Back to Journals » OncoTargets and Therapy » Volume 11
Enzalutamide-resistant castration-resistant prostate cancer: challenges and solutions
Authors Tucci M, Zichi C ![]() , Buttigliero C
, Buttigliero C ![]() , Vignani F, Scagliotti GV, Di Maio M
, Vignani F, Scagliotti GV, Di Maio M ![]()
Received 9 May 2018
Accepted for publication 21 September 2018
Published 24 October 2018 Volume 2018:11 Pages 7353—7368
DOI https://doi.org/10.2147/OTT.S153764
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Faris Farassati
Marcello Tucci,1,* Clizia Zichi,1,* Consuelo Buttigliero,1 Francesca Vignani,2 Giorgio V Scagliotti,1 Massimo Di Maio2
1Division of Medical Oncology, Department of Oncology, University of Turin, San Luigi Gonzaga Hospital, 10043 Orbassano, Turin, Italy; 2Division of Medical Oncology, Ordine Mauriziano Hospital, Torino, Italy
*These authors contributed equally to this work
Abstract: The new-generation hormonal agent enzalutamide has been approved for the treatment of metastatic castration-resistant prostate cancer (CRPC), in both post- and predocetaxel setting, due to the significant improvement in overall survival. More recently, enzalutamide also showed impressive results in the treatment of men with nonmetastatic CRPC. Unfortunately, not all patients with CRPC are responsive to enzalutamide, and even in responders, benefits are limited by the development of drug resistance. Adaptive resistance of metastatic prostate cancer to enzalutamide treatment can be due to the activation of both androgen receptor (AR)-dependent pathways (expression of constitutively active AR splice variants, AR point mutations, gene amplification and overexpression) and mechanisms independent of AR signaling pathway (altered steroidogenesis, upregulation of the glucocorticoid receptor, epithelial–mesenchymal transition, neuroendocrine transformation, autophagy and activation of the immune system). In this review, we focus on resistance mechanisms to enzalutamide, exploring how we could overcome them through novel therapeutic options.
Keywords: prostate cancer, enzalutamide, hormonal treatment, mechanisms of resistance, castration-resistant prostate cancer, androgen receptor
Introduction
Prostate cancer (PC) is the most common nonskin cancer, accounting for almost one in every five new cancer diagnoses, and the second leading cause of cancer-related death among men in the USA.1
At the time of diagnosis, most men have localized disease amenable to curative surgery or radiotherapy (RT). However, ~5%–10% of PC presents with de novo metastatic disease, and in addition, some patients will recur after local treatment. Five-year survival is almost 100% for localized and locally advanced disease, but it is <30% in patients with metastatic disease.2
Because of the crucial role of androgen receptor (AR) in the development and progression of PC, the mainstay of therapy for relapsed or metastatic patients is represented by androgen deprivation therapy (ADT), achieved by either orchiectomy or medical castration with luteinizing hormone-releasing hormone (LHRH) agonists or antagonists. Despite initial high response rates (in the range of 80%–90%), nearly all men eventually develop progressive disease within some years (median 2–3 years) from the start of ADT, entering the so-called castration-resistant phase of the disease.
In the past years, many therapeutic advances have been achieved in metastatic castration-resistant PC (mCRPC), with the approval of several new drugs.3 Two new generation hormonal agents, abiraterone and enzalutamide, have shown an improvement in overall survival (OS) in both post-4,5 and predocetaxel setting.6,7
Cabazitaxel, an innovative taxane, is currently used in patients who have failed docetaxel-based chemotherapy.8,9
Radium-223, an alpha-emitting bone target agent, demonstrated a prolongation of OS compared to placebo, regardless of previous treatment with docetaxel, and it was approved for symptomatic bone mCRPC patients.10
The Food and Drug Administration (FDA) also approved sipuleucel-T, an autologous cellular immunotherapy, for the treatment of asymptomatic or mild symptomatic castration-resistant prostate cancer (CRPC) patients.11
In addition, to delay the establishment of a castration-resistant condition, consequently improving patients’ quality of life and OS, the treatment paradigm of hormone-sensitive PC (HSPC) is changing too. At first, two randomized trials demonstrated a clinically relevant OS benefit, with the addition of six cycles of chemotherapy with docetaxel to standard ADT in metastatic HSPC patients.12–14
In the same setting, two trials have recently shown that adding abiraterone to ADT is associated with a significant OS prolongation in men with metastatic HSPC.15,16
Finally, one of the current therapeutic end points is to anticipate treatment to delay the appearance of metastasis in high-risk nonmetastatic CRPC patients. Based on the results of SPARTAN trial, in which apalutamide reduced the RR of metastasis or death by more than 70% compared to placebo, the FDA has recently approved this drug for patients with nonmetastatic CRPC.17 Overlapping results have been observed with enzalutamide in the PROSPER trial.18
Despite the large list of recent positive studies and therapeutic improvements, all these treatments have limited benefits, and sooner or later acquired drug resistance appears. In this review, we focus on the mechanisms of resistance to enzalutamide.
Enzalutamide
Enzalutamide is an oral AR inhibitor, with affinity for its target eightfold higher than bicalutamide. Unlike bicalutamide, which acts only at the receptor level, enzalutamide also prevents AR nuclear translocation, DNA binding and coactivator recruitment.19
Initially, enzalutamide was evaluated in the postdocetaxel setting, in the context of AFFIRM, a Phase III randomized, double-blind, placebo-controlled study, which enrolled 1,199 CRPC patients. Enzalutamide showed a significant improvement in OS compared to placebo: median OS was 18.4 vs 13.6 months, HR 0.63, 95% CI 0.53–0.75, P<0.0001. Enzalutamide maintained its superiority over placebo in all secondary end points, including time to prostate-specific antigen (PSA) progression (median 8.3 vs 3.0 months, HR 0.25, P<0.001), soft tissue response rate (29% vs 4%, P<0.001) and improvement in the quality of life (43% vs 18%, P<0.001).5
Subsequently, enzalutamide was evaluated in chemotherapy-naïve patients with progressive mCRPC. In the Phase III PREVAIL study, 1,717 men were randomized to receive enzalutamide or placebo. Treatment with enzalutamide, compared to placebo, showed a statistically significant 29% reduction in the risk of death (HR 0.71, 95% CI 0.60–0.84, P<0.001) and an 81% reduction in the risk of radiological progression (HR 0.19, 95% CI 0.15–0.29, P<0.0001).7
Enzalutamide is usually well tolerated, and the most common adverse events (AEs; occurring in ≥5% of patients) are fatigue, hot flashes, diarrhea, hypertension and musculoskeletal pain.5,7
Based on these data, the FDA and the European Medicines Agency (EMA) granted approval for enzalutamide for the treatment of both docetaxel-naïve and docetaxel-pretreated CRPC patients.
More recently, enzalutamide showed impressive results in the treatment of men with nonmetastatic CRPC, namely enzalutamide decreased the risk of distant metastasis or death by 71% (HR 0.29, 95% CI 0.24–0.35, P<0.0001) with an improvement of 21.9 months (36.6 months vs 14.7 months) in median metastasis-free survival.18
AR: structure and mechanism of action
AR is a nuclear ligand-activated transcriptional factor, a member of the steroid hormone nuclear receptor family, whose coding gene is located on chromosome 10. The AR consists of four functional domains: the N-terminal transactivation domain (NTTD), the DNA-binding domain (DBD), the hinge region and the ligand-binding domain (LBD). Main ligands are the male sexual hormones such as testosterone and dihydrotestosterone; adrenal androgens bind the AR to lower affinity. AR binding to androgens induces a conformational change of the AR. The androgen–AR complex may translocate to the nucleus, dimerize and bind to specific androgen response elements (AREs), regulating target gene transcription. This process and the cross talk with other pathways are modulated by many coactivator and suppressor proteins. Regardless of the DNA binding, the androgen–AR complex may lead to a rapid phosphorylation of the second messenger pathways, such as ERK, AKT and MAPK.20 The AR normally promotes epithelial differentiation, but in PC, this pathway can modulate the expression of genes that regulate proliferation and survival of tumor cells.21
The adaptive response to androgen deprivation pressure
In recent years, scientific knowledge about PC biology and growth has substantially improved. Nowadays, it is well established that the progression from HSPC to CRPC is not determined by a true ADT resistance, but by an adaptation of PC cells to a microenvironment with reduced testosterone levels. In fact, notwithstanding the deep decrease in serum androgen levels caused by ADT, PC cells could synthesize androgens and could modify AR in a way that even low androgen levels could activate it.
