Back to Journals » Drug Design, Development and Therapy » Volume 14
Enhancing the Butyrylcholinesterase Activity in HEK-293 Cell Line by Dual-Promoter Vector Decorated on Lipofectamine
Authors Mirzaie V ![]() , Eslaminejad T
, Eslaminejad T ![]() , Babaei H, Nematollahi-Mahani SN
, Babaei H, Nematollahi-Mahani SN ![]()
Received 6 May 2020
Accepted for publication 6 August 2020
Published 4 September 2020 Volume 2020:14 Pages 3589—3599
DOI https://doi.org/10.2147/DDDT.S260419
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Qiongyu Guo
Vida Mirzaie,1 Touba Eslaminejad,2 Homayoon Babaei,3 Seyed Noureddin Nematollahi-Mahani4,5
1Department of Anatomy, Afzalipour School of Medicine, Kerman University of Medical Sciences, Kerman, Iran; 2Pharmaceutics Research Centre, Institute of Neuropharmacology, Kerman University of Medical Sciences, Kerman, Iran; 3Department of Clinical Sciences, Faculty of Veterinary Medicine, Shahid Bahonar University of Kerman, Kerman, Iran; 4Neuroscience Research Centre, Institute of Neuropharmacology, Kerman University of Medical Sciences, Kerman, Iran; 5Afzal Research Institute (NGO), Kerman University of Medical Sciences, Kerman, Iran
Correspondence: Seyed Noureddin Nematollahi-Mahani Tel +983 433257343
Email [email protected]
Purpose: Human butyrylcholinesterase (BChE) serves as a bio scavenger to counteract organophosphate poisoning. It is also a potential drug candidate in several therapeutic fields. Therefore, in the present study, we constructed a new dual-promoter plasmid consisting of Cytomegalovirus (CMV) and human elongation factor 1α (EF-1α) promoters and transfected that into HEK-293 cells using Lipofectamine to enhance the BChE secretion.
Methods: The new dual-promoter construction (pBudCE dual BChE) including two copies of the BChE gene was designed and transfected into cells by liposomal structures. The cloned plasmids were evaluated by enzyme digestion and gel electrophoresis analysis. Experimental groups were categorized into the cells transfected by pBudCE dual BChE (treatment), pCMV (positive control) vectors, and nontransfected cells (negative control). BChE gene expression was evaluated by qRT-PCR and the enzyme activity was assessed using modified Ellman’s method. The freeze-thaw process was carried out for analyzing the stability of the pBudCE dual BChE vector.
Results: Validation examination of the cloned plasmids confirmed the successful cloning process. The gene expression level and Ellman’s method value in pBudCE dual BChE was higher than the other groups. CMV promoter has also increased the enzyme activity, although the difference was not significant compared with the control group. Interestingly, freeze-thaw cycles followed by several passages did not affect the enzyme activity.
Conclusion: The designed construction with CMV and EF-1α promoters could increase BChE gene expression and the activity of the BChE enzyme in HEK-293 cell line. Large-scale production of BChE enzyme can be achieved by using dual-promoter plasmid construction compared to a single-promoter vector to be used in clinical trials.
Keywords: dual-promoter, vector, recombinant protein, drug delivery, Ellman’s method, HEK-293
Introduction
Cholinesterases are a range of circulating enzymes present in tissues or in body fluids and plasma. They are divided into two main groups of acetylcholinesterase and butyrylcholinesterase (BChE) based on their behavior in the presence of specific substrates and susceptibility to the limiting agents.1 Human BChE is a tetrameric blood globulin glycoprotein consisting of 4 subunits that are stabilized with PRAD peptide and each subunit contains 574 amino acids and 9 carbohydrate chains.2,3 The tetramer form of BChE has a long half-life, while monomer and dimer forms will remove from the blood after a few minutes and have less pharmacological effect.4 Serum BChE is pharmacologically and toxicologically important, since the enzyme has the ability to hydrolyze ester drugs such as succinylcholine and cocaine and converts bambuterol into bronchodilating drug terbutaline.5 BChE can be used either prophylactically or as a treatment drug. One of the most important applications of BChE is detoxifying organophosphate toxins. Organophosphates or carbamate esters are used in insecticide, pesticides, chemical weapons of mass destruction, and some medicines to treat diseases such as glaucoma, Alzheimer’s, and parasitic infections.1,6 According to the World Health Organization (WHO), 200,000 people die annually due to poisoning from organophosphate pesticides in developing countries.7–9 BChE is the only bio scavenger known as a “new development drug” in the FDA in 2006 which can prevent poisoning from organophosphate toxins.10,11 The use of external BChE 200 mg per 70 kg body weight has been effective in preventing acetylcholinesterase inactivation.12 However, BChE could be isolated from the blood plasma, and several investigations have been done in the past 20 years in order to produce higher amounts of the enzyme. According to studies, a high volume of human plasma (100 liters) is