")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Enhanced oral absorption and therapeutic effect of acetylpuerarin based on D-α-tocopheryl polyethylene glycol 1000 succinate nanoemulsions
Authors Sun D, Wei X, Xue X, Fang Z, Ren M, Lou H, Zhang X
Received 8 March 2014
Accepted for publication 27 April 2014
Published 18 July 2014 Volume 2014:9(1) Pages 3413—3423
DOI https://doi.org/10.2147/IJN.S63777
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Deqing Sun,1,2 Xinbing Wei,1 Xia Xue,2 Zengjun Fang,3 Manru Ren,1 Haiyan Lou,1 Xiumei Zhang1
1Department of Pharmacology, School of Medicine, Shandong University, Jinan, People’s Republic of China; 2Department of Pharmacy, 3Department of Clinical Pharmacology, Second Hospital of Shandong University, Jinan, People’s Republic of China
Background: Acetylpuerarin (AP), because of its lower water solubility, shows poor absorption that hinders its therapeutic application. Thus, the aim of this study was to prepare nanoemulsions for AP, enhance its oral bioavailability, and thus improve the therapeutic effect.
Methods: The nanoemulsions stabilized by D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) were prepared by high-pressure homogenization and characterized in terms of particle size, drug loading, morphology, and in vitro drug release. A lipid digestion model was used to predict in vivo drug solubilization in the gastrointestinal environment. The pharmacokinetics of AP formulations were performed in rats; meanwhile, a chylomicron flow-blocking rat model was used to evaluate the lymphatic drug transport. Moreover, the therapeutic effects of AP nanoemulsions on the model of focal cerebral ischemia-reperfusion for brain injury were also assessed.
Results: The nanoemulsions with a droplet size of 150 nm were well stabilized by TPGS and showed a high loading capacity for AP. In the digestion model, the distribution of AP in aqueous phase/pellet phase was about 90%/10% for nanoemulsions and 5%/95% for oil solution, indicating that the drug encapsulated in nanoemulsions would present in solubilized form after transportation into the gastrointestinal tract, whereas drug precipitation would occur as the oil solution was orally administered. The area under the curve value of AP nanoemulsions was 5.76±0.56 µg·hour·mL-1, or was about 2.6 and 1.7 times as great as that of suspension and oil solution, respectively, indicating enhanced drug absorption and thus achieving a better neuroprotection effect on cerebral ischemic reperfusion injury. The values of peak plasma concentration and area under the curve from the blocking model were significantly less than those of the control model, suggesting that the lymphatic transport performed a very important role in absorption enhancement.
Conclusion: Enhanced oral bioavailability in nanoemulsions was achieved via the mechanism of the maintenance of drug solubilization in the gastrointestinal tract and the enhancement of lymphatic transport, which resulted in therapeutic improvement of cerebral ischemic reperfusion injury.
Keywords: absorption, nanoemulsions, TPGS, lipid digestion, lymphatic transport, chylomicron flow-blocking rat model, transient middle cerebral artery occlusion rat model
Introduction
At this time, cardiovascular diseases are among the top health problems of the Chinese people. A fifth of adults are suffering from the disease, and both morbidity and mortality remain at a high level.1 Of the subtypes, ischemic cerebrovascular disease, which accounts for 80% of all cases, is a leading cause of disability and death in both rural residents and urban citizens.1,2 Thus, the measures for treatment of ischemic cerebrovascular disease need to be further enforced without delay.
In an ischemic cerebrovascular disease, blood cannot reach the brain, making the brain cells suffer from a lack of the nutrients and oxygen that they normally get.2 Thrombolytic and protective therapies of drug are two major measures in the treatment of ischemic brain vascular diseases at this time. Acetylpuerarin (AP; Figure 1) is one of the effective drugs that can improve blood circulation, treat coronary heart disease and arrhythmia, and decrease myocardial oxygen consumption in both animals and human patients suffering from cardiovascular disease.3,4 Our previous reports indicate that AP could exert protective effects against brain ischemia-reperfusion injury by reducing the damage of oxygen-free radicals, increasing the activity of antioxidase, and inhibiting the cell apoptosis in an ischemia area in vivo.2,5,6 Parenteral administration of AP produces significant neuroprotective effects on rats against focal cerebral ischemia-reperfusion injury because the drug goes directly into the systemic circulation and rapidly reaches the site of action; moreover, the onset of action is quick. Nevertheless, looking at the quality of life and need for follow-up therapy after diagnosis of the disease, the oral route has gained a major focus compared with the parenteral route.7 However, as a result of its lower water solubility, the oral administration of AP is limited by its lower bioavailability (only 5%–6%) and the resultant poor therapeutic effects.8–11 Thus, a new formulation that can enhance its solubility and oral absorption is highly desirable.
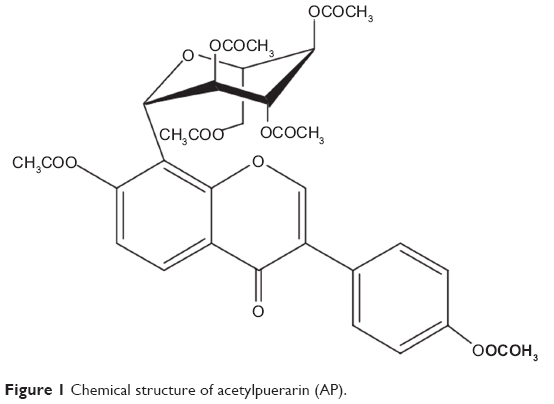 | Figure 1 Chemical structure of acetylpuerarin (AP). |
Lipid-based nanoemulsions are nonequilibrium, heterogeneous systems consisting of two immiscible phases in which one phase is dispersed in the other phase as droplets with a mean diameter of 1–1,000 nm.12,13 Oil-in-water nanoemulsion is a promising delivery system for the safe and effective delivery of oral insoluble drugs with the potential for application to human therapy by increasing the efficiency of oral administration.13,14 The drug can be incorporated into the oil droplets with high drug loading, enhancing its oral bioavailability and stability; meanwhile, the increased surface-to-volume ratio of nanoemulsions facilitates drug transport across the cell membrane, achieving sustained and targeted drug delivery.15 Moreover, the nanoemulsions are easy to prepare and scale up, having great promise for being industrialized and applied in the clinic.16,17
To stabilize the nanoemulsions, an emulsifier must be involved to decrease the interface tension of oil droplets. D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS), which is formed by the esterification of vitamin E succinate with polyethylene glycol, is a water-soluble derivative of natural vitamin E, which has been used in pharmaceutical formulations approved by the US Food and Drug Administration.10,18 It possesses much higher emulsification effects than other emulsifiers such as Tween 80 (BASF Corporation, Florham Park, NJ, USA), Cremophor EL (BASF Corporation, Florham Park, NJ, USA), and egg lecithin;18 moreover, it can increase the membrane permeability of insoluble drugs including paclitaxel, docetaxel, cisplatin, and probucol by inhibiting p-glycoprotein and thereby improving oral absorption.19,20 It is believed that TPGS is an ideal emulsifier for nanoemulsion-based oral delivery systems for insoluble drugs.21
The present study develops and characterizes the TPGS-stabilized nanoemulsions loaded with AP, evaluates in vitro drug release and in vitro lipid digestion, assesses the oral bioavailability and mechanism of enhanced absorption in rats, and studies the therapeutic effects on cerebral ischemic reperfusion injury in rats.
