Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Enhanced oral bioavailability of fenofibrate using polymeric nanoparticulated systems: physicochemical characterization and in vivo investigation
Authors Yousaf AM, Kim D, Oh Y, Yong CS, Kim JO, Choi H
Received 8 December 2014
Accepted for publication 24 January 2015
Published 5 March 2015 Volume 2015:10(1) Pages 1819—1830
DOI https://doi.org/10.2147/IJN.S78895
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Abid Mehmood Yousaf,1 Dong Wuk Kim,1 Yu-Kyoung Oh,2 Chul Soon Yong,3 Jong Oh Kim,3 Han-Gon Choi1
1College of Pharmacy and Institute of Pharmaceutical Science and Technology, Hanyang University, Ansan, 2College of Pharmacy, Seoul National University, Seoul, 3College of Pharmacy, Yeungnam University, Gyongsan, South Korea
Background: The intention of this research was to prepare and compare various solubility-enhancing nanoparticulated systems in order to select a nanoparticulated formulation with the most improved oral bioavailability of poorly water-soluble fenofibrate.
Methods: The most appropriate excipients for different nanoparticulated preparations were selected by determining the drug solubility in 1% (w/v) aqueous solutions of each carrier. The polyvinylpyrrolidone (PVP) nanospheres, hydroxypropyl-β-cyclodextrin (HP-β-CD) nanocorpuscles, and gelatin nanocapsules were formulated as fenofibrate/PVP/sodium lauryl sulfate (SLS), fenofibrate/HP-β-CD, and fenofibrate/gelatin at the optimized weight ratios of 2.5:4.5:1, 1:4, and 1:8, respectively. The three solid-state products were achieved using the solvent-evaporation method through the spray-drying technique. The physicochemical characterization of these nanoparticles was accomplished by powder X-ray diffraction, differential scanning calorimetry, scanning electron microscopy, and Fourier-transform infrared spectroscopy. Their physicochemical properties, aqueous solubility, dissolution rate, and pharmacokinetics in rats were investigated in comparison with the drug powder.
Results: Among the tested carriers, PVP, HP-β-CD, gelatin, and SLS showed better solubility and were selected as the most appropriate constituents for various nanoparticulated systems. All of the formulations significantly improved the aqueous solubility, dissolution rate, and oral bioavailability of fenofibrate compared to the drug powder. The drug was present in the amorphous form in HP-β-CD nanocorpuscles; however, in other formulations, it existed in the crystalline state with a reduced intensity. The aqueous solubility and dissolution rates of the nanoparticles (after 30 minutes) were not significantly different from one another. Among the nanoparticulated systems tested in this study, the initial dissolution rates (up to 10 minutes) were higher with the PVP nanospheres and HP-β-CD nanocorpuscles; however, neither of them resulted in the highest oral bioavailability. Irrespective of relatively retarded dissolution rate, gelatin nanocapsules showed the highest apparent aqueous solubility and furnished the most improved oral bioavailability of the drug (~5.5-fold), owing to better wetting and diminution in crystallinity.
Conclusion: Fenofibrate-loaded gelatin nanocapsules prepared using the solvent-evaporation method through the spray-drying technique could be a potential oral pharmaceutical product for administering the poorly water-soluble fenofibrate with an enhanced bioavailability.
Keywords: gelatin nanocapsules, hydrophilic polymeric matrix, crystallinity, ameliorated oral bioavailability
Introduction
Fenofibrate is clinically used to normalize the abnormally elevated plasma titer of low-density lipoproteins and cholesterol in patients with hypercholesterolemia and hypertriglyceridemia.1,2 Fenofibrate, a Biopharmaceutics Classification System (BCS) class II drug, is very lipophilic (log P=5.24)3 and virtually insoluble in water.4 An orally administered agent possessing aqueous solubility <0.1 mg/mL usually demonstrates very poor absorption, owing to impaired aqueous solubility and dissolution rate.5 Similarly, fenofibrate furnishes poor oral bioavailability, owing to insufficient absorption from the aqueous environment of the gastrointestinal tract.1 Thus, the oral bioavailability of fenofibrate can be raised by increasing its aqueous solubility. Several conventional formulation techniques, such as salt formation, the use of surface-active agents, pH modification, complex formation, micronization, formation of liposomes, and solid dispersion formulations, have been employed to enhance the oral bioavailability of various hydrophobic drugs by promoting their aqueous solubility and dissolution rates.6–11
Nanoparticle formation is a promising strategy to ameliorate the aqueous solubility, dissolution rate, and absorption of poorly water-soluble entities.12 In pharmaceutical nanotechnology, either the drug itself is converted to nano-sized particles or the nanoparticles are formed from carriers, for example, polymers.12 Natural polymeric carriers possess unique benefits in nanoparticulated drug delivery because of their excellent safety, efficacy, biocompatibility, and biodegradation.13 Pharmaceutical polymeric nanoparticles, a class of popular drug carriers in pharmaceutical nanotechnology,14 are drug-loaded particles with diameters <1,000 nm.15,16 Depending upon the nanoparticle matrix and method of preparation, the drug can be entrapped, encapsulated, or attached to the polymeric material to develop nanospheres, nanocapsules, or nanocorpuscles, respectively.16 In drug delivery, nanoparticulated systems are preferred over micro-sized particles, owing to their smaller size and larger surface area, which improve dissolution rate, absorption, and bioavailability.17 Pharmaceutical nanoparticulated systems can be developed using a variety of techniques, such as solvent evaporation, emulsification, spray drying, phase inversion, self-assembly, and solvent displacement.18,19 Spray drying is one of the most frequently employed techniques in the preparation of a dry powder from a liquid phase,20 because of the simple operation, good yield, and the fact that it is easy to scale up.21,22 In the process of nanoparticle manufacture, all of the components, completely dissolved in suitable solvents, are sprayed into a stream of hot air to evaporate the solvent.23
A polymeric nanosphere is a nano-sized, round-shaped structure in which the drug is uniformly distributed in the matrix.14,15 Nanoparticulated solid dispersion is a promising way to overcome the problems pertaining to poor aqueous solubility, impaired dissolution rate, and low bioavailability of BCS class II drugs.10,24,25 Solid dispersion consists of an amorphous or crystalline hydrophobic drug dispersed in a hydrophilic polymeric matrix.24 It enormously improves the aqueous solubility, dissolution rate, and oral bioavailability of hydrophobic drugs. Nanospheres of solid dispersions can be prepared by spray drying a clear solution formed by completely dissolving all components in suitable solvents.23 The term “nanocorpuscle” can be given to a nanoparticle having an irregular shape and/or surface.