The mechanisms responsible for this phenomenon are related to the selective pressure of ADT and include AR gene overexpression, AR gene mutation, AR variant of splicing (ARsv) expression, upregulation of transcriptional coactivators and overexpression of enzymes able to induce the production of androgens.19,21
Mechanisms of resistance to enzalutamide
Despite the therapeutic effect of enzalutamide in CRPC patients, a significant percentage of patients are primarily resistant or acquire resistance within a variable period of time following the start of enzalutamide treatment.19
Primary resistance is defined as the treatment failure within the first 3 months after the start of therapy, because of the deterioration of clinical conditions, with or without radiological imaging progression.22 On the other hand, acquired resistance consists in the treatment failure that arises later during treatment.
A comprehensive knowledge of the pathways associated with the development of both primary and acquired resistance remains a key factor to identify predictive factors of treatment efficacy and to investigate new therapeutic options.
Adaptive resistance of PC to enzalutamide treatment can be due to the activation of both AR-dependent pathways and mechanisms not dependent on AR signaling pathways.
Mechanisms of resistance dependent on AR signaling pathway
AR amplification and overexpression
Up to 80% of CRPC patients show AR protein overexpression, and in 20%–30% of patients, this is related to AR gene amplification.23 In vitro studies demonstrate that this alteration is more frequent in patients progressing on enzalutamide than in treatment-naïve patients,24 and consequently, it is considered as a potential mechanism of resistance. In a cohort of patients previously treated with either enzalutamide or orteronel, a CYP17A inhibitor, 50% showed the evidence of AR amplification and only 13% had a clinically significant response when treated with abiraterone.24,25 These observations have been confirmed in a correlative biomarker study, in a cohort of 94 patients treated with enzalutamide in the context of the PREMIERE trial; AR-amplified tumors had a poor response to treatment and a largely shorter OS (HR 11.08, 95% CI 2.16–56.95, P<0.004).26
A promising strategy to block AR overexpression consists in the administration of supraphysiologic doses of testosterone, that in the preclinical models prevented PC cell growth.27,28 Unfortunately, this effect vanishes away in a short time, and PC cells progressively downregulate the AR, restoring cell growth in the presence of testosterone. A potential viable therapeutic approach consists in the so-called “bipolar androgen therapy”, namely the swinging from supraphysiologic levels of androgens to near-castrate levels. The results of a Phase II trial testing this strategy in mCRPC patients after progression on enzalutamide have been recently published.29 Nine of the 30 enrolled patients (30%; 95% CI 15%–49%) achieved a >50% PSA reduction, and 21 proceeded to enzalutamide rechallenge, with 15 patients (52%; 95% CI 33%–71%) experiencing >50% PSA reduction.29 Two Phase II trials are currently recruiting patients: the first (NCT02286921) aims to confirm the therapeutic benefits of the bipolar androgen therapy, while the second (NCT02090114) is designed to test the activity and efficacy of sustained supraphysiologic testosterone concentration (Table 1).
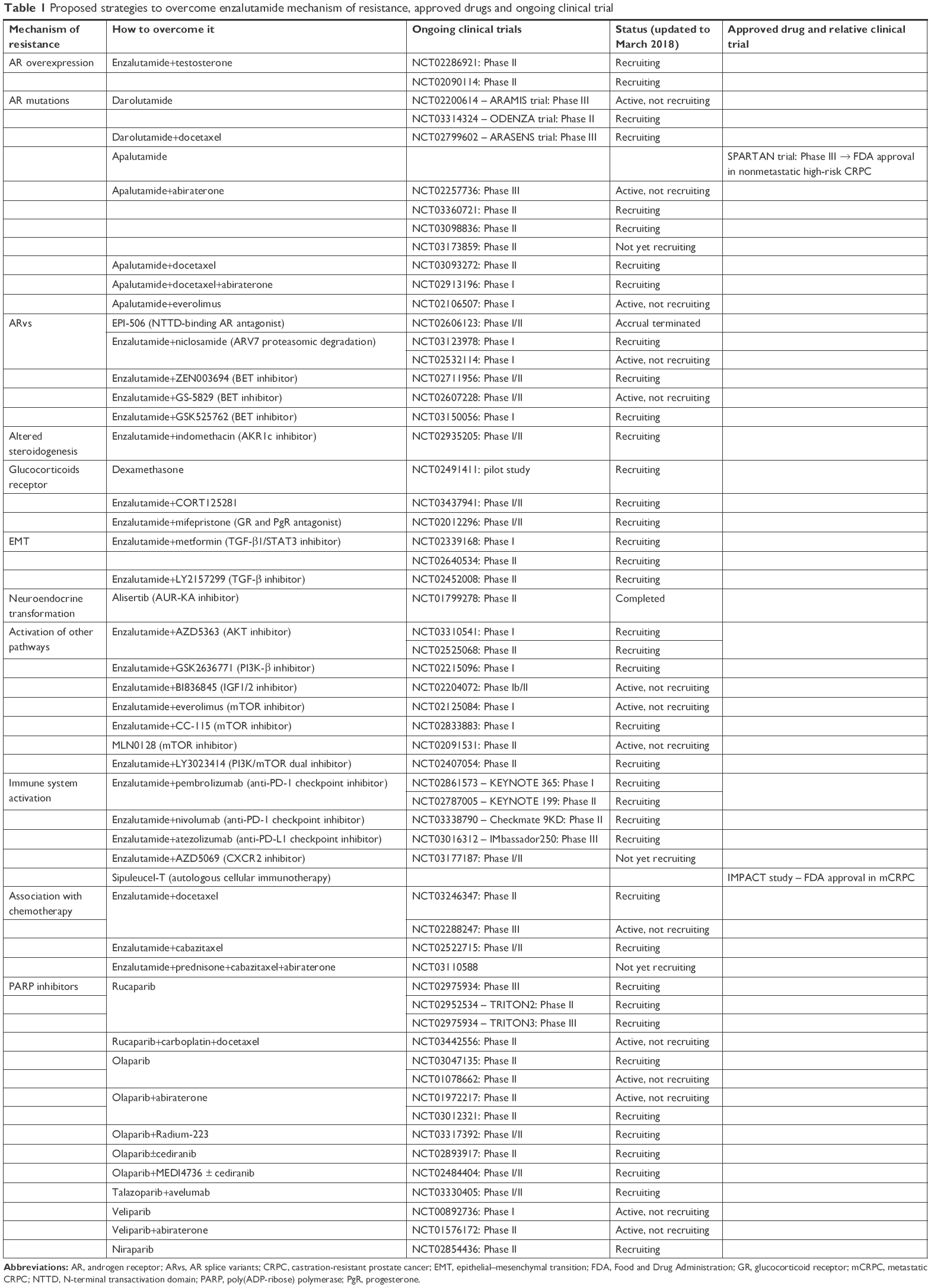 | Table 1 Proposed strategies to overcome enzalutamide mechanism of resistance, approved drugs and ongoing clinical trial |
AR mutations
AR mutations, that are quite rare in untreated PC, can be detected in 5%–30% of CRPC patients treated with ADT, and their incidence may become relevantly higher during treatment with abiraterone and enzalutamide.
Most of the clinically relevant AR somatic point mutations are located in the LBD, thus altering the steroid-binding pocket. This biochemical change leads to AR activation by alternative ligands, such as progesterone, hydrocortisone, oestradiol and some AR antagonists.24,30 One of the most frequently reported somatic mutations is F877L (previously reported in the literature as F876L), which has been identified in patients treated with enzalutamide and apalutamide (a new AR inhibitor). Both these drugs, binding to the mutated AR, act as partial agonists. Unexpectedly, sensitivity to first-generation agents such as bicalutamide is maintained.31,32
Other mutations associated with poor response to enzalutamide are L702H, conferring acquired responsiveness to glucocorticoids,33 and T878A (previously reported in the literature as T877A), resulting in progesterone-mediated activation of the AR.30
Preclinical data show that darolutamide, a new oral AR antagonist, is able to significantly inhibit the transcriptional activity of two of the following mutated forms: T878A and F877L.34
AR splice variants
Constitutively, transcriptionally activated ARvs have emerged as critical players in the development and progression of CRPC. ARvs lack the LBD and remain constitutively active even in the absence of binding to androgenic hormones: their expression is significantly increased during ADT and correlates with PC progression. The most common variants are ARV7 and ARV567.35–37
Enzalutamide is able to induce ARvs expression in PC cell lines and CRPC xenograft models: as ARvs levels increase, drug resistance increases.38 A recent study by Antonarakis et al,39 using a circulating tumor cell (CTC) assay, showed a significant relationship between ARV7 mRNA expression and resistance to enzalutamide: ARV7-positive patients had lower PSA response rates (0% vs 53%, P=0.004) and shorter OS (median 5.5 months vs not reached; P=0.002) than ARV7-negative patients. However, some recent evidences show contrasting results: at the 2018 American Association for Cancer Research Meeting, results of an Australian research have been presented. ARV7 and ARV9 were detected using a whole blood assay, but no significant differences in PSA response rate were found between ARV-positive (66%) and ARV-negative patients (64%) treated with enzalutamide or abiraterone.40
Because of the clinical relevance of ARvs, several innovative pharmacological strategies are currently under development. In detail, a potential strategy is targeting the NTTD of the AR, that is, critical for its transcriptional activity. EPI compounds are a new class of drugs that prevent the binding of transcriptional activator proteins to the AR N-terminal domain and decrease the expression of genes regulated by both full-length AR and ARV7.41 EPI-506 (ralaniten acetate) is a member of EPI compound family that has been tested in a Phase I/II study (NCT02606123) in mCRPC patients progressed to enzalutamide, abiraterone or both (Table 1). Unfortunately, a suboptimal compliance to the high number of daily pills (18 capsules/day) was reported at the end of Phase I; therefore, it is currently unclear if the clinical development of this agent will continue.
