Back to Journals » International Journal of Nanomedicine » Volume 12
Enhanced oral bioavailability of valsartan using a polymer-based supersaturable self-microemulsifying drug delivery system
Authors Yeom DW, Chae BR, Son HY, Kim JH, Chae JS, Song SH ![]() , Oh D, Choi YW
, Oh D, Choi YW ![]()
Received 9 March 2017
Accepted for publication 11 April 2017
Published 8 May 2017 Volume 2017:12 Pages 3533—3545
DOI https://doi.org/10.2147/IJN.S136599
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Dong Woo Yeom,1,* Bo Ram Chae,2,* Ho Yong Son,1 Jin Han Kim,1 Jun Soo Chae,1 Seh Hyon Song,2 Dongho Oh,2 Young Wook Choi1
1College of Pharmacy, Chung-Ang University, Seoul, 2Daewon Pharm. Co., Ltd, Seoul, Republic of Korea
*These authors contributed equally to this work
Abstract: A novel, supersaturable self-microemulsifying drug delivery system (S-SMEDDS) was successfully formulated to enhance the dissolution and oral absorption of valsartan (VST), a poorly water-soluble drug, while reducing the total quantity for administration. Poloxamer 407 is a selectable, supersaturating agent for VST-containing SMEDDS composed of 10% Capmul® MCM, 45% Tween® 20, and 45% Transcutol® P. The amounts of SMEDDS and Poloxamer 407 were chosen as formulation variables for a 3-level factorial design. Further optimization was established by weighting different levels of importance on response variables for dissolution and total quantity, resulting in an optimal S-SMEDDS in large quantity (S-SMEDDS_LQ; 352 mg in total) and S-SMEDDS in reduced quantity (S-SMEDDS_RQ; 144.6 mg in total). Good agreement was observed between predicted and experimental values for response variables. Consequently, compared with VST powder or suspension and SMEDDS, both S-SMEDDS_LQ and S-SMEDDS_RQ showed excellent in vitro dissolution and in vivo oral bioavailability in rats. The magnitude of dissolution and absorption-enhancing capacities using quantity-based comparisons was in the order S-SMEDDS_RQ > S-SMEDDS_LQ > SMEDDS > VST powder or suspension. Thus, we concluded that, in terms of developing an effective SMEDDS preparation with minimal total quantity, S-SMEDDS_RQ is a promising candidate.
Keywords: valsartan, SMEDDS, supersaturation, factorial design, optimization, bioavailability
Introduction
Valsartan (VST), a highly selective antagonist of the angiotensin II receptor, has been widely used to treat high blood pressure and congestive heart failure.1 It is a poorly water-soluble (3.08 μg/mL) and highly permeable drug, belonging to class II of the biopharmaceutical classification system (BCS).1 After oral administration of a capsule (VST 80 mg), absolute bioavailability (BA) of VST was relatively low at ~23%, which might be due to the limited solubility in water and acidic pH of the stomach.2 In order to enhance the solubility and oral absorption of VST, various formulation approaches, such as solid dispersion, spray-dried emulsion, spray-dried nanosuspension, and supercritical antisolvent process, have been studied.3–6
Self-microemulsifying drug delivery systems (SMEDDS) are widely known to overcome low solubility and poor oral absorption of water-insoluble drugs. In the gastrointestinal (GI) tract, they can form oil-in-water (o/w) microemulsions spontaneously, thus allowing rapid dissolution of drugs and enhancing permeation of the drugs across the intestinal membrane.7 In addition, a formed o/w microemulsion could be delivered by lymphatic transport to avoid first-pass metabolism in the liver, thus improving BA of the drug.8 Nevertheless, SMEDDS have several shortcomings for practical drug product development. First, the volume of solubilizing vehicle, including oil and surfactants, is generally too large to accommodate the usual dose of the drug in a unit dosage form. Studies indicated that high amounts of SMEDDS (800–1,920 mg) were required to solubilize VST 80 mg, a recommended unit dose, although SMEDDS increased the oral BA 2- or 3-fold compared with VST powder and/or commercial product.1,2,9–11 Regarding patient compliance, the large quantity of SMEDDS required makes fabrication of an appropriate drug dosage into a soft gelatin capsule impractical. Thus, reducing the volume is one of the major concerns for developing a SMEDDS-based drug product. Moreover, with repetitive dosing, severe GI irritation could be induced due to the high content of surfactant (~60%) in SMEDDS formulations.12
The concept of supersaturation has been proposed to overcome these limitations of SMEDDS formulations.12,13 If the volume of the solubilizing vehicle is reduced to a certain magnitude, drugs are prone to precipitate. Thus, several supersaturating agents, including a polymeric precipitation inhibitor and an auxiliary surfactant or cosolvent, have been employed to stabilize the drug in a temporary, supersaturated state of solubilization.12,14 The supersaturating agent could improve the solubility of drugs in SMEDDS and stabilize the micelle structure by mingling with the existing surfactant.13,15 The polymeric precipitation inhibitors thermodynamically and/or kinetically prolong the supersaturated state of active molecules by reducing the rate of drug nucleation and crystal growth through physical interaction with the drugs or by changing the medium properties.16 Simultaneously, polymeric inhibitors also synergize to enhance the initial drug dissolution and protract the supersaturated state of the drug in aqueous medium.13,17
The present study was performed to develop a novel, supersaturable SMEDDS (S-SMEDDS) for improving dissolution and oral absorption of VST, while minimizing the total volume of the preparation. The compositions of SMEDDS were determined by physical characterization and in vitro dissolution. The S-SMEDDS were formulated by screening various types of hydrophilic and/or amphiphilic polymers. Both, to minimize the total volume of S-SMEDDS and to maximize the oral absorption of VST, we used a 3-level factorial design (3-LFD) with a smaller number of trials, and statistical optimization based on response surface methodology. Subsequently, in vivo BA of VST following oral administration of the optimized S-SMEDDS in rats was compared with that of drug suspension and SMEDDS. Finally, in vitro–in vivo correlations (IVIVCs) were evaluated.
Materials and methods
Materials
VST was supplied by Daewon Pharm. Co. Ltd. (Seoul, Republic of Korea). Capryol™ 90, Lauroglycol™ 90, Labrasol®, and Transcutol® P were supplied by Gattefossé (Saint Priest, France). Capmul® MCM was supplied by Abitec Co. (Janesville, WI, USA). Cremophor® EL (Kolliphor® EL), polyvinylpyrrolidone K90 (PVP K90; Kollidon® K90), d-α-tocopheryl polyethylene glycol 1000 succinate (Vitamin E TPGS; Kolliphor® TPGS), poloxamer 407 (Pluronic® F-127; POL), and Soluplus® were supplied by BASF (Ludwigshafen, Germany). Hypromellose E2910 (hydroxypropyl methylcellulose, [HPMC]) was supplied by Shin-Etsu Chemical Co. (Tokyo, Japan). Olive oil, corn oil, Tween® 20, Tween 80, and tetraglycol were purchased from Sigma-Aldrich (St Louis, MO, USA). Polyethylene glycol and polyethylene glycol 6000 PEG 6000) were purchased from Duksan Pure Chemical Co. Ltd. (Ansan, South Korea), and high-performance liquid chromatography (HPLC)-grade acetonitrile and methanol were purchased from JT Baker (Phillipsburg, NJ, USA). All other chemicals used were of analytical grade.
Animals
Male Sprague–Dawley rats (200–250 g, 7–9 weeks old) were purchased from Orient Bio (Kyungki-Do, Republic of Korea). The rats were fasted for ~12–18 h prior to drug administration, with free access to water. All animal experiments were performed in accordance with the National Institute of Health guidelines “Principles of laboratory animal care” and were approved by the Institutional Animal Care and Use Committee of Chung-Ang University, Seoul, Republic of Korea.
