Back to Journals » International Journal of Nanomedicine » Volume 12
EpCAM aptamer-functionalized polydopamine-coated mesoporous silica nanoparticles loaded with DM1 for targeted therapy in colorectal cancer
Authors Li Y, Duo Y, Bao SY, He L, Ling K, Luo J, Zhang Y, Huang H, Zhang H, Yu X
Received 6 June 2017
Accepted for publication 26 July 2017
Published 26 August 2017 Volume 2017:12 Pages 6239—6257
DOI https://doi.org/10.2147/IJN.S143293
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Dongwoo Khang
Yang Li,1,* Yanhong Duo,2,3,* Shiyun Bao,1 Lisheng He,4 Kai Ling,5 Jinfeng Luo,4 Yue Zhang,1 Hao Huang,2 Han Zhang,2 Xiaofang Yu1
1Department of Hepatobiliary and Pancreas Surgery, Second Clinical Medical College of Jinan University, Shenzhen People’s Hospital, 2Shenzhen Engineering Laboratory of Phosphorene and Optoelectronics, Collaborative Innovation Center for Optoelectronic Science and Technology, Key Laboratory of Optoelectronic Devices and Systems of Ministry of Education and Guangdong Province, College of Optoelectronic Engineering, Shenzhen University, Shenzhen, 3Key Laboratory of Plant Cell Activities and Stress Adaptation, Ministry of Education, School of Life Sciences, Lanzhou University, Lanzhou, 4Department of Pathology, 5Institute of Respiratory Diseases, Second Clinical Medical College of Jinan University, Shenzhen People’s Hospital, Shenzhen, China
*These authors contributed equally to this work
Abstract: DM1, a maytansine derivative, is a highly potential cytotoxic agent but with severe side effects; therefore, its application in clinical cancer therapy is limited. Here, in order to mitigate this intrinsic drawback of DM1, we developed mesoporous silica nanoparticles (MSNs) loaded with DM1 and surface-decorated with hydrochloride dopamine (PDA), polyethylene glycol (PEG), and epithelial cell adhesion molecule (EpCAM) aptamer (APt) for the targeted treatment of colorectal cancer (CRC). In this system, the PDA coating could be used as pH-sensitive gatekeepers to control the release of DM1 from MSNs in response to the pH stimulus and EpCAM APt-guided active targeting enables the increased delivery of DM1 to CRC as well as a reduction in toxicity and side effects by minimizing the exposure of normal tissues to DM1. Results demonstrated that DM1 inhibited the formation of microtubules and induced apoptosis in tumor cells via caspase signaling. In comparison with the control groups, the MSNs-DM1@PDA-PEG-APt bioconjugates exhibited increased binding ability and much higher cytotoxicity to the CRC SW480 cell line. Furthermore, in vivo assays confirmed the advantages of such a strategy. These findings suggested that MSNs-DM1@PDA-PEG-APt could represent a promising therapeutic platform for EpCAM-positive CRC.
Keywords: DM1, EpCAM aptamer, mesoporous silica nanoparticles, colorectal cancer
Introduction
Colorectal cancer (CRC) is the third most common cancer and the fourth cause of oncological death worldwide,1 with estimated 376,300 new cases and 191,000 deaths that occurred in China in 2015.2 Most patients with CRC are not diagnosed until the advanced stage, and surgery associated with (neo)adjuvant chemotherapeutic agents remains the main treatment. However, traditional chemotherapy often causes severe adverse systemic effects due to the inability to selectively target the tumor sites.3
Maytansine, discovered in 1972,4 has been isolated from higher plants, mosses, and microorganisms.5 This potent anti-microtubule agent binding to tubulin at the vinca binding site depolymerizes microtubules and exhibits 100–1,000 times higher cytotoxicity than vinca alkaloids, which act through a similar mechanism.6 However, severe side effects involving neuronal and gastrointestinal toxicities along with a narrow therapeutic window displayed by maytansine in clinical trials limit its application in cancer therapy.7 Consequently, novel targeted drug delivery systems are required to channel chemotherapeutic agents to tumor cells to enhance efficacy while reducing the side effects.
Recently, the development of targeted drug delivery techniques, especially antibody drug conjugates, has revived interest in maytansine. Antibody-maytansinoid conjugates, such as SAR3419 (anti-CD19mAb-DM4),8 MLN2704 (MLN591-DM1),9 and AVE9633 (anti-CD33mAb-DM4),10 are currently in different phases of clinical trials. Interestingly, Kadcyla (trastuzumab-DM1) was approved by the US Food and Drug Administration (FDA) in 2013 for late-stage breast cancer after treatment with conventional chemotherapeutics.11 Despite this encouraging development, several limitations related to antibodies, such as large size, immunogenicity, and production cost, limit their applications.12
With the development of nanotechnology, nanomaterials attract considerable attention for their applications in drug delivery, diagnostic and medical imaging, and engineering. Among the available nanomaterials, mesoporous silica nanoparticles (MSNs) are excellent candidates for drug delivery systems due to unique properties, including large surface area–volume ratio, tailored mesoporous structure, high chemical and mechanical stability, and favorable biocompatibility.13 Besides enhanced permeability and retention (EPR) effect-mediated passive targeting, MSNs can be engineered to selectively deliver various therapeutic agents to cancer cells of interest when combined with active targeting ligands such as antibodies, peptides, small molecules, or aptamers (APts).14 Hydrochloride dopamine (PDA) coating, a well-documented gatekeeper on the surface of MSNs, is highly sensitive to pH.15 Polyethylene glycol (PEG), approved by FDA, is known to reduce uptake by the reticuloendothelial system (RES), providing good dispersibility in aqueous solvents and decreased association with proteins.
Nucleic acid APts, isolated through SELEX, are single-stranded RNA or DNA oligonucleotides, which can fold into a three-dimensional structure to bind target molecules with high affinity and selectivity.16 APts have distinct advantages over traditional antibodies, including smaller size, lower immunogenicity, higher ratio of target accumulation, easier production, and higher in vivo stability.17 Given their properties, APts have attracted much attention as molecular probes for cancer cell detection and targeted cancer therapy.18–21 Epithelial cell adhesion molecule (EpCAM) APt is one of the DNA APts can specifically bind to EpCAM with high affinity.22 EpCAM (CD326), a 40 kDa transmembrane glycoprotein, is overexpressed in most rapidly proliferating tumors of epithelial origin, while at a variable but generally lower level in the normal epithelium.23 For example, high expression of EpCAM is detected in 97.7% patients with colon adenocarcinoma.24 EpCAM could be considered an ideal therapeutic target for the treatment of colorectal adenocarcinoma.22
In this study, we report the development of MSNs loaded with DM1 and surface-decorated with PDA, PEG, and EpCAM APt for the targeted treatment of CRC. PDA coating on the surface of MSNs could be used as a pH-sensitive gatekeeper to control the release rate of loaded drugs,15 while PEGylation provides an effective steric hindrance to improve particle dispersion in saline, increase the circulatory half-life, reduce MSN uptake by RES, and enhance the EPR effect.25 Meanwhile, EpCAM APt selectively binds to EpCAM, which is considered an ideal therapeutic target for the treatment of colorectal adenocarcinoma.
Characterization of the developed MSNs-DM1@PDA-PEG-APt bioconjugates, including particle size, morphology, and zeta potential, was also carried out. Then, cellular uptake efficiency and effects on microtubule and apoptosis of the novel MSNs-DM1@PDA-PEG-APt bioconjugates were assessed in the EpCAM overexpressing colon cancer SW480 cell line. Further, biodistribution, antitumor activity, and systematic toxicity were evaluated in xenograft tumor mice. In this work, the construction of MSNs-DM1@PDA-PEG-Apt bioconjugates, which incorporates the targeted efficacy of EpCAM APt with the potent cytotoxic effects of DM1, allows selective release of DM1 within the targeted tumor cells, and could be of great value in medical use.
Materials and methods
Materials
Hexadecyltrimethylammonium bromide (CTAB), tetraethyl orthosilicate (TEOS), 4′-6′-diamino-2-phenylindole hydrochloride (DAPI), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and IR783 were purchased from Sigma-Aldrich (St Louis, MO, USA). α-Maleimidyl-W-N-hydroxy-succinimidyl PEG (NH2-PEG-MAL, MW3500) was obtained from JenKem Technology Co, Ltd (Beijing, China). The microtubule fluorescence staining kit (cat no GMS 10278) was purchased from GenMed Scientific Inc (Arlington, TX, USA). Fluorescein isothiocyanate (FITC)-conjugated annexin V and propidium iodide (PI) kit for apoptosis detection was purchased from Thermo Fisher Scientific (Waltham, MA, USA). DM1 was obtained from BrightGene Bio-Medical Technology Co, Ltd (Jiangsu, China). RIPA lysis buffer was from Beyotime Biotechnology (Jiangsu, China). RPMI 1640, Dulbecco’s Modified Eagle’s Medium (DMEM), and fetal bovine serum (FBS) were purchased from Thermo Fisher Scientific.
Primary antibodies used in this work included anti-caspase 3 (9665), anti-caspase 9 (9504), anti-cleaved caspase 3 (9664), anti-cleaved caspase 9 (9509), anti-poly (ADP-ribose)polymerase (anti-PARP; 9532; Cell Signaling Technology, Danvers, MA, USA), anti-α-tubulin (Abcam, Cambridge, UK; ab70672), and anti-β-tubulin (Abcam; ab70672). Secondary antibodies (KPL), Alexa Fluor® 568 goat-anti rabbit IgG (Thermo Fisher Scientific; A-11011), and anti-β-actin (Sigma-Aldrich; clone AC-15, A5441) were used as well. EpCAM DNA APt (5′-amino-CAC TAC AGA GGT TGC GTC TGT CCCACG TTG TCA TGG GGG GTT GGC CTG-SH-3′) was synthesized by Sangon Biotech (Shanghai, China). All cell lines were provided by Nanfang Hospital, which purchased them from the American Type Culture Collection (Manassas, VA, USA), including SW480 (EpCAM+ human colon cancer cell line), NCM460 (human normal colon epithelial cell line), and 293T (EpCAM− human embryonic kidney cell line) cells.26 All other chemicals and reagents of highest grade were commercially available and used as directed.
Fabrication of different nanoparticles (NPs)
MSNs were synthesized as previously described.27 Briefly, CTAB (1.82 g, 5 mmol) and NH4F (3 g, 81 mmol) were dissolved in 500 mL of water and heated to 80°C. Then, TEOS (9 mL, 8.41 g) was added dropwise into the mixture and stirred for 6 hours. The products were refluxed in HCl/methanol for 24 hours at 80°C in order to completely remove the template, collected by centrifugation, and vacuum dried.
To obtain the DM1@MSNs, 100 mg MSNs were added into 5 mL of aqueous solution of DM1 (10 mg/mL) and stirred for 24 hours at room temperature. Next, DM1@MSNs were separated by centrifugation (12,000 rpm, 10 minutes) and washed with deionized water to remove unencapsulated DM1 from the external surface of MSNs.
Polydopamine-coated NPs (MSNs-DM1@PDA) were synthesized by incubating 50 mg of MSNs-DM1 with 0.5 mg/mL dopamine hydrochloride in Tris buffer (10 mM, pH 8.5) for 3 hours at room temperature with shaking. Then, MSNs-DM1@PDA was centrifuged (12,000 rpm, 10 minutes) and washed with water to remove the unpolymerized dopamine.15,28
For PEG-APt preparation, 10 OD EPCAM APt-SH was dispersed in 1 mL Tris–HCl buffer (pH 7.4, 10 mM), followed by addition of 1 mg NH2-PEG-MAL and 20 μL TCEP solution (1 mg mL−1). The mixture was stirred for 6 hours to obtain PEG-APt.