Materials and methods
Materials
Puerarin (batch 110752-200912) was purchased from the National Institutes for Food and Drug Control (Beijing, People’s Republic of China). AP was gifted by Professor Sun (Shandong Academy of Medical Science, Jinan, People’s Republic of China). TPGS was obtained from Eastman Chemical Company (Kingsport, TN, USA). Ethyl linoleate was from Energy Chemical Co., Ltd. (Shanghai, People’s Republic of China). Polysorbate 80 (Tween 80) was purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, People’s Republic of China). 4-bromophenylboronic acid and 2,3,5-triphenyltetrazolium chloride were from Aldrich Chemicals Co. (St Louis, MO, USA). Male Wistar rats (Jinan, Shandong, People’s Republic of China; clean grade, certificate SCXK [LU] 2013009) were purchased from the Laboratory Animal Center, Shandong University, People’s Republic of China. Other reagents were of analytical grade and used as received.
Preparation of AP nanoemulsions and stability study
The AP nanoemulsions were prepared by high-pressure homogenization.22,23 Briefly, the oil phase was prepared by dissolving 100 mg AP in ethyl linoleate (2 mL) under sonication at room temperature; the aqueous phase was prepared by dispersing 0.48 g TPGS in distilled water (30 mL) under the same conditions. The aqueous phase and oil phase were combined and then mixed by high-shear homogenization at 10,000 rpm for 5 minutes, using a FSH-2A blender (Jintan Medical Instrument, Jiangsu, People’s Republic of China) to form the coarse emulsions. Nanoemulsions were obtained by passing these coarse emulsions through a high-pressure homogenization (Niro-Soavi, S.p.A., Parma, Italy) for 30 cycles at 12,000 psi.
Reference formulations were also prepared in which 61 mg AP was dispersed in 19.9 mg 0.1% sodium carboxymethyl starch water solution or ethyl linoleate to prepare AP suspension or oil solution.
Short-term stability of the optimized formulations (100 mg AP, 2 mL ethyl linoleate, 0.48 g TPGS, 30 mL water) for 30 days was maintained at room temperature by assessing the particle size and drug loading.
Particle size determination
After diluting 100-fold with distilled water, the mean particle size and polydispersity index (PI) of nanoemulsions were determined by Zetasizer 3000 HS (Malvern Instruments, Malvern, United Kingdom). Three parallel measurements were performed for each sample.
Transmission electron microscope
The morphology of nanoemulsions was analyzed by an H-7000 transmission electron microscope (Hitachi, Tokyo, Japan). After dilution (1/100) in water, the sample was stained with 2% (weight/volume [w/v]) uranyl acetate for 5 minutes at 25°C and was then placed on copper film grids and observed by transmission electron microscopy after drying for 10 minutes at 25°C.
In vitro drug release
The AP release from nanoemulsions and suspension was determined using a dialysis method described by Doh et al.24 Briefly, a 0.2 mL sample was added to a dialysis bag (12,000–14,000 MW cutoff) that was incubated in 100 mL release medium that contained 1% Tween 80 (w/v) to maintain a sink condition. The experiment was performed in a controlled temperature water bath with a shaking speed of 100 rpm at 37°C. At predetermined time intervals, 1 mL external medium was removed for analysis and replaced with 1 mL release medium. After being filtered through a 0.22 μm cellulose acetate membrane, the concentration of AP was determined by a high-performance liquid chromatography method described in the section of “Analytical methods”. Each sample was carried out in triplicate.
In vitro digestion
The digestion experiment was carried out at 37°C, as described in previous reports.25–27 Briefly, 50 mg nanoemulsions or an ethyl linoleate-containing drug was dispersed in 20 mg digestion medium containing a mixture of 50 mM tris-maleate (pH, 7.5), 150 mM sodium chloride, 5 mM calcium chloride, 5 mM sodium taurocholate, and 1.25 mM soy phosphatidylcholine. The reaction was initiated by dispersing 1 mL porcine pancreatic lipase solution (4,000 tributyrin units/mL) into the digestive system. A solution of sodium hydroxide (1M) was added into the digestive system to maintain the pH at 7.5.25–27 At the end of the experiment, the lipolysis was terminated by the addition of 1 mL lipase inhibitor (1 M 4-bromophenylboronic acid in methanol). Two milliliters of the postdigestion mixtures were withdrawn and ultracentrifuged at 310,000 g for 30 minutes at 4°C (Optima L-80XP ultracentrifuge; Beckman Coulter Inc., Brea, CA, USA) to separate the mixtures into both an aqueous phase, containing the drug solubilized in micellar structures making up endogenous bile salts and phospholipid and exogenous lipid digestion products, and a pellet phase containing precipitated drug and insoluble (calcium) soaps of fatty acid.28 The aqueous phase or pellet phase was dissolved in 2 mL methanol and then centrifuged at 5,000 g for 5 minutes to remove the undissolved substances. The drug in supernatant was measured by a high-performance liquid chromatography method described in the section of “Analytical methods”.
Pharmacokinetics and mechanism of absorption in rats
The rats (200–250 g) used in the experiments received care according to Shandong University Unit for Laboratory Animal Medicine guidelines. The animal experiments were conducted using protocols approved by the Shandong University Institutional Animal Care and Use Committee.
The pharmacokinetics of test formulation (optimized nanoemulsions of AP) and reference formulations (AP suspension containing 0.1% sodium carboxymethyl starch and oil solution of AP) were conducted in Wistar rats. Eighteen rats fasted for 12 hours and were randomly divided into three groups (six rats for each group). The AP formulations were orally administered to rats in each group at an AP dose of 30 mg/kg. At predetermined points, a blood sample of 0.3 mL was collected from jugular vein in heparinized tubes. Plasma samples were separated by centrifugation for 10 minutes at 3,000 g and stored at −20°C until analysis.
To evaluate the mechanism of enhanced absorption, a chylomicron flow-blocking rat model was used.29,30 Twenty-four rats were divided into four groups (six rats in each group), which were treated with either an intraperitoneal injection of 3 mg/kg cycloheximide in saline (0.6 mg/mL) or with an equal volume of saline 1 hour before the experiment. One hour after the injection, the formulations of oil solution and nanoemulsions were orally administered to the rats at an AP dose of 30 mg/kg. The blood samples were collected as described in the above paragraph.
Sample preparation
Fifty microliters plasma sample, 25 μL methanol-acetonitrile solution (1:1, volume/volume [v/v]), and 150 μL an internal standard solution (100 ng/mL of genistein in methanol) were placed into a plastic tube and vortexed for 1 minute. After being centrifuged at 10,000 g for 10 minutes, 15 μL of supernatant was injected for analysis, as described in the section of “Analytical methods”.