Solubility study
The solubility of VST in various excipients was determined using the equilibrium method. An excess amount of VST was added to 1 mL of selected excipients. Test tubes containing the mixtures were sealed and kept in ambient conditions with intermittent shaking (Cute Mixer CM-1000; Eyela, Tokyo, Japan) for 24 h to achieve equilibrium. The mixtures were then centrifuged (Micro 17TR; Hanil Science, Incheon, Republic of Korea) at 16,000 × g for 10 min to remove excess VST. The supernatant was filtered through a 0.45 μm polyvinylidene difluoride syringe filter (Whatman GmbH, Dassel, Germany), and the concentration of VST in the filtrate was measured using HPLC after appropriate dilution with methanol.
HPLC analysis of VST
The concentration of VST was determined using HPLC. The HPLC system included a pump (W2690/5; Waters Corporation, Milford, MA, USA), ultraviolet detector (W2489; Waters Corporation), data station (Empower 3; Waters Corporation), and chromatographic C18 column (250×4.6 mm, 5 μm; Shiseido, Tokyo, Japan) that was maintained at a flow rate of 1.0 mL per min at 25°C. Isocratic mobile phase included acetonitrile and distilled water (DW) (60:40 [v/v]). The pH was adjusted to 3.0 using 10% phosphoric acid. Finally, 20 μL of each sample was injected into the column, and VST concentration was measured with ultraviolet detection at 247 nm.
Construction of pseudo-ternary phase diagram
The boundaries of the microemulsion domains were determined with the aid of a pseudoternary phase diagram. The phase diagram of oil, surfactant/cosurfactant (S/CoS), and water was constructed using a water titration method in the drug-free condition. Based on the results of the solubility test, Capmul MCM, Tween 20, and Transcutol P were selected as the oil, surfactant, and cosurfactant, respectively. While the blend ratios of S/CoS (Km) were maintained as 1:2 (0.5), 1:1 (1), and 2:1 (2) (v/v), respectively, the proportion of oil in the mixture with the S/CoS blend was varied from 9:1 to 1:9. Water was added dropwise, under magnetic stirring at 25°C, to the oily mixture. Following the addition of an aliquot of the water phase, the mixture was visually examined for transparency. Regions that were transparent and/or bluish white were microemulsions.
Determination of droplet size
A photon correlation spectrometer (Zetasizer Nano ZS; Malvern Instruments, Malvern, UK) was employed to determine the size of the emulsion droplets. An aliquot of test formulation (10 μL) was added to 10 mL of DW and then gently stirred to obtain a homogenous dispersion. The samples were loaded into a cuvette placed in a thermostatic chamber, and light scattering was monitored at a 90° angle at 25°C.
Preparation of VST-loaded SMEDDS and S-SMEDDS
Based on the phase diagram, the composition of 10% Capmul MCM, 45% Tween 20, and 45% Transcutol P was selected as the blank SMEDDS formulation. VST-loaded SMEDDS was prepared by adding 80 mg VST to different quantities of the blank SMEDDS (200–600 mg). The components were mixed by vortexing at 25°C until the VST was completely dissolved. Separately, to prepare S-SMEDDS, several supersaturating agents (5% [w/w]) were added to the SMEDDS. VST dissolution profiles were compared to select a good supersaturating agent, and the selected S-SMEDDS was subjected to further optimization.
Optimization of VST-loaded S-SMEDDS using 3-LFD
The 3-LFD was used to optimize the composition of the S-SMEDDS formulation to minimize the quantity of S-SMEDDS and to maximize the drug release. Design-Expert Software version 7 (Stat-Ease Inc, Minneapolis, MN, USA) was used for developing and evaluating the experimental design. The experiment was designed using the two components as independent variables. Based on the results of the maximum solubility of SMEDDS and the dissolution test, the amount of SMEDDS (X1) was set from 100 to 400 mg, and the content of Poloxamer 407 (POL) (weight percentage of SMEDDS; X2) as a supersaturating agent was set from 1% to 10%. Mean droplet size (nm; Y1), percentage of drug released in 15 min (%; Y2), and the total quantity of S-SMEDDS (mg; Y3) were evaluated as response variables to determine the optimal formulation with excellent physiochemical characteristics.
In vitro dissolution test
In vitro dissolution tests were performed using the USP apparatus II (paddle) method with Vision® Classic 6™ Dissolution Tester and Vision heater (Hanson Research, Chatsworth, CA, USA). A pH 1.2 medium was prepared by dissolving 2 g of sodium chloride in 7 mL of hydrochloric acid and diluting with DW to 1,000 mL. A pH 6.8 medium was prepared by mixing 250 mL of 0.2 M potassium phosphate monobasic solution and 118 mL of 0.2 M sodium hydroxide solution and diluting with DW to 1,000 mL. Each formulation, containing 80 mg of VST, was introduced into the pH 1.2 or 6.8 dissolution media (500 mL) at 37°C±0.5°C, and stirred at 50 or 125 rpm. Samples (5 mL) were taken at predetermined sampling points (5, 15, 30, 60, 90, and 120 min) and filtered through a 0.45-μm polyvinylidene difluoride membrane. After appropriate dilution of the filtrate with methanol, the concentration of VST in each sample was assayed by HPLC.
In vivo oral absorption study
Oral administration and plasma sampling
After overnight fasting of the rats for 12–18 h, test samples were administered via oral gavage, as reported elsewhere.3,18 The rats were randomly divided into four groups (n=5–7): group 1 received VST suspension (reference), group 2 received SMEDDS (320 mg), group 3 received S-SMEDDS in large quantity (S-SMEDDS_LQ) containing SMEDDS 320 mg equivalent, and group 4 received S-SMEDDS in reduced quantity (S-SMEDDS_RQ) containing SMEDDS 131.5 mg equivalent. In all treatments, a dose of 10 mg/kg equivalent of VST was accurately weighed and diluted with 1 mL DW prior to administration. ~0.3 mL blood samples were collected from the retro-orbital plexus into heparinized tubes at predetermined time points (0.33, 0.66, 1, 1.5, 2, 4, 7, 12, and 24 h) and were centrifuged at 16,000 × g for 15 min. Plasma samples were stored at −80°C until analysis by liquid chromatography-tandem mass spectrometry (LC-MS/MS).
Whole plasma samples (50 μL) were mixed with 700 μL of methanol and 20 μL of internal standard (IS) solution (10,000 ng/mL VST-d3 in 50% methanol) and were vortexed for 3 min. After centrifugation at 16,000 × g for 5 min, 20 μL of the supernatant was carefully transferred to a test tube and was evaporated to dryness under nitrogen. The dried residue was reconstituted in 480 μL of DW, and the mixture was vortexed and centrifuged at 16,000 × g for 5 min. Finally, 100 μL of the supernatant was transferred to autosampling vials for introduction into the LC-MS/MS system.
Determination of VST in plasma samples by LC-MS/MS
Liquid chromatographic separation was performed using an Agilent 1260 autosampler (Agilent Technologies Inc, Santa Clara, CA, USA). The temperature of the autosampler was maintained at 7°C, and 5 μL components of each reconstituted sample were separated using a Waters Atlantis dC18 column (50×2.1 mm, 3 μm; Milford, MA, USA) at 35°C. An isocratic mobile phase, containing 10 mM ammonium formate (pH 2.7) and methanol (20:80, [v/v]), was used at a flow rate of 0.3 mL per min.