EpCAM APt-functionalized MSNs (MSNs-DM1@PDA-PEG-APt) were prepared by incubating MSNs-DM1@PDA in Tris buffer (10 mM, pH 8.5) containing NH2-PEG-APt for 2 hours at room temperature with shaking, followed by centrifugation. The purified MSNs-DM1@PDA-PEG-APt sample was resuspended in phosphate-buffered saline (PBS) or HyPure water for further use, stored at 4°C.
Fluorescent-labeled MSNs-FITC@PDA-PEG-APt and MSNs-IR783@PDA-PEG-APt were prepared by the same procedure, except that FITC-MSNs29 and MSNs-IR783 were used instead of MSNs, avoiding light for MSNs-FITC@PDA-PEG-APt.
Characterization of the synthesized NPs
Sizes and surface zeta potential values of the resultant nanomaterials were determined by dynamic light scattering (DLS; Zetasizer Nano ZS90; Malvern Instruments, Malvern, UK). Particle sizes and morphological features of the synthesized NPs were characterized by transmission electron microscopy (TEM; Tecnai G2 20; FEI Company, Hillsboro, OR, USA). Conjugation of the EpCAM DNA APt on the surface of MSNs-DM1@PDA-PEG was determined by agarose gel electrophoresis. All samples (DNA ladder, free APt, MSNs-DM1@PDA-PEG, and MSNs-DM1@PDA-PEG-APt) were loaded onto a 1% agarose gel prepared in TBE solution (89 mM Tris buffer at pH 8.6, 89 mM boric acid, 2.5 mM EDTA). After electrophoresis (80 V, 1.5 hours), the gel was stained with ethidium bromide and photographed on a MultiImage™ Light Cabinet (Alpha Innotech Corporation, San Leandro, CA, USA).
Cell culture
SW480 and NCM460 cells were cultured in RPMI 1640. 293T cells were cultured in DMEM. All cell culture media were supplemented with 10% FBS and 1% streptomycin/penicillin. Cells were cultured at 37°C in a humid environment containing 5% CO2.
In vitro drug release
In vitro release experiments were carried out in PBS (pH 7.4 or 5.5), with doxorubicin (DOX) selected as the model drug. DOX-loaded NPs were dispersed in 1.5 mL PBS in a dialysis membrane bag (MWCO =3,500), which was immersed in 15 mL PBS (pH 7.4 or 5.5) and gently stirred at 100 rpm at 37°C. At various time points, 1.0 mL of the external buffer was collected and replaced by 1 mL fresh PBS, to maintain a constant volume. DOX concentration was measured by high performance liquid chromatography.
Targeted binding ability and cellular uptake of MSNs-DM1@PDA-PEG-APt
The targeting capability in vitro was assessed for FITC-labeled MSNs-FITC@PDA-PEG and MSNs-FITC@PDA-PEG-APt. EpCAM+ human colon cancer SW480 cells, EpCAM− noncancerous colon epithelial NCM460 cells, and EpCAM− human embryonic kidney 293T cells were employed to evaluate the cell selectivity of NPs. Cells were seeded into 8-well chambered cover glasses in 12-well plates and allowed to attach for 12 hours. Then, the cells were incubated with medium containing equivalent amounts of MSNs-FITC@PDA-PEG and MSNs-FITC@PDA-PEG-APt for 6 hours. After two washes with PBS, the cells were fixed with 4% paraformaldehyde and counterstained with DAPI. Images were acquired by confocal laser scanning microscopy (CLSM; Olympus Fluoview FV-1000; Olympus Corporation, Tokyo, Japan). The findings were also confirmed by flow cytometry. Cells were harvested at different time points (6 and 12 hours) and the fluorescence histograms of FITC were recorded on a flow cytometer (BD Accuri C6; BD Biosciences, San Jose, CA, USA).
In vitro cell viability assay
The cytotoxic effects of MSNs@PDA-PEG, free DM1, MSNs-DM1@PDA-PEG, and MSNs-DM1@PDA-PEG-APt on the human colon cancer SW480 cell line were evaluated by the MTT and bromodeoxyuridine (BrdU) assays.
For the MTT assay, SW480 cells were seeded in 96-well plates at a density of 5×104 viable cells per well and allowed to attach overnight. Then, the medium was replaced with 100 μL of fresh medium containing MSNs@PDA-PEG, free DM1, MSNs-DM1@PDA-PEG, and MSNs-DM1@PDA-PEG-APt, respectively. At designated time points (12, 24, and 48 hours), the medium was supplemented by 10 μL of MTT solution (5 mg/mL) and further incubated for 4 hours. After careful removal of the cell culture medium, 100 μL of dimethyl sulfoxide was added to each well for 15 minutes. Absorbance was measured at 490 nm on a microplate reader.
For BrdU assay, SW480 cells (1×106 per well) were seeded in 6-well plates containing coverslips and treated with MSNs@PDA-PEG, free DM1, MSNs-DM1@PDA-PEG, and MSNs-DM1@PDA-PEG-APt for 6 hours, respectively. After addition of 1 mL DMEM and 50 μL 5-fluoiouridine (200 mM) to each well, incubation was performed for about 15–30 minutes at 37°C. The cell medium was removed, and cells were fixed with 4% formaldehyde in PBS for 15 minutes, and permeabilized in PBS containing 0.1% Triton X-100 for another 10 minutes. Blocking was performed with 3% bovine serum albumin (BSA) in PBS for 2 hours at room temperature followed by incubation with BrdU mouse monoclonal antibody at 4°C overnight. The coverslips were incubated with rhodamine-conjugated mouse secondary antibodies for 1 hour. After staining with 0.5 μg/mL DAPI for 10 minutes, fluorescence of rhodamine or DAPI was observed under an Olympus FV1000 confocal microscope.
Immunofluorescent staining
SW480 cells were seeded in 12-well plates with cover glasses (diameter 18 mm) and incubated at 37°C with 5% CO2 for 24 hours to allow attachment. The medium was replaced with 100 μL of fresh medium containing MSNs@PDA-PEG, free DM1, MSNs-DM1@PDA-PEG, and MSNs-DM1@PDA-PEG-APt, respectively, and further incubated for 6 or 12 hours. Then, the cells were washed with ice cold PBS and fixed in 4% paraformaldehyde at room temperature for 15 minutes, followed by three PBS washes. Subsequently, the cells were permeabilized by incubation with 0.1% Triton X-100–PBS for 10 minutes, and blocked with 3% BSA for 1 hour at room temperature. Next, cells were incubated with appropriately diluted mouse anti-BrdU and α-tubulin monoclonal antibodies, respectively, at 4°C overnight. After three washes with PBS, fluorescent dye-linked secondary antibodies were applied for 1 hour at room temperature. Table S1 lists the antibody concentrations. Then, cells were washed twice with PBS and incubated with PBS containing DAPI (10 μg/mL) for 10 minutes. Finally, fluorescent images were captured on a Leica confocal microscope (Leica Microsystems, Wetzlar, Germany).
Microtubule staining
To assess the effect of DM1 on microtubule morphology, a microtubule fluorescence staining kit (GenMed Scientific Inc) containing anti-α-tubulin monoclonal antibodies and FITC-conjugated secondary antibodies was used, according to the manufacturer’s instructions.
Western blot
Cells or tissue samples were lysed in RIPA buffer. Equal amounts of total protein were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels and transferred onto polyvinylidene fluoride membranes. The membranes were blocked with 5% nonfat dried milk for 2 hours at room temperature, and incubated at 4°C overnight with specific primary antibodies (Table S1 shows the antibody concentrations). After three washes with Tris-buffered saline–Tween 20, the membranes were incubated with secondary antibodies at room temperature for 1 hour and detected using an enhanced chemiluminescence detection system. Protein levels were normalized to β-actin.
Apoptosis analysis
Cell apoptosis was determined with an annexin V-FITC/PI apoptosis detection kit according to the manufacturer’s instructions. Briefly, SW480 cells were seeded in 6-well plates and incubated with MSNs@PDA-PEG, free DM1, MSNs-DM1@PDA-PEG, and MSNs-DM1@PDA-PEG-APt, respectively, for 12 hours. After washing with PBS, the cells were resuspended in binding buffer, incubated with annexin V-FITC and PI at room temperature for 10 minutes, and analyzed immediately by flow cytometry (FACS Influx SE; BD Biosciences).
SW480 xenograft tumor model
Animal care and procedures were carried out in strict accordance with the Guide for the Care and Use of Laboratory Animals (Ministry of Science and Technology of China, 2006) and approved by the Administrative Committee on Animal Research of the Second Clinical Medical College of Jinan University. Female nude mice (4–5 week old) were obtained from the Medical Experimental Animal Center of Guangdong Province (Guangdong, China). The animals were allowed free access to sterile food pellets and water. SW480 (2×106) cells in 100 μL culture medium were implanted subcutaneously in the flank region of each mouse. The tumor size was measured by calipers, and the tumor volume was calculated as follows:
|
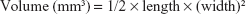
In vivo imaging and biodistribution
When the tumor volume reached ~100–150 mm3, the mice were randomly divided into three groups (n=3), and injected with 100 μL of free IR783 (0.7 mg/kg), MSNs-IR783@PDA-PEG, or MSNs-IR783@PDA-PEG-APt (0.7 mg/kg IR780 equivalent for NPs) via the tail vein. In vivo fluorescent images were acquired at 1, 6, 12, 24, and 48 hours postinjection on a Maestro™ Automated In vivo Imaging system (CRi Maestro, CRI Inc., Woburn, MA, USA). Excitation wavelength of IR783 was 748 nm with an emission wavelength of 780 nm. The mice were euthanized 48 hours after injection, and the heart, liver, spleen, lung, kidney, and tumors were collected for semiquantitative biodistribution analysis and imaging on a Maestro Automated In vivo Imaging system.
In vivo antitumor efficacy and systemic toxicity
When the tumor volume reached about 100 mm3, the mice were randomly divided into five groups (n=4). Then, free DM1, MSNs-DM1@PDA, MSNs-DM1@PDA-PEG, and MSNs-DM1@PDA-PEG-APt were intravenously injected via the tail vein at 0, 3, and 6 days, respectively, at an equivalent dosage of 0.5 mg/kg DM1. The control group was treated with saline. Tumor volume and body weights were recorded every other day. Fourteen days later, the mice were sacrificed, and tumors were photographed and cut into two pieces. The first part was lysed for Western blot as described above; the other tumor piece as well as samples of main tissues (heart, liver, spleen, lung, and kidney) were fixed in formalin and paraffin embedded for histological analysis.
TUNEL assay
To detect apoptotic cells in tumor tissues, a TUNEL assay (One-Step Fluorometric TUNEL System; KeyGEN Biotech, Nanjing, China) was performed following the manufacturer’s instructions. Dewaxed sections were stained with streptavidin-FITC, while cell nuclei were stained with DAPI. Samples were imaged by CLSM (Olympus Fluoview FV-1000).
Statistical analysis
Data are expressed as mean ± SEM from three independent experiments and were analyzed with the GraphPad Prism software (version 5.0; GraphPad Software Inc, La Jolla, CA, USA). Groups were compared by Student’s t-test (group pairs) or one-way analysis of variance (multiple groups). P<0.05 was considered statistically significant. *P<0.05; **P<0.01.
Results and discussion
Synthesis procedure and characteristics of the NPs
The fabrication of EpCAM APt-functionalized MSNs (MSNs-DM1@PDA-PEG-APt) is summarized in Figure 1. In weak alkaline conditions (pH 8–8.5), the catechol group of dopamine, similar in composition to mussel adhesive proteins, is oxidized to quinone and reacts with other catechols/quinones to form polymerized dopamine (PDA), which can bind on the surface of MSNs via covalent and non-covalent interactions. PDA coating, in turn, serves as a versatile platform for reactions with amine- or thiol-terminated functional ligands via Michael addition or Schiff base reactions.28,30 Then, PEG-APt was synthesized due to the interaction between −MAL in PEG and −SH in EpCAM APt.31 Eventually, NH2-PEG-APt was conjugated to the PDA surface of NPs via a Schiff base reaction under oxidizing conditions.