Analytical methods
The drug concentration of AP in both the release and digestion medium was measured by a high-performance liquid chromatography method (Agilent 1200; Agilent Technologies, Santa Clara, CA, USA) described in our previous report with minor modification.11 The separation was performed on an Eclipse XDB C18 column (250×4.6 mm, 5 μm; Agilent Technologies) at 25°C. The mobile phase consisted of a mixture of 0.1% phosphoric acid solution and methanol (40:60, v/v). The flow rate of the mobile phase was 0.7 mL/minute. The eluent was detected by an ultraviolet detector at 250 nm.
AP would be metabolized to puerarin after oral administration; thus, the pharmacokinetic evaluation was based on the quantification of puerarin in plasma.9,11 The drug concentration of puerarin in plasma samples was determined by an liquid chromatography–mass spectrometry/mass spectrometry system that included an Agilent 1100 system (consisting of a binary pump with a vacuum degasser, a thermostated column compartment, and an autosampler) and a 4000QTRAP triple–quadrupole mass tandem spectrometer (AB Sciex Instruments, Framingham, MA, USA) equipped with an electrospray ionization source for mass analysis and detection. Analyst 1.3 software (Thermo Fisher Scientific, Waltham, MA, USA) was used for data collection and analysis.
Mass spectrometric analysis was performed in positive ion mode and set up in a multiple reactions monitoring mode by monitoring ion transitions at m/z 417.2→297.2 for puerarin and m/z 271.2→153.0 for the internal standard, genistein. The conditions of the mass spectrum were set as turbo ion spray temperature, 450°C; ion spray voltage, 5,000 V; declustering potential, 80 V; and collision energy, 34 V for puerarin and 39 V for genistein. Nitrogen served as nebulizer gas, auxiliary gas, curtain gas, and collision gas.
Chromatographic separation was carried out on a CAPCELL PAK C18 MG III column (150×2.0 mm, 5 μm; Shiseido, Kanagawa, Japan) by isocratic elution at a flow rate of 0.35 mL/minute. The mobile phase consisted of a mixture of 5 mM ammonium formate/methanol (35:65, v/v) containing 0.1% formic acid solution. The column temperature was maintained at 35°C.
Pharmacodynamics in rats
Focal cerebral ischemia–reperfusion model and oral administration
The rats (280–320 g) were randomly divided into five groups (eight rats for each group): the sham-operated (Sham) group and groups treated with blank nanoemulsions without drug loading, AP formulations (suspension, oil solution, and nanoemulsions). The rats were orally administrated for 7 days before middle cerebral artery occlusion (MCAO) procedure and 0.5 hours before the onset of ischemia at an AP dose of 30 mg/kg.
The model of brain ischemia–reperfusion injury was prepared by MCAO in rats. The MCAO rats were produced by intraluminal occlusion of the left middle cerebral artery with a nylon monofilament, as described previously.31 Briefly, the rats were anesthetized with 10% chloral hydrate at a dose of 350 mg/kg. After separating the external carotid artery and internal carotid artery, a nylon monofilament was introduced into the external carotid artery lumen and advanced into the internal carotid artery until resistance was encountered. After 1.5 hours of ischemia, the reperfusion was initiated by withdrawing the nylon monofilament. In the Sham group, animals were prepared in the same procedure with the exception of the insertion of the nylon suture into the left internal carotid artery.
Neurologic deficit score
The neurological deficit score of each rat was assessed at 24 hours after reperfusion. The rats were scored on a 5-point scale: 0, no observable neurological deficit; 1, failure to extend the right forepaw fully; 2, circling to the right; 3, falling to the right; and 4, absence of spontaneous motor activity.
Infarct volume
To calculate the infarct volume (percentage), the separated brain tissues were cut into 2 mm thick coronal sections that were then incubated in the dark at 37°C for 30 minutes in phosphate-buffered saline (pH, 7.4) containing 2% 2,3,5-triphenyltetrazolium chloride and photographed by a digital camera. The unstained areas that appeared pale were defined as infarct regions, whereas the normal regions appeared red. The area of infarct regions and each hemisphere were measured by a blinded observer using digital imaging (Digital Camera, Olympus MDF-382E; Olympus Corporation, Tokyo, Japan) and morphological image-analysis system (JD-801, Jiangsu, People’s Republic of China). Infarction volume was calculated according to the following formula and expressed as the percentage of infarction in the ipsilateral hemisphere.
| (1) |
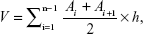
where V is the volume of fraction, Ai is the infarct area of each slice, and h is the slice thickness.
Hematoxylin and eosin staining
The rats were perfused intracardially with ice-cold normal saline and 4% paraformaldehyde after anesthesia. The separated brain tissues were fixed in 4% paraformaldehyde for 24 hours at 4°C, placed in 70% ethanol for 24 hours at room temperature, dehydrated, and finally embedded in paraffin. Hematoxylin and eosin staining was carried out to observe the morphological changes of injured neurons in the cerebral cortex.
Data analysis and statistics
Pharmacokinetic parameters were calculated by noncompartmental analysis based on statistical moment theory using Drug And Statistics professional software version 2.0 (Anhui, People’s Republic of China). The maximum plasma concentration (Cmax) and time of maximum concentration (Tmax) were obtained directly from the plasma concentration–time plots.
The results were expressed as mean ± standard deviation. Statistical significance was determined by one-way analysis of variance followed by a least significant difference test using SPSS 19.0 (IBM Corporation, Armonk, NY, USA). Results with P<0.05 were considered statistically significant.
Results and discussion
Preparation and characterization of AP nanoemulsions
AP could be easily solubilized in ethyl linoleate with a solubility of 50 mg/mL. The nanoemulsions were prepared with a procedure of high-pressure homogenization, in which no drug precipitation was observed and from which a yellowish milk-like suspension was obtained. The particle size of the nanoemulsion was about 150 nm, with a narrow size distribution (PI <0.3; Figure 2A). The size and morphology of the droplets were further confirmed by transmission electron microscope analysis, as shown in the representative image of the AP nanoemulsion (Figure 2B), in which the droplets with a size of 100–150 nm were spherical and uniform. There was no significant change in particle size and encapsulation efficiency after the nanoemulsions were stored for 30 days at 25°C (Figure 2C); thus we determined that the nanoemulsions were stable during the storage period.