The components eluted from the column were delivered into an API 4500 triple quadrupole mass spectrometer (Applied Biosystems/MDS SCIEX, Foster City, CA, USA) with electrospray ionization in positive ion mode for ion production. The ion spray voltage was set at 5.5 kV, and the source temperature was set at 550°C. Multiple reaction monitoring was performed using nitrogen as the collision gas. The analytes were detected by monitoring the transitions 436.2 (Q1)291.0 (Q3) and 439.2 (Q1)294.0 (Q3) m/z, with a declustering potential of 28 V and collision energies of 23 V, for VST and IS, respectively. Other mass spectrometer conditions were as follows: nebulizer gas (gas 1) at 40°C and heater gas (gas 2) at 70°C. For quantifying VST in the plasma samples, each peak area of VST was divided by that of the IS, and the ratio was compared with a calibration curve obtained using VST standard solution in the same manner.
Pharmacokinetic assessment
Data analysis was performed using the BA Calc 2007 pharmacokinetic analysis program (Ministry of Food and Drug Safety [formerly Korea Food and Drug Administration], Chungcheongbuk-do, Korea). Area under the curve (AUC) from 0 to 24 h was calculated using the linear trapezoidal rule. Maximum plasma concentration (Cmax) and the time to reach the maximum plasma concentration (Tmax) were determined directly from the concentration–time data. Relative BA (RBA) was calculated by dividing the AUCs of the test samples with those of the VST suspension.
Statistical analysis
All data are expressed as mean ± standard deviation (SD). Statistical significance was determined using Student’s t-test, with P<0.05 considered statistically significant. Design-Expert software was used to determine the simultaneously assigned statistical values of all responses.
Results and discussion
Preparation and characterization of SMEDDS
To develop a nanodispersion system with the minimal amount of SMEDDS to solubilize the unit dose of VST (80 mg), suitable components that show high capacity to solubilize VST need to be selected. As shown in Table 1, Capmul MCM, Tween 20, and Transcutol P showed the highest capacity among the oils, surfactants, and cosurfactants, respectively. They possessed high solubilizing potential for VST at >200 mg/mL. In addition, because this combination has shown excellent self-emulsification efficiency in an earlier study, it was selected as SMEDDS formulation.19
 | Table 1 Solubility of VST in selected excipients |
Pseudoternary phase diagrams composed of water, Capuml MCM (oil), and mixed surfactants of Tween 20 (S) and Transcutol P (CoS) were constructed to determine the appropriate ratio of S/CoS by macroscopic observation of phase behavior. As shown in Figure 1, the light gray area represented the self-microemulsifying region with a clear and/or bluish white appearance, when the dispersion time was measured within 1 min. The self-microemulsifying region that occurred with an S/CoS ratio of 1:1 was quite broad compared with others. However, the microemulsion area varied considerably as the ratio of oil changed. To determine the best SMEDDS formulation, the effect of the percentage of oil in the SMEDDS formulation (S/CoS =1) was observed by evaluating the droplet size and polydispersity index (PDI). The particle size increased as the content of oil increased (Figure 2). SMEDDS formulation containing 10% oil had small droplet sizes (<200 nm) and low PDI (<0.4), thus were selected for further experiments. The droplet size and PDI are crucial factors for determining self-microemulsification performance to enhance oral absorption of poorly water-soluble drugs. A small droplet size provides an increased interfacial surface area and allows faster drug release. A narrow and unimodal size distribution of microemulsion is preferable for reproducible BA.10
 | Figure 1 Pseudoternary phase diagram of Capmul® MCM (oil), Tween® 20 (surfactant), and Transcutol® P (cosurfactant). |
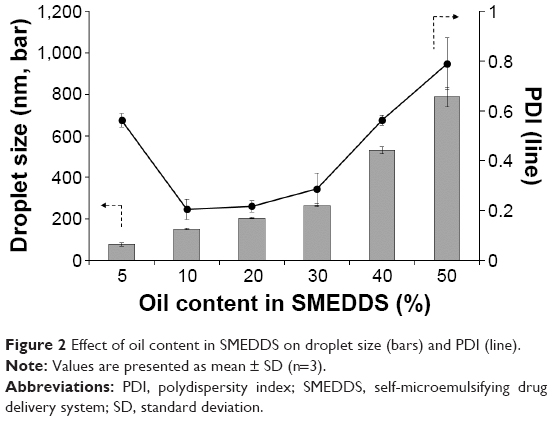 | Figure 2 Effect of oil content in SMEDDS on droplet size (bars) and PDI (line). |
To find a suitable quantity of SMEDDS, dissolution profiles were compared based on the amount of SMEDDS added (Figure 3A). As the amount of SMEDDS to load VST 80 mg increased, the cumulative percentage of drug released increased. The lower quantity of SMEDDS (200 mg) showed a limited dissolution of <10%, but the doubled quantity of SMEDDS (400 mg) increased the dissolution to ~50%. With a greater quantity of SMEDDS (500–600 mg), VST dissolution was increased, showing a mild oversaturation effect: increase to over 65% (500 mg) or 80% (600 mg) at 15 min, but gradually decreasing to below 60% (500 mg) or 70% (600 mg) at 2 h. For further comparison, the dissolution enhancing capacity per unit quantity (DEC) was calculated as follows: DEC = (DESMEDDS − DEVST)/VSMEDDS, where DESMEDDS and DEVST are the dissolution efficiency of SMEDDS and VST, respectively, and VSMEDDS is the amount (g) of SMEDDS formulation. Dissolution efficiency (DE) was calculated by the trapezoidal rule as follows: 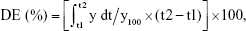 where y is the percentage of dissolved product.20,21 As shown in Figure 3B, DEC proportionally increased up to 400 mg SMEDDS, but reached a plateau afterward. Therefore, we decided the maximum quantity of SMEDDS to be 400 mg for further studies.
where y is the percentage of dissolved product.20,21 As shown in Figure 3B, DEC proportionally increased up to 400 mg SMEDDS, but reached a plateau afterward. Therefore, we decided the maximum quantity of SMEDDS to be 400 mg for further studies.