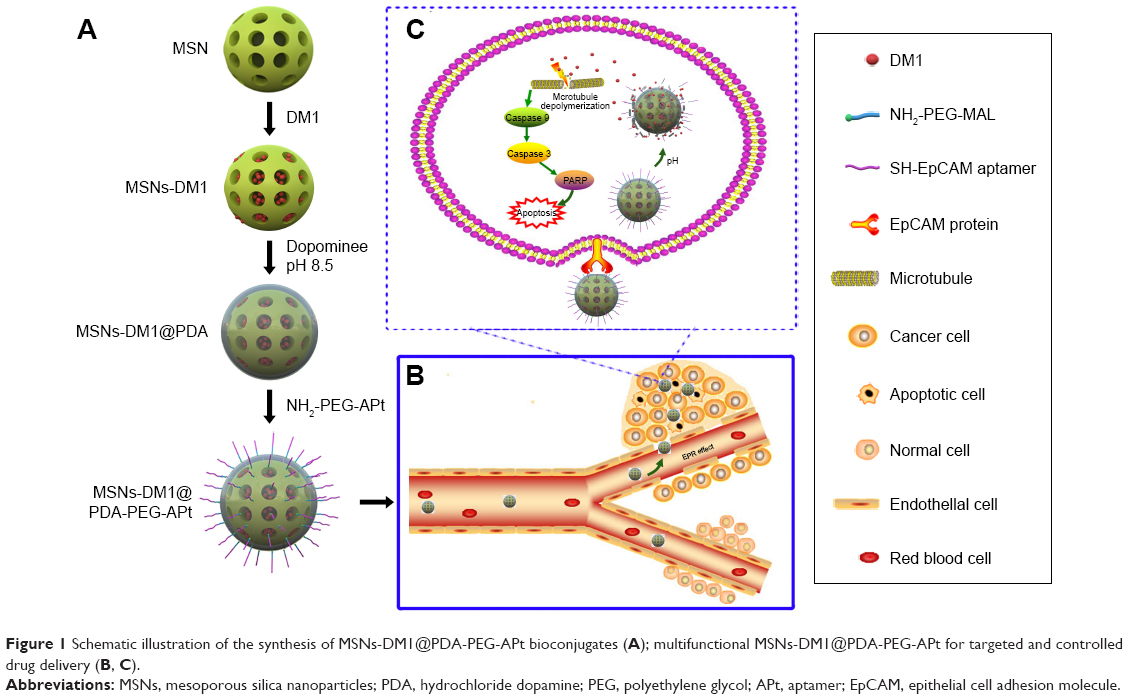 | Figure 1 Schematic illustration of the synthesis of MSNs-DM1@PDA-PEG-APt bioconjugates (A); multifunctional MSNs-DM1@PDA-PEG-APt for targeted and controlled drug delivery (B, C). |
The size and surface peculiarities of NPs have a vital role in cellular uptake, drug release, in vivo pharmacokinetics, and biodistribution.32 As shown in Table 1, the average particle size of MSNs-DM1 was 141.45±5.64 nm, while those of MSNs-DM1@PDA and MSNs-DM1@PDA-PEG-APt were 169.43±4.65 and 203.75±2.37 nm, respectively. The size increase (~25 nm) of MSNs-DM1@PDA or MSNs-DM1@PDA-PEG-APt may be attributed to the thin PDA films that form around MSNs-DM1, and reflects the successful coating of PDA through oxidative polymerization. The mean hydrodynamic size of DM1-loaded MSNs was about 140~150 nm in diameter, which is an appropriate size range for easy accumulation in the tumor vasculature under the influence of the EPR effect.33 Table 1 showed that the absolute value of zeta potential of MSNs-DM1@PDA-PEG-APt (−10.84±5.43 mV) was slightly decreased compared with or similar to those of MSNs-DM1 (−7.68±4.32 mV) and MSNs-DM1@PDA (−26.77±4.54 mV). The specific surface area, pore volume, and the most probable pore size of MSNs, MSNs-DM1, MSNs-DM1@PDA-PEG, and MSNs-DM1@PDA-PEG-APt are also presented in Table 1. Compared to MSNs and MSNs-DM1, all the pore parameters of MSNs-DM1@PDA-PEG and MSNs-DM1@PDA-PEG-APt were found to significantly decrease. The Brunauer–Emmett–Teller (BET) surface area was 257.36 m2/g, the pore volume was 0.58 cm3/g, and, as evaluated by the Barrett–Joyner–Halenda method, the most probable pore size was about 2.65 nm. Moreover, the pore size distribution of MSNs was rather narrow. With loading of DM1, the BET surface area and the most probable pore size decreased to 125.25 m2/g and 2.23 nm, respectively. The BET surface area of MSNs-DM1@PDA-PEG and MSNs-DM1@PDA-PEG-APt was 50.58 and 52.78 m2/g, respectively. However, the pore volumes were 0.14 and 0.12 cm3/g due to the coating of PDA, PEG, and APt onto the surface of DM1-loaded MSNs. Overall, the structural parameters of MSNs, MSNs-DM1, MSNs-DM1@PDA-PEG, and MSNs-DM1@PDA-PEG-APt suggested that DM1 occupied the pore space of MSNs and DM1-loaded MSNs were coated with PDA, PEG, and APt. The encapsulation efficiency of DM1 in MSNs is about 94.43±1.95.
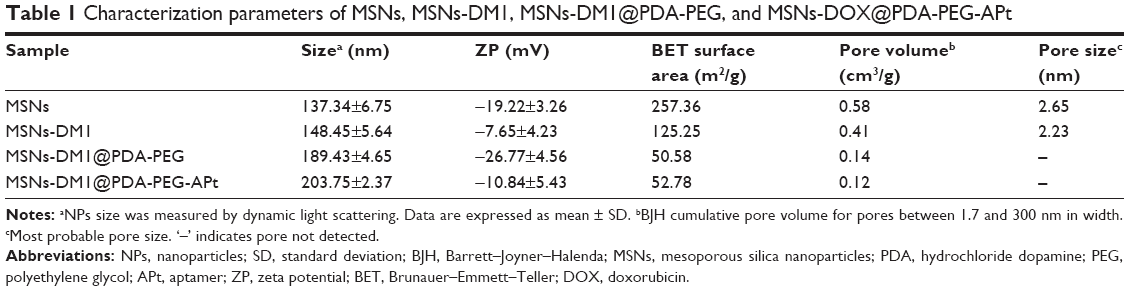 | Table 1 Characterization parameters of MSNs, MSNs-DM1, MSNs-DM1@PDA-PEG, and MSNs-DOX@PDA-PEG-APt |
As a result of the slightly negative charge of MSNs, needless clearance by the RES (eg, in the liver) could be reduced, followed by the perfecting of blood compatibility and NP accumulation in the tumor sites.34 In order to access the morphology of NPs, TEM was performed in this study. Figure 2A depicts the TEM images of MSNs. MSNs were spherical with a single surface, while thin films or membranes were visualized on the surface of MSNs-DM1@PDA-PEG-APt, suggesting that PDA, PEG, and APt films were successfully deposited on the surface of MSNs via oxidative polymerization. The average particle size was around 100 nm in TEM images, a smaller size than that obtained in DLS experiments. This difference may be due to a tendency to shrink and collapse, while NPs are in the dry state.35
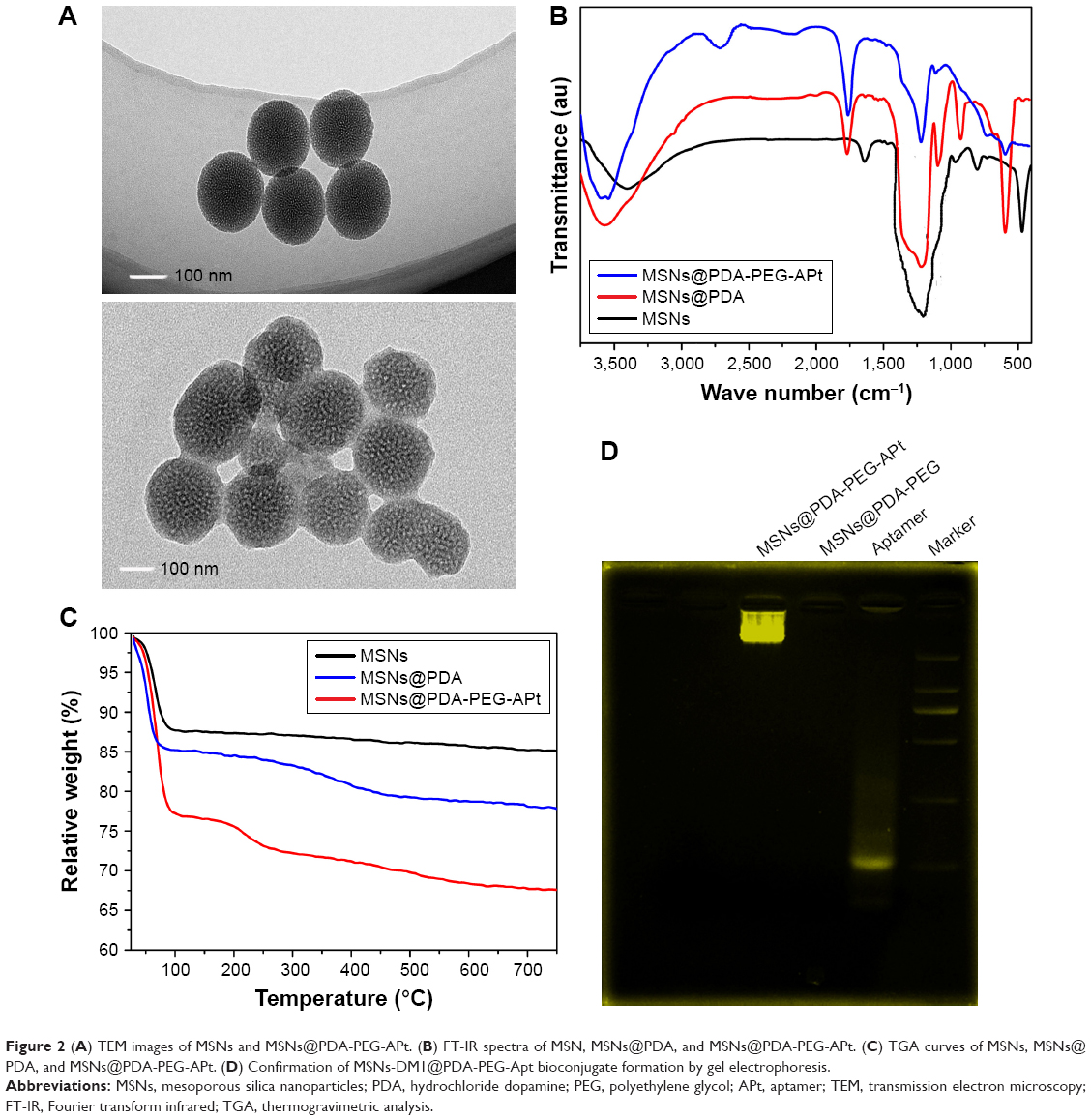 | Figure 2 (A) TEM images of MSNs and MSNs@PDA-PEG-APt. (B) FT-IR spectra of MSN, MSNs@PDA, and MSNs@PDA-PEG-APt. (C) TGA curves of MSNs, MSNs@PDA, and MSNs@PDA-PEG-APt. (D) Confirmation of MSNs-DM1@PDA-PEG-Apt bioconjugate formation by gel electrophoresis. |
Fourier transform infrared (FT-IR) spectroscopy was employed to verify the successful modification of PDA film and surface chemical groups of NPs. As shown in Figure 2B, several new absorption signals appeared after surface modification. The band at 1,276 cm−1 in the unconjugated polymer spectra (MSNs), reflecting the C=O stretching vibration of carboxylic acid, was assigned to C=H stretching and C=H bending vibrations from CTAB.36 The absorption peaks at 1,776 and 1,630 cm−1 were from PDA layers, indicating the successful incorporation of the PDA layer on the surface of NPs.37 After PEG conjugation, typical absorption peaks of the C=H stretching vibration in PEG emerged at 2,389 and 2,253 cm−1, further confirming the successful functionalization of PEG.38 Upon EpCAM APt conjugation, a distinct peak emerged at 1,139 cm−1 and was mainly assigned to the C=H bending vibration in deoxyribose of DNA oligonucleotides. Thus, the successful decoration of cell targeting EpCAM APt on the NPs was verified.31
Taken together, TEM and FT-IR spectroscopy studies indicated a successful incorporation of the PDA film and APt conjugation on NPs. For quantitative analysis, MSNs@PDA-PEG-APt NPs were further verified by thermogravimetric analysis (TGA; Figure 2C). In this work, the organic material-modified MSNs well inherited the NIR absorption property of MSNs. The weight reduction of PDA, PEG-coated, and EpCAM APt-modified NPs MSNs@PDA-PEG-APt was 32.4%. Only the PDA-coated NPs MSNs-PDA showed 22.2% reduction when heated in air atmosphere to 750°C, while that of the bare MSNs was only 15% in the same temperature range.27 TGA data further demonstrated that MSNs were successfully modified with PDA, the content of which was 7.2%, also indicating a successful conjugation of PEG and EpCAM to MSNs@PDA NPs.