Other studies have indicated that targeting coactivators and suppressors of AR-mediated transcription may bypass secondary resistance. As detailed below, one of these possible targets is the coactivator bromodomain containing protein (BRD)4: preclinical studies demonstrated that preventing its interaction with the NTTD of the AR is associated with reduction in ARV7 and ARV567 mRNA and protein expression and with induction of cell cycle arrest and apoptosis of cancer cells.24,42
Another interesting molecule is niclosamide, an FDA-approved anthelmintic drug, that may induce ARV7 protein degradation through a proteasome-dependent pathway.43 In enzalutamide-resistant in vitro models, the combination of niclosamide and enzalutamide inhibits tumor cell growth and induces apoptosis.44 Two Phase I studies are currently ongoing to evaluate recommended dose and tolerability of niclosamide given together with enzalutamide in CRPC patients (NCT03123978 and NCT02532114; Table 1).
Another targeted approach is represented by the heterogeneous nuclear ribonucleoproteins (hnRNPAs), whose downregulation reduces ARV7 expression and resensitizes enzalutamide-resistant cells. Quercetin, a natural polyphenolic compound, reduced the expression of hnRNPAs in PC cells.45
Another interesting procedure is based on the use of antisense oligonucleotides to target both the full-length AR and its splice variant ARv7. Yamamoto et al46 demonstrated that this strategy is able to suppress the growth of enzalutamide-resistant cell lines and xenografts. Subsequently, the same group of authors evaluated the combination of AR antisense oligonucleotides with siRNA targeting clusterin, a chaperone protein involved in the mechanisms of resistance to AR inhibition. They used an innovative lipid nanoparticle formulation of siRNA to facilitate tumor cell permeation and retention. Results demonstrated a significantly decreased tumor cell growth, both in vivo and in vitro, in enzalutamide-resistant CRPC models.47
The observation that a long noncoding RNA, metastasis-associated lung adenocarcinoma transcript 1 (Malat1), is requested for enzalutamide-induced ARV7 production paved the way to a new approach to overcome resistance. Wang et al48 demonstrated that treatment with Malat1 small interfering RNA inhibits ARV7 expression in cell lines and significantly reduces tumor growth in enzalutamide-resistant xenografts.
New promising targets include the BRD2, BRD3 and BRD4, chromatin readers of the bromodomain and extra-terminal (BET) family. Particularly, BRD4 is a co-regulator of AR, and the inhibition of this family of proteins significantly decreased AR signaling and growth of a patient-derived xenograft model of CRPC with both ARV7 expression and AR amplification.49 The efficacy of BET inhibitors has been recently demonstrated in enzalutamide-resistant PC models: these molecules markedly repressed ARV7 expression in vitro and inhibited tumor cell growth in vivo.50 Three clinical trials are currently ongoing to evaluate the combination of enzalutamide with different BET inhibitors: ZEN-3694 (NCT02711956), GS-5829 (NCT02607228) and GSK525762 (NCT03150056; Table 1).
Finally, several natural substances from traditional Chinese medicine have recently shown interesting effects against PC cells both in vitro and in vivo. For example, Han et al51 demonstrated that low dose of triptolide, an active compound extracted from the herb “Thunder God Vine”, inhibited phosphorylation of both full-length and truncated AR. A synergistic effect was noted in the combination with enzalutamide. Other natural products with promising anti-PC activity are urolithins (metabolites of human microflora), rhizochalinin (a marine compound), sintokamide (a chlorinated peptide), all targeting AR and ARvs with several mechanisms.51
Altered steroidogenesis
Testosterone and 5α-dihydrotestosterone are sex steroid hormones, mainly synthesized from cholesterol in the testes. Only 5%–10% of their synthesis take place in the adrenal glands.52
The intratumoral synthesis of these hormones, quite limited in untreated primary PC, increases in CRPC patients, and both cholesterol and weak androgens, such as androstenedione and dehydroepiandrosterone, synthesized in the adrenal glands, may act as precursors.53 The increased intratumoral level of androgens promotes both paracrine and autocrine activation of the AR, regardless of systemic hormone levels.54
Preclinical studies, conducted on both cell lines and xenografts, have suggested that enzalutamide resistance may be related to an overexpression of genes encoding for enzymes involved in androgen biosynthesis: SRD51A, HSD3β1 and AKR1C3.55 One of these enzymes, AKR1C, is a potential target for pharmacological approaches. In both in vitro and in vivo preclinical models, indomethacin, an NSAID, inhibits AKR1C activity and resensitizes CRPC to enzalutamide.56 A Phase I/II trial, evaluating the combination of indomethacin and enzalutamide in CRPC patients, is currently recruiting patients (NCT02935205; Table 1).
AR-independent mechanisms of resistance
Overexpression and upregulation of glucocorticoid receptor (GR)
The GR is a member of the steroid hormone nuclear receptor family, expressed in almost all human tissues. Like AR, the GR consists of four functional domains: the DNA and the ligand binding, the N-terminal and the hinge region. Moreover, these two receptors share response elements in target genes and have overlapping transcriptomes.57
The GR resides in the cytoplasm, complexed with heat shock proteins. The binding of glucocorticoids promotes its homo-dimerization and translocation in the nucleus, where GR mediates transcriptional activation, influencing target gene expression.57
Puhr et al58 recently investigated GR expression and its functional significance in both PC cell lines and human tissues obtained from 177 PC patients. It was observed that GR expression is low in primary PC tissue and significantly increased during long-term treatment with enzalutamide. These results confirm previous findings of Arora et al59 which demonstrated how GR overexpression conferred resistance to enzalutamide. Particularly, AR inhibition may determine GR overexpression and upregulation and able to restore the expression of certain AR target genes. In the same study, dexamethasone, that is a potent GR agonist, conferred resistance to enzalutamide, whereas GR antagonists restored sensitivity.
Unexpectedly, in a randomized Phase II trial comparing daily administration of dexamethasone vs prednisone in men with chemotherapy-naïve CRPC, dexamethasone induced a PSA response rate of 47% vs 24% (P=0.05).60 A pilot study (NCT02491411) is currently recruiting patients with mCRPC, who were previously treated with enzalutamide and docetaxel, to study how dexamethasone may contrast GR overexpression, allowing the retreatment with enzalutamide (Table 1). Another trial is evaluating the safety and efficacy of the GR antagonist CORT125281 in combination with enzalutamide in men with mCRPC (NCT03437941; Table 1).
Recently, mifepristone, a synthetic steroidal antagonist of both GR and progesterone receptor, has gained great interest as a potential agent against PC. Unfortunately, although a clinical trial evaluating the combination of mifepristone and enzalutamide (NCT02012296) is still ongoing (Table 1), recent reports questioned the real efficacy of this association.61 Moreover, in preclinical models of PC, mifepristone significantly reduced the efficacy of enzalutamide, probably because of its partial AR agonist activity.62
Epithelial–mesenchymal transition (EMT)
The transition of epithelial into mesenchymal cells is an essential physiological mechanism for both embryonic development and wound healing. Unfortunately, this process also contributes pathologically to fibrosis and cancer metastasis.63
Epithelial phenotype is characterized by apical–basal cell polarity, intercellular adhesion and cohesive interaction and lack of mobility of individual cells. Mesenchymal cells are completely different, being neither adherent nor apically polarized.64
EMT may be induced by ADT in metastatic PC, and increasing evidence shows that this process promotes tumor progression and drug resistance. The activation of different pathways, such as TGF-β and SMAD, modulates the activity of some transcriptional factors such as Snail and Twist, reducing the expression of e-cadherin, a key event in EMT.65 Twist is a transcriptional factor that is upregulated during ADT and enzalutamide treatment. This mechanism, together with protein kinase C (PKC) activation, is responsible for the occurrence of resistance. Up to now, no direct Twist inhibitors are available; therefore, Shiota et al66 tested the small molecule Ro318220, a PKC inhibitor, to revert resistance in PC cell lines. The combination of Ro318220 with enzalutamide produced promising results, increasing the therapeutic effects of androgen depletion. Another transcriptional factor, Snail, can promote resistance to enzalutamide and increase migration and invasion of PC cells. Indeed, an elevated expression of Snail was documented in enzalutamide-resistant PC cells, while its downregulation resensitized cells to the pharmacological treatment.67
Recent studies by Liu et al68 showed that metformin, a well-known oral hypoglycaemic agent, reverts resistance in PC cell lines and mice xenografts. In detail, metformin exerts its effects repressing TGF-β1/STAT3 pathway, whose activation instead induced EMT. Currently, there are two clinical trials recruiting patients to evaluate the role of metformin as anticancer agent in combination with enzalutamide (NCT02339168 and NCT02640534; Table 1).
