Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Enhanced local bioavailability of single or compound drugs delivery to the inner ear through application of PLGA nanoparticles via round window administration
Authors Cai H, Wen X, Wen L, Tirelli N, Zhang X, Zhang Y, Su H, Yang F, Chen G
Received 11 August 2014
Accepted for publication 11 October 2014
Published 1 December 2014 Volume 2014:9(1) Pages 5591—5601
DOI https://doi.org/10.2147/IJN.S72555
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Hui Cai,1 Xingxing Wen,1 Lu Wen,1 Nicola Tirelli,2,3 Xiao Zhang,1 Yue Zhang,1 Huanpeng Su,1 Fan Yang,1 Gang Chen1,4
1School of Pharmacy, Guangdong Pharmaceutical University, Guangzhou, People’s Republic of China; 2School of Materials, 3School of Biomedicine, University of Manchester, Manchester, United Kingdom; 4Department of Clinical pharmacy, Guangdong Pharmaceutical University, Guangzhou, People’s Republic of China
Abstract: In this paper, the potential of poly(D,L-lactide-co-glycolide acid) (PLGA) nanoparticles (NPs) for carrying single or compound drugs traversing the round window membrane (RWM) was examined after the round window (RW) administration of different NPs to guinea pigs. First, coumarin-6 was incorporated into PLGA NPs as a fluorescent probe to investigate its ability to cross the RWM. Then, PLGA NPs with salvianolic acid B (Sal B), tanshinone IIA (TS IIA), and total panax notoginsenoside (PNS) including notoginsenoside R1 (R1), ginsenoside Rg1 (Rg1), and ginsenoside Rb1 (Rb1) were developed to evaluate whether NPs loaded with compound drugs would pass through the RWM and improve the local bioavailability of these agents. PLGA NPs loaded with single or compound drugs were prepared by the emulsification solvent evaporation method, and their particle size distribution, particle morphology, and encapsulation efficiency were characterized. In vitro release study showed sustained-release profiles of Sal B, TS IIA, and PNS from the NPs. The pharmacokinetic results showed that NPs applied to the RWM significantly improved drug distribution within the inner ear. The AUC0–t of coumarin-6 in the perilymph (PL) following RW administration of NPs was 4.7-fold higher than that of coumarin-6 solution, and the Cmax was 10.9-fold higher. Furthermore, the AUC0–t of R1, Rg1, and Rb1 were 4.0-, 3.1-, and 7.1-fold greater, respectively, after the application of NPs compared to the compound solution, and the Cmax were, respectively, 14.4-, 10.0-, and 16.7-fold higher. These findings suggest that PLGA NPs with unique properties at the nanoscale dimensions have a powerful ability to transport single or compound drugs into the PL through the RWM and remarkably enhance the local bioavailability of the encapsulated drugs in the inner ear. The use of PLGA NPs as nanoscale delivery vehicles to carry drugs across the RWM may be a promising strategy for the treatment of inner ear diseases.