required to produce 200 mg (one dose) of the BChE enzyme, which is expensive, and has the potential of contamination.13,14 As an alternative method, it is possible to produce BChE as a recombinant protein in prokaryotic or eukaryotic cells.15,16 The expression of protein in prokaryotic cells is relatively simple and economically affordable. However, scientists could not successfully produce active BChE in Escherichia coli bacteria.17 A recent study has shown successful recombination of BChE in bacteria, although the activity of the produced enzyme for detoxification remains to be evaluated.18 The expression of recombinant proteins requires the use of special expression vectors, promoters, and cell lines.19 As an appropriate cell line, HEK-293 has been used extensively as a standard host for the cloning process since 1974.20 Cytomegalovirus immediate-early promoter (CMV) and human elongation factor 1α (EF-1α) are two of the strongest promoters in gene expression. Jane et al showed that EF-1α could be strong in all types of cells consistently, while CMV is slightly weaker than EF-1α, and also has variable effects that are very strong in some cell types and rather weak in others.21 Although many studies worked on the enhancement of BChE production using different methods, introducing two copies of the BChE gene in front of the two mentioned promoters, simultaneously in one plasmid, with the aim of increasing the enzyme production has not been studied yet. The present study investigated the effect of the simultaneous use of two promoters of CMV and EF-1α in a single plasmid on the BChE gene expression level and enzyme activity in the HEK-293 cell line. The transient stability of the new construction in HEK-293 cells was also evaluated in several freeze–thaw cycles.
Materials and Methods
Materials
The research was approved by the ethical committee of Kerman University of Medical Sciences and the Ethics Approval Code is IR.KMU.REC.1398.294. Restriction enzymes, T4 DNA ligase, GeneJET Plasmid Miniprep® kit (#K0502, #K0503), and Taq DNA Polymerase Master Mix RED were purchased from Thermo Scientific (Germany) and Ampliqon® (Denmark) respectively. Plasmids pCMV3-BChE (Cat No., HG12010-UT) and pBudCE4.1 (Cat No., V532-20) were provided by Sino Biological (China) and Invitrogen (USA). RiboEx™ (Cat No., 301–001) was purchased from GeneAll (Portugal). Parstous cDNA synthesis kit (Cat No., A101161) was provided by Parstous Biotechnology company (Mashhad, Iran). SinaSYBR Blue HS-qPCRMix, 2x (Cat No., MM2171) was provided by Sinaclone (Tehran, Iran). 5.5′-Dithiobis(2-nitrobenzoic acid) DTNB (Cat No., D218200), Hyamine 1622 (Cat No., 121–54-0), and butyrylthiocholine iodide (Cat No., B3253) were purchased from Sigma Aldrich (Saint Louis, MO, USA). Dulbecco’s modified Eagle’s medium (DMEM) was purchased from Gibco (Grand Island, USA). Fetal bovine serum (FBS) and penicillin streptomycin were purchased from Hangzhou Sijiqing Biological Engineering Materials Co. Ltd (Hangzhou, China). The HEK-293 cell line was provided from Pasteur Institute (Tehran, Iran).
Cloning Process
Overview of the Vector
pCMV-BChE was used as the standard plasmid in the positive control group and the parent plasmid was in high-copy number plasmid pBudCE4.1, which has the SV40 ori and BleoR gene (Figure 1).
 |
Figure 1 The schematic overview of the plasmid pBudCE4.1, containing two promoters of CMV and EF-1α. |
Plasmid Construction and Validation (Digestion and Ligation)
Specific sense and antisense primers flanked by SalI and KpnI, ScaI and XhoI sequences, respectively, were designed by NEBcutter® V2.0. Restriction enzyme sites were selected based on the pBudCE4.1 plasmid sequence (Figure 2).
 |
Figure 2 Restriction sites on the forward (F) and reverse (R) BChE gene primers. |
cDNA of the BChE gene (gene ID: 590, 1809 bp) was synthesized by Takapozist company (Tehran, Iran). PCR amplification of the BChE gene was done using the mentioned primers according to the PCR program; 95ºC for 7 min, followed by 40 cycles of 95ºC for 15 s, 65ºC for 20 s, and 72ºC for 35 s. PCR product and pBudCE4.1 plasmid were double digested by SalI, ScaI, and KpnI, XhoI enzymes, in separate tubes according to the manufacturer’s protocol. Briefly, 1 µL of each enzyme was added to 1 µL DNA (concentration 1 µg/µL), 2 µL tango buffer (33 mM Tris-acetate (pH 7.9 at 37°C), 10 mM magnesium acetate, 66 mM potassium acetate, 0.1 mg/mL BSA) and 15 µL sterile water, the final volume reaching 20 µL. Tubes were incubated at 37ºC for 1 h and inactivated at 65ºC for 15 min. The ligation process was done according to the manufacturer’s protocol as follows: genes and plasmids which were digested by the same restriction enzymes were mixed and ligated by using the T4 ligation enzyme and buffer, and incubated at 22ºC for 1 h. The resulting products are two tubes containing two cloned plasmids, one with using SalI, ScaI and the other with KpnI, XhoI. Cloning validation was confirmed by digestion of each cloned plasmid with related digesting enzymes and the products were run on the agarose gel (Figure 3).