Another Phase II trial (NCT02452008) is currently ongoing to evaluate the efficacy of the TGF-β inhibitor, galunisertib, given in combination with enzalutamide (Table 1).
Neuroendocrine transformation
Prostate adenocarcinoma may transdifferentiate into neuroendocrine PC (NEPC) as an adaptive response to intense androgen signaling inhibition. Infrequently present at the time of diagnosis, NEPC usually develops at later stages of treatment and is characterized by rapid growth and poor prognosis. When transformation to NEPC occurs, AR expression is low or absent, so these tumors are typically resistant to AR-directed therapies such as enzalutamide.69
In these cases, even without robust clinical evidence, cisplatin or carboplatin combined with etoposide is the most commonly used regimen. The Phase II trial GETUG P01 demonstrated an overall response rate (ORR) of <9% and a median OS of 9.6 months (95% CI 8.7–12.7) in a cohort of 60 patients with either NEPC or visceral metastasis treated with the combination of carboplatin and etoposide every 3 weeks.70 The combination of platinum and taxane has been tested in patients with mixed high-grade adenocarcinoma and NEPC, reporting an ORR of 41% and a median OS of 12 months.71
Ongoing genomic and biochemical research aims to better characterize these tumors, looking for potential new targets.
One of the most promising targets is Aurora Kinase A (AUR-KA), an oncogene whose overexpression/amplification has been found in 40% of NEPC patients. AUR-KA stabilizes N-myc and cooperatively promotes neuroendocrine transdifferentiation.72 When evaluated in 59 metastatic NEPC patients, the AUR-KA inhibitor alisertib demonstrated modest activity. Preliminary results of this Phase II clinical trial (NCT01799278) were presented at the 2016 European Society of Medical Oncology (ESMO) meeting: the 6-month progression-free rate was 11.1% (16.3% for patients with pathologically confirmed NEPC; 95% CI 5%–31.6%). Of note, two patients obtained an exceptional response, with complete regression of liver metastasis. The identification of biomarkers predictive of response would be crucial for future developments (Table 1).
Another target is the enhancer of zeste homolog 2 (EZH2), an epigenetic reprogramming gene, involved in NEPC development. Different EZH2 inhibitors, including GSK-126/343/503, have recently been developed, and in preclinical studies they showed growth inhibition of CRPC cell lines. Moreover, the block of EZH2 may resensitize to enzalutamide PTEN and Rb double knockout mice.73
Activation of other pathways
The acquired resistance to enzalutamide may also be mediated by the activation of parallel signaling pathways, including PI3K, HER2 and NF-κB.
The PI3K–AKT–mTOR pathway regulates all major cellular processes: cell growth, proliferation, apoptosis and protein synthesis. Its aberrant activation is implicated in PC development and progression.74 The cross talk between the AR and PI3K was demonstrated in preclinical models by Carver et al.75 While the inhibition of one pathway activated the other, in murine models and human xenografts, the concomitant inhibition of AR and PI3K induced PC regression. These results were confirmed by Thomas et al,76 both in vitro and in vivo, testing the combination of the AKT inhibitor AZD5363 with the antiandrogen, bicalutamide. Two trials are ongoing to evaluate the association of the AZD5363 with enzalutamide: one is a Phase I study enrolling patients with specific AKT mutations (NCT03310541), whereas the other is enrolling men with mCRPC already treated with one or two lines of chemotherapy and at least 12 weeks of abiraterone, without molecular selection (NCT02525068; Table 1).
There are different classes and subtypes of PI3K: class IA includes two isoforms, p110 α and β, both ubiquitously expressed. The former is associated with insulin and growth factor pathway, and the latter regulates cell mitosis and survival. Several attempts have been made to develop isoform β-specific inhibitors, including the recent synthesis of BL140. This agent effectively suppressed PC growth in all PC lines tested77 and exhibited a stronger inhibitory effect than GSK2636771, the only PI3K-β inhibitor currently assessed in a clinical trial (NCT02215096; Table 1).
Because of the cross talk between PI3K and the insulin-like growth factor (IGF) pathway, a Phase Ib/II trial (NCT02204072) is ongoing to investigate the combination of enzalutamide with BI836845, a humanized IgG1 monoclonal antibody targeting IGF1/2.
The direct inhibition of mTOR is another potential therapeutic strategy, and two Phase I trials are evaluating the combination of enzalutamide, respectively, with everolimus (NCT02125084) and CC-115 (NCT02833883), while the single-agent mTOR inhibitor, MLN0128, is evaluated in a Phase II trial (NCT02091531; Table 1). Another Phase II trial (NCT02407054) is assessing the PI3K/mTOR dual inhibitor, LY3023414, in men with mCRPC (Table 1).
The HER2/HER3 pathway, through the activation of PI3K/AKT, may represent another mechanism of resistance to enzalutamide. The cross talk between the HER2/HER3 and the AR has been extensively demonstrated. Increased HER2 expression was documented in enzalutamide-resistant PC, and enzalutamide may induce HER2 expression in PC cell lines. The combination of enzalutamide and lapatinib, a tyrosine kinase inhibitor (TKI) of both HER2 and EGFR receptors, improved the response rate compared to enzalutamide alone.78
The activation of NF-κB/p52 pathway also contributes to the development of enzalutamide resistance. In vitro studies demonstrated an activation of NF-κB/p52 and overexpression of ARvs in PC cell lines chronically treated with enzalutamide. Conversely, the downregulation of NF-κB/p52 reduced ARV7 expression and resensitized PC cells to enzalutamide.79 Moreover, NF-κB regulates the expression of several cytokines, in both malignant and normal tissues. IL-6 is highly expressed in CRPC, and it modulates transcriptional activity of the AR. Up to now, experimental therapies against IL-6 have not been successful.80
Autophagy
Autophagy is an adaptive process to maintain cellular homeostasis through degradation and recycling of cellular components. There is a physiological balance between autophagy and apoptosis, which may be lost in cancer development, where dysfunction of apoptosis is a common phenomenon. The consequent upregulation of autophagy may be used as a source of nourishment by high-proliferative tumor cells.81
The AR is a key modulator of autophagy. Treatment with bicalutamide of PC cell lines induced autophagy,82 and a similar effect was reported by Nguyen et al,83 within in vitro and in vivo PC models. The autophagy modulators such as clomipramine and metformin significantly improved enzalutamide activity reducing tumor growth within in vivo mouse models.
Autophagy may also be chaperone mediated. Clusterin, a heat-shock chaperone-like protein is induced by AR antagonists such as enzalutamide. The coadministration of enzalutamide and OXG-011, a clusterin inhibitor, enhanced apoptosis and delayed progression, within both in vitro and in vivo PC models.84
The immune system activation
Despite immunotherapy is an intriguing therapeutic option and the initial preclinical data with immune checkpoint inhibitors are encouraging, its clinical use in PC is still very limited. In preclinical mouse models with PC resistant to enzalutamide, Bishop et al85 demonstrated an increased expression of both intratumoral PD-L1 and circulating PD-L1/2-positive dendritic cells. These data were confirmed in vivo, comparing blood samples of patients progressing during enzalutamide with samples of treatment-naïve or treatment-responsive patients. However, the current evidence about the predictive role of both PD-1 and PD-L1 as biomarkers of response to specific immunotherapy is still not defined. Moreover, PC is characterized by minimal immune cell infiltrate and a relatively low mutational burden, both known as negative predictive factors of response to immunotherapy in other tumors.86
Having all those “caveats” in mind, a Phase II single-arm clinical trial demonstrated a meaningful activity of pembrolizumab, an anti-PD-1 checkpoint inhibitor already approved for the treatment of other solid tumors, in CRPC patients after progression with enzalutamide. Of the 10 patients enrolled, three experienced a biochemical response and two a radiological response, including a patient with microsatellite instability (MSI), which has been shown to be a predictive factor for this therapeutic approach.87 Combination therapy of pembrolizumab and enzalutamide is currently under clinical investigation in the Phase I KEYNOTE 365 (NCT02861573) and in the Phase II KEYNOTE 199 (NCT02787005; Table 1). Other trials are evaluating the anti-PD1 checkpoint inhibitor, nivolumab (NCT03338790), and the anti-PD-L1, atezolizumab (NCT03016312; Table 1).