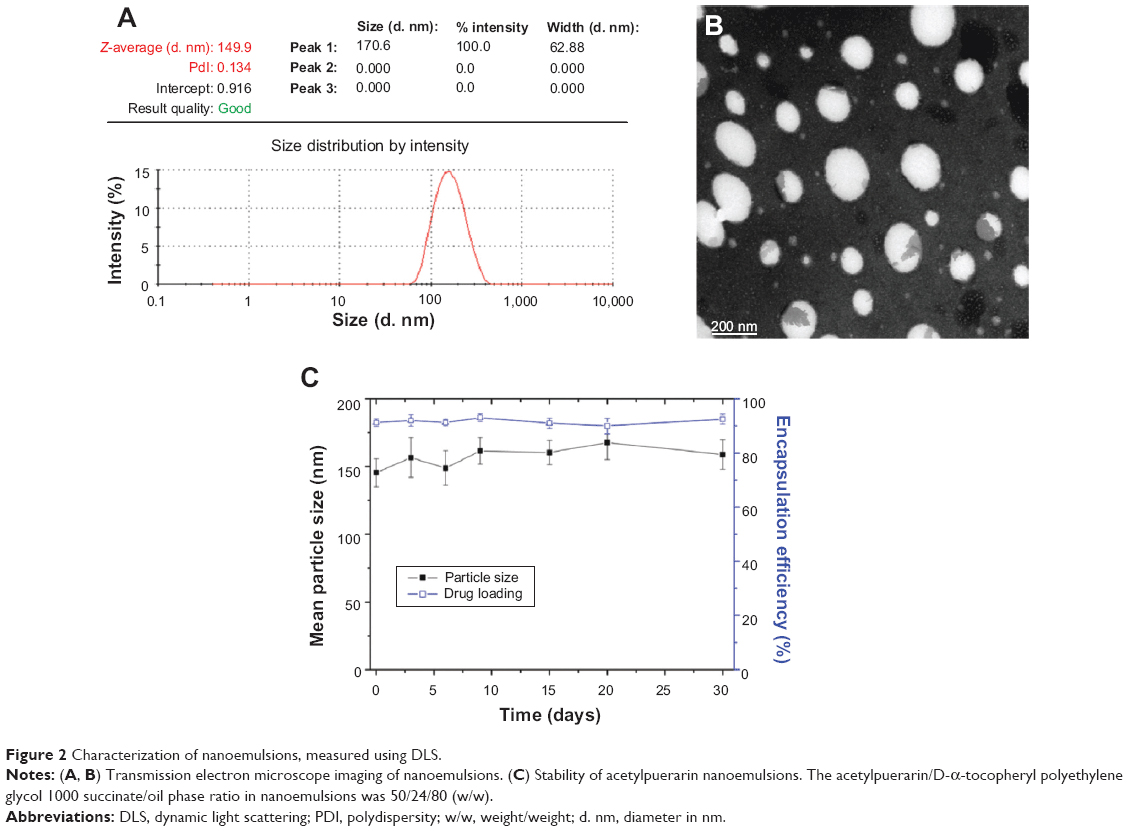 | Figure 2 Characterization of nanoemulsions, measured using DLS. |
The influence of the amounts of TPGS (based on the oil phase, weight/weight [w/w]) on the mean particle size and PI of oil droplets is shown in Figure 3A. Both particle size and PI values decreased as the amounts of TPGS increased from 1% to 20%; however, the increase from 20% to 50% produced no effect on the particle size. It was reported that the droplet size decreased with an increase in the amounts of emulsifier.32,33 The droplets were not covered completely at a low concentration of emulsifier, leading to a coalescence of droplets and thus producing large droplets. An increase in emulsifier not only decreased the coalescence of droplets but also made the emulsifier absorb onto the droplets more rapidly, leading to decreasing the droplet size and improving the size distribution. However, droplet size was not affected by the emulsifier concentration when all the surface of droplets was covered completely.
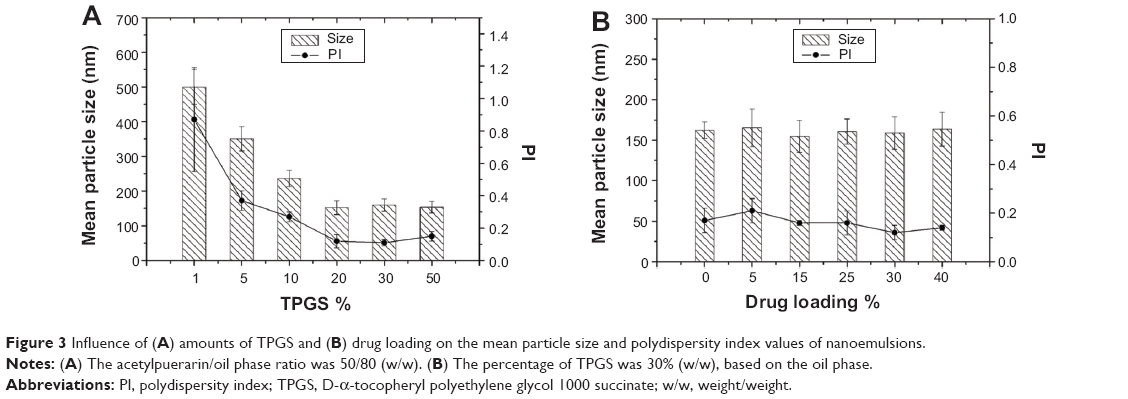 | Figure 3 Influence of (A) amounts of TPGS and (B) drug loading on the mean particle size and polydispersity index values of nanoemulsions. |
To evaluate the drug-loading capacity of TPGS-stabilized nanoemulsions, the influence of the drug loading (based on the oil phase, w/w) on the mean particle size and PI of oil droplets was performed. As shown in Figure 3B, the effects of drug loading on the particle size and PI of droplets were negligible, as the amounts of drug ranged from 0%–40%, indicating that drug loading would not affect the stability of nanoemulsions. A similar report by Akkar and Müller indicated that the incorporation of drug into the droplets enhanced the dispersion of nanoemulsions and thus resulted in decrease in droplet size.34 It was because the presence of drug molecules in the surfactant layer led to a further reduction in the interfacial tension of oil/water.22
In vitro drug release
The AP release from nanoemulsions and suspension is shown in Figure 4. More than 90% of the drug was released from the suspension within 2 hours as a result of the solubilization effects of the surfactant in release medium. In contrast, the drug release from nanoemulsions was less than 20% after 24 hours, indicating that the drug release was retarded and a controlled release profile was achieved. Moreover, no burst release was observed, thereby suggesting there was no drug precipitation in the system and that the drug was completely encapsulated in the oil cores of droplet. A combination mechanism (diffusion through the oil-droplet core and diffusion across the droplet interfacial film) was ascribed to the drug release from the droplets;35,36 diffusion across the droplet interfacial film was a rate-limiting step for the release. The drug molecules must simultaneously overcome the two barriers before they present in release medium, thereby decreasing the rate of drug release from nanoemulsions.