 |
Figure 3 Cloning validation was confirmed by gel electrophoresis; (A) cloned plasmid, (B) 25kb ladder, (C) SalI, ScaI digested products, (D) KpnI, XhoI digested products. |
After determining that the gene was inserted into pBudCE4.1 plasmid, then pBudCE dual BChE vector was constructed as follows:
pBudCE4.1 plasmid was digested by SalI, ScaI, KpnI and XhoI restriction enzymes: 1 µL of plasmid, 1 µL of each restriction enzyme (4 µL), 6 µL tango buffer, and 19 µL sterile water, final volume 30 µL. Double-digested genes (gene digested by SalI, ScaI and KpnI, XhoI, separately) were added to the plasmid which is digested by all four enzymes afterward. The ligation process was done to construct the final form of the vector (pBudCE dual BChE) (Figure 4).
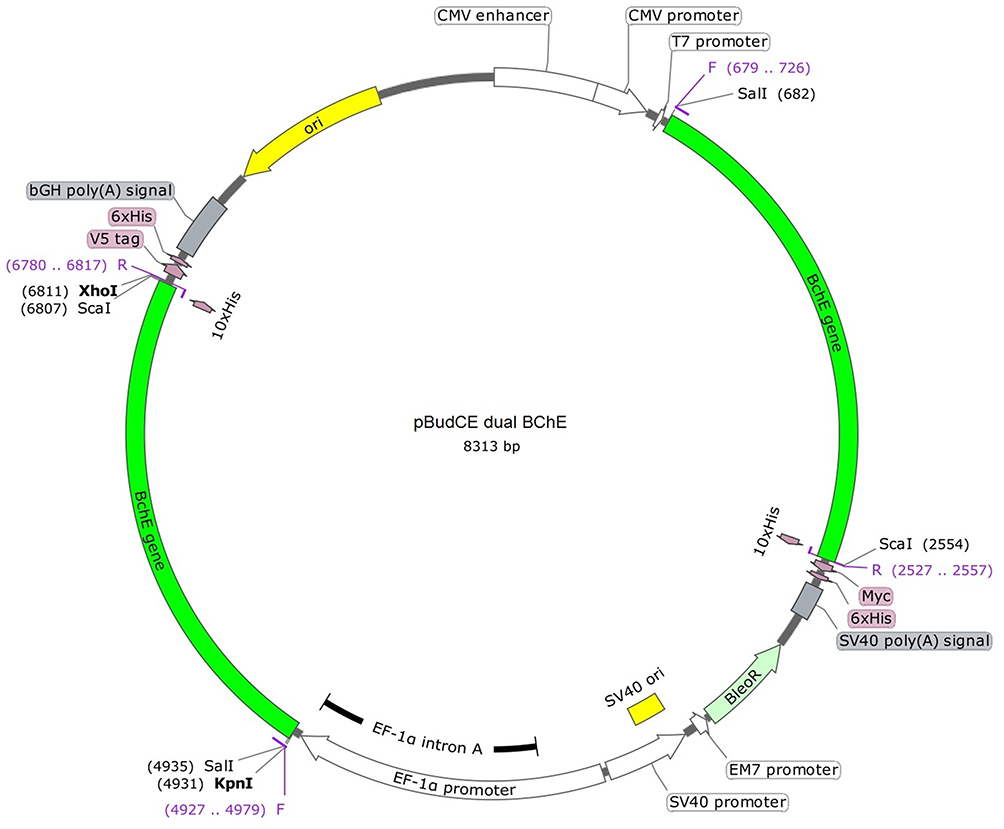 |
Figure 4 The final construction of the pBudCE dual BChE vector. |
E. coli strain TOP10 competent cells prepared using the CaCl2 method and were transformed with the recombinant pBudCE dual BChE vector by the heat shock method. Transformed E. coli was cultivated at 37°C overnight in LB medium supplemented with Zeocin™ (50 μg/mL concentration). Plasmid extraction was done by GeneJET Plasmid Miniprep® Kit. The fresh LB/Zeocin™ medium was inoculated with the transformed bacteria and incubated at 37°C for 16 h. Bacterial cells were harvested by centrifugation at 6800 × g for 2 min and resuspended in lysis buffer (50 mM NaH2PO4, 300 mM NaCl, pH 8.0). Then, the plasmids were purified according to the protocol.