A polymeric nanocapsule is a vesicular system in which the drug-loaded reservoir (core material) is enwrapped in a polymeric coating (wall material), forming a typical core–shell structure.14,16 Pharmaceutical nanoencapsulation facilitates the bioavailability of various hydrophobic drugs.26 Gelatin, a heterogeneous mixture of polypeptides,27 can promote the aqueous solubility, dissolution, and bioavailability of numerous water-insoluble drugs.28 In the preparation of gelatin nanocapsules, hydroalcoholic solutions containing dissolved lipophilic drugs and gelatin can be converted to dry powders through the spray-drying technique, in which water is evaporated considerably and drug-loaded ethanol is encapsulated within a water-soluble gelatin shell.29,30 Encapsulated drugs can exist in the amorphous state28 or in the crystalline form.31
The present work is a unique study, which involves the physicochemical characterization and in vivo assessment of various solubility-enhancing polymeric nanoparticulated systems, prepared with optimized ratios of fenofibrate/polyvinylpyrrolidone (PVP)/sodium lauryl sulfate (SLS), fenofibrate/hydroxypropyl-β-cyclodextrin (HP-β-CD), and fenofibrate/gelatin, in order to determine a nanoparticulated formulation with the most improved oral bioavailability of the poorly water-soluble fenofibrate. The selection of the most appropriate hydrophilic excipients for the preparation of various polymeric nanoparticulated drug-delivery systems was performed after the appropriate screening of numerous hydrophilic polymers and surfactants in terms of drug solubility. Thus, PVP, HP-β-CD, gelatin, and SLS were chosen as the most suitable hydrophilic components for different nanoparticulated formulations. Physicochemical physiognomies of the nanoparticles were perused using powder X-ray diffraction (PXRD), differential scanning calorimetry (DSC), scanning electron microscopy (SEM), and Fourier-transform infrared (FTIR) spectroscopy. Moreover, their aqueous solubility, dissolution rates, and pharmacokinetics in rats were studied in comparison with the drug powder.
Materials and methods
Materials
Fenofibrate was supplied by Hanmi Pharm. Co. (Suwon, South Korea). Hydroxypropyl methylcellulose, hydroxypropyl cellulose, and polyvinyl alcohol were procured from Shin-Etsu Co. (Tokyo, Japan). Carboxymethylcellulose sodium and dextran were obtained from Duksan Chemical Co. (Ansan, South Korea) and Sigma-Aldrich (St Louis, MO, USA), respectively. PVP K30, Solutol HS15, poloxamer 407, cremophore A25, cremophore RH40, cremophore EL, and cremophore ELP were purchased from BASF (Ludwigshafen, Germany). Polyethylene glycol (400 and 6000) was acquired from Duksan Chemical Co. Gelatin, polysorbate 80 (Tween 80), and sorbitan monooleate 80 (Span 80) were supplied by Daejung Chemical Co. (Siheung, South Korea). Fenofibric acid, HP-β-CD, SLS, and docusate sodium were obtained from Hanmi Pharm. Co. Clofibric acid was purchased from Sigma-Aldrich. All other solvents and chemical substances were of reagent grade.
Selection of suitable carriers for various nanoparticulated formulations
For selecting the different components appropriate for the development of various nanoparticulated systems, the saturation solubility of fenofibrate was determined in 1% (w/v) aqueous solution of each candidate. An excess amount of fenofibrate powder was transferred to 3 mL of 1% (w/v) aqueous solution of each carrier in a 15 mL conical tube. After mixing with a vortex, the tubes were held on a shaker in a water bath (25°C) and agitated at 100 rpm for 7 days. Then, 1 mL of each sample was centrifuged at 10,000× g for 10 minutes (Smart 15; Hanil Science Industrial Co., Gangneung, South Korea). The supernatant was diluted with acetonitrile and filtered (0.45 μm). The concentration of fenofibrate in the filtrate (50 μL) was quantified by high-performance liquid chromatography (HPLC) (Agilent 1260 Infinity; Agilent Technologies, Santa Clara, CA, USA), which was equipped with a Capcell Pak C18 column (4.6 mm ID ×250 mm, 5 μm; Shiseido, Tokyo, Japan). The column temperature was set at 30°C. The mobile phase, consisting of acetonitrile and 0.1% (v/v) aqueous phosphoric acid at a volume ratio of 75:25, was eluted at a flow rate of 2 mL/min. The pH value of 0.1% (v/v) aqueous phosphoric acid was 2.1. The eluent was monitored at 286 nm for the quantification of fenofibrate.32
Preparation of various nanoparticulated systems
A Büchi B-290 nozzle-type mini spray dryer (Büchi Co., Flawil, Switzerland) was used for the development of different nanoparticulated formulations. Each transparent solution, containing completely dissolved components in an optimal ratio, was sprayed into the drying chamber at a flow rate of 3 mL/min. The pressure of the spraying air was 4 kg/cm2. The flow of the drying air was maintained at the aspirator setting of 100%.
The PVP nanospheres and HP-β-CD nanocorpuscles were prepared with fenofibrate/PVP/SLS at a weight ratio of 2.5:4.5:1 (w/w/w) and fenofibrate/HP-β-CD at the weight ratio of 1:4. The constituents, completely dissolved in 95% ethanol, were spray dried at an inlet temperature of 100°C and an outlet temperature of 60°C–65°C. Likewise, gelatin nanocorpuscles were developed with fenofibrate/gelatin at the weight ratio of 1:8. The components were dissolved in 40% ethanol at 50°C. The final clear solution was sprayed into the drying chamber. The inlet and outlet temperatures were 120°C and 65°C, respectively.
Aqueous solubility
An excess amount of each nanoparticulated formulation (about 10 mg) was poured into a 15 mL conical tube containing 3 mL of distilled water, separately. After vortex mixing, the tubes were shaken (100 rpm) in a water bath (37°C) for 7 days. Then, 1 mL of sample was centrifuged at 10,000× g for 10 minutes (Smart 15; Hanil Science Industrial Co.). The supernatant was diluted with acetonitrile and filtered (0.45 μm). The concentration of fenofibrate in the filtrate (50 μL) was quantified using the HPLC method described above.32 The test was performed in triplicate for each formulation.
Dissolution
The dissolution test was carried out using USP dissolution apparatus II (paddle apparatus) (Vision® Classic 6™; Hanson Research Co., Los Angeles, CA, USA). Each fenofibrate-loaded nanoparticulated formulation or drug powder equivalent to 80 mg of fenofibrate was placed in an empty, hard gelatin capsule. The filled capsule was inserted into the sinker and immersed in the dissolution medium. The dissolution medium consisted of 900 mL of 2% (w/v) Tween 80.33,34 The dissolution test was accomplished at 37°C±0.5°C using a paddle rotation of 100 rpm. The dissolution medium-containing vessel was surrounded by an outer water bath in order to maintain a specific temperature. At predetermined time intervals, 1 mL of the dissolution medium was withdrawn, filtered (0.45 μm), and assayed using the HPLC method described above.32
Structural aspects
The crystallinity of the samples was assessed using a Rigaku X-ray diffractometer (D/MAX-2500 PC; Rigaku Corporation, Tokyo, Japan) equipped with a miniflex goniometer. The PXRD analysis was performed at room temperature using a Cu Kα1 monochromatic radiation source with 40 mA of current and 40 kV voltage. The X-ray diffraction patterns were obtained in the range of 10°C–80°C with a 2θ scanning mode, a step size of 0.02°/sec, and a scan speed of 5°/min.35
Thermal characteristics
The thermal features of fenofibrate powder and fenofibrate-loaded nanoparticulated preparations were examined using a differential scanning calorimeter (DSC Q20; TA Instruments, New Castle, DE, USA). Approximately 5 mg of each sample, enclosed in an aluminum pan, was scanned over a range of 30°C–130°C at a heating rate of 10°C/min and a nitrogen flow of 25 mL/min.
FTIR spectroscopic analysis
In the prepared nanoparticulated formulations, possible chemical interactions between the drug and the carriers were investigated by FTIR spectroscopic analysis. A Nicolet-6700 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) was used for scanning the samples in the range of 400–4,000 cm−1. Infrared spectra of the drug powder and fenofibrate-loaded nanoparticles were obtained using OMNIC software. The infrared spectrum of each nanoparticulated formulation was compared with that of the corresponding physical mixture.