Keywords: inner ear administration, nanoparticles, perilymph, local bioavailability, poly(D,L-lactide-co-glycolide acid)
Introduction
The effective medical treatment of the inner ear diseases depends on achieving therapeutic concentrations of medications within the inner ear. However, the existence of the blood–labyrinthine barrier (BLB) and limited blood flow to the cochlea prevent the delivery of drugs from the blood to the inner ear and represent a fundamental obstacle to systemic application.1–4 The BLB is a major barrier separating the inner ear from systemic circulation with tight junctions, made up of capillary endothelial cells that line blood vessels located in the stria vascularis.5 Recent inner ear cellular biology findings have suggested several possibilities for the novel medical treatment of certain inner ear diseases and have subsequently directed increasing interest toward the topical administration of drugs to the inner ear.6
At present, there are two main routes for topical drug delivery to the inner ear: 1) intracochlear or intravestibular drug delivery and 2) intratympanic (extracochlear) drug application to the round window membrane (RWM). The most widely used approach is round window (RW) application mainly because it is a safe and feasible way to deliver drugs to the inner ear, especially to minimize hearing damage.7 In intratympanic delivery, drugs in formulations are delivered through the tympanic membrane to the middle ear, from where the drugs permeate through the RWM into the inner ear by passive diffusion.8 Local application onto the RWM is a promising way of targeting drugs to the structures of the inner ear, which can directly deliver chemotherapeutic agents into the inner ear, bypassing the BLB and avoiding systemic side effects.9,10 It is noteworthy that single or repeated intratympanic injections of gentamicin11 are now commonly used in the treatment of Ménière’s Disease. In addition to aminoglycosides and anesthetics, a variety of drugs have been applied extracochlearly to the RW niche in humans, including neurotransmitters and neurotransmitter antagonists for tinnitus, steroids12 and monoclonal antibodies for autoimmune inner ear disease, and apoptosis inhibitors (AM-111) for noise-induced hearing loss.7
The RWM, a semipermeable membrane and physical barrier, is the most important interface between the middle and inner ear. The passage of substances through the RWM is affected by the membrane’s thickness, morphologic integrity, the presence and duration of inflammation, and the physicochemical characteristics of the substances.13,14 Although the RWM is accessible in the middle-ear space, some potential therapeutic agents are unable to readily cross the RWM to the cochlea due to their properties,15 which limits the treatment of inner ear disorders. Moreover, the direct contact of some therapeutic agents, such as hydrocortisone, with middle ear structures can elicit morphological changes in the RWM.16 Thus, sophisticated approaches are required to enhance the ability of drugs to access the inner ear, and nanoparticles (NPs) can compensate for the insufficient molecular properties of some drugs to cross the RWM.17
The use of novel NPs made from biodegradable and biocompatible polymers as promising drug delivery vehicles may complement existing therapeutic strategies. NP-based drug delivery systems preferentially manipulate drug distribution and improve their bioavailability. Poly(D,L-lactide-co-glycolide acid) (PLGA), a biodegradable and nontoxic polymer approved by the US Food and Drug Administration (FDA) for human use, is thought to be an ideal particulate drug carrier for NPs.18 The encapsulation of drugs in PLGA nanocarriers can reduce the undesirable shortcomings of therapeutic agents, such as short circulation half-life or non-site-specific targeting, resulting in undesired systemic side effects.19 Based on nanoscale size, PLGA NPs can permeate through the RWM easily. Several articles have reported on the local application of PLGA NPs onto the RWM for drug delivery to the cochlea. PLGA NPs encapsulating rhodamine, a red fluorescent dye, placed on the RWM were detected in the scala tympani.20 Similarly, iron oxide-loaded PLGA NPs applied to the RWM were found in the cochlea with or without exposure to magnetic forces.21 PLGA NPs loaded with multiple agents and applied to the RWM have scarcely been reported. Because the different physicochemical characteristics of drugs lead to their inconsistent absorption through the RWM, the compound drugs cannot achieve the best effects. This will be circumvented by the application of PLGA NPs incorporating the combined drugs to treat inner ear diseases.
Coumarin-6, a kind of lipophilic fluorescent dye with high sensitivity, is nontoxic and releases minimally from PLGA NPs at acidic pH, and was therefore chosen as an appropriate marker for in vivo tracking of NPs and related mechanism studies.22,23 Salvianolic acid B (Sal B) and tanshinone IIA (TS IIA) are the major hydrophilic and lipophilic bioactive components, respectively, in the extract of Salvia miltiorrhiza (Danshen in Chinese) and together have been proven to be responsible for the neuroprotective effects against apoptosis and antioxidation effects.24–26 Total panax notoginsenoside (PNS), including notoginsenoside R1 (R1), ginsenoside Rg1 (Rg1), ginsenoside Rb1 (Rb1), and others, from Panax notoginseng (Sanqi in Chinese) protects spiral ganglion cells from cochlear ischemia.27 Danshen in combination with Sanqi as a traditional Chinese herb-combined prescription has been successfully prescribed to patients for the treatment of inner ear diseases, such as sudden deafness and Ménière’s Disease.28,29
To investigate the potential use of PLGA NPs to facilitate the migration of pharmacological agents across the RWM, coumarin-6-loaded PLGA NPs were prepared and applied onto guinea pig RWM as a fluorescent probe to examine the ability of PLGA NPs to cross the RWM. Subsequently, a polymeric NP delivery system for TS IIA, Sal B, and PNS was developed to evaluate whether the NPs would carry payloads of compound drugs onto the RWM and improve the local bioavailability of these therapeutic agents.