Preparation of Plasmids Decorated on Lipofectamine
For each group (pCMV and pBudCE dual BChE), 14 µL Lipofectamine™ (2.5%) was added to 250 µL serum-free medium in a microtube for liposomal formation, and 5 µg of DNA plasmid (1%) was diluted in 250 µL serum-free medium in a separate microtube, simultaneously. The DNA suspension was gently added to the liposomal suspension afterward to the final volume of 500 µL. There were nine wells for each group; therefore, 50 µL of the liposomal DNA was added to each well. The final DNA concentration was 0.5 µg/well.
Transfection and Cell Culture
HEK-293 cells were grown in DMEM-F12 supplemented with 10% FBS and were maintained in a humidified incubator with 5% CO2 at 37°C to attain 90–95% confluency. Cells were trypsinized and re-suspended in the fresh serum-free (2% FBS) medium cell culture at a density of 2×105 cells/mL one day before transfection. After incubating the cells for 24 h, the medium was changed with 50 µL of the prepared liposomal DNA suspension without antibiotics. The experimental groups were divided into transfected with pBudCE dual BChE (treatment), pCMV (positive control), and nontransfected cells (negative control) groups that have been followed for 3, 6, and 9 days. The culture medium of each group was collected for Ellman’s method and the cells were lysed for RNA extraction and gene expression, separately.
mRNA Extraction and Gene Expression
Cells were lysed by adding 1 mL of RiboEx™ reagent following the manufacturer’s instructions. Proteins were removed by phenol/chloroform extraction to obtain an A260:A280 ratio of 1.81 ± 0.06. The concentration and purity of isolated RNA were assessed by absorbance (A) readings on a UV spectrophotometer (Thermo Scientific™ NanoDrop 2000) at the wavelengths of 260 and 280 nm. Synthesis of cDNA was carried out using a Parstous cDNA synthesis kit in 20 µL of the final volume, according to the manufacturer’s recommendations. For every first-strand cDNA synthesis, 0.1 µg of total RNA was used, based on the following PCR program temperature as described below (Table 1).
 |
Table 1 RT-PCR Program for Synthesis of cDNA |
Real-time PCR was performed using the SinaSYBER blue reaction mix without lowROX (SinaClon) in a magnetic induction cycler (mic) real-time PCR system, using 1.5 μL of each cDNA sample, and 10 pmol of each primer. The reaction was run as follows: 95ºC for 7 min, followed by 40 cycles of 95ºC for 15 s, 65ºC for 20 s, and 72ºC for 35 s. The samples were electrophoresed to check the appearance of the PCR products. The CT (threshold cycle) values were analyzed using 2−∆∆CT methods.22 The fold change in gene expression was normalized to an endogenous reference gene (β-actin) using the following primers and expressed relative to the control (Table 2). micPCR software (Bio Molecular Systems, Australia) was used to evaluate the expression stability of the candidate reference gene.
 |
Table 2 Primer Sequence of β-Actin Gene |
BChE Activity Assay
BChE activity was determined using a modified Ellman’s method. Briefly, 100 μL of the supernatant was mixed with 3 mL of sterile 1 × PBS. Then, 100 μL of the resulting solution was transferred into a well of a 96-well culture plate. Butyrylcholinesterase isolated from human plasma (Sigma, USA) was used as a control to build the calibration curve. Then, 100 μL of DTNB (0.28 mmol) and 100 μL of butyrylcholine iodide (3.2 mmol) were introduced into the wells containing the samples and controls and incubated at 37°C for 10 min. The measurements were carried out at a wavelength of 412 nm.23,24
Stability Validation
Transfected cells by pBudCE dual BChE vector were entered in the six freeze-thaw and subculture cycles consistently in order to analyze the stability of the cloning process. Transfection was done as explained before and after 24 h the cells were trypsinized and divided into two parts: frozen and cultured cells. The cultured cells were incubated under standard culture condition for six days, the medium was then collected as the first passage. The frozen cells were thawed the next day and again divided into the same two parts as mentioned above and continued the process until six passages. Finally, the collected medium of the six passages of the cells was analyzed with the modified Ellman’s method.
Freezing Procedure
The cells were trypsinized and transferred to a sterile centrifuge tube, centrifuged at 1000 RPM and 4°C for 3–5 min. The supernatant was removed and the cell pellet was re-suspended by adding 1 mL freezing medium (10% dimethylsulfoxide, and 90% FBS) per vial. The procedure was continued by keeping the cryovial at 4°C for 1 h, −20°C for 2 h, and −80°C overnight. The next day cells were transferred to liquid N2 tank for 15 min and thawed afterward.
Thawing Procedure
Cryovial was removed from liquid N2 tank and hold in 37°C incubator until sides were thawed but the center remained frozen. Warm tissue culture medium (0.5 mL) was added to the vial and after pipetting gently, the cells were poured into a 3 cm plate. The medim was changed after 24 h and 1.5 mL fresh medium was added to the plate and maintained until 6 days.