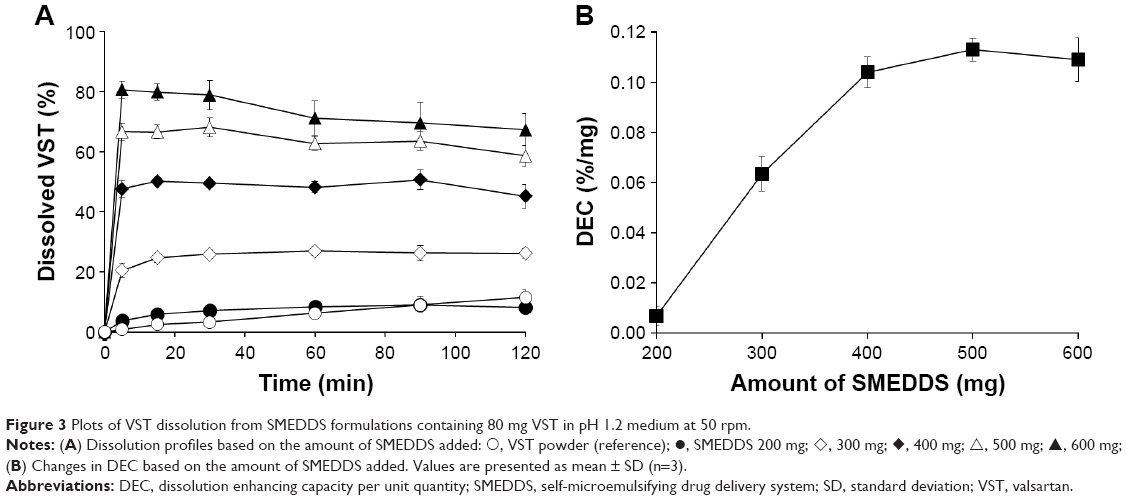 | Figure 3 Plots of VST dissolution from SMEDDS formulations containing 80 mg VST in pH 1.2 medium at 50 rpm. |
Screening of supersaturating agents
To potentiate the degree and duration of enhanced solubility of VST, various hydrophilic and amphiphilic polymers were screened as supersaturating agents. Based on VST solubility in 1% (w/v) aqueous polymer solutions (Table 1), amphiphilic polymers produced greater solubility than hydrophilic polymers did. Hydrophilic polymers could induce and maintain a supersaturated drug solution by inhibiting nucleation and crystal growth through steric stabilization, and/or specific interactions with the drug, such as hydrogen bonding, hydrophobic interactions, and complex formation.17,22 PVP, PEG, and HPMC have been found to be effective drug precipitation inhibitors in various formulations, including lipid-based formulations.23–26 Meanwhile, amphiphilic polymers have been widely used not only to prevent drug precipitation but also to act as surfactants for solubilization via micelle formation. Poloxamers, Vitamin E TPGS, and Soluplus® have been incorporated into aqueous formulations to increase solubility and improve dissolution and BA of poorly water-soluble drugs.21,27,28 They are usually used above the critical micelle concentration (CMC) to solubilize drugs by micelle formation or below the CMC to inhibit drug precipitation.12,29
The supersaturating agents were added at 5% (w/w) to the VST (80 mg)-containing SMEDDS (400 mg) to prepare S-SMEDDS, and the dissolution profiles were compared to select a good supersaturating agent, which could increase the high-energy form of the drug molecule in the solution. As shown in Figure 4, VST dissolution was significantly increased by adding supersaturating agents, although the enhancing effect was dependent on the type of polymer used. Except for Soluplus, amphiphilic polymer-added S-SMEDDS formulations showed significantly higher dissolution than hydrophilic polymer-added S-SMEDDS did. Unlike the structure of hydrophilic polymers, amphiphilic polymers have both hydrophilic and hydrophobic groups in their structure, possessing the potential to act like surfactants. Hydrophobic tails with large molecular weights carry high transfer free energies to intermingle with surfactants that constitute the SMEDDS droplets, subsequently stabilizing the micelle structure.13 The free energy to solvate small hydrophobic molecules correlates linearly with solute volume, whereas that to solvate large hydrophobic species correlates linearly with surface area.30 The most effective amphiphilic polymer was POL, followed by Vitamin E TPGS, and then Soluplus, which corresponds with the solubility test results. Maximizing the solubilization should be accomplished in such a way that the solvent capacity of S-SMEDDS upon aqueous dilution is sufficient to keep the full dose of poorly water-soluble drug in solution.13 Thus, POL was used as the selective supersaturating agent for further optimization studies.
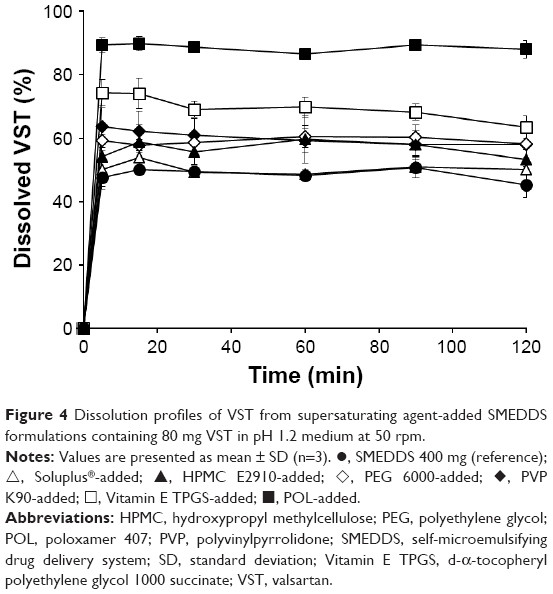 | Figure 4 Dissolution profiles of VST from supersaturating agent-added SMEDDS formulations containing 80 mg VST in pH 1.2 medium at 50 rpm. |
Experimental design of S-SMEDDS using 3-LFD
The 3-LFD was applied to find an optimal composition of S-SMEDDS that minimized the quantity of S-SMEDDS and maximized the oral absorption of VST. The quantity of SMEDDS (mg; X1) and the content of POL (wt % of SMEDDS; X2) were chosen as independent variables, as listed in Table 2. The mean droplet size (nm; Y1) and the percentage of drug released in 15 min (%; Y2) were introduced as response variables that play crucial roles in determining self-emulsification to improve oral absorption of poorly water-soluble drugs.19 The total quantity of S-SMEDDS (mg; Y3) was additionally chosen as a response variable to fulfill the goal of minimizing S-SMEDDS formulations. As shown in Table 3, for the eleven experimental runs, Y1, Y2, and Y3 were in the ranges of 130.6–658.1 mg, 7.08%–95.2%, and 101–440 mg, respectively. All responses were simultaneously fitted to linear, two-factor interaction (2FI), quadratic, and cubic models, using the Design-Expert software (Table 4). As several statistical parameters, such as sequential P-value, lack of fit P-value, SD, squared correlation coefficient (R2), and adjusted R2 value, were compared to find the best-fit mathematical model, Y1, Y2, and Y3 were selected for the