Materials and methods
Materials
Coumarin-6 (98%) was purchased from Sigma-Aldrich (St Louis, MO, USA). Sal B (95%) and TS IIA (98%) were purchased from Shenzhen Medherb Bio-Tech Co, Ltd (Shenzhen, People’s Republic of China). PNS was obtained from Chengdu Must Bio-Tech Co, Ltd (Chengdu, People’s Republic of China). The contents of R1, Rg1, and Rb1 were determined to be 26.1%, 36.5%, and 31.8% (w/w), respectively, by high-performance liquid chromatography. Sephadex G-50 was purchased from Pharmacia Co, Ltd (Switzerland). Span-80 was purchased from Shanghai Shenyu Medical Chemical Industry (Shanghai, People’s Republic of China). LABRAFILM 1944 CS was kindly supplied by GATTEFOSSE Co, Ltd (Shanghai, People’s Republic of China). PLGA (LA/GA =50:50 and 75:25, MW 10 kDa) was supplied by Shandong Medical Institute, People’s Republic of China. Polyvinyl alcohol 4–88 (PVA, MW 31 kDa) was purchased from Sigma-Aldrich.
NP preparation
Coumarin-6-loaded PLGA NPs were prepared by the emulsion/solvent evaporation method. Briefly, 30 mg of PLGA (LA/GA =75:25) and 60 μg of coumarin-6 in 1 mL of dichloromethane/ethyl acetate (7:3) was added into 5 mL of 3% (w/v) PVA aqueous solution on ice using a probe sonicator set at 190 W of energy output (Scientz Biotechnology Co, Ltd, People’s Republic of China) for 100 seconds with pulses of 5 seconds on and 5 seconds off to form an oil-in-water (O/W) emulsion. Then this emulsion was diluted with a 0.5% (w/v) PVA aqueous solution, followed by the evaporation of organic solvents under magnetic stirring for 3 h to create a coumarin-6 nanosuspension.
PLGA NPs loaded with TS IIA, Sal B, and PNS were prepared by a double emulsion/solvent evaporation method. PLGA (LA/GA =50:50, 125 mg), TS IIA (10 mg), and Sal B (18 mg) were dissolved in 1 mL of dichloromethane/ethyl acetate (7:3) containing span-80 (0.05 g/mL) and LABRAFILM 1944 CS (0.05 g/mL) as a mixture emulsifier to form the oil phase. The primary emulsion was formed between the oil phase and 0.1 mL of PNS aqueous solution (400 mg/mL) using a probe sonicator (190 W) for 1 minute with pulses of 5 seconds on and 5 seconds off on ice. Then 3 mL of aqueous solution containing 2% (w/v) PVA was added to the primary emulsion to obtain the W/O/W emulsion using intermittent sonication on ice for 5 minutes (pulses of 5 seconds on and 5 seconds off, 190 W), which was further diluted with a 1% (w/v) PVA aqueous solution under magnetic stirring for 3 h to remove organic solvents. The NPs were collected from the supernatant colloidal suspension using a centrifuge for 30 minutes at 3,000 rpm.
NP characterization
Particle size
The particle size and polydispersity index (PDI) of the polymer NPs were determined using a Mastersizer 2000 laser particle size analyzer (Malvern Instruments, Worcestershire, UK).
Transmission electron microscopy
Morphological characteristics were determined using a transmission electron microscope (TEM, JEOL JEM-100CX II, Tokyo, Japan). Samples of the nanosuspension were placed on a copper grid covered with nitrocellulose and dried at room temperature. After being negatively stained with 1.5% (w/v) phosphotungstic acid, the samples were observed using TEM.