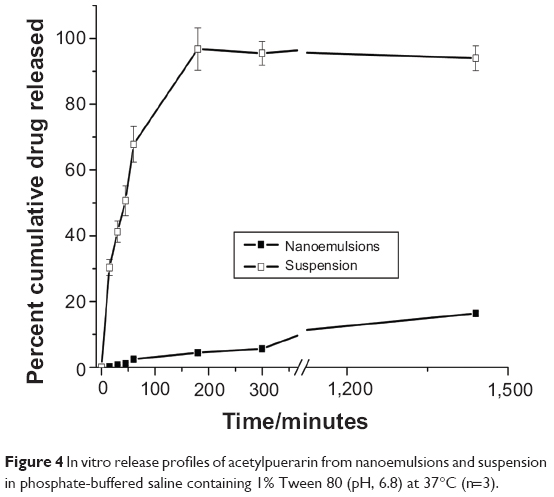 | Figure 4 In vitro release profiles of acetylpuerarin from nanoemulsions and suspension in phosphate-buffered saline containing 1% Tween 80 (pH, 6.8) at 37°C (n=3). |
In vitro digestion
For oral lipid-based formulations, the model of in vitro lipid digestibility was a useful tool to predict in vivo drug solubilization/precipitation patterns in the gastrointestinal environment. As shown in Figure 5A, lipid digestion was rapid for both droplets and oil solution, and digestion was completed within the first 30 minutes. In contrast, the digestion rate of droplets was lower than that of oil solution. It was because the lipases must diffuse across the TPGS films before lipases accessed to the droplet cores, retarding the lipid digestion.37
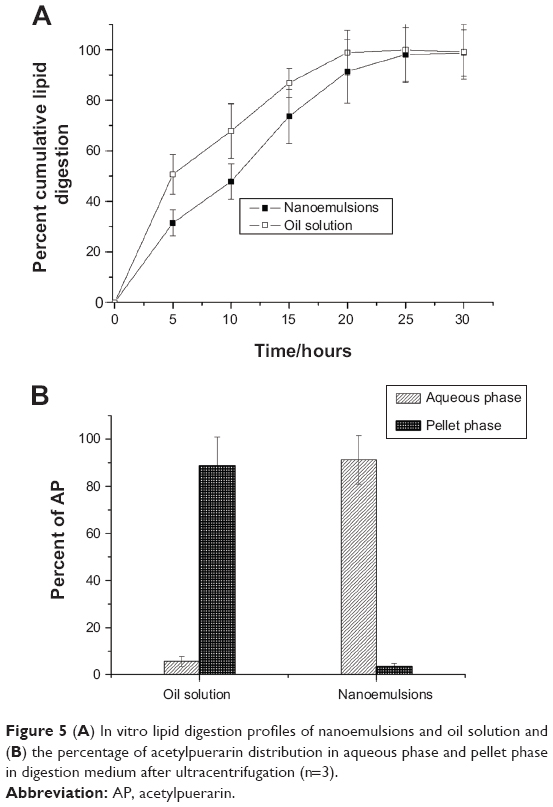 | Figure 5 (A) In vitro lipid digestion profiles of nanoemulsions and oil solution and (B) the percentage of acetylpuerarin distribution in aqueous phase and pellet phase in digestion medium after ultracentrifugation (n=3). |
After ultracentrifugation, the digestion medium was separated into an aqueous phase and a pellet phase; no poorly dispersed oil phase was observed. As shown in Figure 5B, the percentage of AP in the aqueous phase/pellet phase was about 90%/10% for nanoemulsions and 5%/95% for oil solution. The drug containing in aqueous phase from nanoemulsions was much more than that of oil solution. It thus indicated that the drug encapsulated in nanoemulsions would present in a solubilized form after being transported into the gastrointestinal tract, whereas drug precipitation would occur as the oil solution was orally administered.38,39 It was ascribed to the solubilization effects of mixed micelles present in the digestion medium that consisted of exogenous TPGS, lipid digestion products, and the endogenous surfactants (bile salts and phospholipids).28,38 It was believed that the maintenance of the drug solubilized form in the intestinal fluid made the drug molecules diffuse easily across the unstirred water layer, enhancing drug absorption.40
Pharmacokinetics and mechanism of absorption in rats
The mean plasma drug concentration–time profiles of test and reference formulations and pharmacokinetic parameters are shown in Figure 6 and Table 1. The values of Cmax/Tmax of puerarin were 0.73 μg·mL−1/1.83 hours for suspension and 0.95 μg·mL−1/1.67 hours for oil solution. In contrast, the values of Cmax/Tmax of puerarin in nanoemulsions were 2.11 μg·mL−1/1.33 hours, indicating that a more rapid drug absorption with higher peak plasma concentration were achieved. The area under the curve (AUC) value of puerarin in nanoemulsions was 5.76±0.56 μg·h·mL−1, which was about 2.6- and 1.7-fold, respectively, as great as that of suspension and oil solution, thereby indicating enhanced drug absorption. It was also observed that the AUC value from oil solution was significantly greater than that of suspension, because the ethyl linoleate lipid could also improve the solubility and absorption of the insoluble drugs.41
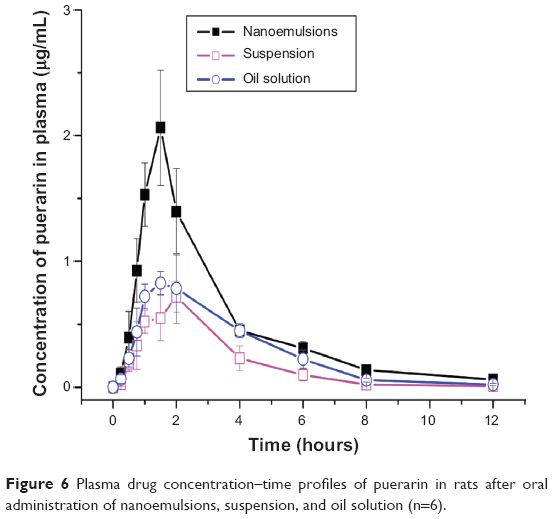 | Figure 6 Plasma drug concentration–time profiles of puerarin in rats after oral administration of nanoemulsions, suspension, and oil solution (n=6). |
 | Table 1 Pharmacokinetic parameters of puerarin after oral administration of nanoemulsions, suspension, and oil solution in rats |
For oral administration, the drug was absorbed into the systemic circulation via the intestinal lymphatic transport and blood transport. For a lipid formulation, intestinal lymphatic transport played a very important role in the absorption enhancement of a lipophilic drug because the drug could be delivered to the systemic circulation without first-passing effect of liver.28,30 The chylomicron flow-blocking rat model was an efficient tool for evaluating lymphatic absorption. Thus, we further assessed the bioavailability of oil solution and nanoemulsions in such a model. The mean plasma drug concentration–time profiles of AP in rats treated with cycloheximide or saline (control) are shown in Figure 7. Compared with the control model, a significant decrease in Cmax from the blocking model was observed (P<0.05); moreover, a 14% Cmax decrease for oil solution and a 76% Cmax decrease for nanoemulsions in AUC were observed. For nanoemulsion formulation, the blood transport and lymphatic absorption were simultaneously ascribed to the drug delivery to systemic circulation, whereas the latter was a determinant. The percentage of lymphatic absorption resulting from the oil phase of nanoemulsions was about 46% (calculated by the ratio of AUC values of the cycloheximide and control model). Thus, the percentage of lymphatic absorption resulting from TPGS was 30%. That percentage was obtained by the difference of percentage (76%) decrease for the nanoemulsions AUC and percentage (46%) lymphatic absorption of the oil phase. It might be explained by the exogenous TPGS promoting chylomicron synthesis and then enhancing lymphatic transport, improving the drug absorption.42
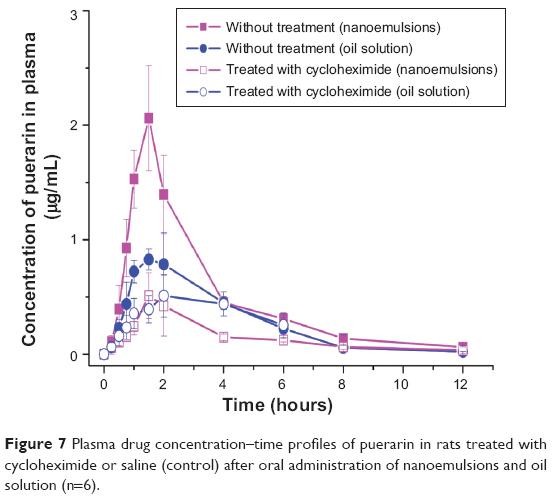 | Figure 7 Plasma drug concentration–time profiles of puerarin in rats treated with cycloheximide or saline (control) after oral administration of nanoemulsions and oil solution (n=6). |
The presence of molecular state of drug in the gastrointestinal tract was a prerequisite for absorption, and the in vivo drug solubilization was well correlated with the drug absorption.39 Thus, the drug absorption was greatly affected by the drug solubilization/precipitation patterns that could be predicted by the lipid digestion model.43 Compared with the oil solution (Figure 5B), almost the entire drug in nanoemulsions was solubilized in the mixed micelles in the digestion medium, which was partly attributed to the enhanced bioavailability.