Morphological physiognomies
The morphological features of the drug powder and fenofibrate-loaded nanoparticles were inspected using a scanning electron microscope (S-4800; Hitachi, Tokyo, Japan). The samples were attached to the open side of a double-sided adhesive carbon tape fixed on a brass disk. Subsequently, the samples were coated in platinum for 4 minutes using the EMI Teck Ion Sputter (K575K) in order to make them electrically conductive for imaging. Coating was performed under conditions of 8×10−3 mbar pressure, 15 mA current, and 100% turbo speed.36
Pharmacokinetics
Care of experimental organisms
The experimental organisms, male Sprague-Dawley rats (250–310 g), were freely provided with regular standard laboratory food and water. They were retained in cages under maintained environmental conditions of temperature (20°C–25°C) and relative humidity (45%–60%). Food was removed about 24 hours before the commencement of pharmacokinetic experimentations; however, they had free access to drinking water.37 The use of experimental organisms in pharmacokinetic studies was performed in accordance with the Guiding Principles in the Use of Animals in Toxicology,38 which were approved by the Institutional Animal Care and Use Committee and adopted by the Institute of Laboratory Animal Resources of Hanyang University.
Oral administration and blood sampling
A polyethylene tube was surgically introduced into the right carotid artery of each anesthetized rat. Then, each rat, appropriately held with an infusion harness, was placed in a separate chamber. The rats were free to move within their chambers and drink water. Twenty-four rats, cannulized in the right carotid artery, were trialed in this investigation. Each drug powder or fenofibrate-loaded nanoparticulated formulation, at a drug dose of 20 mg/kg, was filled in small, hard gelatin capsules (#9, Suheung Capsule Co., Seoul, South Korea) for oral administration.37 Six rats were given the drug powder via the oral route. Similarly, six rats were used for the pharmacokinetic evaluation of each formulation. Four hundred microliters of blood was sampled through the cannula tube with heparinized syringes at the following specified time points:15 minutes, 30 minutes, 1 hour, 1.5 hours, 2 hours, 4 hours, 6 hours, 10 hours, 14 hours, 24 hours, and 40 hours.39 Plasma was immediately separated from blood samples by centrifugation (Smart 15, Hanil Science Industrial Co.) at 10,000× g for 10 minutes. Thereafter, these plasma samples were stored at −20°C until HPLC analysis.39
Sample preparation and HPLC analysis
Fenofibrate is a prodrug that is bio-transformed by tissue and plasma esterases to the active metabolite fenofibric acid; therefore, there is essentially no fenofibrate detectable in the plasma after oral administration. Accordingly, the pharmacokinetic evaluation of fenofibrate is based on the quantification of fenofibric acid in the plasma.40–42 Fenofibric acid in rat plasma samples was extracted by liquid–liquid extraction. Two hundred microliters of each plasma sample was acidified with 200 μL of 1 M HCl in a 2 mL microtube. Then, 40 μL of an internal standard solution (clofibric acid, 400 μg/mL in acetonitrile) was added and vortex mixed for 1 minute. Subsequently, the sample mixture was extracted twice with 2 mL of diethyl ether by vortex mixing for 5 minutes and centrifuging at 10,000× g for 10 minutes (Smart 15; Hanil Science Industrial Co.). The supernatant organic layer was transferred to another clean microtube and evaporated at 40°C. After evaporation, the residue was reconstituted with 200 μL of acetonitrile by vortex mixing and centrifuged at 10,000× g for 10 minutes (Smart 15, Hanil Science Industrial Co.). One hundred and fifty microliters of supernatant was carefully withdrawn using a pipette and poured into a small-volume HPLC vial. A 100 μL of aliquot was assayed for fenofibric acid quantification. The HPLC system (Agilent 1260 Infinity; Agilent Technologies) used in this study was equipped with a Capcell Pak C18 column (4.6 mm ID ×250 mm, 5 μm; Shiseido). The column temperature was set at 30°C. The mobile phase, consisting of acetonitrile and 0.1% (v/v) aqueous solution of phosphoric acid at a volume ratio of 54:46, was eluted at a flow rate of 2 mL/min. The eluent was monitored at 285 nm for fenofibric acid detection. The inter- and intraday variances were within the acceptable limits (R2=0.999).
Results
The results of the saturation solubility of fenofibrate in 1% (w/v) hydrophilic polymer solutions and 1% (w/v) surfactant solutions are shown in Figure 1A and B, respectively. Among the tested hydrophilic polymers, HP-β-CD, gelatin, and PVP exhibited better solubility of fenofibrate; thus, they were selected for the preparation of various polymeric nanoparticulated formulations. Among the surfactants tested in this study, SLS showed the highest solubility of fenofibrate and was selected for the production of nanospheres of ternary solid dispersion in conjunction with PVP. The solubility of fenofibrate in each 1% (w/v) solution of HP-β-CD, PVP, gelatin, and SLS was 6.45±0.64 μg/mL, 1.96±0.12 μg/mL, 3.33±1.09 μg/mL, and 338.25±10.62 μg/mL, respectively. For each formulation, the optimized ratio of the corresponding components, which demonstrated the highest aqueous solubility and dissolution rate of fenofibrate, was selected for the preparation of the nanoparticulated formulations. Thus, the optimized ratios of constituents for PVP nanospheres, HP-β-CD nanocorpuscles, and gelatin nanocapsules were 2.5:4.5:1, 1:4, and 1:8, respectively.
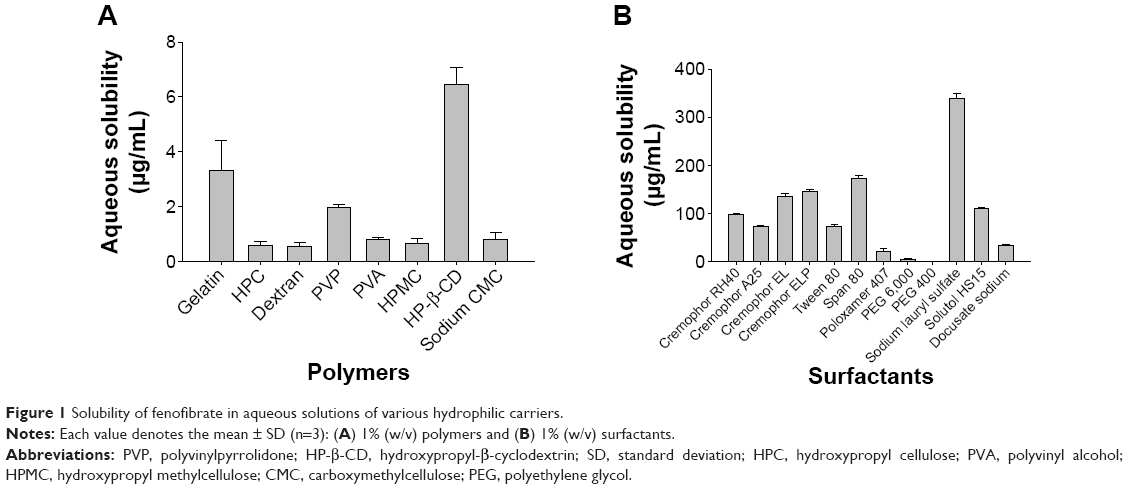 | Figure 1 Solubility of fenofibrate in aqueous solutions of various hydrophilic carriers. |
The influence of different nanoparticulated systems on the aqueous solubility and dissolution rate of fenofibrate is displayed in Figures 2 and 3, respectively. All formulations demonstrated remarkably improved solubility and dissolution rates of the drug compared to the fenofibrate powder (P<0.05). The gelatin nanocapsules produced the highest drug solubility (390.39±88.01 μg/mL); however, it was not significantly different (P>0.05) from the solubility outcomes of the HP-β-CD nanocorpuscles and PVP nanospheres (247.00±63.01 μg/mL and 272.26±58.84 μg/mL, respectively). The initial dissolution rates (up to 10 minutes) of the nanoparticulated formulations were significantly different (P<0.05) from one another; however, they were not significantly different (P>0.05) beyond 30 minutes. The initial dissolution rate was faster in the decreasing order of HP-β-CD nanocorpuscles > PVP nanospheres > gelatin nanocapsules. After 40 minutes, the dissolutions of the drug with the fenofibrate powder, PVP nanospheres, HP-β-CD nanocorpuscles, and gelatin nanocapsule formulations were 54.07%±1.72%, 88.19%±4.32%, 95.06%±5.25%, and 90.75%±6.40%, respectively. Thus, all of the nanoparticulated formulations significantly enhanced drug dissolution (about 80%–95%) compared to the drug powder; however, they were not significantly different from one another after 30 minutes.
