Ouimet JM. Determinants of morbidity and mortality of systemic sclerosis in Canada. Semin Arthritis Rheum. 2010;39(4):269–277. doi:10.1016/j.semarthrit.2008.06.002
37. Ryerson CJ, O’Connor D, Dunne JV, et al. Predicting mortality in systemic sclerosis-associated interstitial lung disease using risk prediction models derived from idiopathic pulmonary fibrosis. Chest. 2015;148(5):1268–1275. doi:10.1378/chest.15-0003
38. Morisset J, Elicker BM, Hu X, et al. Predictors of mortality and risk prediction among patients with scleroderma related interstitial lung disease [abstract]. Eur Respir J. 2015;46(Suppl):59.
39. Hao Y, Husdon M, Carreira P, et al. Early mortality in Australian, Canadian and Spanish scleroderma patients: rationale for establishing a multi-national inception cohort of patients with systemic sclerosis [abstract]. Arthritis Rheum. 2014;66(Suppl 10):S316.
40. Le Guern V, Mahr A, Mouthon L, Jeanneret D, Carzon M, Guillevin L. Prevalence of systemic sclerosis in a French multi-ethnic county. Rheumatology (Oxford). 2004;43(9):1129–1137. doi:10.1093/rheumatology/keh253
41. Assayag D, Hudson M, Tatibouet S, et al. Clinical correlates of lung scan abnormalities in patients with systemic sclerosis-associated interstitial lung disease [abstract]. Am J Respir Crit Care Med. 2012;185:A6616.
42. Mulla E, Shaffu S, Hassan W. A comparative study of the difference in clinical manifestations and disease outcomes between South Asian and Caucasian patients with systemic sclerosis in a large NHS trust, within the United Kingdom [abstract]. Ann Rheum Dis. 2015;74(Suppl 2):1128. doi:10.1136/annrheumdis-2015-eular.1996
43. Hoffmann-Vold AM, Molberg Ø, Midtvedt Ø, Garen T, Gran JT. Survival and causes of death in an unselected and complete cohort of Norwegian patients with systemic sclerosis. J Rheumatol. 2013;40(7):1127–1133. doi:10.3899/jrheum.121390
44. Gaultier JB, Hot A, Cathébras P, Grange C, Ninet J, Rousset H. [Systemic sclerosis in men]. Rev Med Interne. 2008;29(3):181–186. doi:10.1016/j.revmed.2007.07.010
45. Fransen J, Popa-Diaconu D, Hesselstrand R, et al. Clinical prediction of 5-year survival in systemic sclerosis: validation of a simple prognostic model in EUSTAR centres. Ann Rheum Dis. 2011;70(10):1788–1792. doi:10.1136/ard.2010.139832
46. Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: a study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. 2010;69(10):1809–1815. doi:10.1136/ard.2009.114264
47. Ovalles-Bonilla JG, Martínez-Barrio J, López-Longo F, et al. Survival, causes of death and mortality risk factors in systemic sclerosis [abstract]. Ann Rheum Dis. 2013;71(Suppl 3):402. doi:10.1136/annrheumdis-2012-eular.2717
48. Hachulla E, Carpentier P, Gressin V, et al. Risk factors for death and the 3-year survival of patients with systemic sclerosis: the French ItinérAIR-Sclérodermie study. Rheumatology (Oxford). 2009;48(3):304–308. doi:10.1093/rheumatology/ken488
49. Abhishek A, Yazdani R, Pearce F, et al. Outcome of systemic sclerosis associated interstitial lung disease treated with intravenous cyclophosphamide. Clin Rheumatol. 2011;30(8):1099–1104. doi:10.1007/s10067-011-1734-1
50. Fernández-Codina A, Simeón-Aznar CP, Berastegui-García C, et al. Update on lung transplantation in systemic sclerosis in Spain [abstract]. Ann Rheum Dis. 2014;73(Suppl 2):560. doi:10.1136/annrheumdis-2014-205310
51. Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis. 2007;66(7):940–944. doi:10.1136/ard.2006.066068
52. Mendoza F, Derk CT. Systemic sclerosis mortality in the United States: 1999–2002 implications for patient care. J Clin Rheumatol. 2007;13(4):187–192. doi:10.1097/RHU.0b013e318124a89e
53. Hao Y, Hudson M, Carreira P, et al. Early mortality in Australian, Canadian and Spanish scleroderma patients: rationale for establishing a multi-national inception cohort of patients with systemic sclerosis. In: ACR/ARHP Annual Meeting. Boston, MA, USANovember1419.723 ed2014
54. Chung L, Krishnan E, Chakravarty EF. Hospitalizations and mortality in systemic sclerosis: results from the Nationwide Inpatient Sample. Rheumatology (Oxford). 2007;46(12):1808–1813. doi:10.1093/rheumatology/kem045
55. Nietert PJ, Silverstein MD, Silver RM. Hospital admissions, length of stay, charges, and in-hospital death among patients with systemic sclerosis. J Rheumatol. 2001;28(9):2031–2037.