Available results from the Phase III trial, CA184-043, testing the anti-CTLA4 ipilimumab vs placebo after RT in patients with mCRPC who had progressed after docetaxel chemotherapy, showed no difference in OS between treatment arms. However, a post hoc analysis reported a possible benefit in patients with good prognostic factors (median OS 22.7 vs 15.8 months, HR 0.62, 95% CI 0.45–0.86, P=0.0038).88
Current clinical trials are exploring new promising target agents, such as AZD5069, a chemokine receptor 2 (CXCR2) antagonist, in combination with enzalutamide (NCT03177187; Table 1).
Currently, the only FDA-approved immunotherapy remains sipuleucel-T, an autologous cellular treatment for patients with mCRPC with mild or no symptoms.86
Liquid biopsy: a promising tool for recognizing mechanisms of resistance
Liquid biopsy is a noninvasive blood test to detect CTCs or cancer-derived nucleic acids, such as DNA and RNA. It is clearly less invasive than a traditional tissue biopsy and may better characterize the tumor molecular assessment, recognizing mechanisms of resistance and guiding therapeutic choices.89
In PC patients, the number of CTCs correlates with disease outcome. Major efforts have been done to characterize the expression of ARvs in the contest of CTCs. Unfortunately, as previously described in this review, the correlation between ARvs detection in CTCs and resistance to enzalutamide treatment has not been homogeneously confirmed in all the series.39,40 Probably, this could be due to the development of various and heterogeneous methods of isolation, based on different definitions of CTCs. By now, CellSearch® is the only CTC detection platform approved by the FDA, but it is not currently used in common clinical practice.89
Another interesting approach is the whole genomic analysis of pooled CTCs. Gupta et al90 enrolled patients with mCRPC with primary or acquired resistance to new-generation hormonal drugs, identifying potentially actionable genomic alterations, such as BRD4, PI3K and AR.
As mentioned earlier, the direct sequencing of cell-free DNA may also provide useful information. Initially, the attention was focused on the detection of AR gene aberrations, related to both enzalutamide and abiraterone resistance.91 Subsequently, the analysis has also been extended to other genes and their potentially actionable alterations, namely Wyatt et al92 collected plasma samples of 65 patients with mCRPC treated with enzalutamide. Cell-free DNA was isolated and sequenced: not only mutations and amplifications of AR but also PI3K and damage repair genes were detected, at baseline and, even more, at disease progression.
To better understand primary resistances, Annala et al93 collected plasma specimens and analyzed cell-free DNA from patients with mCRPC, before starting treatment with either enzalutamide or abiraterone, within a randomized Phase II trial. Among the genomic alterations detected, mutations of BRCA, ATM and p53 defects were associated with primary resistance and poor outcome. AR gene structural rearrangements were frequently found in patients with primary resistances, but their prognostic role was not confirmed. On the other hand, this analysis showed that a high rate of copy number gene changes had no significant prognostic and predictive value. These data have been confirmed by Belic et al94 that the AR amplification alone is not predictive of resistance to enzalutamide. Interestingly, authors found a correlation between blood levels of lactate dehydrogenase and the detection of cell-free DNA.
Genomic and transcriptomic analyses to identify mechanisms of resistance
To better understand the mechanisms of resistance to enzalutamide and develop new therapeutic strategies, both genomic and transcriptomic analyses have been recently performed. Following the demonstration that multiple genomic alterations may contribute to the development of resistance, King et al95 tried to organize these alterations into different pathways, identifying those potentially actionable: MEK, EGFR, RAS and NF-κB. Similarly, with an integrative analysis of four transcriptome datasets, a group of researchers from the University of Shanghai tried to identify the different expression of key genes in both resistant and sensitive enzalutamide cell lines.96 One of the patterns explored was ubiquitin-mediated proteolysis: its abnormal function may lead to enzalutamide resistance.
Looking forward: a more effective interference with AR pathway
Among the drugs designed to overcome the resistance induced by enzalutamide, those with more advanced clinical data are apalutamide and darolutamide. Their safety, activity and efficacy are being evaluated in both HSPC and CRPC patients, in the metastatic and locally advanced setting. The unexpected negative outcome of the ARMOR 3-SV study led to an early discontinuation of the development program of galeterone, another promising innovative antiandrogen drug (NCT02438007).
Apalutamide
Apalutamide (also known as ARN-509) is an orally available, selective and irreversible AR antagonist, which inhibits AR nuclear translocation, preventing the binding of AREs.97
Apalutamide was shown to reduce the proliferation of PC cells in in vitro models and to induce a higher tumor regression than enzalutamide in murine tumor models of CRPC.98 A first-in-human Phase I study (ARN-501-001) demonstrated that treatment with ARN-509 was safe and well tolerated.99 Promising data of drug activity were confirmed in the Phase II portion of the same trial; apalutamide showed durable PSA response in nonmetastatic and early mCRPC, untreated with new-generation hormonal agents and chemotherapy. In the nonmetastatic CRPC group, median time to PSA progression was 24 months, and the metastasis-free survival at 12 months was 87%. However, results were substantially worse in mCRPC patients pretreated with abiraterone, whose median time to PSA progression was 3.7 months.100
More recently, the Phase III trial, SPARTAN, showed that in patients with high-risk, nonmetastatic CRPC, the addition of apalutamide to ADT reduced the risk of distant metastasis or death by 72% (HR 0.28, 95% CI 0.23–0.35, P<0.0001) and prolonged median metastasis-free survival by more than 20 months compared to placebo.17
Another Phase III trial (NCT02257736) is currently comparing the combination of apalutamide and abiraterone vs abiraterone alone in chemotherapy-naïve patients (Table 1). Other Phase I/II trials are testing the combination of apalutamide with docetaxel (NCT03093272), abiraterone (NCT03360721, NCT03098836, NCT0317385911), both docetaxel and abiraterone (NCT02913196) and even everolimus (NCT02106507; Table 1).
Darolutamide
Darolutamide (also known as ODM-201) is a novel, nonsteroidal highly selective AR inhibitor, with a superior AR-binding affinity than enzalutamide. Unlike bicalutamide, which showed significant agonism in AR-overexpressing cells, darolutamide demonstrated full AR antagonist properties, even against clinically relevant AR mutations associated with enzalutamide resistance: AR F877L, W742L and T877A.101 Based on in vitro studies, darolutamide has a low potential for drug–drug interaction with other substances metabolized by CYP enzymes.102
It is well known that enzalutamide and its active metabolites cross the blood–brain barrier, even if the recent UPWARD study has shown that the incidence of seizures, initially considered as a typical AE potentially associated with the administration of enzalutamide, is actually similar in patients with mCRPC and seizure risk factors, regardless of enzalutamide exposure.103 In any case, there is no evidence that darolutamide significantly penetrates the blood–brain barrier, and it could be consequently associated with a lower risk of inducing seizures.101
The first-in-man study was an open-label, Phase I/II dose escalation–expansion cohort trial.104 Darolutamide showed a favorable safety profile, and no dose-limiting toxic effects were reported. Moreover, clinical activity was reported, mainly in patients who not previously treated with CYP17 inhibitors. The Phase I ARAFOR trial evaluated both the bioavailability and activity of darolutamide in men with chemotherapy-naïve and CYP17 inhibitor-naïve CRPC: a PSA decline ≥50% was shown in 83% of the 30 patients enrolled. Disease control was observed in 78% of bone and 91% of soft tissue lesions.105
Two randomized, double-blind, placebo-controlled, Phase III trials are ongoing: ARAMIS (NCT02200614, active but not recruiting) and ARASENS (NCT02799602, active and recruiting patients; Table 1). In detail, ARAMIS is evaluating efficacy and safety of darolutamide in men with high-risk nonmetastatic CRPC. The risk of developing metastasis was defined by confirmed rising PSA levels and short PSA doubling time. ARASENS is testing the same drug in addition to standard ADT and docetaxel in metastatic HSPC. The primary end point of the study is OS.
Because of the growing value of patient-reported outcomes in the assessment of the balance between symptom control and the impact on the quality of life of side effects, the Phase II ODENZA trial, not yet recruiting, will assess patients’ preference between darolutamide and enzalutamide through specific questionnaires (NCT03314324; Table 1).