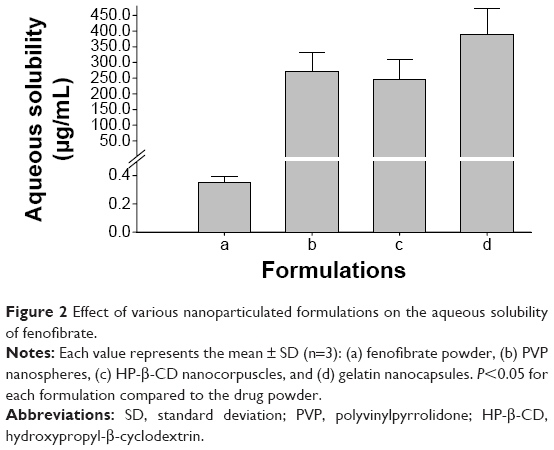 | Figure 2 Effect of various nanoparticulated formulations on the aqueous solubility of fenofibrate. |
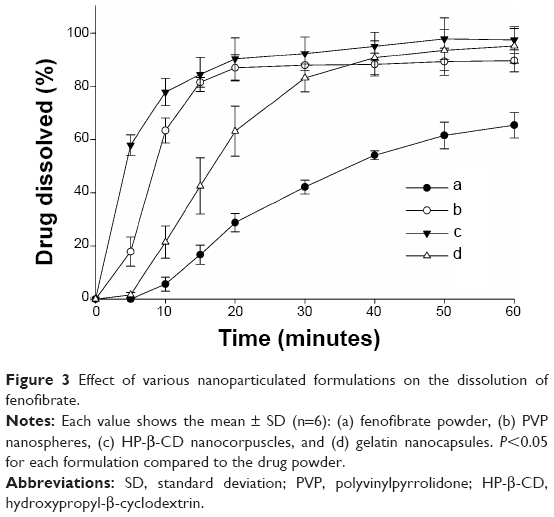 | Figure 3 Effect of various nanoparticulated formulations on the dissolution of fenofibrate. |
Figure 4A shows the X-ray diffraction patterns of the drug with fenofibrate powder and various nanoparticulated systems. Fenofibrate furnished sharp characteristic peaks at diffraction angles suggestive of the typical crystalline nature of the drug (Figure 4A, a). In the PVP nanospheres, the intensity of fenofibrate crystallinity was diminished (Figure 4A, b). However, crystalline fenofibrate peaks did not appear in the pattern obtained with HP-β-CD nanocorpuscles (Figure 4A, c). This suggested that the drug was changed into the amorphous state in the HP-β-CD nanocorpuscles. Furthermore, after nanoencapsulation with gelatin, the intensity of drug crystallinity was diminished remarkably (Figure 4A, d).
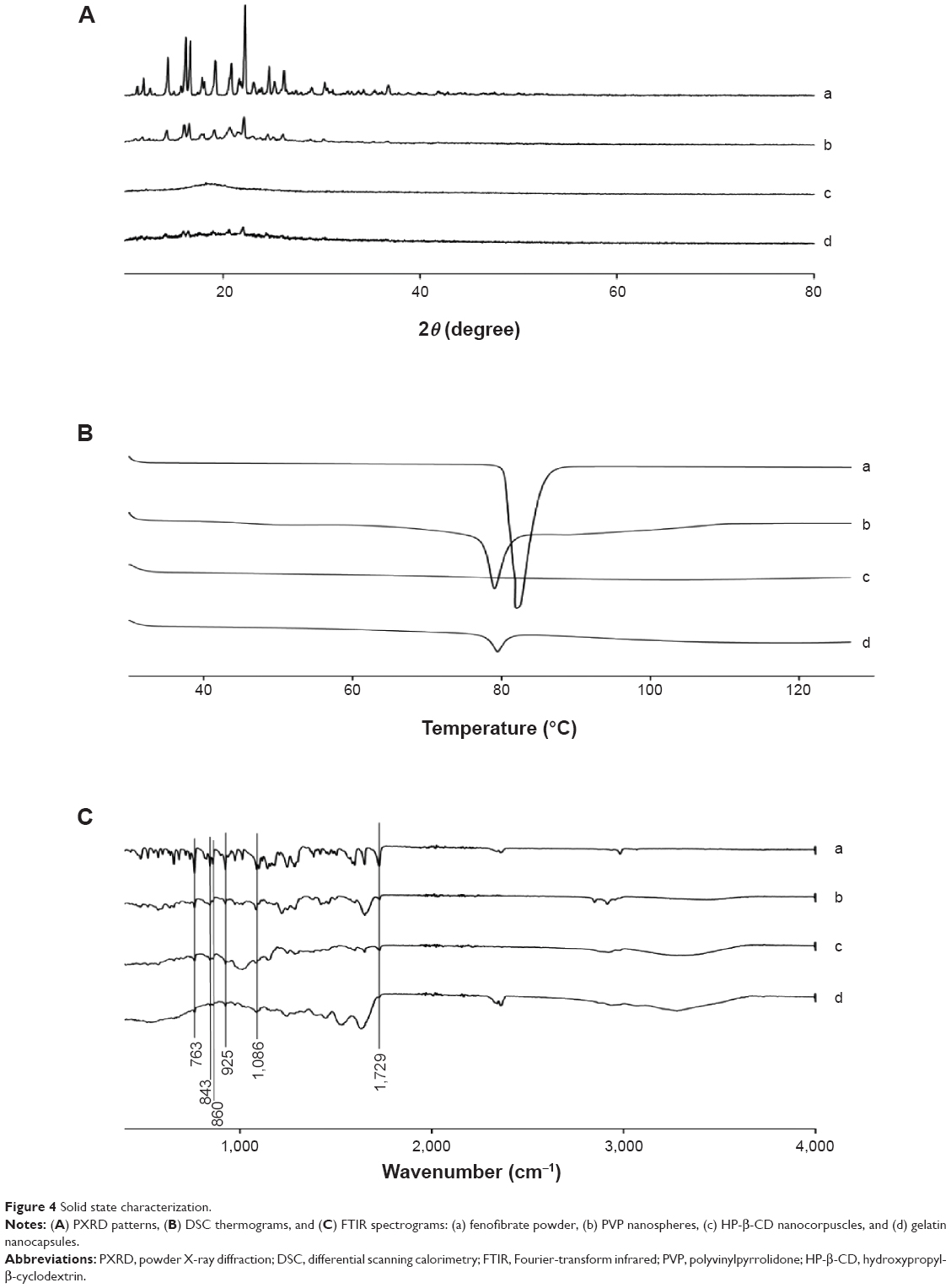 | Figure 4 Solid state characterization. |
Thermal characteristics of fenofibrate powder and different nanoparticles are displayed in Figure 4B. Fenofibrate powder generated a deep endotherm, corresponding to its melting point at approximately 82°C, confirming its characteristic crystalline nature (Figure 4B, a). Similarly, PVP nanospheres (Figure 4B, b) also produced a sharp endotherm at the melting point of fenofibrate, confirming that the drug was present in the crystalline state in the PVP nanospheres. Conversely, the HP-β-CD nanocorpuscles (Figure 4B, c) did not show an endotherm, meaning that the drug was present in an amorphous state in them. A very small endotherm was observed in case of the gelatin nanocapsules; therefore, the nanoencapsulated drug was in the crystalline state (Figure 4B, d).