quadratic, linear, and 2FI models, respectively. The sequential P-values for Y1, Y2, and Y3 were <0.05, indicating that the model terms were significant up to a 95% confidence level. The lack of fit P-values of the responses Y1 and Y2 were >0.1, suggesting the adequacy of the model fit.31 The lack of fit P-value for Y3 was not evaluated since it revealed no difference between residual error and pure error from the replicated design points. To validate the analysis, R2 and adjusted R2 represented multiple regression analyses of the responses.19 All R2 and adjusted R2 values of Y1, Y2, and Y3 were >90%, indicating satisfactory analysis quality. Similar values of R2 and adjusted R2 have been reported as desirable for a good model fit.32 Substituting the regression coefficients, we can derive the model equations, regarding the level of factors and responses as follows:
 | Table 2 Variables used in the 3-LFD |
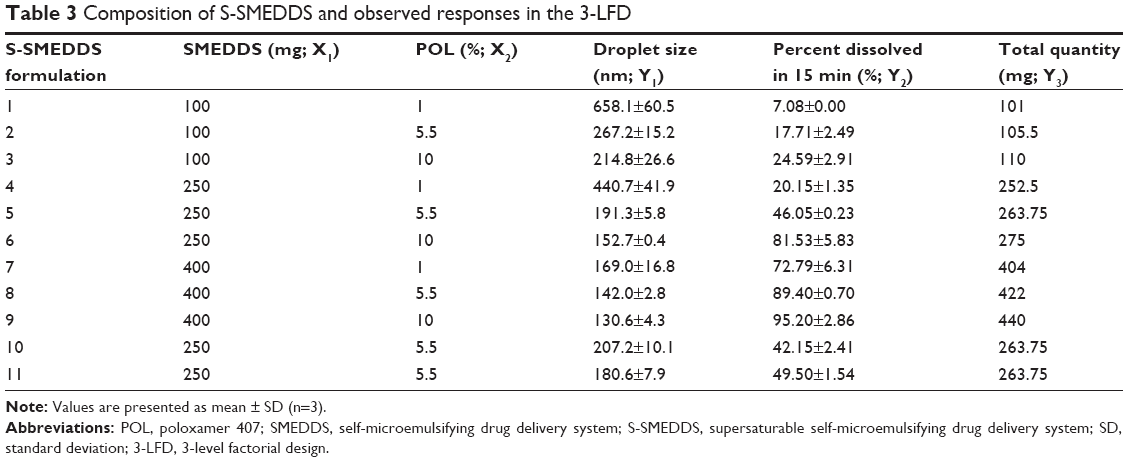 | Table 3 Composition of S-SMEDDS and observed responses in the 3-LFD |
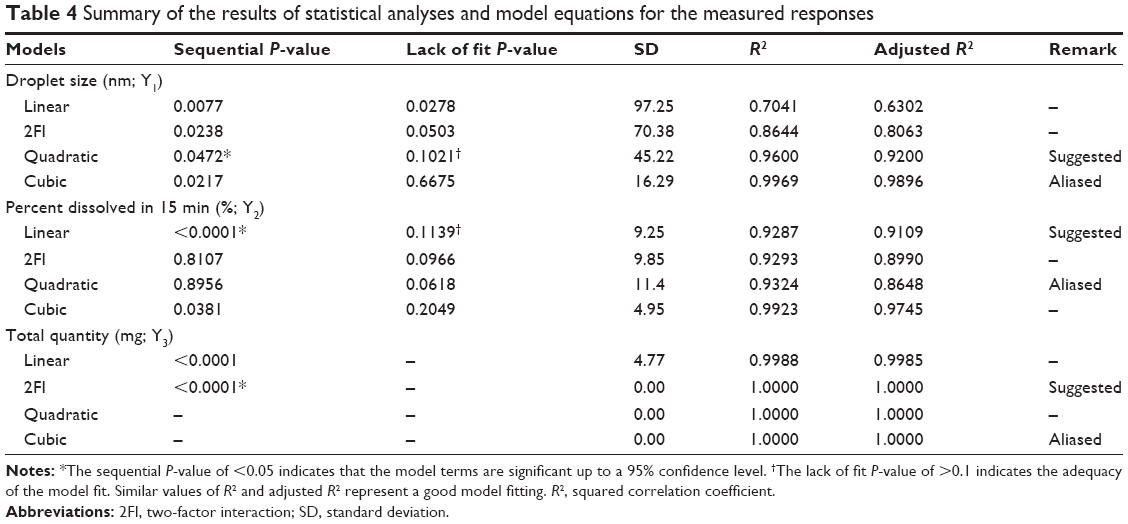 | Table 4 Summary of the results of statistical analyses and model equations for the measured responses |
|
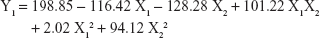
|
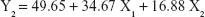
|
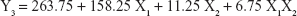
Figure 5 shows the effects of X1 and X2 on the responses. Droplet size decreased from 658.1 to 130.1 nm as X1 and X2 increased. X2 significantly interacted with X1. If the lines of the interaction effects plot are not parallel, it indicates interactions between the two factors.33,34 When compared with S-SMEDDS containing 10% POL, S-SMEDDS containing 1% POL showed a greater decrease in droplet size with an increasing amount of SMEDDS (X1) from 100 to 400 mg. Regardless of the SMEDDS quantity, the droplet sizes of S-SMEDDS containing 10% POL were smaller, from 100 to 200 nm, compared with those of other S-SMEDDS formulations that contained 1% or 5.5% POL. This result indicates that 10% POL effectively stabilizes the dispersion by sufficient adsorption on the surface of the oily droplet or free drug molecules. On the contrary, the dissolution rate (Y2) increased from 7.08% to 95.2% as X1 and X2 increased. There was no significant interaction effect between X1 and X2 on drug release, suggesting that both X1 and X2 affect drug release in a similar manner. Increased X1 and X2 could improve the solubilizing capacity and offer high thermodynamic activity of the drug.
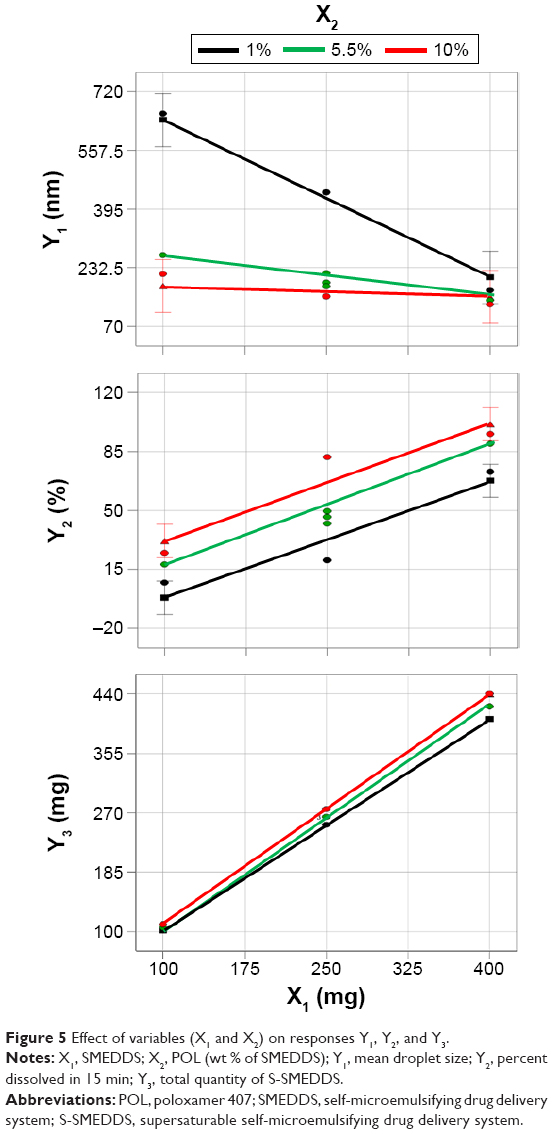 | Figure 5 Effect of variables (X1 and X2) on responses Y1, Y2, and Y3. |
Optimization of S-SMEDDS using desirability function
The independent variables were simultaneously optimized for responses by using the desirability function. As shown in Table 2, Y1 and Y3 were set to be minimized, whereas Y2 was set to be maximized. Furthermore, these variables should establish different levels of importance to achieve the objectives of both improving drug dissolution rate and reducing the volume of S-SMEDDS. When the importance of response variables is changed from “+” to “++++,” it is feasible to attribute different desirability to the responses and, consequently, to increase or decrease the range of acceptable values in the optimization process.35 As shown in Table 5, optimization was performed, placing different weights on responses: 1) considering >80% dissolution of the drug in pH 1.2 medium, the importance of Y2 for S-SMEDDS_LQ was set to “++++” and the others to “+”; 2) considering a total quantity similar to the unit dose of a commercial product (Diovan®), the importance of Y3 for S-SMEDDS_RQ was set to “++” and the others to “+.” As a result, the corresponding desirability values were 0.715 and 0.708 for S-SMEDDS_LQ and S-SMEDDS_RQ, respectively. When an equal importance of “+” was given to all responses, the corresponding desirability value was 0.689. Desirability values lie between zero and one, where one is the most desirable and zero is completely undesirable.36 Thus, we successfully optimized S-SMEDDS_LQ and S-SMEDDS_RQ using suitable desirability functions. Meanwhile, to determine the accuracy of prediction and confirm the validity of the optimized formulations, the predicted and experimental values were compared using percentage prediction errors. The percentage prediction errors associated with both S-SMEDDS formulations were low (<10%), indicating that the 3-LFD used to optimize the S-SMEDDS formulations was accurate and reliable.