Determination of encapsulation efficiency
The encapsulation efficiency (EE) of the coumarin-6 NPs was determined by Sephadex filtration method.30 A gel chromatographic column (1.5 cm ×20 cm) filled with Sephadex G-50 was used to separate the NPs and the free drugs. One milliliter of the sample of the coumarin-6 nanosuspension was added to the column, and eluted using distilled water to collect the NPs. The original coumarin-6 nanosuspension (1 mL) and the eluted coumarin-6 nanosuspension (0.5 mL) were ruptured using adequate acetonitrile, and then diluted with methanol. The total drug content and the drug entrapped in the nanosuspension were then analyzed using a Shimadzu HPLC system (Shimadzu, Japan) consisting of a Dikma Diamonsil C18(2) column (150 mm ×4.6 mm, 5 μm), a LC-10ATvp pump, and a RF-10AXL fluorescence detector. The emission wavelength and excitation wavelength of this detector were 504 and 466 nm, respectively, and the injection volume was 20 μL. The mobile phase, consisting of acetonitrile and distilled water (80:20, v/v), was kept at a flow rate of 1.0 mL/min. The EE of the coumarin-6 NPs was calculated as follows:
| (1) |
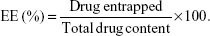
The EE of the NPs loaded with TS IIA, Sal B, and PNS was determined by the reverse dialysis method described by Fei.31 A dialysis bag (MW cutoff value of 8–14 kDa, Guangzhou Qiyun Biological Technology Co, Ltd, People’s Republic of China) containing 2 mL of saline with 0.2% sodium dodecyl sulfate (SDS, w/v) was placed directly into 25 mL of the nanosuspension. After equilibrating for 12 hours, the dialysis bag was withdrawn from the nanosuspension, and then the free dissolved drugs in the dialysis bag were measured using HPLC. Then, the encapsulation efficiency was calculated using the following formula:
| (2) |
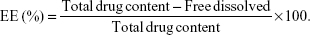
To determine the total drug content in the nanosuspension, 1 mL of the nanosuspension was dissolved in adequate acetonitrile to destruct its packaged status. The drugs were then extracted using methanol and filtered through a 0.22-μm Millipore filter (EMD Millipore, Billerica, MA, USA). The concentrations of TS IIA, Sal B, and PNS were simultaneously measured by HPLC. The Shimadzu HPLC system was equipped with an Amethyst C18-P column (150 mm ×4.6 mm, 5 μm), two LC-10ATvp pumps, an SCL-10Avp controller, and an SPD-M10Avp detector. The detection wavelength was set to 203 nm for PNS and 280 nm for TS IIA and Sal B. The analytical column was maintained at 25°C and eluted with a mobile phase consisting of acetonitrile (A) and 0.05% phosphoric acid aqueous solution (B) using the following gradient program: 20%–25% A at 0–10 minutes; 25% A at 10–15 minutes; 25%–35% A at 15–35 minutes; 35%–60% A at 35–40 minutes; 60% A at 40–70 minutes; 60%–20% A at 70–71 minutes; and 20% A at 71–80 minutes at a flow rate of 1.0 mL/min. The injection volume of the sample solution was 20 μL.
In vitro release study
The release test of coumarin-6 in vitro was performed by incubating a dialysis bag containing 3.0 mL of coumarin-6 nanosuspension and 2.0 mL PBS (pH 7.4) in 15 mL of PBS (pH 7.4). The entire system was maintained at 37°C with continuous stirring at 100 rpm. At predetermined time points, 1 mL samples were withdrawn from the medium and an equal volume of fresh medium was added. The coumarin-6 content in each of release samples was determined by HPLC. The in vitro release of NPs loaded with TS IIA, Sal B, and PNS was evaluated in saline containing 0.2% (w/v) SDS using the dialysis method. A sealed dialysis bag containing 2.0 mL of nanosuspension (harboring an equivalent of 1.16 mg of R1, 2.81 mg of Rg1, 2.82 mg of Rb1, 1.03 mg of Sal B, and 7.42 μg of TS IIA) dispersed in 2.0 mL of saline supplemented with 0.2% (w/v) SDS was fully immersed into 30 mL of release medium, and incubated at 37°C with horizontal shaking at 100 rpm. Then, 1 mL of release medium was withdrawn at predetermined time intervals and assayed for drug content by HPLC.