The FTIR spectra of fenofibrate powder and various nanoparticles are shown in Figure 4C. Fenofibrate produced major distinguishing peaks at 1,729 cm−1, 1,086 cm−1, 925 cm−1, 860 cm−1, 843 cm−1, and 763 cm−1 (Figure 4C). These specific peaks also appeared in the FTIR patterns of the PVP nanosphere (Figure 4C, b), HP-β-CD nanocorpuscle (Figure 4C, c), and gelatin nanocapsule (Figure 4C, d) formulations.
The SEM images of the drug powder and the fenofibrate-loaded nanoparticulated formulations are displayed in Figure 5. The micrograph of the drug powder (Figure 5A) exhibited the presence of a crystalline solid. Figure 5B shows very small, round-shaped particles of the PVP nanospheres. However, HP-β-CD nanocorpuscles had irregular shapes and surfaces (Figure 5C). The fenofibrate-loaded gelatin nanocapsules (Figure 5D) appeared as smooth-surfaced spherical particles.
 | Figure 5 Scanning electron micrographs. |
The mean plasma concentration–time profiles of fenofibric acid with various nanoparticles, equivalent to a 20 mg/kg fenofibrate dose, are shown in Figure 6. At all time points, each fenofibrate-loaded nanoparticulated formulation showed a significantly higher plasma titer of fenofibric acid than the drug powder (P<0.05). The pharmacokinetic parameters are shown in Table 1. The area under the curve (AUC) and Cmax of each formulation were significantly enhanced compared to those of the drug powder (P<0.05). In addition, the gelatin nanocapsule formulation showed significantly higher AUC and Cmax values compared to the other formulations (P<0.05); however, AUC and Cmax values of PVP nanospheres and HP-β-CD nanocorpuscles were not significantly different from each other (P>0.05). The AUC values of the drug powder, PVP nanospheres, HP-β-CD nanocorpuscles, and gelatin nanocapsules were 17.72±1.52 h μg/mL, 61.01±14.66 h μg/mL, 42.59±11.55 h μg/mL, and 96.80±15.42 h μg/mL, respectively, whereas the Cmax values were 2.36±0.34, 6.73±1.75, 6.46±1.83, and 9.14±2.47, respectively. Furthermore, the half-life (t1/2), elimination rate constant (Kel), and mean residence time (MRT) were not significantly different (P>0.05) from one another.
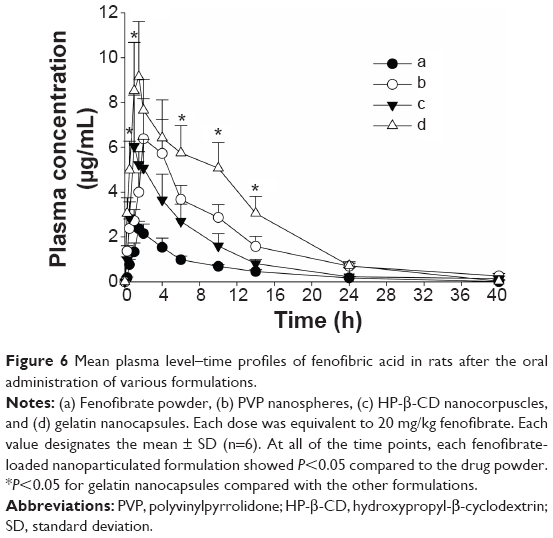 | Figure 6 Mean plasma level–time profiles of fenofibric acid in rats after the oral administration of various formulations. |
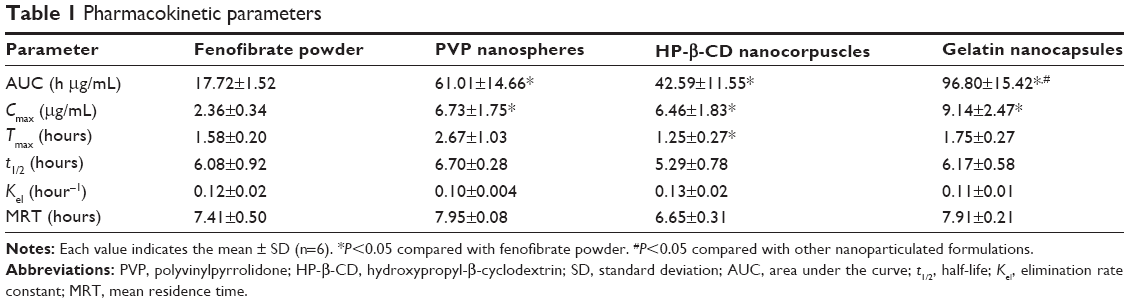 | Table 1 Pharmacokinetic parameters |
Discussion
Fenofibrate is highly lipophilic (log P=5.24)3 and practically insoluble in water;4 therefore, it is poorly absorbed from the gastrointestinal tract, resulting in impaired oral bioavailability.5 Nanoparticle formation is a promising way to ameliorate the aqueous solubility, dissolution rate, and absorption of poorly water-soluble drugs.12 In this study, the purpose was to determine a nanoparticulated formulation with the most improved oral bioavailability of poorly water-soluble fenofibrate among the tested nanoparticulated systems.
At first, to choose the appropriate carriers for the preparation of various nanoparticulated systems, the saturation solubility of fenofibrate in 1% (w/v) aqueous solutions of various hydrophilic polymers and surfactants was determined. Among the tested polymers, HP-β-CD, gelatin, and PVP demonstrated better solubility of fenofibrate (Figure 1A). Likewise, SLS exhibited the highest solubility of the drug among the surfactants tested in this study (Figure 1B). In the past, the concomitant use of PVP and SLS, as carriers in various formulations, has improved the aqueous solubility of several poorly water-soluble drugs;43–47 therefore, they were preferred as carriers for preparing nanospheres of a ternary solid dispersion in our study. Moreover, changing to the amorphous state can be a useful approach to remarkably improve the dissolution rate and bioavailability of poorly water-soluble active pharmaceutical ingredients.10,48 This can be accomplished by use of cyclodextrins (CDs).49–51 All previous toxicity studies have proven that orally administered CDs are practically nontoxic, owing to the lack of absorption from the gastrointestinal tract.50,52 HP-β-CD was selected for making fenofibrate/HP-β-CD nanocorpuscles because of its better solubilization ability for many lipophilic molecules.53 Furthermore, gelatin, a heterogeneous mixture of polypeptides,27 can promote the aqueous solubility, dissolution, and bioavailability of numerous water-insoluble drugs;28 thus, it was chosen for the preparation of fenofibrate/gelatin nanocapsules.