 | Table 5 Experimental and predicted values for two optimal S-SMEDDS |
In vitro dissolution profiles
Dissolution profiles of VST from various samples, such as VST powder, SMEDDS (320 mg), S-SMEDDS_LQ, and S-SMEDDS_RQ, were determined in pH 1.2 and 6.8 media (Figure 6). VST dissolution was pH-dependent, revealing a very low dissolution of <20% at pH 1.2 but complete dissolution of ~100% at pH 6.8 in 2 h. The equilibrium solubility of VST was measured separately to find a pH-solubility profile, resulting in the values of 0.07±0.01 mg/mL at pH 1.2 and 5.75±0.37 mg/mL at pH 6.8. VST possesses two weakly ionizable groups, a tetrazole derivative (pKa=4.73) and carboxylic acid (pKa=3.9), which may contribute pH-dependent solubility characteristics.37 In pH 1.2 medium at 50 rpm, S-SMEDDS_ LQ drastically enhanced the dissolution of VST, with >80% dissolution within 5 min, while S-SMEDDS_RQ and SMEDDS (320 mg) formulations showed ~40% dissolution by 2 h. Compared with that of VST powder, SMEDDS formulations of S-SMEDDS_LQ, S-SMEDDS_RQ, and SMEDDS (320 mg) increased drug release by 7.5-, 3.8-, and 3.4-fold, respectively. This indicates that the SMEDDS and S-SMEDDS formulations improved the dissolution of VST by producing nano-sized dispersions for solubilizing VST. Meanwhile, in the pH 6.8 medium at 50 rpm, S-SMEDDS_LQ, SMEDDS (320 mg), and VST powder revealed a high dissolution of >90%, whereas S-SMEDDS_RQ showed delayed drug release, resulting in >90% dissolution after 90 min. In the early stage of the dissolution test at 50 rpm, S-SMEDDS_RQ formed a highly viscous mass that was not instantly dispersed throughout the medium. As the rotating speed increased to 125 rpm, it dispersed faster, with >90% dissolution within 5 min. In the pH 1.2 medium at 125 rpm, the dissolution of VST from S-SMEDDS_RQ and SMEDDS (320 mg) increased by 1.5- and 1.6-fold, respectively, compared with those in the same medium at 50 rpm.
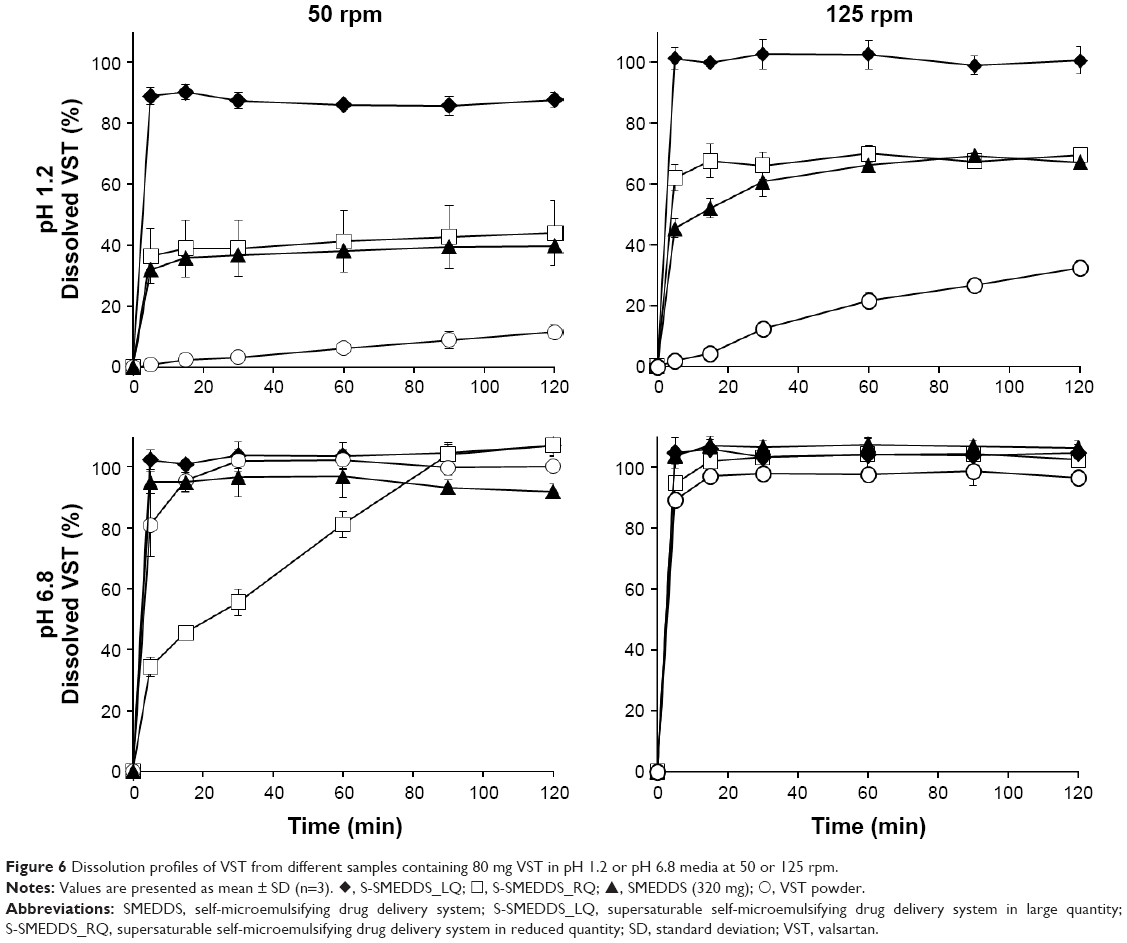 | Figure 6 Dissolution profiles of VST from different samples containing 80 mg VST in pH 1.2 or pH 6.8 media at 50 or 125 rpm. |
In vivo oral absorption and pharmacokinetic behavior
The pharmacokinetic behavior of VST in rats was investigated after oral administration of S-SMEDDS_LQ, S-SMEDDS_RQ, SMEDDS (320 mg), and VST suspension. Plasma levels of VST were measured and are plotted against time in Figure 7. Especially for the period from 0 to 2 h, SMEDDS (320 mg), S-SMEDDS_LQ, and S-SMEDDS_RQ showed higher plasma levels than VST suspension. This initial greater absorption with SMEDDS formulations might be due to the improvement of VST dissolution in acidic stomach conditions. The pharmacokinetic parameters are listed in Table 6. The Tmax values were identical for all samples. However, the AUC and Cmax values of VST suspension were much lower than those of the others, specifically S-SMEDDS_LQ > S-SMEDDS_RQ > SMEDDS (320 mg) > VST suspension. The Cmax values of SMEDDS_LQ, S-SMEDDS_RQ, and SMEDDS (320 mg) were 3.6, 2.5, and 1.9 times higher, respectively, than that of VST suspension. Based on AUC values, compared with VST suspension, the RBAs of SMEDDS (320 mg), S-SMEDDS_LQ, and S-SMEDDS_RQ were 207%, 263%, and 219%, respectively. These results were consistent with the findings in the dissolution study. There are numerous reports that BA enhancement generally correlates with the increase in dissolution rate of a drug.21,38 Meanwhile, when compared with SMEDDS (320 mg), the RBAs of S-SMEDDS_LQ and S-SMEDDS_RQ were 127% and 106%, respectively. Addition of supersaturating agents further increased the oral BA, which is consistent with an earlier report that incorporation of 10% polymer to a SMEDDS formulation resulted in a supersaturated solution of a poorly soluble drug that presented higher amounts for absorption throughout the experimental timeframe.39 It is noteworthy that SMEDDS (320 mg) and S-SMEDDS_RQ had a similar BA, despite the total quantity of SMEDDS being reduced by 45% in the latter. Thus, we suggest that an adequate amount of POL effectively affords a supersaturated state to improve the oral absorption of VST. In terms of developing an effective SMEDDS preparation of minimal quantity, S-SMEDDS_RQ is promising. To verify the advantage of this system, in terms of reduced GI irritation, further safety evaluations are still needed.