Statistical Analysis
Data were presented as mean ± SD while experiments were performed in triplicate. SPSS software 20 for Windows (SPSS Inc., Chicago) was utilized for statistical analysis. The univariate test was applied to determine the difference between the groups and Tukey’s multiple range test was used to find the significant difference among means at the probability level of ≤0.05. The nonparametric Kruskal–Wallis test was used to analyze stability data. Microsoft Office Excel 2010 was also used as appropriate software when required.
Results
Cloning Validation
E. coli bacteria were transfected with the recombinant pBudCE dual BChE vector. Colonies of the transformed bacteria were selected by the Zeocin™ (50 μg/mL) supplemented liquid broth (LB) agar medium that confirmed the validity of the cloning and transformation process. Gel electrophoresis results of digestion of the recombinant vectors showed that each digested product had two bands, one for the gene of interest (1860 bp) and the other one for the remaining part of the plasmid (4595 bp) (specific sites: SalI, ScaI and KpnI, XhoI). The cloned plasmid had a 6455 bp band and the control plasmid was 4595 bp (Figure 3).
mRNA Extraction and Gene Expression
Cells were lysed using RiboEx™ reagent and the purified RNA was used for cDNA synthesis. After PCR amplification of the gene with the cDNA, BChE gene expression was assessed using micPCR software (Bio Molecular Systems, Australia). Overall, results showed that the difference between groups was significant at p≤0.05 and the gene expression level in pBudCE dual BChE group was higher than in other groups in all days. Three days after inoculation the gene expression level in the negative control group was significantly lower than the pBudCE dual BChE and positive control (pCMV) groups. The gene expression level in pBudCE dual BChE was higher than the pCMV group, although the increase was not significant. The expression level fold changes were significantly different among all three groups at day six of inoculation, although on the ninth day, only the pBudCE dual BChE group showed a significant increase compared to the negative control group (Figure 5).
 |
Figure 5 Expression levels of the BChE gene in the different experimental groups at different days of inoculation (mean± SD); (*p≤0.05). |
Determining the BChE Enzyme Activity by Modified Ellman’s Method
The culture medium of each group was collected for Ellman’s method. The results showed that the BChE activity in the pBudCE dual BChE group was significantly higher than that of the positive control (pCMV) and negative control groups. The difference of negative control, positive control, and pBudCE dual BChE groups was significant on third and sixth days of inoculation, while on the ninth day only the pBudCE dual BChE group was significantly different to the others. Although the enzyme activity in the pBudCE dual BChE group nonsignificantly decreased on the ninth day of inoculation compared with its level in the third and sixth days, it was higher than what was observed in other groups at any time point (Figure 6).
 |
Figure 6 The BChE enzyme activity in different experimental groups during different days of inoculation. Data are presented as mean±SD with dilution factor = 100 folds; (*p≤0.05). |
Stability of the Transfected Cells
The culture medium of each freeze-thaw cycle (6 cycles) of the transfected cells was harvested after six days in order to analyze the BChE enzyme activity by Ellman’s method. The results showed that there was no significant difference among all passages of the cells (p≤0.05). This means that the cloning process could be stable in the subculturing and freeze-thaw process (Table 3).
 |
Table 3 Stability Examination of the pBudCE Dual BChE Vector in the Transfected Cells During Six Freeze-Thaw Cycles and Passages |
Discussion
Since the BChE enzyme is one of the main treatments in organophosphate toxin (OPT) exposures, and is introduced as a drug in several therapeutic procedures, the aim of our study was to construct an efficient recombinant human BChE expression system to increase enzyme production. It is well documented that CMV and EF-1α promoters are separately potent in recombinant protein production. However, we constructed a new dual-promoter vector to promote the plasmid’s characteristics. The designed dual-promoter vector was capable of increasing the BChE gene expression level as well as the activity of the enzyme. Composition of a plasmid, choosing the best cell line and the most suitable vector are some of the essential factors that influence transgene expression magnitude, stability, and efficiency. Promoters have different ectopic gene expression patterns and potentials in various cell lines. EF-1α and CMV are two well-studied and strong promoters in producing recombinant proteins in mammalian cell lines. They may affect the gene transcription, gene expression, and protein synthesis in the cloning processes.25 CMV is mostly used in many biotechnological studies as a standard plasmid.21 Although CMV is a suitable promoter in terms of expression magnitude and stability, there are reports that with the extended cultivation periods, the productivity decreases by using this promoter.26 It has been reported that DNA methylation results in transcriptional silencing of the CMV promoter. By contrast, several strong promoters like EF-1α have been exploited with the aim of long-term and potent expression of a gene. EF-1α promoter tends to be more stable in long-term culture, even in the absence of selection pressure. It is constitutively active in a broad range of cell types and is able to increase the transgene expression, the transfection efficiency, the copy number of the vector, and the proportion of expression-positive clones.27 The EF-1a promoter is often active in cells in which viral promoters failed to express the controlled genes or are gradually silenced.28 In 2012 an expression system was designed comparing CMV and EF promoters separately