Pharmacodynamics in rats
The therapeutic effects of AP formulations on MCAO rats are depicted in Figure 8. Compared with the Sham group, both the pale cortex and striatum in the left parietal lobe were observed from the group without drug treatment (Figure 8A). However, the symptoms from the groups treated with AP formulations were alleviated, and a more profound effect from the group treated with nanoemulsions was observed. The values of infarct volume (percentage) and neurological deficit score were 39.05%±4.02% and 2.75±0.46 for the group without drug treatment, respectively. In contrast, the groups treated with AP formulations significantly reduced the infarct volume and scores. Moreover, more significant reduction from the group treated with nanoemulsions was obtained compared with the other two formulations (Figure 8B and C). It thus indicated that the nanoemulsion formulation provided better neuroprotection against focal cerebral ischemia–reperfusion injury.
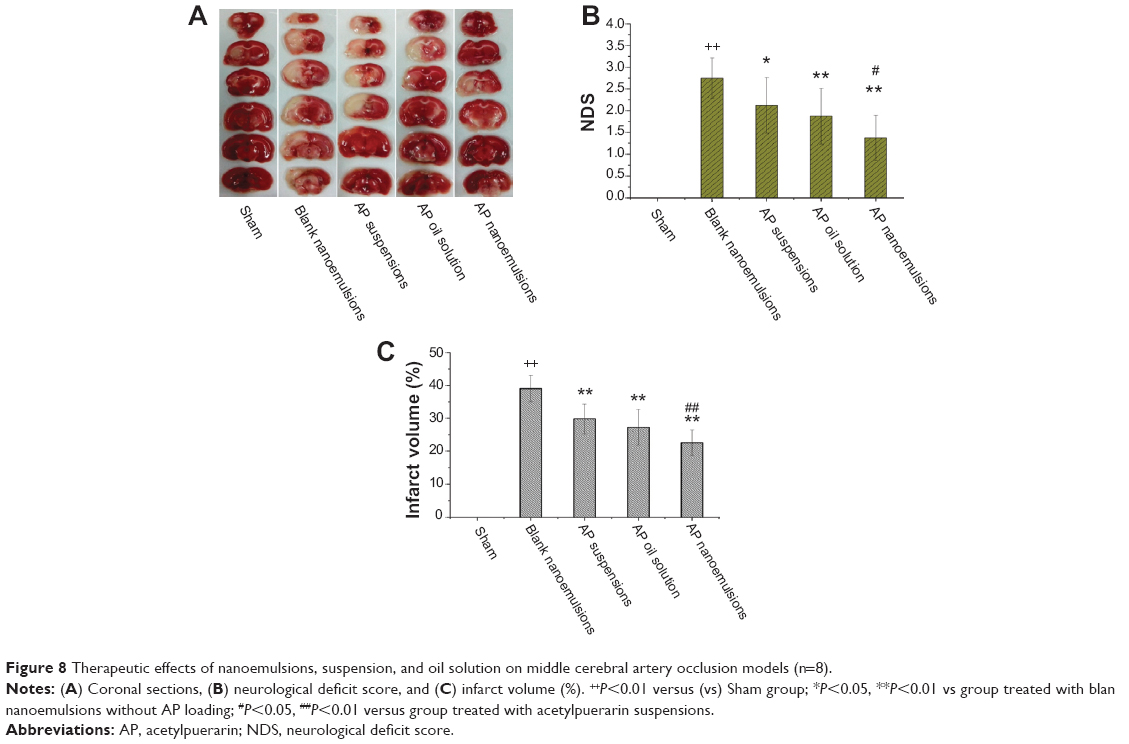 | Figure 8 Therapeutic effects of nanoemulsions, suspension, and oil solution on middle cerebral artery occlusion models (n=8). |
To further evaluate the neuroprotection of AP formulations, the histopathology change of cerebral cortex was examined with hematoxylin and eosin staining. As shown in Figure 9A and B, no histopathological abnormalities were observed in Sham rats, whereas in groups treated with blank nanoemulsions without AP loading, most neurons in the infarct core appeared shrunken with eosinophilic cytoplasm and a triangulated pyknotic nucleus. The infarct core was surrounded with necrotic neurons that exhibited a pyknotic shape and condensed nucleus in peri-infarct zone. In contrast, the number of necrotic neurons in brain tissue from rats treated with AP formulations was markedly decreased, and the infarction area was significantly reduced, indicating that the ischemic injury was attenuated after AP treatment (Figure 9C–E). Furthermore, a more significant reduction in the number of necrotic and condensed neurons was observed from the group treated with AP nanoemulsions compared with the group treated with AP suspension and oil solution. It is thus suggested that AP nanoemulsions produce better protective effects on ischemic reperfusion injury, which is in line with the results of infarct volume and neurological deficit score.