Pharmacokinetic and tissue distribution studies
Guinea pigs weighing 250–300 g were obtained from the Experimental Animal Center at Guangzhou University of Chinese Medicine (Guangzhou, People’s Republic of China). The animal protocols were approved by and performed in accordance with the Animal Ethical Committee (Guangdong Pharmaceutical University, Guangzhou, People’s Republic of China). The animals had free access to food and water. Ninety guinea pigs were randomly assigned to two groups. Groups 1 and 2 received RW administration of the coumarin-6 NPs (equivalent to 0.186 μg/kg) and coumarin-6 solution (equivalent to 0.185 μg/kg), respectively, and saline was used as a blank control in each group. At time points of 0.5 hours, 1 hour, 2 hours, 3 hours, 4 hours, 5 hours, 6 hours, 8 hours, 12 hours, 18 hours, 24 hours, 36 hours, 48 hours, 72 hours, and 96 hours post administration in guinea pigs under anesthesia with ethyl carbamate (1 g/kg), the inner ear perilymph (PL) was collected by piercing the RMW with a microsyringe. Blood was withdrawn from the heart and centrifuged for 10 minutes at 3,500 rpm, and then the plasma was separated. The plasma and PL were subjected to methanol extraction for HPLC analysis.
In addition, 78 guinea pigs were randomly assigned to two groups for pharmacokinetic investigation: 1) RWM application with PLGA NPs loaded with TS IIA, Sal B, and PNS (equivalent to 12.43 μg/kg TS IIA, 766.38 μg/kg Sal B, 1.05 mg/kg R1, 2.08 mg/kg Rg1, and 2.31 mg/kg Rb1) and 2) RWM application with compound solution (equivalent to 0.73 mg/kg TS IIA, 8 mg/kg Sal B, 3.13 mg/kg R1, 4.38 mg/kg Rg1, and 3.82 mg/kg Rb1). The compound solution was prepared using mixed solvents (ethanol/glycerol/PEG 400 =2:1:2), and saline was used as a blank control in each group. At time points of 0.17 hours, 0.5 hours, 1 hour, 2 hours, 3 hours, 4 hours, 6 hours, 8 hours, 10 hours, 12 hours, 18 hours, 24 hours, and 36 hours after RW administration under anesthesia, plasma and PL were separately collected as described earlier. The PL was diluted in sufficient methanol for HPLC analysis. All the plasma samples for each point were separated into two parts to analyze the various components. One part (100 μL) was acidified with 10% HCl (20 μL) and vortex-mixed with 1 mL acetic ether for 5 minutes, followed by centrifugation for 10 minutes at 10,000 rpm to collect the supernatant. The underlayer was extracted again as before. The two supernatants were amalgamated and evaporated to dryness under a stream of nitrogen. The residue was dissolved in 100 μL of methanol. A 20-μL sample was injected into the HPLC system to assay TS IIA and Sal B. To extract the plasma PNS (R1, Rg1, and Rb1), the other part (100 μL) was vortex-mixed with methanol (300 μL) for 5 minutes and centrifuged at 10,000 rpm for 10 minutes. The separated supernatant was collected to determine PNS (R1, Rg1, and Rb1) by HPLC.
Results and discussion
Preparation and characterization of PLGA NPs
Several factors, such as the composition and molecular weight of PLGA, surfactant content, w/o ratio, and drug/PLGA ratio, have been shown to affect the characteristics of PLGA nanoparticles prepared by the emulsion-solvent evaporation method. Glycolic acid is more hydrophilic than lactic acid, and therefore glycolide-rich PLGA copolymers are more compatible with water-soluble drugs and have a higher entrapment efficiency.32 Similarly, lactide-rich PLGA copolymers have a higher affinity with lipid-soluble drugs. The interaction or affinity of coumarin-6 with PLGA contributes to the increased encapsulation efficiency. In our previous study, we found that PLGA polymers with a lactide/glycolide (L:G) ratio of 75:25 in NP formations achieved a higher entrapment efficiency of coumarin-6 than PLGA polymers with an L:G ratio of 50:50. Thus, PLGA polymers with 75:25 ratio of L:G were used to formulate the coumarin-6-loaded NPs. PNS and Sal B are water-soluble micromolecule drugs (MW: R1-933, Rg1-801, Rb1-1109, and Sal B-719), which are difficult to encapsulate into NPs.32 Therefore, PLGA polymers with an L:G ratio of 50:50 were applied in the preparation of PLGA NPs loaded with TS IIA, Sal B, and PNS. The TEM image and size distribution of coumarin-6 loaded PLGA NPs are shown in Figure 1. It can be seen that the copolymers formed small, spherical particles. The average size of the NPs was 135 nm with a PDI of 0.17. The mean entrapment efficiency of coumarin-6 NPs was 51.6%.