Galeterone
Galeterone is a selective, multitargeted antiandrogen agent, characterized by at least three different mechanisms of action: inhibition of the enzyme CYP17, antagonism and degradation of both full-lenght AR and ARvs, including ARV7. Preclinical studies both in vitro and in vivo have demonstrated that galeterone reduces the androgen-dependent proliferation of human PC cells.106
The Phase I multicenter dose-escalation clinical trial, ARMOR 1, included 49 patients with progressive CRPC not treated with chemotherapy or the other new generation hormonal agents.107 The study evaluated safety and activity of oral galeterone, whereas the first part of the Phase II trial ARMOR 2 tested a spray dry dispersion formulation of the drug. Overall, galeterone was well tolerated, with an acceptable safety profile in all cohorts. PSA responses were observed at all doses in ARMOR 1, particularly 42% of patients treated at the highest dose of 2,600 mg/die showed a PSA reduction of ≥50%.
The dose expansion cohort of ARMOR 2 trial enrolled 95 patients treated with galeterone at 2,550 mg/die. Promising efficacy data were reported with PSA reduction of ≥50% observed in 70% of evaluable patients.106
Based on these clinical results, a Phase III study was conducted (NCT02438007). The ARMOR 3-SV was an open-label, randomized trial of galeterone compared to enzalutamide in men with ARV 7-positive mCRPC. Unfortunately, according to the Independent Data Monitoring Committee recommendation, the study was prematurely closed. A careful analysis of all safety and efficacy data concluded that the trial would hardly have reached the primary end point.
Beyond the AR: the contemporary role of AR-independent pathway inhibition
PC is a heterogeneous disease in which both AR-positive and AR-negative neoplastic cells coexist, and both contribute to cancer progression.19,21
Adaptive response to hormonal therapy is mediated by the activation of AR-dependent pathways, but at the same time, it can be attributable to crucial mechanisms of progression bypassing AR signaling.19,21 Endocrine therapy, in fact, blocking AR-positive cells can induce treatment resistance causing clonal selection and the consequent growth of cellular clones independent of AR signalling.19
These biological evidences represent the basis for the administration of chemotherapy in association with endocrine therapy, in the attempt to interfere also with AR-independent survival pathways, inhibiting AR-negative cells; such approach could delay the development of hormonal treatment resistance mechanisms blocking the growth of a larger amount of neoplastic cells.19,21
In this context, recent evidence showed that adding docetaxel to ADT in patients with high-risk or high-volume hormone-sensitive metastatic disease is able to induce a significant improvement in OS compared to ADT alone.12–14
These preclinical and clinical findings provide the rationale to combine new generation hormonal therapies such as enzalutamide with chemotherapy. A Phase II study testing the combination of ADT and docetaxel with the addition of enzalutamide in metastatic HSPC patients is ongoing. The aim of the trial is to evaluate whether ADT plus docetaxel plus enzalutamide may increase treatment efficacy compared to ADT plus docetaxel (NCT03246347; Table 1). Another trial is investigating the activity of docetaxel added to enzalutamide in overcoming enzalutamide resistance in mCRPC patients (NCT02288247; Table 1). Furthermore, two studies are evaluating the efficacy of the combination of enzalutamide with cabazitaxel in mCRPC patients (NCT02522715, NCT03110588; Table 1).
Recently, Robinson et al108 prospectively investigated the whole-exome and transcriptome sequencing from soft tissue or bone secondary lesions in 150 mCRPC patients. Aberrations of AR were detected at higher rate than expected (63%). Furthermore, among non-AR-related alterations, DNA repair gene pathways (BRCA2, BRCA1 and ATM) were commonly altered (19.3%).
In a Phase II trial, 50 mCRPC patients were treated with the PARP inhibitor, olaparib.109 Next-generation sequencing detected DNA repair gene aberrations in 16 patients; among these patients, 14 patients had objective response and the response rate was higher than in those patients who were not harboring DNA repair gene alterations.
These biological and clinical data confirm the crucial role of DNA repair gene mutations as a tumor cell survival pathway in CRPC patients and provide a rationale for the use of PARP inhibitors. Several studies are ongoing, testing the activity of PARP inhibitors alone or in combination with new generation hormonal therapies in metastatic prostate carcinoma (Table 1).
Future perspectives and conclusion
In recent years, several researches regarding CRPC showed resistance mechanisms to ADT mainly due to AR amplification or mutation.19,21 The identification of these escape mechanisms allowed to understand that AR pathway activity persists despite low levels of serum testosterone and favored the development of new generation AR antagonists such as enzalutamide.19,21
The advent of new generation hormonal therapies set the stage for a new era in the treatment landscape of CRPC. Although these agents showed to increase OS, a relevant proportion of patients do not benefit at all or acquire resistance during treatment.
Primary and secondary resistance to enzalutamide is a multifactorial phenomenon which depends on not only ligand-dependent and ligand-independent AR activation but also pathways bypassing AR signaling.
Taking into account the future shift for enzalutamide in the early disease stages, in the next years, physicians will face several therapeutic challenges. The most important challenge will be a more complete knowledge of heterogeneity of PC. Nowadays, we know that metastatic PC is a heterogeneous disease, in which AR-positive and AR-independent neoplastic cells coexist19 and in which AR aberrations responsible for enzalutamide resistance could be present at various degrees.19,21 This heterogeneity is the main reason for different biological behaviors and particularly for different responses to new-generation hormonal therapies among various PCs.
Future in vitro and in vivo studies should enable us to obtain a better understanding of biological PC heterogeneity to identify patients who will not respond to enzalutamide therapy or those who will likely develop acquired resistance in a few months. The identification of predictive biomarkers of response, as well as the detection of PC molecular subtypes based on proteomic and genomic assessment, will be critical to improve the activity of enzalutamide and to identify new approaches for bypassing resistance to personalize treatment on the basis of PC biology.
Disclosure
Massimo Di Maio received honoraria from Astellas and Janssen for lectures in meeting and participation in advisory boards. The other authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. | ||
American Cancer Society. Cancer Facts and Figures 2016. Atlanta: American Cancer Society; 2016:10. | ||
Tucci M, Scagliotti GV, Vignani F. Metastatic castration-resistant prostate cancer: time for innovation. Future Oncol. 2015;11(1):91–106. | ||
de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med Overseas Ed. 2011;364(21):1995–2005. | ||
Scher HI, Fizazi K, Saad F, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med Overseas Ed. 2012;367(13):1187–1197. | ||
Ryan CJ, Smith MR, Fizazi K, et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2015;16(2):152–160. | ||
Beer TM, Armstrong AJ, Rathkopf DE, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med Overseas Ed. 2014;371(5):424–433. | ||
de Bono JS, Oudard S, Ozguroglu M, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376(9747):1147–1154. | ||
Oudard S, Fizazi K, Sengeløv L, et al. Cabazitaxel versus docetaxel as first-line therapy for patients with metastatic castration-resistant prostate cancer: a randomized phase III trial-FIRSTANA. J Clin Oncol. 2017;35(28):3189–3197. | ||
Parker C, Nilsson S, Heinrich D, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med Overseas Ed. 2013;369(3):213–223. | ||
Kantoff PW, Higano CS, Shore ND. for the IMPACT Study Investigators. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. | ||
Sweeney CJ, Chen Y-H, Carducci M, et al. Chemohormonal therapy in metastatic hormone-sensitive prostate cancer. N Engl J Med Overseas Ed. 2015;373(8):737–746. | ||
James ND, Sydes MR, Clarke NW, et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet. 2016;387(10024):1163–1177. | ||