to express the butyrylcholinesterase and the proline-rich region of the ColQ gene simultaneously by using two plasmids24 or one plasmid29 in CHO cells. Their results demonstrated that the EF promoter produced higher levels of the active form of the BChE gene compared to the CMV promoter. Gene expression results in our study showed that the pCMV and pBUD dual BChE plasmids had comparable levels and they both could enhance the expression of the BChE gene significantly compared to the control; however, pBUD dual BChE plasmid showed higher levels. Although the pCMV vector increased the BChE gene expression, its level was significantly different from the control just at day three and six of inoculation. Meanwhile, the level of gene expression in new construction (pBUD dual BChE plasmid) was significantly higher at all three times of measurement which may be the effect of EF promoter. Sinae Kim and colleagues in 2007 evaluated the efficacy of promoters in the lentiviral gene delivery system in some mammalian ES cells to express EGFP. They used CMV and EF-1α promoters, and, similarly to our results, they showed that the EF-1α promoter had higher expression than that of the CMV promoter in all ES cells tested.30 Research investigating the importance of promoters for the generation of stable colon carcinoma cell lines showed that the level of stable clones generated with EF1-α promoter was much more than clones generated with CMV promoter.31 Another study in 2017 comparing various promoters to improve transgene expression, transgene stability, and copy number showed that the EF1-α promoter is a more potent promoter because compared to the other promoters including CMV,27 which their results were comparable to ours that indicates; besides the effect of the presence of two copies of the gene in a single plasmid, EF-1α promoter could play an important role in the protein production.
The HEK-293 cell line was selected as a candidate host in our study for BChE recombinant protein expression system. CHO cell lines are being used in many investigations as an effective transcription system while our results showed that HEK-293 could also be efficient as a cloning host. Epithelial origin and the biochemical machinery of the HEK-293 cell line make it capable to carry out most of the post-translational folding and processings which are required to generate functional and mature proteins from a wide range of both mammalian and non-mammalian nucleic acids. This cell line has many characteristics that make it appropriate for gene expression, eg, easy reproduction, high efficiency of transfection and protein production, and faithful translation and processing of proteins.20 Chilukuri et al investigated an AdenoVATOR system containing recombinant human butyrylcholinesterase in the Hek-293 cell line. Their results showed that most rHu BChE expressed in these cells was in tetrameric form by the co-expression of the proline-rich attachment domain.32
Many studies tried to produce recombinant BChE with several different systems, eg, transgenic animals,33 transgenic plants,25,34 and the CHO cell line.35 Although some of these studies reported some degrees of success, large quantity production of the BChE enzyme is a major limitation so far.33,34,36,37 BChE enzyme activity was measured by a modified Ellman’s method in the present study. Results showed BChE enzyme activity among pCMV, control and pBUD dual BChE groups was significantly different in all three periods of inoculation, and pBUD dual BChE group had the highest production level. Thus, the pBudCE dual BChE vector containing two copies of the BChE gene under the control of the EF-1α and CMV promoters was the most promising construct. On the third day of inoculation, the activity level in the pCMV group was lower than the control group, while it increased in subsequent days. This may be due to the fact that the optimum function of this vector may begin from four days after transfection, as other studies mentioned.31 The most functional time for enzyme production with new construction is six days after transfection as the level of the enzyme activity decreased on day nine, although the difference was not significant. Nevertheless, the minimum level of the enzyme in the pBudCE dual BChE group at day nine was higher than the maximum level of the other groups.
The inoculation date did not significantly alter the results, although it is obvious that in both gene expression and enzyme activity, the highest level was observed six days after transfection. This result was comparable to the results of Jasmine et al that acclaimed the maximum expression level of the BChE gene is around day five of transfection.13 For this reason, this time (six days after inoculation) was selected for the stability test.
An inevitable factor in recombinant protein production is the durability and stability of the introduced method or the new construction. The production needs to be continued after some freeze-thaw or passages to become usable in a period of time. In order to examine the transient stability of the designed structure, the transfected cells with pBudCE dual BChE vector were entered into several freeze-thaw cycles. The transfected HEK-293 cells proved to be a stable BChE producer in six freeze-thaw cycles. Neither several generations nor the freeze-thaw process altered the BChE enzyme activity level, confirming the stability of the constructed vector.24
Conclusion
We can conclude that the designed construction containing EF-1α and CMV promoters is a more efficient system than a single-promoter vector in protein and enzyme production. However, co-expression of the BChE gene and PRAD needs to be evaluated to achieve a high level of the tetrameric form of the enzyme. The HEK-293 cell line is an appropriate host in the cloning process that remains stable in several freeze-thaw and subculture procedures. Other cell lines than HEK-293 need to be evaluated in order to choose the most appropriate host for higher production of BChE.