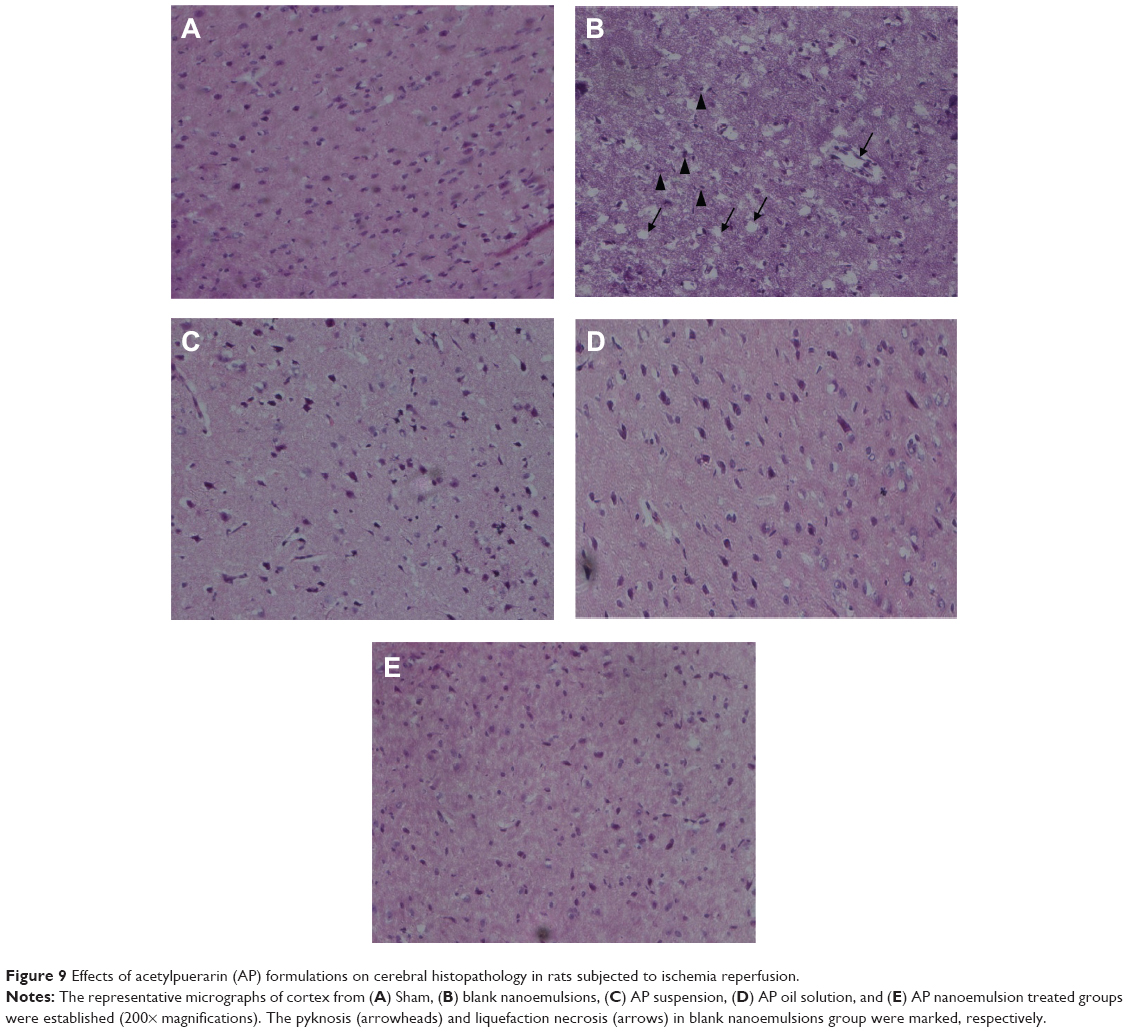 | Figure 9 Effects of acetylpuerarin (AP) formulations on cerebral histopathology in rats subjected to ischemia reperfusion. |
It was reported that AP provided protective effects on cerebral ischemia reperfusion injury via its antioxidant and anti-inflammatory properties.31 However, its clinical application was limited by its low water solubility.9,11 The formulation of AP nanoemulsions had better neuroprotection effects on brain injury compared with the formulations of suspension and oil solution, indicating a correlation between enhanced bioavailability and improved therapeutic outcome.
Conclusion
The TPGS-stabilized nanoemulsions with a droplet size of 150 nm and high AP loading were prepared successfully. Enhanced oral bioavailability in nanoemulsions was achieved via the mechanism of the maintenance of drug solubilization in the gastrointestinal tract and the enhancement of lymphatic transport, which resulted in therapeutic improvement on cerebral ischemic reperfusion injury.
Acknowledgments
This work was supported by Shandong Provincial Natural Science Foundation (No. Q2007C06) and Shandong Provincial Healthy Department Foundation (2007QZ010).
Disclosure
The authors report no conflicts of interest in this work.
References
Hu SS, Kong LZ, Gao RL, et al; Editorial Board. Outline of the report on cardiovascular disease in China, 2010. Biomed Environ Sci. 2012;25(3):251–256. | ||
Hou L, Wei X, Li X, et al. Protective effects of acetylpuerar in on foca brain ischemia-reperfusion in jury in rats. China Pharm J. 2007;42(19):1469–1472. | ||
Weiping S, Changqing Y, Yanjing C, Zhiguo Z, Xiangzheng K. Self-assembly of an amphiphilic derivative of chitosan and micellar solubilization of puerarin.Colloids Surf B Biointerfaces. 2006;48(1):13–16. | ||
Zeng X, Chen H, Zheng Y, et al. Enhanced adsorption of puerarin onto a novel hydrophilic and polar modified post-crosslinked resin from aqueous solution. J Colloid Interface Sci. 2012;385(1):166–173. | ||
Hou L, Zhang XM, Wei XB. Protective effects of Compounds N-2211 on focal brain ischemia–reperfusion injury in rats. Qilu Pharm Affairs. 2004;3:52–54. | ||
Liu R, Wei XB, Zhang XM. Effects of acetylpuerarin on hippocampal neurons and intracellular free calcium subjected to oxygen-glucose deprivation/reperfusion in primary culture. Brain Res. 2007;1147:95–104. | ||
Thanki K, Gangwal RP, Sangamwar AT, Jain S. Oral delivery of anticancer drugs: challenges and opportunities. J Control Release. 2013;170(1):15–40. | ||
Wang Y, Ma Y, Zheng Y, et al. In vitro and in vivo anticancer activity of a novel puerarin nanosuspension against colon cancer, with high efficacy and low toxicity. Int J Pharm. 2013;441(1–2):728–735. | ||
Guo D, Yang D, Chen S. Pharmacokinetic investigation of Pur and derivatives in rats. J Shaanxi Tradit Chin Med. 2008;31(52):52–54. | ||
Guo Y, Luo J, Tan S, Otieno BO, Zhang Z. The applications of Vitamin E TPGS in drug delivery. Eur J Pharm Sci. 2013;49(2):175–186. | ||
Xiang Y, Liu S, Wei C, et al. In vivo pharmacokinetic study of hexaacetylpurerarin in rats. Chin Tradit Herbal Drugs. 2012;43(11):2247–2249. | ||
Mattheolabakis G, Rigas B, Constantinides PP. Nanodelivery strategies in cancer chemotherapy: biological rationale and pharmaceutical perspectives. Nanomedicine (Lond). 2012;7(10):1577–1590. | ||
Nazarzadeh E, Anthonypillai T, Sajjadi S. On the growth mechanisms of nanoemulsions. J Colloid Interface Sci. 2013;397:154–162. | ||
Mei L, Zhang Z, Zhao L, et al. Pharmaceutical nanotechnology for oral delivery of anticancer drugs. Adv Drug Deliv Rev. 2013;65(6):880–890. | ||
An HZ, Helgeson ME, Doyle PS. Nanoemulsion composite microgels for orthogonal encapsulation and release. Adv Mater. 2012;24(28):3838–3844, 3895. | ||
Lim SB, Banerjee A, Önyüksel H. Improvement of drug safety by the use of lipid-based nanocarriers. J Control Release. 2012;163(1):34–45. | ||
Hirsjärvi S, Dufort S, Gravier J, et al. Influence of size, surface coating and fine chemical composition on the in vitro reactivity and in vivo biodistribution of lipid nanocapsules versus lipid nanoemulsions in cancer models. Nanomedicine. 2013;9(3):375–387. | ||
Zhang Z, Tan S, Feng SS. Vitamin E TPGS as a molecular biomaterial for drug delivery. Biomaterials. 2012;33(19):4889–4906. | ||
Collnot EM, Baldes C, Schaefer UF, Edgar KJ, Wempe MF, Lehr CM. Vitamin E TPGS P-glycoprotein inhibition mechanism: influence on conformational flexibility, intracellular ATP levels, and role of time and site of access. Mol Pharm. 2010;7(3):642–651. | ||