56. Su R, Bennett M, Jacobs S, et al. An analysis of connective tissue disease-associated interstitial lung disease at a US Tertiary Care Center: better survival in patients with systemic sclerosis. J Rheumatol. 2011;38(4):693–701. doi:10.3899/jrheum.100724
57. Andréasson K, Saxne T, Bergknut C, Hesselstrand R, Englund M. Prevalence and incidence of systemic sclerosis in southern Sweden: population-based data with case ascertainment using the 1980 ARA criteria and the proposed ACR-EULAR classification criteria. Ann Rheum Dis. 2014;73(10):1788–1792. doi:10.1136/annrheumdis-2014-205310
58. Coral-Alvarado P, Pardo AL, Castaño-Rodriguez N, Rojas-Villarraga A, Anaya J-M. Systemic sclerosis: a world wide global analysis. Clin Rheumatol. 2009;28(7):757–765. doi:10.1007/s10067-009-1144-9
59. Allcock RJ, Forrest I, Corris PA, Crook PR, Griffiths ID. A study of the prevalence of systemic sclerosis in northeast England. Rheumatology (Oxford). 2004;43(5):596–602. doi:10.1093/rheumatology/keh124
60. El Adssi H, Cirstea D, Virion JM, Guillemin F, de Korwin JD. Estimating the prevalence of systemic sclerosis in the Lorraine region, France, by the capture-recapture method. Semin Arthritis Rheum. 2013;42(5):530–538. doi:10.1016/j.semarthrit.2012.10.001
61. Radic M, Martinovic Kaliterna D, Fabijanic D, Radic J. Prevalence of systemic sclerosis in Split-Dalmatia county in Southern Croatia. Clin Rheumatol. 2010;29(4):419–421. doi:10.1007/s10067-009-1341-6
62. Eaton WW, Pedersen MG, Atladóttir HO, Gregory PE, Rose NR, Mortensen PB. The prevalence of 30 ICD-10 autoimmune diseases in Denmark. Immunol Res. 2010;47(1–3):228–231. doi:10.1007/s12026-009-8153-2
63. Helmick CG, Felson DT, Lawrence RC, et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Part I. Arthritis Rheum. 2008;58(1):15–25. doi:10.1002/art.23794
64.
65. ACR 1980: Subcommittee for scleroderma criteria of the American Rheumatism Association diagnostic and therapeutics criteria committee. Arthritis Rheum. 1980;23:581–590
66. LeRoy EC, Black C, Fleischmajer R, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol. 1988;15(2):202–205
67. LeRoy EC, Medsger TA Jr. Criteria for the classification of early systemic sclerosis. J Rheumatol. 2001;28(7):1573–1576
68. Van Den Hoogen F, Khanna D, Fransen J, et al. 2013 Classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 2013;65(11):2737–2747
69. Lo Monaco A, Bruschi M, La Corte R, Volpinari S, Trotta F. Epidemiology of systemic sclerosis in a district of northern Italy. Clin Exp Rheumatol. 2011;29(2 Suppl 65):S10–S14.
70. Kaliterna DM, Radic M, Pavic A. [Incidence, prevalence and disease characteristics of systemic sclerosis in Split-Dalmatia County]. Reumatizam. 2010;57(2):94–98.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.