Acknowledgments
This work was a part of the PhD thesis that was submitted under the ethics approval code: IR.KMU.REC.1398.294 in the Kerman University of Medical Sciences (KMU). M. Abolhassani from the department of clinical biochemistry at Afzalipour School of Medicine is acknowledged for his warm support.
Disclosure
The authors declare no conflict of interests for this work.
References
1. Çokuğraş AN. Butyrylcholinesterase: structure and physiological importance. Turk J Biochem. 2003;28(2):54–61.
2. Lockridge O, Bartels CF, Vaughan TA, Wong CK, Norton SE, Johnson LL. Complete amino acid sequence of human serum cholinesterase. J Biol Chem. 1987;262(2):549–557.
3. Østergaard D, Viby–Mogensen J, Hanel H, Skovgaard L. Half–life of plasma cholinesterase. Acta Anaesthesiol Scand. 1988;32(3):266–269. doi:10.1111/j.1399-6576.1988.tb02727.x
4. Duysen EG, Bartels CF, Lockridge O. Wild-type and A328W mutant human butyrylcholinesterase tetramers expressed in Chinese hamster ovary cells have a 16-hour half-life in the circulation and protect mice from cocaine toxicity. J Pharmacol Exp Ther. 2002;302(2):751–758. doi:10.1124/jpet.102.033746
5. Bosak A, Knežević A, Gazić Smilović I, Šinko G, Kovarik Z. Resorcinol-, catechol-and saligenin-based bronchodilating β2-agonists as inhibitors of human cholinesterase activity. J Enzyme Inhib Med Chem. 2017;32(1):789–797. doi:10.1080/14756366.2017.1326109
6. Ashani Y. Prospective of human butyrylcholinesterase as a detoxifying antidote and potential regulator of controlled‐release drugs. Drug Dev Res. 2000;50(3‐4):298–308. doi:10.1002/1098-2299(200007/08)50:3/4<298::AID-DDR13>3.0.CO;2-X
7. Doctor BP, Saxena A. Bioscavengers for the protection of humans against organophosphate toxicity. Chem Biol Interact. 2005;157–158:167–171. doi:10.1016/j.cbi.2005.10.024
8. Eddleston M, Buckley NA, Eyer P, Dawson AH. Management of acute organophosphorus pesticide poisoning. Lancet. 2008;371(9612):597–607. doi:10.1016/S0140-6736(07)61202-1
9. Gupta RD, Goldsmith M, Ashani Y, et al. Directed evolution of hydrolases for prevention of G-type nerve agent intoxication. Nat Chem Biol. 2011;7(2):120. doi:10.1038/nchembio.510
10. Cerasoli D, Griffiths E, Doctor B, et al. (07) in vitro and in vivo characterization of recombinant human butyrylcholinesterase (Protexia™) as a potential nerve agent bioscavenger. Chem Biol Interact. 2005;157–158:362–365. doi:10.1016/j.cbi.2005.10.052
11. Lenz DE, Yeung D, Smith JR, Sweeney RE, Lumley LA, Cerasoli DM. Stoichiometric and catalytic scavengers as protection against nerve agent toxicity: a mini review. Toxicology. 2007;233(1–3):31–39. doi:10.1016/j.tox.2006.11.066
12. Ashani Y, Pistinner S. Estimation of the upper limit of human butyrylcholinesterase dose required for protection against organophosphates toxicity: a mathematically based toxicokinetic model. Toxicol Sci. 2004;77(2):358–367. doi:10.1093/toxsci/kfh012
13. Lockridge O, Schopfer LM, Winger G, Woods JH. Large scale purification of butyrylcholinesterase from human plasma suitable for injection into monkeys; a potential new therapeutic for protection against cocaine and nerve agent toxicity. J Med Chem Biol Radiol Def. 2005;3:nihms5095.
14. Corbin JM, Hashimoto BI, Karuppanan K, et al. Semicontinuous bioreactor production of recombinant butyrylcholinesterase in transgenic rice cell suspension cultures. Front Plant Sci. 2016;7:412. doi:10.3389/fpls.2016.00412
15. Griffiths AJ, Wessler SR, Lewontin RC, Gelbart WM, Suzuki DT, Miller JH. An Introduction to Genetic Analysis. Macmillan; 2005.
16. Khan S, Ullah MW, Siddique R, et al. Role of recombinant DNA technology to improve life. Int J Genomics. 2016;2016.
17. Masson P, Adkins S, Pham-Trong P, Lockridge O. Expression and refolding of functional human butyrylcholinesterase from E. Coli. In: Shafferman A., Velan B. (eds) Multidisciplinary Approaches to Cholinesterase Functions. Springer; 1992:49–52.