 | Table 6 Pharmacokinetic parameters of VST suspension and various VST-loaded SMEDDS formulations in rats |
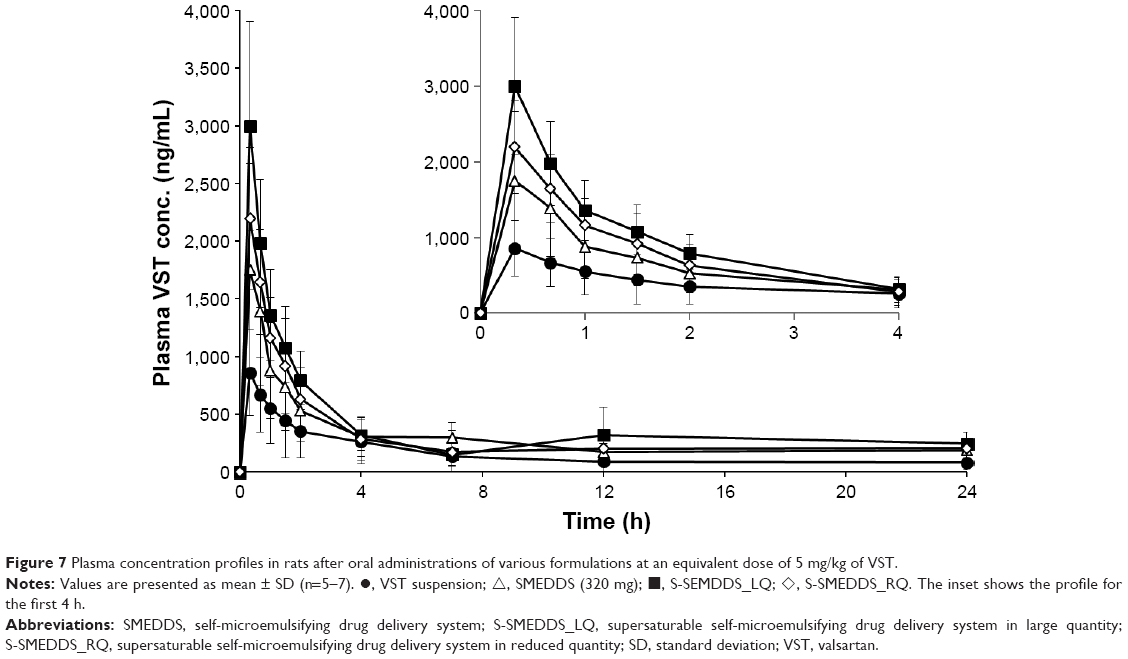 | Figure 7 Plasma concentration profiles in rats after oral administrations of various formulations at an equivalent dose of 5 mg/kg of VST. |
IVIVCs
Application of IVIVC has been recently suggested in the pharmaceutical field for development of novel formulations to reduce the time and cost of the manufacturing process.40 The FDA has developed a regulatory guidance for both immediate- and modified-release dosage forms, thus minimizing the need for BA studies as part of the formulation design and optimization process.41 IVIVC could be employed to serve as a surrogate for in vivo BA studies and to support biowaivers.41 For validating and justifying a biowaiver for VST in S-SEMDDS formulations, we estimated the correlation between in vitro dissolution (pH 1.2, 125 rpm) and in vivo oral BA for various VST formulations. As shown in Figure 8A, the in vitro DE values for 2 h were linearly correlated with the in vivo AUC in rats (R2=0.9714). Biorelevant dissolution can be used successfully to predict in vivo behavior of poorly water-soluble drugs in SMEDDS formulations.21,42 Among poorly water-soluble drugs, BCS class II drugs could be applied to IVIVC, since dissolution is the rate-limiting step for drug absorption in the gut.43 However, since the present study was aimed at developing a novel S-SMEDDS with minimal quantity, while improving dissolution and oral absorption of VST, unit quantity-based correlation was further investigated. Similar to the DEC calculation, absorption enhancing capacity per unit quantity (AEC) was calculated as follows: (AUC0–24 [SMEDDS or S-SMEDDS] − AUC0–24[VST]/V[SMEDDS or S-SMEDDS]), where the AUC difference between the specific SMEDDS formulation and VST suspension was divided by the quantity of the relevant SMEDDS formulation. As shown in Figure 8B, excellent correlation (R2=0.9711) between DEC and AEC was found. The magnitude of DEC and AEC was in the order of S-SMEDDS_RQ > S-SMEDDS_LQ > SMEDDS (320 mg) > VST powder or suspension, indicating that S-SMEDDS_RQ was the most effective in enhancing the dissolution rate and oral absorption of VST.
 | Figure 8 Correlations between in vitro dissolution and in vivo oral BA. |
Conclusion
We successfully optimized a novel formulation of VST-loaded S-SMEDDS using the 3-LFD method to minimize the total quantity and maximize the drug dissolution. POL was added as a selectable supersaturating agent to VST-containing SMEDDS composed of 10% Capmul MCM, 45% Tween 20, and 45% Transcutol P. Further optimization was established by weighting different levels of importance on response variables for dissolution (S-SMEDDS_LQ) and total quantity (S-SMEDDS_RQ), revealing percentage prediction errors of <10%. Consequently, compared with VST powder or suspension and SMEDDS, both S-SMEDDS_LQ and S-SMEDDS_RQ showed excellent in vitro dissolution and in vivo oral BA in rats. From the unit quantity-based analysis for IVIVCs, good correlation was found between dissolution and absorption enhancements. Thus, in terms of developing an effective SMEDDS preparation of minimal quantity, S-SMEDDS_RQ is promising.
Acknowledgment
This work was supported by the Advanced Technology Center program (10051950) funded by the Ministry of Trade, Industry & Energy (MI, Republic of Korea).
Disclosure
The authors report no conflicts of interest in this work.