Finally, the conjugation of EpCAM APt to MSNs@PDA-PEG was assessed by agarose gel electrophoresis. As shown in Figure 2D, migration of free EpCAM APt matched that of the 19 base pairs size marker, while both positive and control MSNs@PDA-PEG-APt bioconjugates almost remained at the origin. To confirm solid conjugation, free APt, MSNs@PDA-PEG, and MSNs@PDA-PEG-APt were mixed without any conjugation process and subjected to 3% gel electrophoresis; these two were well separated. Taken together, these data revealed the efficient conjugation between MSNs@PDA-PEG and EpCAM APt.
pH-sensitive drug release in vitro
pH-responsive systems attract increasing attention in the cancer therapy field, since the human body exhibits changes in pH. The pH value of normal tissues and blood is ~7.4, while that of the tumor microenvironment is between 5.5 and 7.0, mainly due to high levels of CO2 and glycolysis rates. The pH value decreases further in cancer cell organelles such as endosomes (pH 5.5–6.0) and lysosomes (pH 4.5–5.0).13 To assess whether the MSNs-DM1@PDA-PEG-APt prodrug NPs constitute an effective drug delivery system for sustainable release of DM1, we performed drug release assays at pH 5.5 and 7.4 at different time points. In this study, DOX was also selected as an ideal drug for in vitro drug release studies, for the three drug carriers MSNs-DOX, MSNs-DOX@PDA, and MSNs-DOX@PDA-PEG-APt. As shown in Figure 3, the release rates of DOX were different at distinct pH values, indicating pH-sensitivity. Compared with MSNs-DOX that showed a rapid release, both MSNs-DOX@PDA and MSNs-DOX@PDA-PEG-APt exhibited similar sustained release profiles for DOX at pH 7.4 and 5.5. The release rates of MSNs-DOX@PDA and MSNs-DOX@PDA-PEG-APt at pH 5.5 were significantly higher than those obtained at pH 7.4. Particularly, MSNs-DOX@PDA showed the most prominent changes among the three drug carriers, as shown in Figure 3B. This is due to PDA related pH-sensitivity, while PEG-coated and APt-modified NPs have tighter bonds between MSNs and PDA, which are harder to disrupt. As a result, the drug release rate was slightly reduced than in samples only coated with PDA, although MSNs-DOX@PDA-PEG-Apt NPs can achieve targeted drug delivery. By comprehensively considering the targeted drug delivery and release rates of the three drug carriers, we finally selected MSNs-DOX@PDA-PEG-APt for subsequent experiments.
 | Figure 3 Release profile of DOX from MSNs-DOX (A), MSNs-DOX@PDA (B), and MSNs-DOX@PDA-PEG-APt (C). The release experiment was performed in PBS at pH 7.4 and 5.5 in sequence. |
Cell recognition and uptake
The application of MSNs in the field of drug delivery has aroused much interest in recent years. Immense advances in morphology control and surface modification of inorganic delivery vehicles, as well as increased knowledge regarding physiological factors affecting drug delivery systems, have opened new possibilities for more efficient treatments in this burgeoning area of research. Indeed, MSN based materials have been widely used for controlling the intracellular targeted delivery of anticancer drugs. Taking advantage of these features, the feasibility of targeted drug delivery by a guided APt was tested.39 In the current study, SW480 cells seeded into a 6-well dish containing coverslips were incubated at 37°C, after treatment with MSNs@PDA-PEG, free DM1, MSNs-DM1@PDA-PEG, and MSNs-DM1@PDA-PEG-APt, respectively. To confirm the CRC targeting ability of FITC-MSNs-DM1@PDA-PEG-APt in vitro, we carried out confocal microscopy analysis to compare the cellular uptake efficiencies of FITC-MSNs-DM1@PDA-PEG-APt and FITC-MSNs-DM1@PDA-PEG (Figure 4A). The results suggested that cellular uptake of FITC-MSNs-DM1@PDA-PEG-APt in SW480 cells was significantly higher than that of MSNs-DM1@PDA-PEG. Furthermore, cellular uptake of FITC-MSNs-DM1@PDA-PEG-APt in SW480 cells was remarkably higher than in MCN460 and HEK239 cells, which scarcely show any EpCAM factor. Flow cytometry analysis also confirmed the enhanced cellular uptake of MSNs-DM1@PDA-PEG by EpCAM APt modification (Figure 4B). This increase was probably due to APt recognition by EpCAM expressed on SW480 cells, resulting in more NPs attached to SW480 cells. Previous studies have reported similar findings wherein the targeted APt modification increases the cellular uptake of NPs.40 Therefore, MSNs-DM1@PDA-PEG-APt administered intravenously could specifically recognize EpCAM overexpressing cancer cells, and might help treat metastatic cancers.
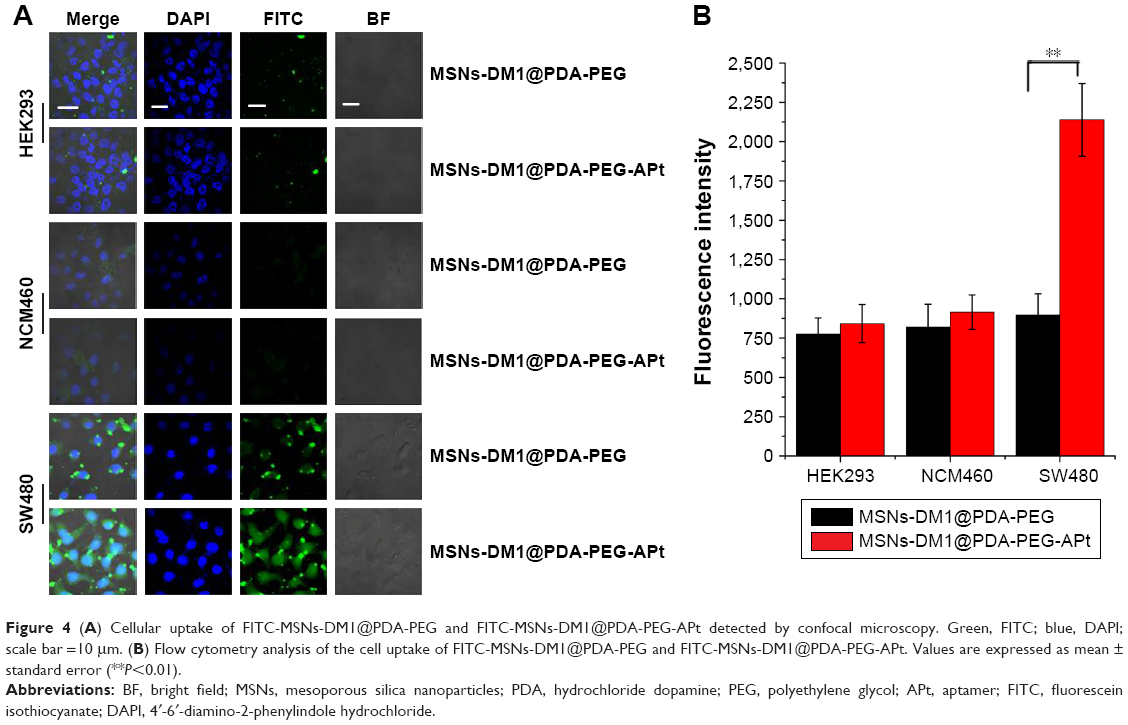 | Figure 4 (A) Cellular uptake of FITC-MSNs-DM1@PDA-PEG and FITC-MSNs-DM1@PDA-PEG-APt detected by confocal microscopy. Green, FITC; blue, DAPI; scale bar =10 μm. (B) Flow cytometry analysis of the cell uptake of FITC-MSNs-DM1@PDA-PEG and FITC-MSNs-DM1@PDA-PEG-APt. Values are expressed as mean ± standard error (**P<0.01). |
In vitro cell viability
In order to evaluate the cytotoxicity of free DM1, MSNs-DM1@PDA-PEG, and MSNs-DM1@PDA-PEG-APt, MTT assay was performed on SW480 and NCM460 cells. The control group was treated with saline, and the cytotoxicity of drug-free NPs was also investigated to exclude any nonspecific effect. NCM460 and SW480 cells were treated with free DM1, MSNs-DM1@PDA-PEG, and MSNs-DM1@PDA-PEG-APt suspensions at an equivalent DM1 concentration of 10 μM for 12, 24, and 48 hours, respectively. As shown in Figure 5A and B, none of the drug-free NPs exhibited obvious cytotoxicity to SW480 and NCM460 cells, declaring both the copolymer and the modification of NPs (PDA coating and conjugation of PEG) biocompatible and nontoxic to cells. As reported previously, extremely high concentrations of MSNs (about 25 mg/mL) at some sizes exhibit cytotoxicity.41 However, the required maximum concentration of MSNs herein was 500 μg/mL, a concentration at which nearly no cytotoxicity could be observed. Similar to our result, PDA and PEG coating was found to be nontoxic in various cell models and in various in vivo studies.28 MSNs-DM1@PDA-PEG-APt (with the specific targeting effect of EpCAM APts) exhibited better in vitro antitumor efficacy than free DM1, and MSNs-DM1@PDA-PEG on SW480 cells (Figure 5B); however, for normal colonic epithelial cell line NCM460, MSNs-DM1@PDA-PEG-APt and MSNs-DM1@PDA-PEG exhibited almost identical cytostatic effects (Figure 5A), suggesting that MSNs-DM1@PDA-PEG-APt could be used as a promising and high-efficiency drug delivery system for CRC therapy.