For the identification of the optimal ratio of selected constituents for the PVP nanospheres, a number of formulations were prepared with a constant ratio of drug/carriers (1:1) and varying polymer/surfactant quantities. Then, these nanosphere formulations were tested for the aqueous solubility and dissolution rate of the loaded drug (data not shown). Among the tested PVP nanosphere formulations, the nanospheres prepared with fenofibrate/PVP/SLS at the weight ratio of 2.5:4.5:1 exhibited the highest solubility and dissolution; therefore, this ratio was chosen for further investigation. Similarly, HP-β-CD nanocorpuscles and gelatin nanocapsules were prepared with the optimized drug/carrier (w/w) ratios of 1:4 and 1:8, respectively.
The hydrophilic carriers present in the nanoparticulated preparations improved the aqueous solubility and dissolution rate of the loaded drug compared with the drug powder. In particular, gelatin nanocapsules ameliorated solubility by about 1,100 times the solubility of the drug powder (390.39±88.01 μg/mL vs 0.35±0.04 μg/mL; Figure 2). However, the solubility results of the formulations were not significantly different from one another (P>0.05). The Food and Drug Administration discourages the use of only water as a dissolution medium for highly lipophilic agents, and recommends the use of a slight quantity of a surfactant in the dissolution medium to improve the dissolution efficiency of such chemical entities.34 Therefore, 900 mL of 2% (w/v) polysorbate 80 was used as a dissolution medium in our study.33 The reduction in the degree of crystallinity might be caused by improvements in the aqueous solubility and dissolution rate of the drug.54 HP-β-CD nanocorpuscles furnished the highest initial dissolution (up to 10 minutes; Figure 3C), owing to the conversion of the crystalline drug into the amorphous state. However, the initial dissolution rates of PVP nanospheres (Figure 3B) and gelatin nanocapsules (Figure 3D) were improved through the diminution of the crystalline intensity of the drug to the nanocrystalline state. The relatively retarded dissolution rate of the gelatin nanocapsules was attributed to the delayed rupture of the gelatin coat before release of the drug into the dissolution medium. Gelatin is soluble in water at temperatures of approximately 40°C and above,55,56 but the dissolution test was performed at 37°C.
The characteristic crystalline peaks in the PXRD pattern (Figure 4A, a) and deep endotherm corresponding to drug’s melting point (~82°C) in the DSC thermograms (Figure 4B, a) suggested that the drug was typically crystalline in nature. Moreover, the SEM image of the fenofibrate powder revealed crystals with irregular shapes and surfaces (Figure 5A).
During the preparation of the PVP nanospheres, all ingredients were dissolved completely prior to spray drying. The spray-dried PVP nanosphere formulation was expected to contain the amorphous form of the drug;22 however, the PXRD pattern showed that only the intensity of the crystallinity was decreased (Figure 4A, b). Furthermore, the DSC trace confirmed that the drug did not change into the amorphous state completely (Figure 4B, b). This suggested that the drug was recrystallized during the spray-drying process. The PVP suppressed the recrystallization of the drug from solution, thereby reducing the degree of crystallinity.57–60 The PXRD, DSC, and SEM (Figure 5B) results collectively advocate that the drug was present in the nanocrystalline state in the smooth-surfaced spherical nanoparticles. Thus, the remarkable enhancement in the solubility and dissolution rate of the drug loaded in the PVP nanospheres might be ascribed to the reduced degree of crystallinity and particle size.
On the other hand, the HP-β-CD nanocorpuscles presented the highest initial dissolution rate among the tested formulations. The PXRD and DSC results showed that the drug was present in the amorphous state in the HP-β-CD nanocorpuscles (Figure 4A, c and B, c).49–51 Moreover, the SEM image showed the reduced particle size of the formulation (Figure 5C). Thus, the improvement in solubility and dissolution rate was attributed to the conversion of the crystalline drug into the amorphous state.10,48
Gelatin is soluble in water ≥40°C.55,56 Moreover, it precipitates from the aqueous ethanol solution at concentrations of about 50% or above. Therefore, in the preparation of the gelatin nanocapsules, all of the components were completely dissolved in 40% ethanol at 50°C. Subsequently, a clear resultant solution was spray dried. The PXRD and DSC results depicted that the encapsulated drug was present in the crystalline form (Figure 4A, d and B, d). Thus, as with the PVP nanospheres, recrystallization also occurred during the spray-drying process of gelatin nanocapsules. The SEM results revealed that the gelatin nanocapsules were smooth-surfaced round nanoparticles (Figure 5D). The amelioration in the aqueous solubility and dissolution can be attributed to improved wetting, owing to the hydrophilic gelatin and a reduction in the degree of crystallinity.28,31
The FTIR spectrum of fenofibrate showed main distinctive peaks at 1,729 cm−1, 1,086 cm−1, 925 cm−1, 860 cm−1, 843 cm−1, and 763 cm−1 (Figure 4C). These particular peaks were also seen in the FTIR spectra of all formulations (Figure 4C). Furthermore, the FTIR spectrum of each formulation and its corresponding physical mixture (data not shown) was exactly the same; accordingly, the drug had no strong interaction with the excipients.
All of the fenofibrate-loaded nanoparticulated systems exhibited significantly higher plasma levels of fenofibric acid compared to the drug powder (Figure 6 and Table 1). The improvement in bioavailability was attributed to enhanced solubility and dissolution rates. The bioavailability of the PVP nanospheres compared to the HP-β-CD nanocorpuscles was not significantly different (P>0.05). As compared with the HP-β-CD nanocorpuscles, the PVP nanospheres positively influenced the oral bioavailability (AUC and Cmax) of fenofibrate, owing to the presence of SLS in the formulation. SLS is a permeation enhancer;61 therefore, it enhanced drug absorption through the gastrointestinal tract of rats.62,63 The bioavailability of gelatin nanocapsules was higher than those of the other formulations (P<0.05). The bioavailability enhancement through gelatin nanoencapsulation was approximately 5.5-fold compared to the drug powder. This might be credited to the slightly higher apparent aqueous solubility of the gelatin nanocapsules.
Conclusion
All of the developed polymeric nanoparticulated systems presented improved aqueous solubility, dissolution rate, and oral bioavailability of fenofibrate compared to that of the plain drug powder (P<0.05). The accelerated dissolution rates of the nanoparticles can be attributed to the following: (a) an ameliorated solubility of fenofibrate, owing to its conversion into the amorphous form or nanocrystalline state (Kelvin’s law);64–66 (b) a comparatively large surface area available for dissolution;67,68 (c) facilitated wetting of fenofibrate, owing to the presence of hydrophilic polymeric matrix in the formulation;69,70 and (d) reduction in the crystalline intensity.54 Among the different nanoparticles studied in this research, including PVP nanospheres, HP-β-CD nanocorpuscles, and gelatin nanocapsules, the latter technique of nanoencapsulation of fenofibrate with gelatin at a ratio of 1:8 (w/w) showed the most improved apparent solubility and oral bioavailability in rats. Accordingly, gelatin nanocapsule might be a promising device to deliver poorly water-soluble fenofibrate with enhanced oral bioavailability.
Acknowledgment
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MEST) (number NRF-2012R1A2A2A01045658).