References
Beg S, Swain S, Singh HP, Patra CN, Rao MB. Development, optimization, and characterization of solid self-nanoemulsifying drug delivery systems of valsartan using porous carriers. AAPS PharmSciTech. 2012;13(4):1416–1427. | ||
Dixit AR, Rajput SJ, Patel SG. Preparation and bioavailability assessment of SMEDDS containing valsartan. AAPS PharmSciTech. 2010;11(1):314–321. | ||
Yan YD, Sung JH, Kim KK, et al. Novel valsartan-loaded solid dispersion with enhanced bioavailability and no crystalline changes. Int J Pharm. 2012;422(1):202–210. | ||
Baek IH, Kim JS, Ha ES, et al. Oral absorption of a valsartan-loaded spray-dried emulsion based on hydroxypropylmethyl cellulose. Int J Biol Macromol. 2014;69:222–228. | ||
Kim MS, Baek IH. Fabrication and evaluation of valsartan-polymer-surfactant composite nanoparticles by using the supercritical antisolvent process. Int J Nanomedicine. 2014;9(1):5167–5176. | ||
Ma Q, Sun H, Che E, et al. Uniform nano-sized valsartan for dissolution and bioavailability enhancement: influence of particle size and crystalline state. Int J Pharm. 2013;441(1):75–81. | ||
Dangre PV, Gilhotra RM, Dhole SN. Formulation and development of solid self micro-emulsifying drug delivery system (S-SMEDDS) containing chlorthalidone for improvement of dissolution. J Pharm Invest. 2016;46(7):633–644. | ||
Porter CJ, Charman WN. Lipid-based formulations for oral administration: opportunities for bioavailability enhancement and lipoprotein targeting of lipophilic drugs. J Recept Signal Transduct Res. 2001;21(2–3):215–257. | ||
Rao BP, Baby B, Durgaprasad Y, et al. Formulation and evaluation of SMEDDS with Capmul MCM for enhanced dissolution rate of valsartan. RGUHS J Pharm Sci. 2013;3(2):33–40. | ||
Poudel BK, Marasini N, Tran TH, Choi HG, Yong CS, Kim JO. Formulation, characterization and optimization of valsartan self-microemulsifying drug delivery system using statistical design of experiment. Chem Pharm Bull. 2012;60(11):1409–1418. | ||
Zhao K, Yuan Y, Wang H, Li P, Bao Z, Li Y. Preparation and evaluation of valsartan by a novel semi-solid self-microemulsifying delivery system using Gelucire 44/14. Drug Dev Ind Pharm. 2016;42(10):1545–1552. | ||
Lee DH, Yeom DW, Song YS, et al. Improved oral absorption of dutasteride via Soluplus®-based supersaturable self-emulsifying drug delivery system (S-SEDDS). Int J Pharm. 2015;478(1):341–347. | ||
Song WH, Park JH, Yeom DW, et al. Enhanced dissolution of celecoxib by supersaturating self-emulsifying drug delivery system (S-SEDDS) formulation. Arch Pharm Res. 2013;36(1):69–78. | ||
Gao P, Rush BD, Pfund WP, et al. Development of a supersaturable SEDDS (S-SEDDS) formulation of paclitaxel with improved oral bioavailability. J Pharm Sci. 2003;92(12):2386–2398. | ||
Bevernage J, Brouwers J, Brewster ME, Augustijns P. Evaluation of gastrointestinal drug supersaturation and precipitation: strategies and issues. Int J Pharm. 2013;453(1):25–35. | ||
Chauhan H, Hui-Gu C, Atef E. Correlating the behavior of polymers in solution as precipitation inhibitor to its amorphous stabilization ability in solid dispersions. J Pharm Sci. 2013;102(6):1924–1935. | ||
Xu S, Dai WG. Drug precipitation inhibitors in supersaturable formulations. Int J Pharm. 2013;453(1):36–43. | ||
Cao QR, Liu Y, Xu WJ, Lee BJ, Yang M, Cui JH. Enhanced oral bioavailability of novel mucoadhesive pellets containing valsartan prepared by a dry powder-coating technique. Int J Pharm. 2012;434(1):325–333. | ||
Yeom DW, Song YS, Kim SR, et al. Development and optimization of a self-microemulsifying drug delivery system for atorvastatin calcium by using D-optimal mixture design. Int J Nanomedicine. 2015;10:3865–3877. | ||
Khan KA. The concept of dissolution efficiency. J Pharm Pharmacol. 1975;27(1):48–49. | ||
Song WH, Yeom DW, Lee DH, et al. In situ intestinal permeability and in vivo oral bioavailability of celecoxib in supersaturating self-emulsifying drug delivery system. Arch Pharm Res. 2014;37(5):626–635. | ||
Kojima T, Higashi K, Suzuki T, Tomono K, Moribe K, Yamamoto K. Stabilization of a supersaturated solution of mefenamic acid from a solid dispersion with EUDRAGIT® EPO. Pharm Res. 2012;29(10):2777–2791. | ||
Chen ZQ, Liu Y, Zhao JH, Wang L, Feng NP. Improved oral bioavailability of poorly water-soluble indirubin by a supersaturatable self-microemulsifying drug delivery system. Int J Nanomedicine. 2012;7(1):1115–1125. | ||
Zhang N, Zhang W, Jin Y, Quan DQ. Studies on preparation of carbamazepine (CBZ) supersaturatable self-microemulsifying (S-SMEDDS) formulation and relative bioavailability in beagle dogs. Pharm Dev Technol. 2011;16(4):415–421. | ||
Gao P, Guyton ME, Huang T, Bauer JM, Stefanski KJ, Lu Q. Enhanced oral bioavailability of a poorly water soluble drug PNU-91325 by supersaturatable formulations. Drug Dev Ind Pharm. 2004;30(2):221–229. | ||
Dash RN, Mohammed H, Humaira T. Design, optimization, and evaluation of ezetimibe solid supersaturatable self-nanoemulsifying drug delivery for enhanced solubility and dissolution. J Pharm Invest. 2016;46(2):153–168. | ||
Chutimaworapan S, Ritthidej GC, Yonemochi E, Oguchi T, Yamamoto K. Effect of water-soluble carriers on dissolution characteristics of nifedipine solid dispersions. Drug Dev Ind Pharm. 2000;26(11):1141–1150. | ||
Sethia S, Squillante E. Physicochemical characterization of solid dispersions of carbamazepine formulated by supercritical carbon dioxide and conventional solvent evaporation method. J Pharm Sci. 2002;91(9):1948–1957. | ||
Dai WG, Dong LC, Li S, Deng Z. Combination of Pluronic/Vitamin E TPGS as a potential inhibitor of drug precipitation. Int J Pharm. 2008;355(1):31–37. | ||
Maibaum L, Dinner AR, Chandler D. Micelle formation and the hydrophobic effect. J Phys Chem B. 2004;108(21):6778–6781. | ||
Kim S, Gupta B, Moon C, et al. Employing an optimized spray-drying process to produce ezetimibe tablets with an improved dissolution profile. J Pharm Invest. 2016;46(6):583–592. | ||
Atikler U, Demir H, Tokatli F, Tihminlioğlu F, Balköse D, Ülkü S. Optimisation of the effect of colemanite as a new synergistic agent in an intumescent system. Polym Degrad Stab. 2006;91(7):1563–1570. | ||
Zu Y, Zhang Y, Zhao X, Zhang Q, Liu Y, Jiang R. Optimization of the preparation process of vinblastine sulfate (VBLS)-loaded folate-conjugated bovine serum albumin (BSA) nanoparticles for tumor-targeted drug delivery using response surface methodology (RSM). Int J Nanomedicine. 2009;4:321–333. | ||
Kim MS, Yeom DW, Kim SR, et al. Development of a chitosan based double layer-coated tablet as a platform for colon-specific drug delivery. Drug Des Devel Ther. 2017;11:45–57. | ||
Bezerra MA, Santelli RE, Oliveira EP, Villar LS, Escaleira LA. Response surface methodology (RSM) as a tool for optimization in analytical chemistry. Talanta. 2008;76(5):965–977. | ||
Dabbas RM, Fowler JW, Rollier DA, Mccarville D. Multiple response optimization using mixture-designed experiments and desirability functions in semiconductor scheduling. Int J Prod Res. 2003;41(5):939–961. | ||
Flesch G, Müller P, Lloyd P. Absolute bioavailability and pharmacokinetics of valsartan, an angiotensin II receptor antagonist, in man. Eur J Clin Pharmacol. 1997;52(2):115–120. | ||
Dressman JB, Reppas C. In vitro–in vivo correlations for lipophilic, poorly water-soluble drugs. Eur J Pharm Sci. 2000;11(Suppl 2):S73–S80. | ||
Gosangari S, Dyakonov T. Enhanced dissolution performance of curcumin with the use of supersaturatable formulations. Pharm Dev Technol. 2013;18(2):475–480. | ||
Yang SG. Biowaiver extension potential and IVIVC for BCS Class II drugs by formulation design: case study for cyclosporine self-microemulsifying formulation. Arch Pharm Res. 2010;33(11):1835–1842. | ||
Emami J. In vitro–in vivo correlation: from theory to applications. J Pharm Pharm Sci. 2006;9(2):169–189. | ||
Kim MS, Ha ES, Choo GH, Baek IH. Preparation and in vivo evaluation of a dutasteride-loaded solid-supersaturatable self-microemulsifying drug delivery system. Int J Mol Sci. 2015;16(5):10821–10833. | ||
Amidon GL, Lennernäs H, Shah VP, Crison JR. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12(3):413–420. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.