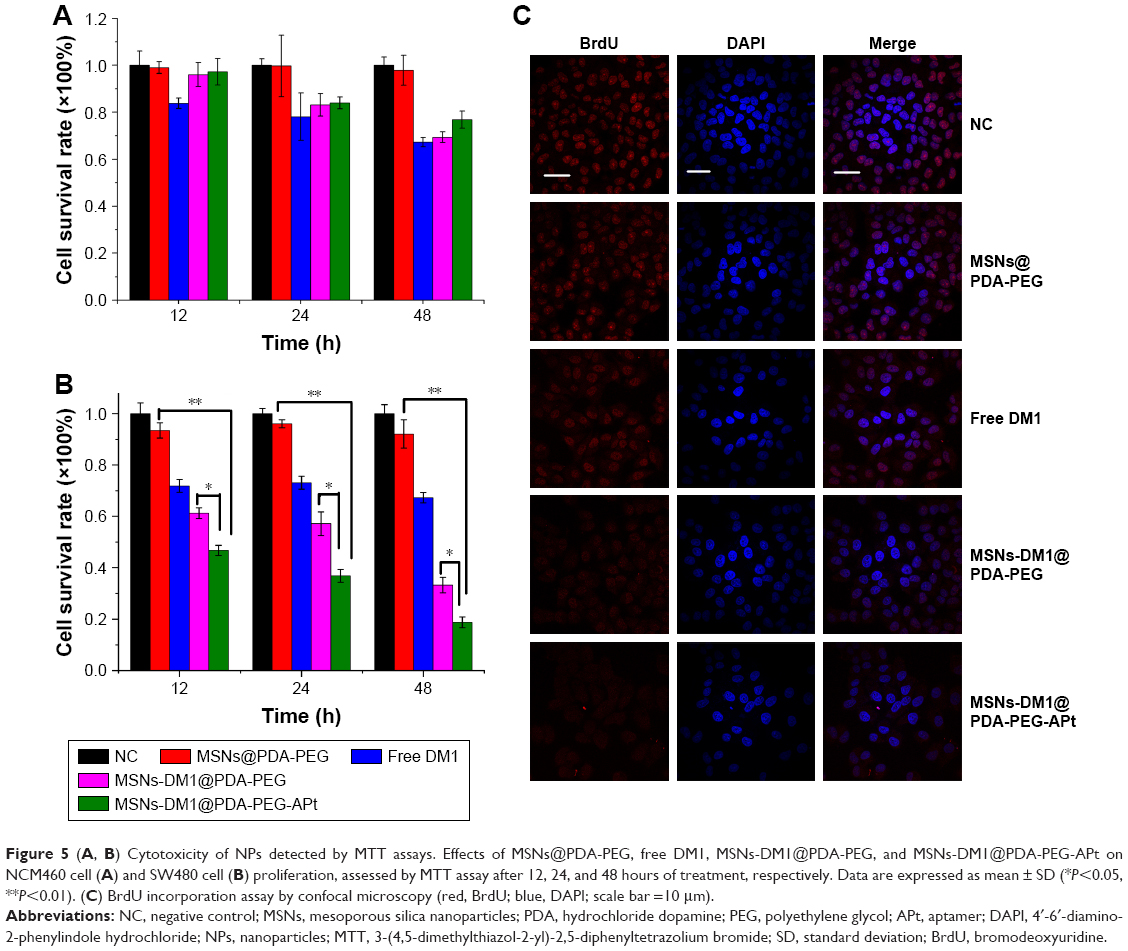 | Figure 5 (A, B) Cytotoxicity of NPs detected by MTT assays. Effects of MSNs@PDA-PEG, free DM1, MSNs-DM1@PDA-PEG, and MSNs-DM1@PDA-PEG-APt on NCM460 cell (A) and SW480 cell (B) proliferation, assessed by MTT assay after 12, 24, and 48 hours of treatment, respectively. Data are expressed as mean ± SD (*P<0.05, **P<0.01). (C) BrdU incorporation assay by confocal microscopy (red, BrdU; blue, DAPI; scale bar =10 μm). |
BrdU labeling allows newly replicated DNA to be distinguished from existing DNA, which is not possible with standard DNA-intercalating dyes such as DAPI or SYBR Green I.42 SW480 cells were treated with MSNs@PDA-PEG, free DM1, MSNs-DM1@PDA-PEG, and MSNs-DM1@PDA-PEG-APt for 6 hours, respectively, and BrdU incorporation was assessed by confocal microscopy. As shown in Figure 5C, MSNs-DM1@PDA-PEG-APt had the best inhibition effect on BrdU incorporation.
Microtubule formation and assembly inhibition by DM1, and related molecular mechanisms
Maytansinoids, including its derivatives, are potent antimitotic agents that exert cytotoxic effects by disrupting microtubule assembly and inducing mitotic arrest.12 The antitumor activity of maytansine has been extensively evaluated in human trials, but its clinical use is limited by general toxicity to proliferating cells, including normal cells; therefore, despite potent in vitro effects, it displays a poor therapeutic window in vivo.43 The high potency of maytansine makes it an attractive candidate for targeted delivery for the selective destruction of tumor cells. The first step was to synthesize a targeted delivery DM1 system, that is, DM1-loaded MSNs@PDA-PEG-EpCAM APt-modified NPs. To identify the mechanisms of the intracellular accumulation and exceptional cytotoxicity of maytansinoids, we analyzed SW480 cells treated with MSNs@PDA-PEG, free DM1, MSNs-DM1@PDA-PEG, and MSNs-DM1@PDA-PEG-APt for 6 and 12 hours, respectively. Microtubule morphology was stained by the microtubule fluorescence staining kit (GenMed Scientific Inc). In agreement with previous reports,44 we demonstrated that DM1 disrupted the organization of interphase microtubules in SW480 cells (Figure 6A and B). The depolymerizing effects increased with the modification of DM1-loaded MSNs, and MSNs-DM1@PDA-PEG-APt produced much stronger depolymerizing effects than other treatments. Meanwhile, to assess whether DM1 reduces α-tubulin expression in colorectal cells, SW480 cells seeded in 6-well plates were treated with MSNs@PDA-PEG, free DM1, MSNs-DM1@PDA-PEG, and MSNs-DM1@PDA-PEG-APt for 6 and 12 hours, respectively. In agreement with previous reports,45 α-tubulin expression was suppressed markedly after treatment with MSNs-DM1@PDA-PEG-APt compared with the remaining groups (Figure 6C and D). Western blot analysis was also used to quantify α-tubulin and β-tubulin levels. As predicted, the expression levels of α-tubulin and β-tubulin were significantly decreased in MSNs-DM1@PDA-PEG-APt-treated cells compared with untreated cells, as well as the MSNs@PDA-PEG, free DM1, and MSNs-DM1@PDA-PEG groups (Figure 6E and F).
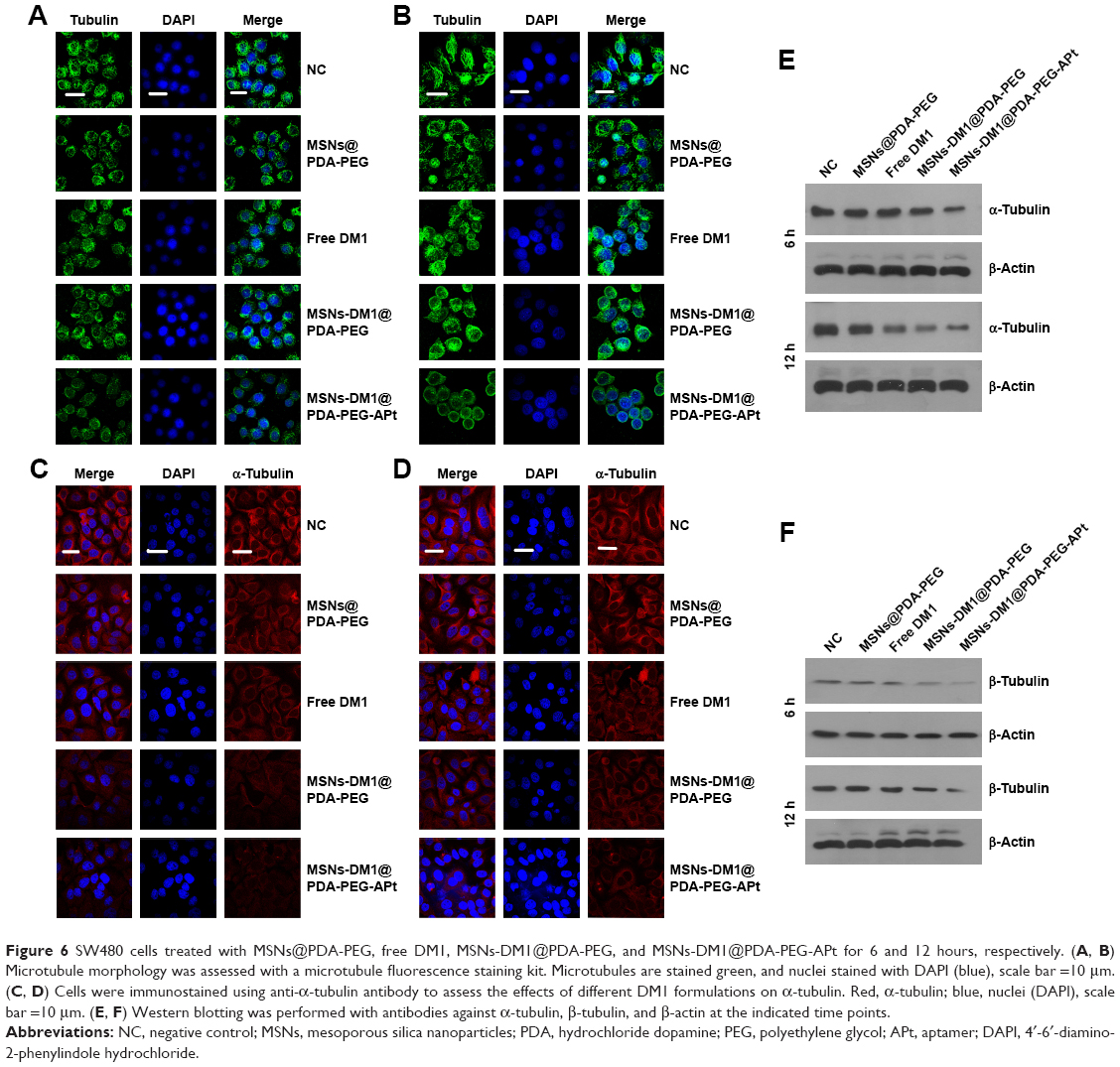 | Figure 6 SW480 cells treated with MSNs@PDA-PEG, free DM1, MSNs-DM1@PDA-PEG, and MSNs-DM1@PDA-PEG-APt for 6 and 12 hours, respectively. (A, B) Microtubule morphology was assessed with a microtubule fluorescence staining kit. Microtubules are stained green, and nuclei stained with DAPI (blue), scale bar =10 μm. (C, D) Cells were immunostained using anti-α-tubulin antibody to assess the effects of different DM1 formulations on α-tubulin. Red, α-tubulin; blue, nuclei (DAPI), scale bar =10 μm. (E, F) Western blotting was performed with antibodies against α-tubulin, β-tubulin, and β-actin at the indicated time points. |
Taken together, these data suggested that DM1 has cytotoxic effects on SW480 cells, characterized by disrupted microtubule assembly, reduced cell viability, and cell cycle arrest.
DM1 induces apoptosis via the caspase pathway by decreasing microtubule formation and assembly disruption
DM1 is a potent antimitotic agent that exerts cytotoxic effects by disrupting microtubule assembly and inducing apoptosis.45 In this study, we explored the effect of DM1 induced apoptosis and explored the detailed molecular mechanisms. SW480 cells treated with MSNs@PDA-PEG, free DM1, MSNs-DM1@PDA-PEG, and MSNs-DM1@PDA-PEG-APt for 12 hours, respectively, were assessed by flow cytometry, after annexin V and PI staining (Figure 7A). For example, the percentage of dead cells increased from 4.13% in controls to 73.09% in MSNs-DM1@PDA-PEG-APt-treated cells (Figure 7B).