Tucci M, Bertaglia V, Vignani F, et al. Addition of docetaxel to androgen deprivation therapy for patients with hormone-sensitive metastatic prostate cancer: a systematic review and meta-analysis. Eur Urol. 2016;69(4):563–573. | ||
Fizazi K, Tran N, Fein L, et al. Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. N Engl J Med Overseas Ed. 2017;377(4):352–360. | ||
James ND, de Bono JS, Spears MR, et al. Abiraterone for prostate cancer not previously treated with hormone therapy. N Engl J Med Overseas Ed. 2017;377(4):338–351. | ||
Smith MR, Saad F, Chowdhury S, et al. Apalutamide treatment and metastasis-free survival in prostate cancer. N Engl J Med Overseas Ed. 2018;378(15):1408–1418. | ||
Hussain M, Fizazi K, Saad F, et al. Enzalutamide in men with nonmetastatic, castration-resistant prostate cancer. N Engl J Med Overseas Ed. 2018;378(26):2465–2474. | ||
Buttigliero C, Tucci M, Bertaglia V, et al. Understanding and overcoming the mechanisms of primary and acquired resistance to abiraterone and enzalutamide in castration resistant prostate cancer. Cancer Treat Rev. 2015;41(10):884–892. | ||
Davey RA, Grossmann M. Androgen receptor structure, function and biology: from bench to bedside. Clin Biochem Rev. 2016;37(1):3–15. | ||
Mostaghel EA, Plymate SR, Montgomery B. Molecular pathways: targeting resistance in the androgen receptor for therapeutic benefit. Clin Cancer Res. 2014;20(4):791–798. | ||
Caffo O, Veccia A, Maines F, Bonetta A, Spizzo G, Galligioni E. Potential value of rapid prostate-specific antigen decline in identifying primary resistance to abiraterone acetate and enzalutamide. Future Oncol. 2014;10(6):985–993. | ||
Edwards J, Krishna NS, Grigor KM, Bartlett JM. Androgen receptor gene amplification and protein expression in hormone refractory prostate cancer. Br J Cancer. 2003;89(3):552–556. | ||
Crona DJ, Whang YE. Androgen receptor-dependent and -independent mechanisms involved in prostate cancer therapy resistance. Cancers. 2017;9(6):67. | ||
Romanel A, Gasi Tandefelt D, Conteduca V, et al. Plasma AR and abiraterone-resistant prostate cancer. Sci Transl Med. 2015;7(312):re10. | ||
Conteduca V, Wetterskog D, Sharabiani MTA, et al. Androgen receptor gene status in plasma DNA associates with worse outcome on enzalutamide or abiraterone for castration-resistant prostate cancer: a multi-institution correlative biomarker study. Ann Oncol. 2017;28(7):1508–1516. | ||
Umekita Y, Hiipakka RA, Kokontis JM, Liao S. Human prostate tumor growth in athymic mice: inhibition by androgens and stimulation by finasteride. Proc Natl Acad Sci U S A. 1996;93(21):11802–11807. | ||
Zhau HY, Chang SM, Chen BQ, et al. Androgen-repressed phenotype in human prostate cancer. Proc Natl Acad Sci U S A. 1996;93(26):15152–15157. | ||
Teply BA, Wang H, Luber B, et al. Bipolar androgen therapy in men with metastatic castration-resistant prostate cancer after progression on enzalutamide: an open-label, phase 2, multicohort study. Lancet Oncol. 2018;19(1):76–86. | ||
Taplin ME, Bubley GJ, Shuster TD, et al. Mutation of the androgen-receptor gene in metastatic androgen-independent prostate cancer. N Engl J Med. 1995;332(21):1393–1398. | ||
Joseph JD, Lu N, Qian J, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013;3(9):1020–1029. | ||
Balbas MD, Evans MJ, Hosfield DJ, et al. Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife. 2013;2:e00499. | ||
Lallous N, Volik SV, Awrey S, et al. Functional analysis of androgen receptor mutations that confer anti-androgen resistance identified in circulating cell-free DNA from prostate cancer patients. Genome Biol. 2016;17(1):10. | ||
Borgmann H, Lallous N, Ozistanbullu D, et al. Moving towards precision urologic oncology: targeting enzalutamide-resistant prostate cancer and mutated forms of the androgen receptor using the novel inhibitor darolutamide (ODM-201). Eur Urol. 2018;73(1):4–8. | ||
Hu R, Dunn TA, Wei S, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69(1):16–22. | ||
Zhang X, Morrissey C, Sun S, et al. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PLoS One. 2011;6(11):e27970. | ||
Ware KE, Garcia-Blanco MA, Armstrong AJ, Dehm SM. Biologic and clinical significance of androgen receptor variants in castration resistant prostate cancer. Endocr Relat Cancer. 2014;21(4):T87–T103. | ||
Hu R, Lu C, Mostaghel EA, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012;72(14):3457–3462. | ||
Antonarakis ES, Lu C, Wang H, et al. AR-V7 and Resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med Overseas Ed. 2014;371(11):1028–1038. | ||
Sq T, Kwan E, Fettke H. AR-V7 and AR-V9 expression is not predictive of response to AR-axis targeting agents in metastatic castration-resistant prostate cancer. Proceedings: AACR Annual Meeting 2018. April 14–18, 2018; Chicago, IL. | ||
Yang YC, Banuelos CA, Mawji NR, et al. Targeting androgen receptor activation function-1 with EPI to overcome resistance mechanisms in castration-resistant prostate cancer. Clin Cancer Res. 2016;22(17):4466–4477. | ||
Chan SC, Selth LA, Li Y, et al. Targeting chromatin binding regulation of constitutively active AR variants to overcome prostate cancer resistance to endocrine-based therapies. Nucleic Acids Res. 2015;43(12):5880–5897. | ||
Liu C, Lou W, Zhu Y, et al. Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin Cancer Res. 2014;20(12):3198–3210. | ||
Armstrong CM, Gao AC. Adaptive pathways and emerging strategies overcoming treatment resistance in castration resistant prostate cancer. Asian J Urol. 2016;3(4):185–194. | ||
Tummala R, Lou W, Gao AC, Nadiminty N. Quercetin targets hnRNPA1 to overcome enzalutamide resistance in prostate cancer cells. Mol Cancer Ther. 2017;16(12):2770–2779. | ||
Yamamoto Y, Loriot Y, Beraldi E, et al. Generation 2.5 antisense oligonucleotides targeting the androgen receptor and its splice variants suppress enzalutamide-resistant prostate cancer cell growth. Clin Cancer Res. 2015;21(7):1675–1687. | ||
Yamamoto Y, Lin PJ, Beraldi E, et al. siRNA Lipid nanoparticle potently silences clusterin and delays progression when combined with androgen receptor cotargeting in enzalutamide-resistant prostate cancer. Clin Cancer Res. 2015;21(21):4845–4855. | ||
Wang R, Sun Y, Li L, et al. Preclinical study using Malat1 small interfering RNA or androgen receptor splicing variant 7 degradation enhancer ASC-J9® to suppress enzalutamide-resistant prostate cancer progression. Eur Urol. 2017;72(5):835–844. | ||
Welti J, Sharp A, Yuan W, et al. Targeting bromodomain and extra-terminal (BET) family proteins in castration-resistant prostate cancer (CRPC). Clin Cancer Res. 2018;24(13):3149–3162. | ||
Asangani IA, Wilder-Romans K, Dommeti VL, et al. BET bromodomain inhibitors enhance efficacy and disrupt resistance to AR antagonists in the treatment of prostate cancer. Mol Cancer Res. 2016;14(4):324–331. | ||
Han Y, Huang W, Liu J, et al. Triptolide inhibits the AR signaling pathway to suppress the proliferation of enzalutamide resistant prostate cancer cells. Theranostics. 2017;7(7):1914–1927. | ||
Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. 2011;32(1):81–151. | ||
Cai C, Balk SP. Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy. Endocr Relat Cancer. 2011;18(5):R175–R182. | ||
Galletti G, Leach BI, Lam L, Tagawa ST. Mechanisms of resistance to systemic therapy in metastatic castration-resistant prostate cancer. Cancer Treat Rev. 2017;57:16–27. | ||
Stanbrough M, Bubley GJ, Ross K, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66(5):2815–2825. | ||
Liu C, Lou W, Zhu Y, et al. Intracrine androgens and AKR1C3 activation confer resistance to enzalutamide in prostate cancer. Cancer Res. 2015;75(7):1413–1422. | ||
Kino T. Glucocorticoid receptor. In: De Groot LJ, Chrousos G, Dungan K, et al, editors. Endotext [Internet]. South Dartmouth: MDText.com, Inc.; 2000. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279171/. Accessed August 15, 2017. | ||
Puhr M, Hoefer J, Eigentler A, et al. The glucocorticoid receptor is a key player for prostate cancer cell survival and a target for improved antiandrogen therapy. Clin Cancer Res. 2018;24(4):927–938. | ||
Arora VK, Schenkein E, Murali R, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155(6):1309–1322. | ||
Venkitaraman R, Lorente D, Murthy V, et al. A randomised phase 2 trial of dexamethasone versus prednisolone in castration-resistant prostate cancer. Eur Urol. 2015;67(4):673–679. | ||
Yang Y, Li X, Mamouni K, Kucuk O, Wu D. Mifepristone has limited activity to enhance the in vivo efficacy of docetaxel and enzalutamide against bone metastatic and castration-resistant prostate cancer. Anticancer Res. 2017;37(11):6235–6243. | ||