Collnot EM, Baldes C, Wempe MF, et al. Mechanism of inhibition of P-glycoprotein mediated efflux by vitamin E TPGS: influence on ATPase activity and membrane fluidity. Mol Pharm. 2007;4(3):465–474. | ||
Mi Y, Zhao J, Feng SS. Targeted co-delivery of docetaxel, cisplatin and herceptin by vitamin E TPGS-cisplatin prodrug nanoparticles for multimodality treatment of cancer. J Control Release. 2013;169(3):185–192. | ||
He W, Lu Y, Qi J, Chen L, Hu F, Wu W. Nanoemulsion-templated shell-crosslinked nanocapsules as drug delivery systems. Int J Pharm. 2013;445(1–2):69–78. | ||
Alayoubi AY, Anderson JF, Satyanarayanajois SD, Sylvester PW, Nazzal S. Concurrent delivery of tocotrienols and simvastatin by lipid nanoemulsions potentiates their antitumor activity against human mammary adenocarcenoma cells. Eur J Pharm Sci. 2012;48(3):385–392. | ||
Doh HJ, Jung Y, Balakrishnan P, Cho HJ, Kim DD. A novel lipid nanoemulsion system for improved permeation of granisetron. Colloids Surf B Biointerfaces. 2013;101:475–480. | ||
Han SF, Yao TT, Zhang XX, et al. Lipid-based formulations to enhance oral bioavailability of the poorly water-soluble drug anethol trithione: effects of lipid composition and formulation. Int J Pharm. 2009;379(1):18–24. | ||
Lian R, Lu Y, Qi J, et al. Silymarin glyceryl monooleate/poloxamer 407 liquid crystalline matrices: physical characterization and enhanced oral bioavailability. AAPS Pharm Sci Tech. 2011;12(4):1234–1240. | ||
Zangenberg NH, Müllertz A, Kristensen HG, Hovgaard L. A dynamic in vitro lipolysis model. II: Evaluation of the model. Eur J Pharm Sci. 2001;14(3):237–244. | ||
Porter CJ, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6(3):231–248. | ||
Dahan A, Hoffman A. Evaluation of a chylomicron flow blocking approach to investigate the intestinal lymphatic transport of lipophilic drugs.Eur J Pharm Sci. 2005;24(4):381–388. | ||
Lind ML, Jacobsen J, Holm R, Müllertz A. Intestinal lymphatic transport of halofantrine in rats assessed using a chylomicron flow blocking approach: the influence of polysorbate 60 and 80. Eur J Pharm Sci. 2008;35(3):211–218. | ||
Wang ZY, Wei XB, Chen L, et al. Neuroprotective effects of hydroxyethylpuerarin against focal cerebral ischemia-reperfusion in rats. Chin J Physiol. 2007;50(5):211–216. | ||
He W, Tan Y, Tian Z, Chen L, Hu F, Wu W. Food protein-stabilized nanoemulsions as potential delivery systems for poorly water-soluble drugs: preparation, in vitro characterization, and pharmacokinetics in rats. Int J Nanomedicine. 2011;6:521–533. | ||
Lee SJ, Choi SJ, Li Y, Decker EA, McClements DJ. Protein-stabilized nanoemulsions and emulsions: comparison of physicochemical stability, lipid oxidation, and lipase digestibility. J Agric Food Chem. 2011;59(1):415–427. | ||
Akkar A, Müller RH. Intravenous itraconazole emulsions produced by SolEmuls technology. Eur J Pharm Biopharm. 2003;56(1):29–36. | ||
Llinàs M, Calderó G, García-Celma MJ, Patti A, Solans C. New insights on the mechanisms of drug release from highly concentrated emulsions. J Colloid Interface Sci. 2013;394:337–345. | ||
Washington C, Evans K. Release rate measurements of model hydrophobic solutes from submicron triglyceride emulsions. J Control Release. 1995;33(3):383–390. | ||
Li Y, Hu M, Du Y, McClements DJ. Controlling lipid nanoemulsion digestion using nanolaminated biopolymer coatings. J Microencapsul. 2011;28(3):166–175. | ||
Porter CJ, Kaukonen AM, Boyd BJ, Edwards GA, Charman WN. Susceptibility to lipase-mediated digestion reduces the oral bioavailability of danazol after administration as a medium-chain lipid-based microemulsion formulation. Pharm Res. 2004;21(8):1405–1412. | ||
Porter CJ, Pouton CW, Cuine JF, Charman WN. Enhancing intestinal drug solubilisation using lipid-based delivery systems. Adv Drug Deliv Rev. 2008;60(6):673–691. | ||
Simovic S, Heard P, Hui H, et al. Dry hybrid lipid-silica microcapsules engineered from submicron lipid droplets and nanoparticles as a novel delivery system for poorly soluble drugs. Mol Pharm. 2009;6(3):861–872. | ||
Wu W, Wang Y, Que L. Enhanced bioavailability of silymarin by self-microemulsifying drug delivery system. Eur J Pharm Biopharm. 2006;63(3):288–294. | ||
Fan Z, Wu J, Fang X, Sha X. A new function of Vitamin E-TPGS in the intestinal lymphatic transport of lipophilic drugs: enhancing the secretion of chylomicrons. Int J Pharm. 2013;445(1–2):141–147. | ||
Dahan A, Hoffman A. Rationalizing the selection of oral lipid based drug delivery systems by an in vitro dynamic lipolysis model for improved oral bioavailability of poorly water soluble drugs. J Control Release. 2008;129(1):1–10. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.