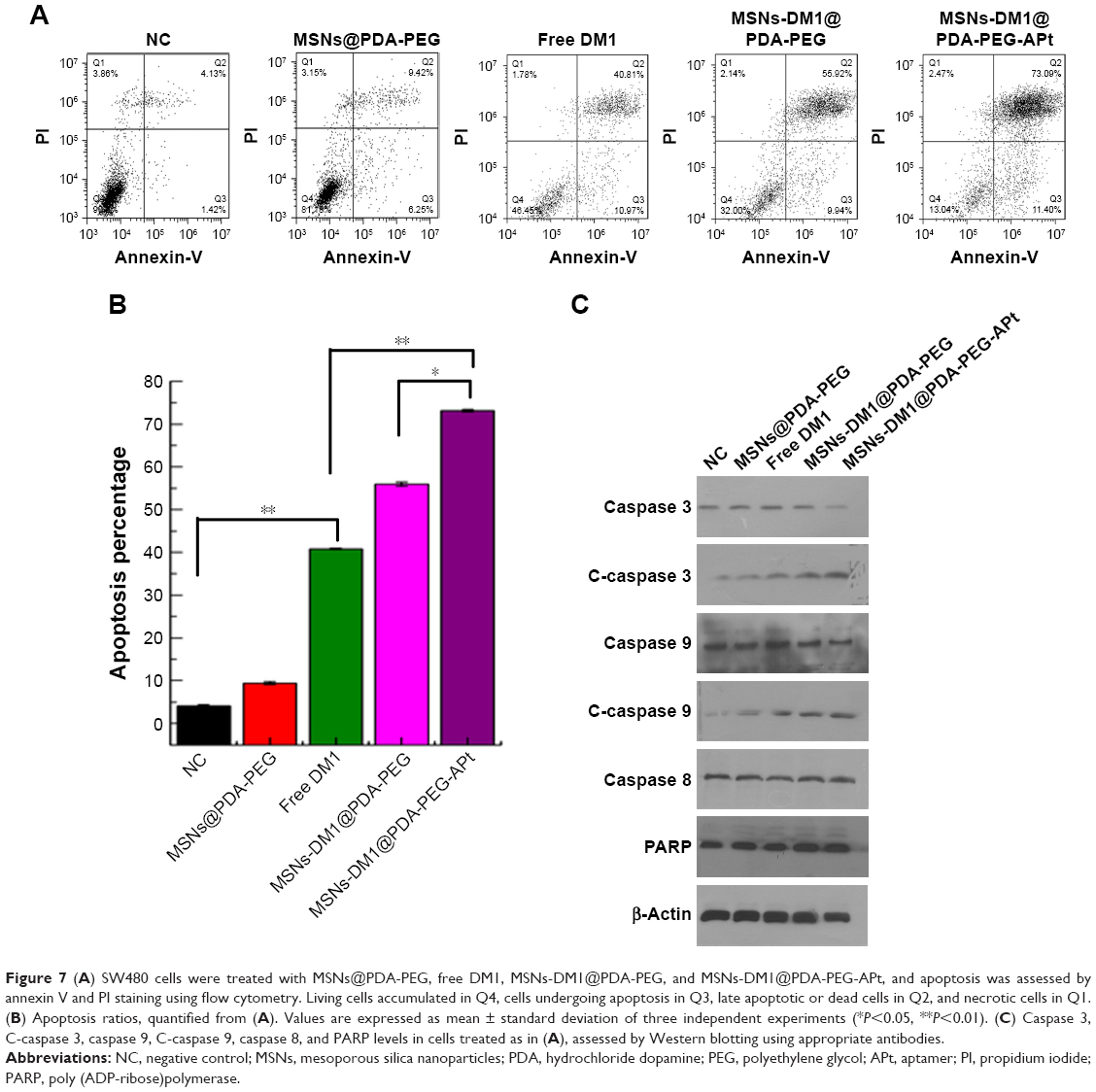 | Figure 7 (A) SW480 cells were treated with MSNs@PDA-PEG, free DM1, MSNs-DM1@PDA-PEG, and MSNs-DM1@PDA-PEG-APt, and apoptosis was assessed by annexin V and PI staining using flow cytometry. Living cells accumulated in Q4, cells undergoing apoptosis in Q3, late apoptotic or dead cells in Q2, and necrotic cells in Q1. (B) Apoptosis ratios, quantified from (A). Values are expressed as mean ± standard deviation of three independent experiments (*P<0.05, **P<0.01). (C) Caspase 3, C-caspase 3, caspase 9, C-caspase 9, caspase 8, and PARP levels in cells treated as in (A), assessed by Western blotting using appropriate antibodies. |
To explore the signaling pathways involved in DM1 induced apoptosis, Western blot was used to quantify the total and cleaved levels of related proteins. As shown in Figure 7C, MSNs-DM1@PDA-PEG-APt treatment significantly reduced the expression levels of caspase 3 and caspase 9, and increased the amounts of cleaved caspase 3, caspase 9, and PARP. PARP is a known marker of apoptosis.46 This result suggested that treatment with MSNs-DM1@PDA-PEG-APt markedly enhanced apoptosis compared with other treatments. Consistent with previous reports, our data indicated that DM1 induces apoptosis via the caspase pathway.
In vivo imaging and biodistribution
To assess the distribution and tumor accumulation of the synthesized NPs in vivo, nude mice bearing SW480 tumors were intravenously injected via tail vein with free IR783, MSNs-IR783@PDA-PEG, and MSNs-IR783@PDA-PEG-APt, respectively. Although significantly higher IR783 accumulation in tumor was observed for both NPs compared with free IR783, the tumor signal intensity of MSNs-IR783@PDA-PEG-APt was much stronger than that of MSNs-IR783@PDA-PEG during the experimental period (Figure 8A and B), further confirming that MSNs-IR783@PDA-PEG-APt can target SW480 tumors, probably by both passive (EPR effect-mediated) and active (EpCAM APt-mediated) mechanisms. To quantitatively compare the biodistribution of MSNs-IR783@PDA-PEG and MSNs-IR783@PDA-PEG-APt, ex vivo fluorescence signals in major organs and tumors were analyzed at 48 hours postinjection. As shown in Figure 8C and D, the amounts of MSNs-IR783@PDA-PEG-APt accumulated in the liver and lung were significantly lower than those of MSNs-IR783@PDA-PEG, suggesting that MSNs-DM1@PDA-PEG-APt could decrease the systemic toxicity of DM1 in normal tissues.
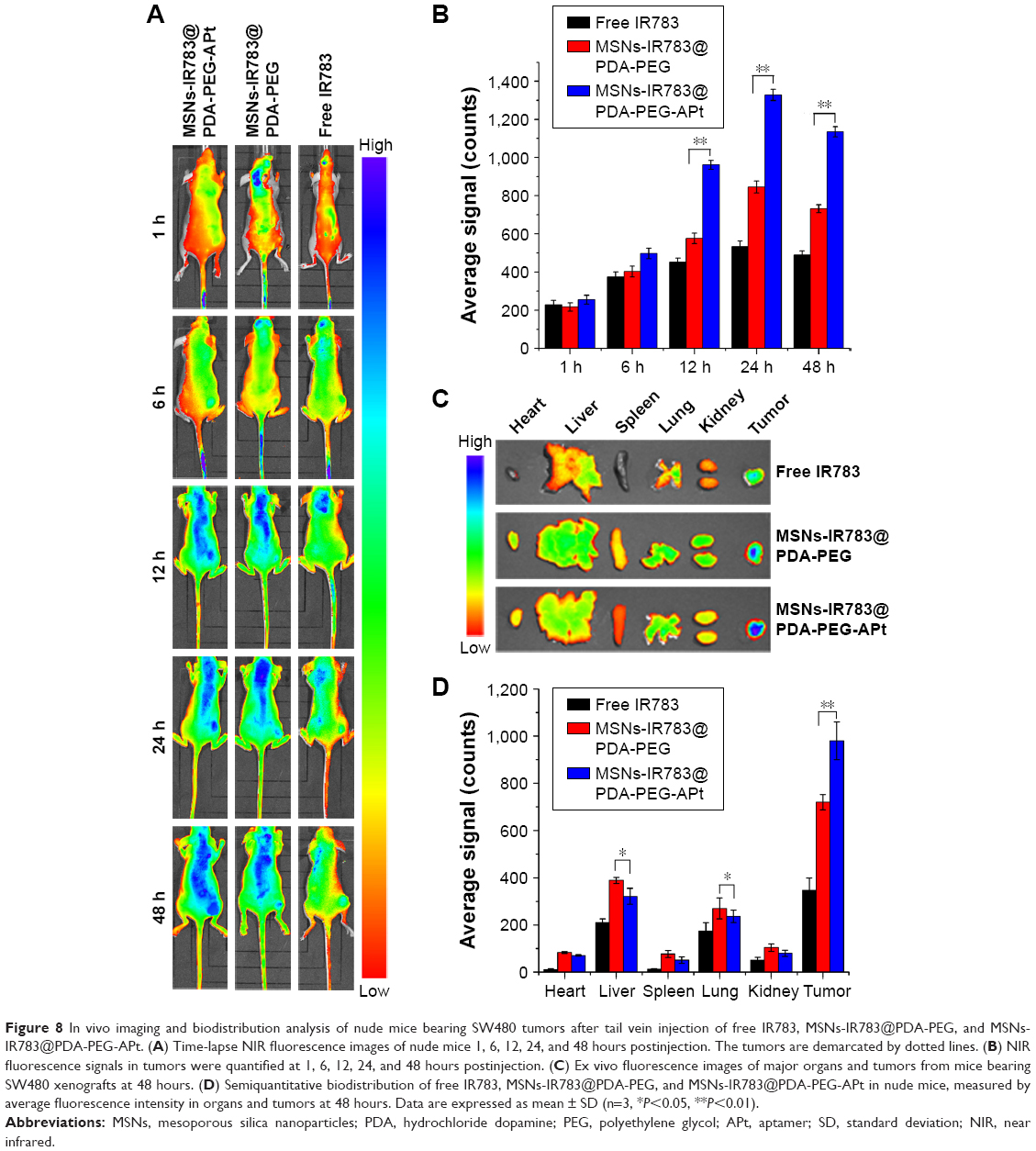 | Figure 8 In vivo imaging and biodistribution analysis of nude mice bearing SW480 tumors after tail vein injection of free IR783, MSNs-IR783@PDA-PEG, and MSNs-IR783@PDA-PEG-APt. (A) Time-lapse NIR fluorescence images of nude mice 1, 6, 12, 24, and 48 hours postinjection. The tumors are demarcated by dotted lines. (B) NIR fluorescence signals in tumors were quantified at 1, 6, 12, 24, and 48 hours postinjection. (C) Ex vivo fluorescence images of major organs and tumors from mice bearing SW480 xenografts at 48 hours. (D) Semiquantitative biodistribution of free IR783, MSNs-IR783@PDA-PEG, and MSNs-IR783@PDA-PEG-APt in nude mice, measured by average fluorescence intensity in organs and tumors at 48 hours. Data are expressed as mean ± SD (n=3, *P<0.05, **P<0.01). |
In vivo anticancer effects and reduced systemic toxicity of MSNs-DM1@PDA-PEG-APt
Encouraged by the promising in vitro results obtained with MSNs-DM1@PDA-PEG-APt (Figure 5), we further assessed tumor growth inhibition by MSNs-DM1@PDA-PEG-APt NPs in nude mice bearing SW480 cells. As shown in Figure 9, treatment with MSNs-DM1@PDA-PEG-APt significantly inhibited tumor growth compared with the remaining four groups (Figure 9A–C); these findings were further supported by hematoxylin and eosin (H&E) staining of tumor sections to evaluate anticancer effects. As shown in Figure 9D, prominent necrosis with decreased tumoral cellularity was observed in tumors treated with MSNs-DM1@PDA-PEG-APt.