Zhou H, Jachan N, Singh M, et al. Abstract 4172: activation of AR signaling by mifepristone enhances prostate cancer growth and impairs enzalutamide response. Cancer Res. 2017;77(13 Suppl):4172. | ||
Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–196. | ||
Larue L, Bellacosa A. Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene. 2005;24(50):7443–7454. | ||
Montanari M, Rossetti S, Cavaliere C, et al. Epithelial-mesenchymal transition in prostate cancer: an overview. Oncotarget. 2017;8(21):35376–35389. | ||
Shiota M, Yokomizo A, Takeuchi A, et al. Inhibition of protein kinase C/Twist1 signaling augments anticancer effects of androgen deprivation and enzalutamide in prostate cancer. Clin Cancer Res. 2014;20(4):951–961. | ||
Ware KE, Somarelli JA, Schaeffer D, et al. Snail promotes resistance to enzalutamide through regulation of androgen receptor activity in prostate cancer. Oncotarget. 2016;7(31):50525–50539. | ||
Liu Q, Tong D, Liu G, et al. Metformin reverses prostate cancer resistance to enzalutamide by targeting TGF-β1/STAT3 axis-regulated EMT. Cell Death Dis. 2017;8(8):e3007. | ||
Akamatsu S, Inoue T, Ogawa O, Gleave ME. Clinical and molecular features of treatment-related neuroendocrine prostate cancer. Int J Urol. 2018;25(4):345–351. | ||
Fléchon A, Pouessel D, Ferlay C, et al. Phase II study of carboplatin and etoposide in patients with anaplastic progressive metastatic castration-resistant prostate cancer (mCRPC) with or without neuroendocrine differentiation: results of the French Genito-Urinary Tumor Group (GETUG) P01 trial. Ann Oncol. 2011;22(11):2476–2481. | ||
Culine S, El Demery M, Lamy PJ, Iborra F, Avancès C, Pinguet F. Docetaxel and cisplatin in patients with metastatic androgen independent prostate cancer and circulating neuroendocrine markers. J Urol. 2007;178(3 Pt 1):844–848. | ||
Beltran H, Rickman DS, Park K, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011;1(6):487–495. | ||
Sy K, Rosario S, Wang Y, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science. 2017;355:78–83. | ||
Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(6320):11–22. | ||
Carver BS, Chapinski C, Wongvipat J, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19(5):575–586. | ||
Thomas C, Lamoureux F, Crafter C, et al. Synergistic targeting of PI3K/AKT pathway and androgen receptor axis significantly delays castration-resistant prostate cancer progression in vivo. Mol Cancer Ther. 2013;12(11):2342–2355. | ||
He C, Duan S, Dong L, et al. Characterization of a novel p110β-specific inhibitor BL140 that overcomes MDV3100-resistance in castration-resistant prostate cancer cells. Prostate. 2017;77(11):1187–1198. | ||
Shiota M, Bishop JL, Takeuchi A, et al. Inhibition of the HER2-YB1-AR axis with Lapatinib synergistically enhances Enzalutamide anti-tumor efficacy in castration resistant prostate cancer. Oncotarget. 2015;6(11):9086–9098. | ||
Nadiminty N, Tummala R, Liu C, et al. NF-κB2/p52 induces resistance to enzalutamide in prostate cancer: role of androgen receptor and its variants. Mol Cancer Ther. 2013;12(8):1629–1637. | ||
Culig Z. Molecular mechanisms of enzalutamide resistance in prostate cancer. Curr Mol Biol Rep. 2017;3(4):230–235. | ||
Farrow JM, Yang JC, Evans CP. Autophagy as a modulator and target in prostate cancer. Nat Rev Urol. 2014;11(9):508–516. | ||
Bennett HL, Stockley J, Fleming JT, et al. Does androgen-ablation therapy (AAT) associated autophagy have a pro-survival effect in LNCaP human prostate cancer cells? BJU Int. 2013;111(4):672–682. | ||
Nguyen HG, Yang JC, Kung HJ, et al. Targeting autophagy overcomes Enzalutamide resistance in castration-resistant prostate cancer cells and improves therapeutic response in a xenograft model. Oncogene. 2014;33(36):4521–4530. | ||
Matsumoto H, Yamamoto Y, Shiota M, et al. Cotargeting androgen receptor and clusterin delays castrate-resistant prostate cancer progression by inhibiting adaptive stress response and AR stability. Cancer Res. 2013;73(16):5206–5217. | ||
Bishop JL, Sio A, Angeles A, et al. PD-L1 is highly expressed in Enzalutamide resistant prostate cancer. Oncotarget. 2014;6(1):234–242. | ||
Schepisi G, Farolfi A, Conteduca V, et al. Immunotherapy for prostate cancer: where we are headed. Int J Mol Sci. 2017;18(12):pii:E2627. | ||
Graff JN, Alumkal JJ, Drake CG, et al. Early evidence of anti-PD-1 activity in enzalutamide-resistant prostate cancer. Oncotarget. 2016;7(33):52810–52817. | ||
Kwon ED, Drake CG, Scher HI, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol. 2014;15(7):700–712. | ||
Hegemann M, Stenzl A, Bedke J, Chi KN, Black PC, Todenhöfer T. Liquid biopsy: ready to guide therapy in advanced prostate cancer? BJU Int. 2016;118(6):855–863. | ||
Gupta S, Li J, Kemeny G, et al. Whole genomic copy number alterations in circulating tumor cells from men with abiraterone or enzalutamide-resistant metastatic castration-resistant prostate cancer. Clin Cancer Res. 2017;23(5):1346–1357. | ||
Azad AA, Volik SV, Wyatt AW, et al. Androgen receptor gene aberrations in circulating cell-free DNA: biomarkers of therapeutic resistance in castration-resistant prostate cancer. Clin Cancer Res. 2015;21(10):2315–2324. | ||
Wyatt AW, Azad AA, Volik SV, et al. Genomic alterations in cell-free DNA and enzalutamide resistance in castration-resistant prostate cancer. JAMA Oncol. 2016;2(12):1598–1606. | ||
Annala M, Vandekerkhove G, Khalaf D, et al. Circulating tumor DNA genomics correlate with resistance to abiraterone and enzalutamide in prostate cancer. Cancer Discov. 2018;8(4):444–457. | ||
Belic J, Graf R, Bauernhofer T. Analysis of circulating tumor DNA reveals genomic alterations in metastatic prostate cancer patients treated with abiraterone acetate plus prednisone or enzalutamide. Proceedings: AACR Annual Meeting 2018. Chicago, IL. | ||
King CJ, Woodward J, Schwartzman J, et al. Integrative molecular network analysis identifies emergent enzalutamide resistance mechanisms in prostate cancer. Oncotarget. 2017;8(67):111084–111095. | ||
Qian S, Xia J, Liu H, Zhang Y, Zhang L, Yu Y. Integrative transcriptome analysis identifies genes and pathways associated with enzalutamide resistance of prostate cancer. Aging Male. 2018;9:1–7. | ||
Rathkopf DE, Antonarakis ES, Shore ND, et al. Safety and antitumor activity of apalutamide (ARN-509) in metastatic castration-resistant prostate cancer with and without prior abiraterone acetate and prednisone. Clin Cancer Res. 2017;23(14):3544–3551. | ||
Clegg NJ, Wongvipat J, Joseph JD, et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res. 2012;72(6):1494–1503. | ||
Rathkopf DE, Morris MJ, Fox JJ, et al. Phase I study of ARN-509, a novel antiandrogen, in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2013;31(28):3525–3530. | ||
Smith MR, Antonarakis ES, Ryan CJ, et al. Phase 2 study of the safety and antitumor activity of apalutamide (ARN-509), a potent androgen receptor antagonist, in the high-risk nonmetastatic castration-resistant prostate cancer cohort. Eur Urol. 2016;70(6):963–970. | ||
Moilanen AM, Riikonen R, Oksala R, et al. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci Rep. 2015;5(1):12007. | ||
Shore ND. Darolutamide (ODM-201) for the treatment of prostate cancer. Expert Opin Pharmacother. 2017;18(9):945–952. | ||
Slovin S, Clark W, Carles J, et al. Seizure rates in enzalutamide-treated men with metastatic castration-resistant prostate cancer and risk of seizure: the UPWARD study. JAMA Oncol. 2018;4(5):702–706. | ||
Fizazi K, Massard C, Bono P, et al. Activity and safety of ODM-201 in patients with progressive metastatic castration-resistant prostate cancer (ARADES): an open-label phase 1 dose-escalation and randomised phase 2 dose expansion trial. Lancet Oncol. 2014;15(9):975–985. | ||
Massard C, Penttinen HM, Vjaters E, et al. Pharmacokinetics, antitumor activity, and safety of ODM-201 in patients with chemotherapy-naive metastatic castration-resistant prostate cancer: an open-label phase 1 study. Eur Urol. 2016;69(5):834–840. | ||
Bastos DA, Antonarakis ES. Galeterone for the treatment of advanced prostate cancer: the evidence to date. Drug Des Devel Ther. 2016;10:2289–2297. | ||
Montgomery B, Eisenberger MA, Rettig MB, et al. Androgen receptor modulation optimized for response (ARMOR) phase I and II studies: galeterone for the treatment of castration-resistant prostate cancer. Clin Cancer Res. 2016;22(6):1356–1363. | ||
Robinson D, van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215–1228. | ||
Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373(18):1697–1708. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.