Disclosure
The authors report no conflicts of interests in this work.
References
Guay D. Micronized fenofibrate: a new fibric acid hypolipidemic agent. Ann Pharmacother. 1999;33(10):1083–1103. | ||
Genest JJ, Nguyen N-H, Theroux P, Davignon J, Cohn JS. Effect of micronized fenofibrate on plasma lipoprotein levels and hemostatic parameters of hypertriglyceridemic patients with low levels of high-density lipoprotein cholesterol in the fed and fasted state. J Cardiovasc Pharmacol. 2000;35(1):164–172. | ||
Munoz A, Guichard JP, Reginault P. Micronised fenofibrate. Atherosclerosis. 1994;110(suppl(0)):S45–S48. | ||
Ming-Thau S, Ching-Min Y, Sokoloski TD. Characterization and dissolution of fenofibrate solid dispersion systems. Int J Pharm. 1994;103(2):137–146. | ||
Dressman J, Butler J, Hempenstall J, Reppas C. The BCS: where do we go from here? Pharm Technol. 2001;25(7):68–77. | ||
Badens E, Majerik V, Horváth G, et al. Comparison of solid dispersions produced by supercritical antisolvent and spray-freezing technologies. Int J Pharm. 2009;377(1–2):25–34. | ||
Cutler L, Howes C, Deeks NJ, Buck TL, Jeffrey P. Development of a P-glycoprotein knockout model in rodents to define species differences in its functional effect at the blood–brain barrier. J Pharm Sci. 2006;95(9):1944–1953. | ||
Yoshihashi Y, Iijima H, Yonemochi E, Terada K. Estimation of physical stability of amorphous solid dispersion using differential scanning calorimetry. J Therm Anal Calorim. 2006;85(3):689–692. | ||
Goddeeris C, Coacci J, Van den Mooter G. Correlation between digestion of the lipid phase of smedds and release of the anti-HIV drug UC 781 and the anti-mycotic drug enilconazole from smedds. Eur J Pharm Biopharm. 2007;66(2):173–181. | ||
Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50(1):47–60. | ||
Perrut M, Jung J, Leboeuf F. Enhancement of dissolution rate of poorly-soluble active ingredients by supercritical fluid processes: part I: micronization of neat particles. Int J Pharm. 2005;288(1):3–10. | ||
Jia Z, Lin P, Xiang Y, et al. A novel nanomatrix system consisted of colloidal silica and pH-sensitive polymethylacrylate improves the oral bioavailability of fenofibrate. Eur J Pharm Biopharm. 2011;79(1):126–134. | ||
Xing F, Cheng G, Yi K, Ma L. Nanoencapsulation of capsaicin by complex coacervation of gelatin, acacia, and tannins. J Appl Polym Sci. 2005;96(6):2225–2229. | ||
Mora-Huertas C, Fessi H, Elaissari A. Polymer-based nanocapsules for drug delivery. Int J Pharm. 2010;385(1):113–142. | ||
Pinto Reis C, Neufeld RJ, Ribeiro AJ, Veiga F. Nanoencapsulation I. Methods for preparation of drug-loaded polymeric nanoparticles. Nanomedicine. 2006;2(1):8–21. | ||
Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE. Biodegradable polymeric nanoparticles as drug delivery devices. J Control Release. 2001;70(1):1–20. | ||
Heng D, Cutler DJ, Chan H-K, Yun J, Raper JA. What is a suitable dissolution method for drug nanoparticles? Pharm Res. 2008;25(7):1696–1701. | ||
Vauthier C, Bouchemal K. Methods for the preparation and manufacture of polymeric nanoparticles. Pharm Res. 2009;26(5):1025–1058. | ||
Quintanar-Guerrero D, Allémann E, Fessi H, Doelker E. Preparation techniques and mechanisms of formation of biodegradable nanoparticles from preformed polymers. Drug Dev Ind Pharm. 1998;24(12):1113–1128. | ||
Broadhead J, Edmond Rouan S, Rhodes C. The spray drying of pharmaceuticals. Drug Dev Ind Pharm. 1992;18(11–12):1169–1206. | ||
Chauhan B, Shimpi S, Paradkar A. Preparation and evaluation of glibenclamide-polyglycolized glycerides solid dispersions with silicon dioxide by spray drying technique. Eur J Pharm Sci. 2005;26(2):219–230. | ||
Joe JH, Lee WM, Park YJ, et al. Effect of the solid-dispersion method on the solubility and crystalline property of tacrolimus. Int J Pharm. 2010;395(1–2):161–166. | ||
Peltonen L, Valo H, Kolakovic R, Laaksonen T, Hirvonen J. Electrospraying, spray drying and related techniques for production and formulation of drug nanoparticles. Expert Opin Drug Deliv. 2010;7(6):705–719. | ||
Van Drooge D, Hinrichs W, Visser M, Frijlink H. Characterization of the molecular distribution of drugs in glassy solid dispersions at the nanometer scale, using differential scanning calorimetry and gravimetric water vapour sorption techniques. Int J Pharm. 2006;310(1):220–229. | ||
Chiou WL, Riegelman S. Pharmaceutical applications of solid dispersion systems. J Pharm Sci. 1971;60(9):1281–1302. | ||
Jizomoto H, Kanaoka E, Sugita K, Hirano K. Gelatin–acacia microcapsules for trapping micro-oil droplets containing lipophilic drugs and ready disintegration in the gastrointestinal tract. Pharm Res. 1993;10(8):1115–1122. | ||
Báez J, Olsen D, Polarek J. Recombinant microbial systems for the production of human collagen and gelatin. Appl Microbiol Biotechnol. 2005;69(3):245–252. | ||
Piao MG, Yang CW, Li DX, et al. Preparation and in vivo evaluation of piroxicam-loaded gelatin microcapsule by spray drying technique. Biol Pharm Bull. 2008;31(6):1284–1287. | ||
Menting L, Hoogstad B, Thijssen H. Diffusion coefficients of water and organic volatiles in carbohydrate-water systems. Int J Food Sci Technol. 1970;5(2):111–126. | ||
Thijssen HAC. Flavour retention in drying preconcentrated food liquids. J Appl Chem Biotechnol. 1971;21(12):372–377. | ||
Li DX, Yan YD, Oh DH, et al. Development of valsartan-loaded gelatin microcapsule without crystal change using hydroxypropylmethylcellulose as a stabilizer. Drug Deliv. 2010;17(5):322–329. | ||
Mehmood Yousaf A, Wuk Kim D, Choi H-G, Oh E. Validation of a highly sensitive RP-HPLC method for quantification of fenofibrate in pure and pharmaceutical dosage forms. Curr Pharm Anal. 2014;10(2):97–104. | ||
Stamm A, Seth P, inventor; Labaratoires Fournier, assignee. Fenofibrate pharmaceutical composition having high bioavailabilty and method for preparing it. Google patents US 6277405. 2001. Available from: http://www.google.com/patents/US6277405 | ||
Shah VP, Noory A, Noory C, et al. In vitro dissolution of sparingly water-soluble drug dosage forms. Int J Pharm. 1995;125(1):99–106. | ||
Sanganwar GP, Gupta RB. Dissolution-rate enhancement of fenofibrate by adsorption onto silica using supercritical carbon dioxide. Int J Pharm. 2008;360(1–2):213–218. | ||