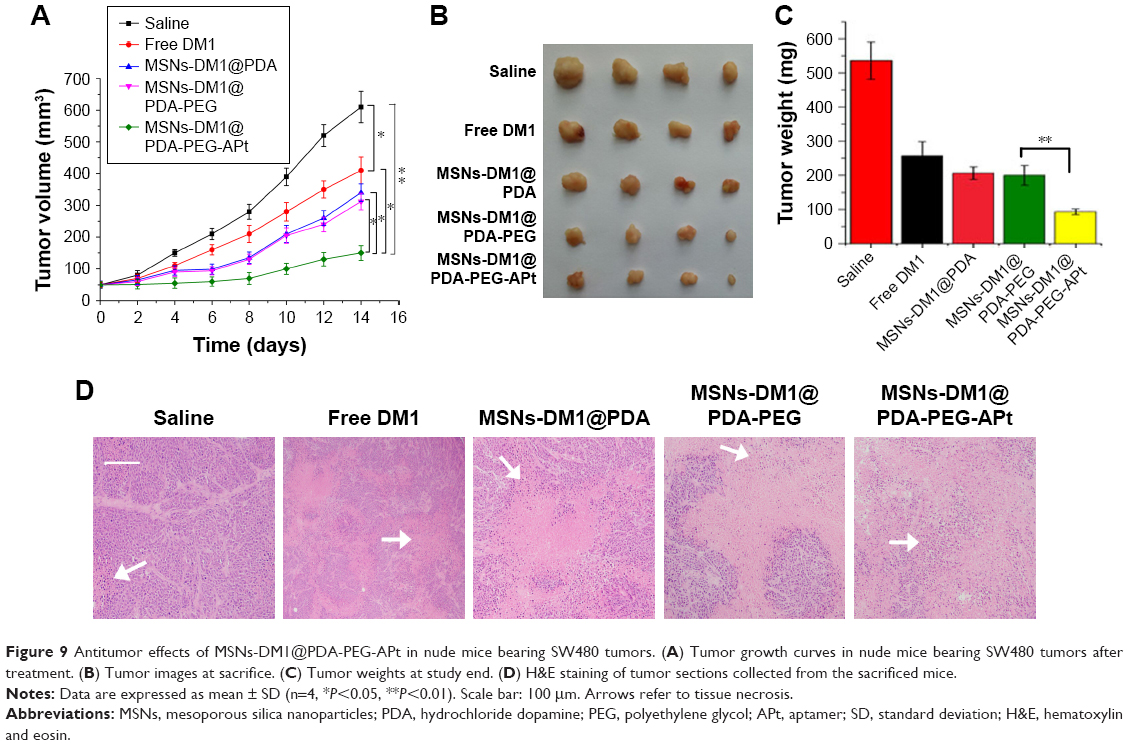 | Figure 9 Antitumor effects of MSNs-DM1@PDA-PEG-APt in nude mice bearing SW480 tumors. (A) Tumor growth curves in nude mice bearing SW480 tumors after treatment. (B) Tumor images at sacrifice. (C) Tumor weights at study end. (D) H&E staining of tumor sections collected from the sacrificed mice. |
We further evaluated whether MSNs-DM1@PDA-PEG-APt could reduce α-tubulin levels and induce apoptosis in tumor tissues after systemic administration, by immunofluorescent staining and Western blot. As shown in Figure 10, MSNs-DM1@PDA-PEG-APt treatment resulted in a drastic inhibition of α-tubulin expression compared with the remaining groups, greatly increasing tumor cell apoptosis as confirmed by TUNEL staining and Western blot analysis of apoptosis related proteins (Figure 11).
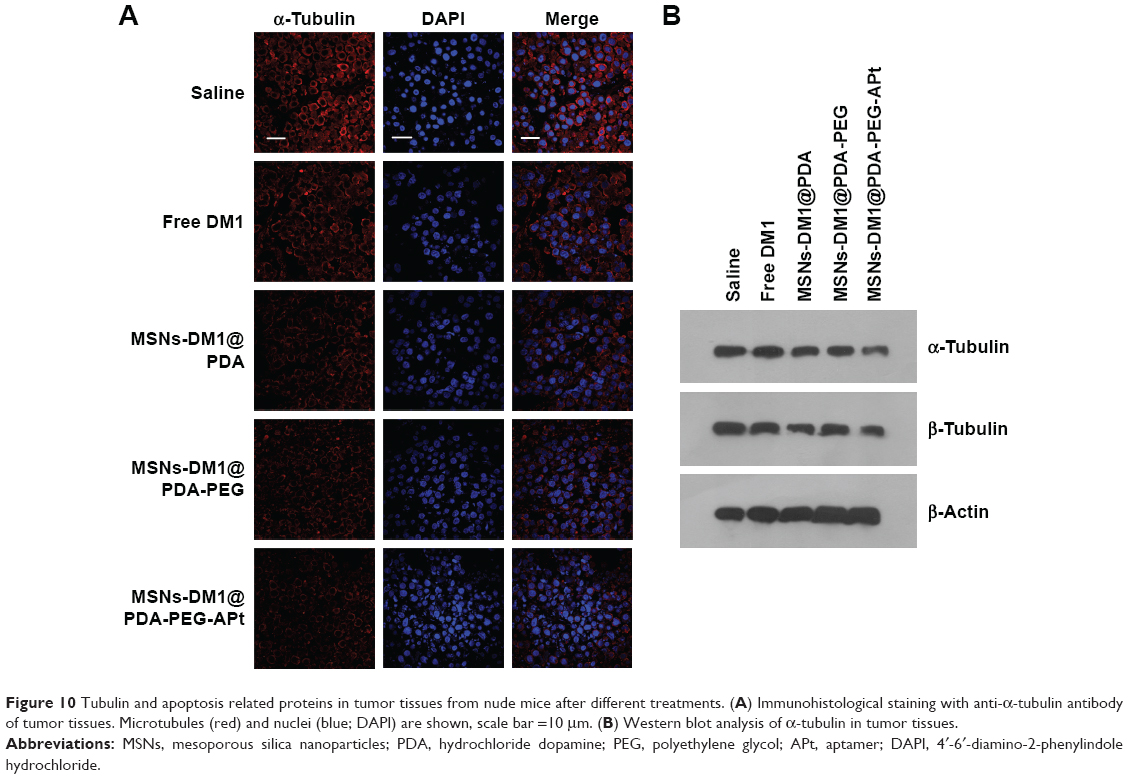 | Figure 10 Tubulin and apoptosis related proteins in tumor tissues from nude mice after different treatments. (A) Immunohistological staining with anti-α-tubulin antibody of tumor tissues. Microtubules (red) and nuclei (blue; DAPI) are shown, scale bar =10 μm. (B) Western blot analysis of α-tubulin in tumor tissues. |
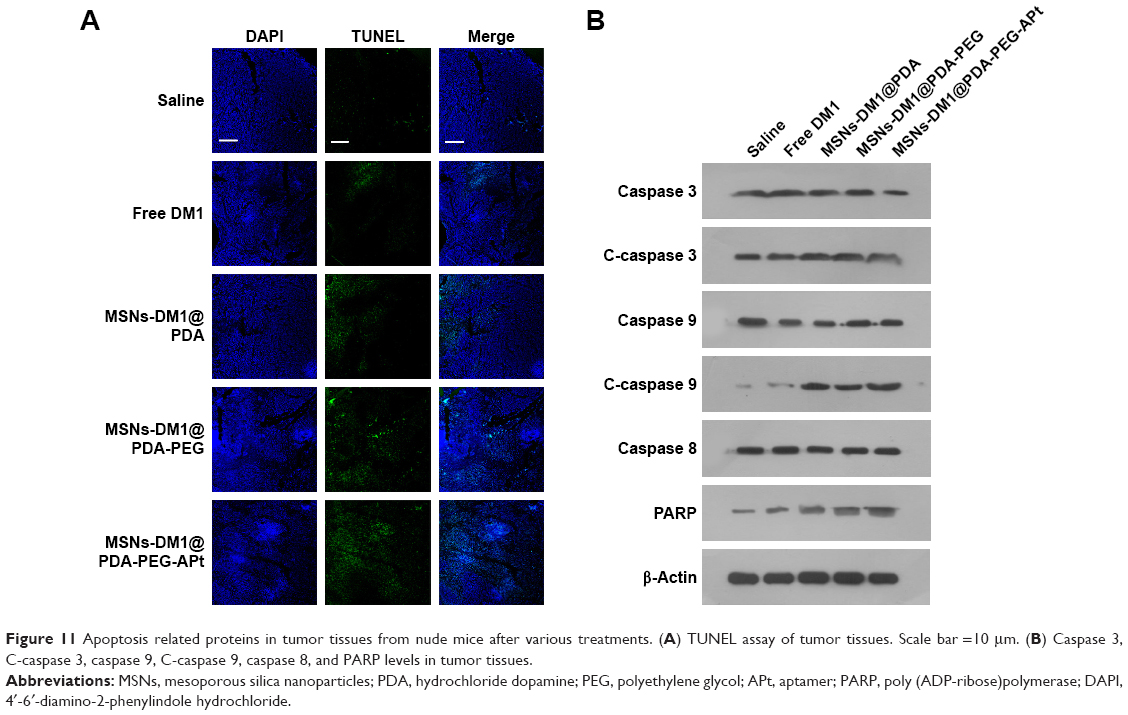 | Figure 11 Apoptosis related proteins in tumor tissues from nude mice after various treatments. (A) TUNEL assay of tumor tissues. Scale bar =10 μm. (B) Caspase 3, C-caspase 3, caspase 9, C-caspase 9, caspase 8, and PARP levels in tumor tissues. |
These results clearly suggest that targeted delivery of DM1 to CRC by MSNs-DM1@PDA-PEG-APt results in much higher anticancer efficiency than other delivery systems, achieving high amounts of DM1 in tumors.
In addition to therapeutic efficacy, we also evaluated systemic toxicity of MSNs-DM1@PDA-PEG-APt versus free DM1, by body weight and H&E staining of organ specimens. As shown in Figure 12, considerable weight loss was found in mice treated with free DM1, while no significant changes were observed in the remaining treatment groups. In nude mice that were administered free DM1, liver and lung damage was observed, suggesting possible liver and lung associated toxicity of DM1.5,7 In contrast, no visible tissue damage in the liver and lung was observed in the MSNs-DM1@PDA-PEG-APt treatment group (Figure 12). The drastically reduced toxicity of MSNs-DM1@PDA-PEG-APt is likely attributed to the decreased accumulation in and more rapid clearance from organs (Figure 8). Collectively, these results confirmed that MSNs-DM1@PDA-PEG-APt can markedly improve DM1 therapeutic effects, while reducing systemic toxicity.
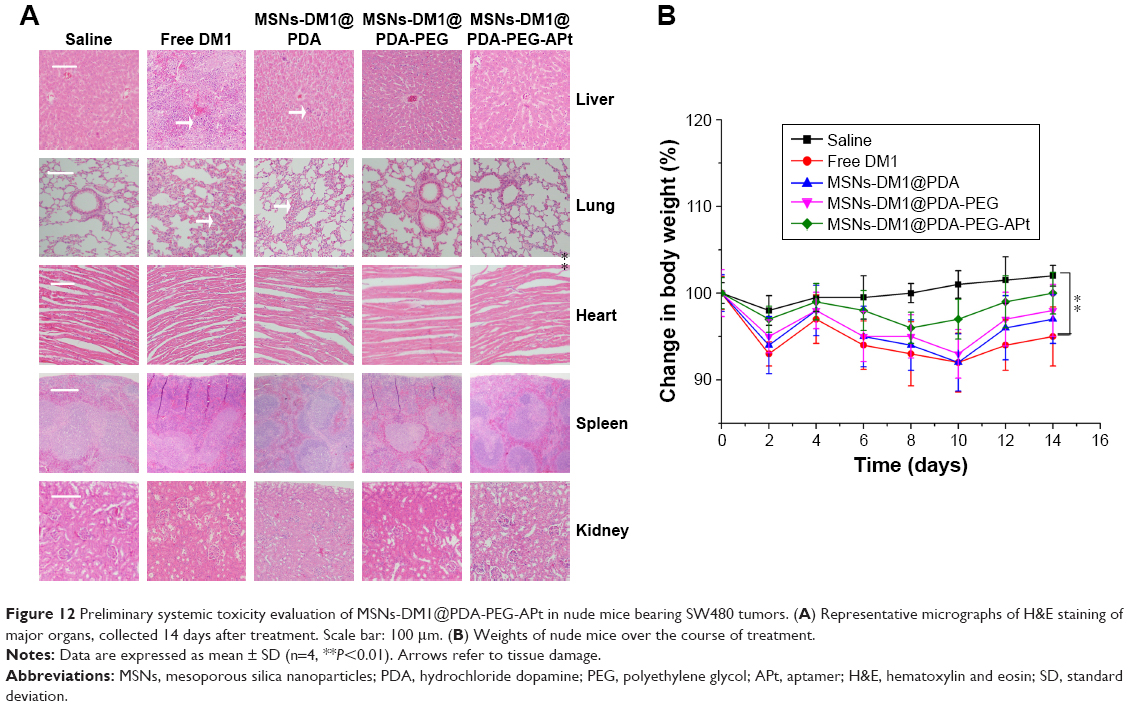 | Figure 12 Preliminary systemic toxicity evaluation of MSNs-DM1@PDA-PEG-APt in nude mice bearing SW480 tumors. (A) Representative micrographs of H&E staining of major organs, collected 14 days after treatment. Scale bar: 100 μm. (B) Weights of nude mice over the course of treatment. |
Conclusion
Overall, we developed a targeted and pH-sensitive drug delivery system based on EpCAM APt-functionalized MSNs, which can effectively deliver the anticancer drug DM1 to human colorectal adenocarcinoma cells and enhance anticancer activity, while minimizing systemic off-target toxicities. These MSNs-DM1@PDA-PEG-APt NPs have several advantages as a therapeutic option in cancer therapy, including 1) high drug loading capacity, 2) enhanced drug release rate at acidic pH, and 3) capability of targeting SW480 tumors by both passive (EPR effect-mediated) and active (EpCAM APt-mediated) mechanisms. The current drug delivery system based on EpCAM APt-functionalized MSN NPs constitutes a concrete promise for the clinical application of DM1 in cancer treatment.