18. Brazzolotto X, Igert A, Guillon V, Santoni G, Nachon F. Bacterial expression of human butyrylcholinesterase as a tool for nerve agent bioscavengers development. Molecules. 2017;22(11):1828. doi:10.3390/molecules22111828
19. Rosano GL, Ceccarelli EA. Recombinant protein expression in Escherichia coli: advances and challenges. Front Microbiol. 2014;5:172. doi:10.3389/fmicb.2014.00172
20. Thomas P, Smart TG. HEK293 cell line: a vehicle for the expression of recombinant proteins. J Pharmacol Toxicol Methods. 2005;51(3):187–200. doi:10.1016/j.vascn.2004.08.014
21. Qin JY, Zhang L, Clift KL, et al. Systematic comparison of constitutive promoters and the doxycycline-inducible promoter. PLoS One. 2010;5(5):e10611. doi:10.1371/journal.pone.0010611
22. Eslaminejad T, Noureddin Nematollahi-Mahani S, Ansari M. Glioblastoma targeted gene therapy based on pEGFP/p53-loaded superparamagnetic iron oxide nanoparticles. Curr Gene Ther. 2017;17(1):59–69. doi:10.2174/1566523217666170605115829
23. Ellman GL, Courtney KD, Andres Jr V, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7(2):88–95. doi:10.1016/0006-2952(61)90145-9
24. Ilyushin D, Haertley O, Bobik T, et al. Recombinant human butyrylcholinesterase as a new-age bioscavenger drug: development of the expression system. Acta Naturae. 2013;5(1):16.
25. Schneider JD, Castilho A, Neumann L, et al. Expression of human butyrylcholinesterase with an engineered glycosylation profile resembling the plasma‐derived orthologue. Biotechnol J. 2014a;9(4):501–510. doi:10.1002/biot.201300229
26. He L, Winterrowd C, Kadura I, Frye C. Transgene copy number distribution profiles in recombinant CHO cell lines revealed by single cell analyses. Biotechnol Bioeng. 2012;109(7):1713–1722. doi:10.1002/bit.24428
27. Wang X, Xu Z, Tian Z, et al. The EF‐1α promoter maintains high‐level transgene expression from episomal vectors in transfected CHO‐K1 cells. J Cell Mol Med. 2017;21(11):3044–3054. doi:10.1111/jcmm.13216
28. Norrman K, Fischer Y, Bonnamy B, Sand FW, Ravassard P, Semb H. Quantitative comparison of constitutive promoters in human ES cells. PLoS One. 2010;5(8):e12413. doi:10.1371/journal.pone.0012413
29. Ilyushin DG, Smirnov IV, Belogurov AA, et al. Chemical polysialylation of human recombinant butyrylcholinesterase delivers a long-acting bioscavenger for nerve agents in vivo. Proc Natl Acad Sci. 2013;110(4):1243–1248. doi:10.1073/pnas.1211118110
30. Kim S, Kim GJ, Miyoshi H, et al. Efficiency of the elongation factor-1α promoter in mammalian embryonic stem cells using lentiviral gene delivery systems. Stem Cells Dev. 2007;16(4):537–546. doi:10.1089/scd.2006.0088
31. Teschendorf C, Warrington JK, Siemann DW, Muzyczka N. Comparison of the EF-1 alpha and the CMV promoter for engineering stable tumor cell lines using recombinant adeno-associated virus. Anticancer Res. 2002;22(6A):3325–3330.
32. Chilukuri N, Parikh K, Sun W, et al. Polyethylene glycosylation prolongs the circulatory stability of recombinant human butyrylcholinesterase. Chem Biol Interact. 2005;157–158:115–121. doi:10.1016/j.cbi.2005.10.013
33. Huang Y-J, Huang Y, Baldassarre H, et al. Recombinant human butyrylcholinesterase from milk of transgenic animals to protect against organophosphate poisoning. Proc Natl Acad Sci. 2007;104(34):13603–13608. doi:10.1073/pnas.0702756104
34. Geyer BC, Kannan L, Cherni I, Woods RR, Soreq H, Mor TS. Transgenic plants as a source for the bioscavenging enzyme, human butyrylcholinesterase. Plant Biotechnol J. 2010;8(8):873–886. doi:10.1111/j.1467-7652.2010.00515.x
35. Terekhov S, Smirnov I, Bobik T, et al. A novel expression cassette delivers efficient production of exclusively tetrameric human butyrylcholinesterase with improved pharmacokinetics for protection against organophosphate poisoning. Biochimie. 2015;118:51–59. doi:10.1016/j.biochi.2015.07.028
36. Brazzolotto X, Wandhammer M, Ronco C, et al. Human butyrylcholinesterase produced in insect cells: huprine‐based affinity purification and crystal structure. FEBS J. 2012;279(16):2905–2916. doi:10.1111/j.1742-4658.2012.08672.x
37. Schneider JD, Marillonnet S, Castilho A, et al. Oligomerization status influences subcellular deposition and glycosylation of recombinant butyrylcholinesterase in N icotiana benthamiana. Plant Biotechnol J. 2014b;12(7):832–839. doi:10.1111/pbi.12184
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.