Windbergs M, Strachan CJ, Kleinebudde P. Understanding the solid-state behaviour of triglyceride solid lipid extrudates and its influence on dissolution. Eur J Pharm Biopharm. 2009;71(1):80–87. | ||
Oh DH, Park Y-J, Kang JH, Yong CS, Choi H-G. Physicochemical characterization and in vivo evaluation of flurbiprofen-loaded solid dispersion without crystalline change. Drug Deliv. 2011;18(1):46–53. | ||
Society of Toxicology. Guiding Principles in the Use of Animals in Toxicology. Reston, VA: Society of Toxicology; 2008. | ||
Li X, Gu L, Xu Y, Wang Y. Preparation of fenofibrate nanosuspension and study of its pharmacokinetic behavior in rats. Drug Dev Ind Pharm. 2009;35(7):827–833. | ||
Straka RJ, Burkhardt RT, Fisher JE. Determination of fenofibric acid concentrations by HPLC after anion exchange solid-phase extraction from human serum. Ther Drug Monit. 2007;29(2):197–202. | ||
Streel B, Hubert P, Ceccato A. Determination of fenofibric acid in human plasma using automated solid-phase extraction coupled to liquid chromatography. J Chromatogr B Biomed Sci Appl. 2000;742(2):391–400. | ||
Tang N, Lai J, Chen Y, Lu Y, Wu W. Fenofibrate solid dispersion pellets prepared by fluid-bed coating: physical characterization, improved dissolution and oral bioavailability in beagle dogs. J China Pharm Sci. 2009;18(2):156–161. | ||
Dave RH, Patel HH, Donahue E, Patel AD. To evaluate the change in release from solid dispersion using sodium lauryl sulfate and model drug sulfathiazole. Drug Dev Ind Pharm. 2013;39(10):1562–1572. | ||
Giri TK, Badwaik H, Alexander A, Tripathi DK. Solubility enhancement of ibuprofen in the presence of hydrophilic polymer and surfactant. Int J Appl Biol Pharm Tech. 2010;1(2):793–800. | ||
Kim MS, Kim JS, Park HJ, Cho WK, Cha K-H, Hwang SJ. Enhanced bioavailability of sirolimus via preparation of solid dispersion nanoparticles using a supercritical antisolvent process. Int J Nanomedicine. 2011;6:2997. | ||
Leane MM, Sinclair W, Qian F, et al. Formulation and process design for a solid dosage form containing a spray-dried amorphous dispersion of ibipinabant. Pharm Dev Technol. 2013;18(2):359–366. | ||
Moes J, Koolen S, Huitema A, Schellens J, Beijnen J, Nuijen B. Pharmaceutical development and preliminary clinical testing of an oral solid dispersion formulation of docetaxel. Int J Pharm. 2011;420(2):244–250. | ||
Hancock B, Parks M. What is the true solubility advantage for amorphous pharmaceuticals? Pharm Res. 2000;17(4):397–404. | ||
Müller BW, Brauns U. Solubilization of drugs by modified β-cyclodextrins. Int J Pharm. 1985;26(1–2):77–88. | ||
Pitha J, Pitha J. Amorphous water-soluble derivatives of cyclodextrins: nontoxic dissolution enhancing excipients. J Pharm Sci. 1985;74(9):987–990. | ||
Antoniadou-Vyza E, Buckton G, Michaleas SG, Loukas YL, Efentakis M. The formation of an inclusion complex of methocarbamol with hydroxypropyl-β-cyclodextrin: the effect on chemical stability, solubility and dissolution rate. Int J Pharm. 1997;158(2):233–239. | ||
Irie T, Uekama K. Pharmaceutical applications of cyclodextrins III. Toxicological issues and safety evaluation. J Pharm Sci. 1997;86(2):147–162. | ||
Miro A, Quaglia F, Giannini L, Cappello B, Immacolata La Rotonda M. Drug/cyclodextrin solid systems in the design of hydrophilic matrices: a strategy to modulate drug delivery rate. Curr Drug Deliv. 2006;3(4):373–378. | ||
Sethia S, Squillante E. Solid dispersion of carbamazepine in PVP K30 by conventional solvent evaporation and supercritical methods. Int J Pharm. 2004;272(1–2):1–10. | ||
Van Den Bulcke AI, Bogdanov B, De Rooze N, Schacht EH, Cornelissen M, Berghmans H. Structural and rheological properties of methacrylamide modified gelatin hydrogels. Biomacromolecules. 2000;1(1):31–38. | ||
Djabourov M, Papon P. Influence of thermal treatments on the structure and stability of gelatin gels. Polymer. 1983;24(5):537–542. | ||
Craig DQ. The mechanisms of drug release from solid dispersions in water-soluble polymers. Int J Pharm. 2002;231(2):131–144. | ||
Pouton CW. Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci. 2006;29(3–4):278–287. | ||
Taylor L, Zografi G. Spectroscopic characterization of interactions between PVP and indomethacin in amorphous molecular dispersions. Pharm Res. 1997;14(12):1691–1698. | ||
Doherty C, York P. Accelerated stability of an X-Ray amorphous frusemide-polyvinylpyrrolidone solid dispersion. Drug Dev Ind Pharm. 1989;15(12):1969–1987. | ||
Rege BD, Yu LX, Hussain AS, Polli JE. Effect of common excipients on Caco-2 transport of low-permeability drugs. J Pharm Sci. 2001;90(11):1776–1786. | ||
Briseid G, Briseid K, Kirkevold K. Increased intestinal absorption in the rat caused by sodium lauryl sulphate, and its possible relation to the cAMP system. Naunyn Schmiedebergs Arch Pharmacol. 1976;292(2):137–144. | ||
Miller JM, Beig A, Krieg BJ, et al. The solubility–permeability interplay: mechanistic modeling and predictive application of the impact of micellar solubilization on intestinal permeation. Mol Pharm. 2011;8(5):1848–1856. | ||
Dai W-G, Dong LC, Song Y-Q. Nanosizing of a drug/carrageenan complex to increase solubility and dissolution rate. Int J Pharm. 2007;342(1):201–207. | ||
Mura P, Cirri M, Faucci M, Ginès-Dorado J, Bettinetti G. Investigation of the effects of grinding and co-grinding on physicochemical properties of glisentide. J Pharm Biomed Anal. 2002;30(2):227–237. | ||
Yonemochi E, Kitahara S, Maeda S, Yamamura S, Oguchi T, Yamamoto K. Physicochemical properties of amorphous clarithromycin obtained by grinding and spray drying. Eur J Pharm Sci. 1999;7(4):331–338. | ||
Kubo H, Mizobe M. Improvement of dissolution rate and oral bioavailability of a sparingly water-soluble drug, (+/−)-5-[[2-(2-naphthalenylmethyl)-5-benzoxazolyl] methyl]-2,4-thiazolidinedione, in co-ground mixture with D-mannitol. Biol Pharm Bull. 1997;20:460–463. | ||
Purvis T, Vaughn JM, Rogers TL, et al. Cryogenic liquids, nanoparticles, and microencapsulation. Int J Pharm. 2006;324(1):43–50. | ||
Chow AH, Hsia CK, Gordon JD, Young JW, Vargha-Butler EI. Assessment of wettability and its relationship to the intrinsic dissolution rate of doped phenytoin crystals. Int J Pharm. 1995;126(1):21–28. | ||
Kawashima Y, Saito M, Takenaka H. Improvement of solubility and dissolution rate of poorly water-soluble salicylic acid by a spray-drying technique. J Pharm Pharmacol. 1975;27(1):1–5. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.