Acknowledgments
This research was supported by the Science, Technology & Innovation Commission of Shenzhen Municipality (No JCYJ20160422170206664) and the Natural Science Foundation of Guangdong Province, China (2014A030310481).
Disclosure
The authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | ||
Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. | ||
Corti A, Pastorino F, Curnis F, Arap W, Ponzoni M, Pasqualini R. Targeted drug delivery and penetration into solid tumors. Med Res Rev. 2012;32(5):1078–1091. | ||
Kupchan SM, Komoda Y, Court WA, et al. Maytansine, a novel antileukemic ansa macrolide from Maytenus ovatus. J Am Chem Soc. 1972;94(4):1354–1356. | ||
Cassady JM, Chan KK, Floss HG, Leistner E. Recent developments in the maytansinoid antitumor agents. Chem Pharm Bull (Tokyo). 2004;52(1):1–26. | ||
Ishitsuka K, Jimi S, Goldmacher VS, Ab O, Tamura K. Targeting CD56 by the maytansinoid immunoconjugate IMGN901 (huN901-DM1): a potential therapeutic modality implication against natural killer/T cell malignancy. Br J Haematol. 2008;141(1):129–131. | ||
Burris HA 3rd, Rugo HS, Vukelja SJ, et al. Phase II study of the antibody drug conjugate trastuzumab-DM1 for the treatment of human epidermal growth factor receptor 2 (HER2)-positive breast cancer after prior HER2-directed therapy. J Clin Oncol. 2011;29(4):398–405. | ||
Kantarjian HM, Lioure B, Kim SK, et al. A phase II study of coltuximab ravtansine (SAR3419) monotherapy in patients with relapsed or refractory acute lymphoblastic leukemia. Clin Lymphoma Myeloma Leuk. 2016;16(3):139–145. | ||
Galsky MD, Eisenberger M, Moore-Cooper S, et al. Phase I trial of the prostate-specific membrane antigen-directed immunoconjugate MLN2704 in patients with progressive metastatic castration-resistant prostate cancer. J Clin Oncol. 2008;26(13):2147–2154. | ||
Lapusan S, Vidriales MB, Thomas X, et al. Phase I studies of AVE9633, an anti-CD33 antibody-maytansinoid conjugate, in adult patients with relapsed/refractory acute myeloid leukemia. Invest New Drugs. 2012;30(3):1121–1131. | ||
Amiri-Kordestani L, Blumenthal GM, Xu QC, et al. FDA approval: ado-trastuzumab emtansine for the treatment of patients with HER2-positive metastatic breast cancer. Clin Cancer Res. 2014;20(17):4436–4441. | ||
Krall N, Pretto F, Decurtins W, Bernardes GJ, Supuran CT, Neri D. A small-molecule drug conjugate for the treatment of carbonic anhydrase IX expressing tumors. Angew Chem Int Ed Engl. 2014;53(16):4231–4235. | ||
Song Y, Li Y, Xu Q, Liu Z. Mesoporous silica nanoparticles for stimuli-responsive controlled drug delivery: advances, challenges, and outlook. Int J Nanomedicine. 2017;12:87–110. | ||
Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25. | ||
Zheng Q, Lin T, Wu H, et al. Mussel-inspired polydopamine coated mesoporous silica nanoparticles as pH-sensitive nanocarriers for controlled release. Int J Pharm. 2014;463(1):22–26. | ||
Song Y, Zhu Z, An Y, et al. Selection of DNA aptamers against epithelial cell adhesion molecule for cancer cell imaging and circulating tumor cell capture. Anal Chem. 2013;85(8):4141–4149. | ||
Ling K, Jiang H, Li Y, Tao X, Qiu C, Li FR. A self-assembling RNA aptamer-based graphene oxide sensor for the turn-on detection of theophylline in serum. Biosens Bioelectron. 2016;86:8–13. | ||
Tang J, Huang N, Zhang X, et al. Aptamer-conjugated PEGylated quantum dots targeting epidermal growth factor receptor variant III for fluorescence imaging of glioma. Int J Nanomedicine. 2017;12:3899–3911. | ||
Fan X, Guo Y, Wang L, Xiong X, Zhu L, Fang K. Diagnosis of prostate cancer using anti-PSMA aptamer A10-3.2-oriented lipid nanobubbles. Int J Nanomedicine. 2016;11:3939–3950. | ||
Jurek PM, Zablocki K, Wasko U, Mazurek MP, Otlewski J, Jelen F. Anti-FGFR1 aptamer-tagged superparamagnetic conjugates for anticancer hyperthermia therapy. Int J Nanomedicine. 2017;12:2941–2950. | ||
Luo S, Wang S, Luo N, Hu FCC, Zhang K. The application of aptamer 5TR1 in triple negative breast cancer target therapy. J Cell Biochem. Epub 2017 Jul 3. | ||
Xie X, Li F, Zhang H, et al. EpCAM aptamer-functionalized mesoporous silica nanoparticles for efficient colon cancer cell-targeted drug delivery. Eur J Pharm Sci. 2016;83:28–35. | ||
Went PT, Lugli A, Meier S, et al. Frequent EpCAM protein expression in human carcinomas. Hum Pathol. 2004;35(1):122–128. | ||
Went P, Vasei M, Bubendorf L, et al. Frequent high-level expression of the immunotherapeutic target EpCAM in colon, stomach, prostate and lung cancers. Br J Cancer. 2006;94(1):128–135. | ||
Chen Y, Chen H, Shi J. In vivo bio-safety evaluations and diagnostic/therapeutic applications of chemically designed mesoporous silica nanoparticles. Adv Mater. 2013;25(23):3144–3176. | ||
Ma F, Bi L, Yang G, et al. ZNF703 promotes tumor cell proliferation and invasion and predicts poor prognosis in patients with colorectal cancer. Oncol Rep. 2014;32(3):1071–1077. | ||
Chang D, Gao Y, Wang L, et al. Polydopamine-based surface modification of mesoporous silica nanoparticles as pH-sensitive drug delivery vehicles for cancer therapy. J Colloid Interface Sci. 2016;463:279–287. | ||
Park J, Brust TF, Lee HJ, Lee SC, Watts VJ, Yeo Y. Polydopamine-based simple and versatile surface modification of polymeric nano drug carriers. ACS Nano. 2014;8(4):3347–3356. | ||
Chen L, She X, Wang T, et al. Overcoming acquired drug resistance in colorectal cancer cells by targeted delivery of 5-FU with EGF grafted hollow mesoporous silica nanoparticles. Nanoscale. 2015;7(33):14080–14092. | ||
Lee H, Dellatore SM, Miller WM, Messersmith PB. Mussel-inspired surface chemistry for multifunctional coatings. Science. 2007;318(5849):426–430. | ||
Wang K, Yao H, Meng Y, Wang Y, Yan X, Huang R. Specific aptamer-conjugated mesoporous silica-carbon nanoparticles for HER2-targeted chemo-photothermal combined therapy. Acta Biomater. 2015;16:196–205. | ||
Rafiei P, Haddadi A. Docetaxel-loaded PLGA and PLGA-PEG nanoparticles for intravenous application: pharmacokinetics and biodistribution profile. Int J Nanomedicine. 2017;12:935–947. | ||
Yang Y, Wang A, Wei Q, et al. Hyperbranched polyglycerol-induced porous silica nanoparticles as drug carriers for cancer therapy in vitro and in vivo. ChemistryOpen. 2017;6(1):158–164. | ||
Ngamcherdtrakul W, Morry J, Gu S, et al. Cationic polymer modified mesoporous silica nanoparticles for targeted siRNA delivery to HER2+ breast cancer. Adv Funct Mater. 2015;25(18):2646–2659. | ||
Feng W, Nie W, He C, et al. Effect of pH-responsive alginate/chitosan multilayers coating on delivery efficiency, cellular uptake and biodistribution of mesoporous silica nanoparticles based nanocarriers. ACS Appl Mater Interfaces. 2014;6(11):8447–8460. | ||
Ma M, Chen H, Chen Y, et al. Hyaluronic acid-conjugated mesoporous silica nanoparticles: excellent colloidal dispersity in physiological fluids and targeting efficacy. J Mater Chem. 2012;22(12):5615–5621. | ||
Iqbal Z, Lai EP, Avis TJ. Antimicrobial effect of polydopamine coating on Escherichia coli. J Mater Chem. 2012;22(40):21608–21612. | ||
Saint-Cricq P, Deshayes S, Zink JI, Kasko AM. Magnetic field activated drug delivery using thermodegradable azo-functionalised PEG-coated core-shell mesoporous silica nanoparticles. Nanoscale. 2015;7(31):13168–13172. | ||
Kempen PJ, Greasley S, Parker KA, et al. Theranostic mesoporous silica nanoparticles biodegrade after pro-survival drug delivery and ultrasound/magnetic resonance imaging of stem cells. Theranostics. 2015;5(6):631–642. | ||
Gao Y, Gu S, Zhang Y, et al. The architecture and function of monoclonal antibody-functionalized mesoporous silica nanoparticles loaded with mifepristone: repurposing abortifacient for cancer metastatic chemoprevention. Small. 2016;12(19):2595–2608. | ||
He Q, Zhang Z, Gao Y, Shi J, Li Y. Intracellular localization and cytotoxicity of spherical mesoporous silica nano- and microparticles. Small. 2009;5(23):2722–2729. | ||
Qi L, Tang Y, He W, et al. Lithium chloride promotes neuronal differentiation of rat neural stem cells and enhances neural regeneration in Parkinson’s disease model. Cytotechnology. 2017;69(2):277–287. | ||
Goldmacher VS, Audette CA, Guan Y, et al. High-affinity accumulation of a maytansinoid in cells via weak tubulin interaction. PloS One. 2015;10(2):e0117523. | ||
Yan H, Endo Y, Shen Y, et al. Ado-trastuzumab emtansine targets hepatocytes via human epidermal growth factor receptor 2 to induce hepatotoxicity. Mol Cancer Ther. 2016;15(3):480–490. | ||
Adams SR, Yang HC, Savariar EN, et al. Anti-tubulin drugs conjugated to anti-ErbB antibodies selectively radiosensitize. Nat Commun. 2016;7:13019. | ||
Venghateri JB, Gupta TK, Verma PJ, Kunwar A, Panda D. Ansamitocin P3 depolymerizes microtubules and induces apoptosis by binding to tubulin at the vinblastine site. PLoS One. 2013;8(10):e75182. |
Supplementary material
 | Table S1 Antibody concentration used in Western blot and immunofluorescent staining experiments |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.