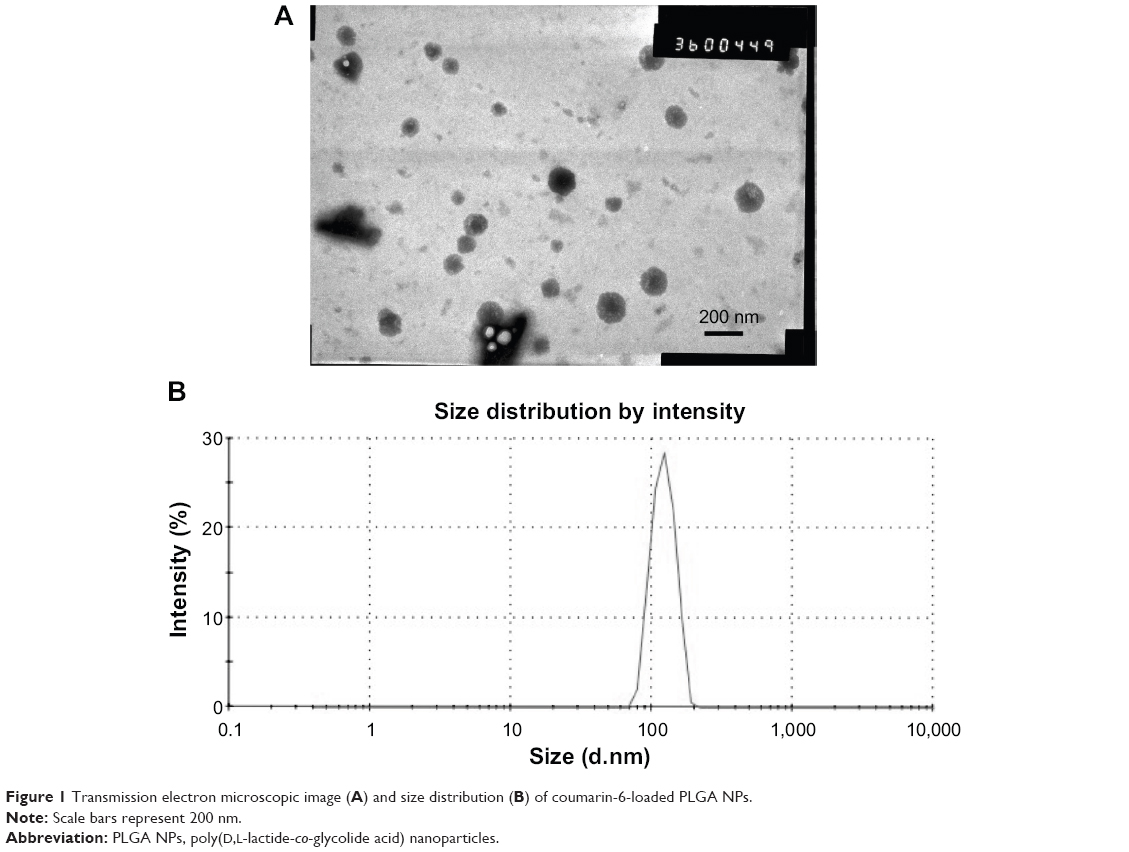 | Figure 1 Transmission electron microscopic image (A) and size distribution (B) of coumarin-6-loaded PLGA NPs. |
Using the double emulsion-solvent evaporation technique, TS IIA, Sal B, and PNS were successfully incorporated into PLGA NPs. The TEM image and size distribution of the NPs were shown in Figure 2. The morphology of the NPs was observed by TEM, and the image showed that particles were spherical. The results from the laser particle size analyzer indicated that the NPs had a mean diameter of 154 nm with a PDI of 0.007 and displayed a narrow distribution.
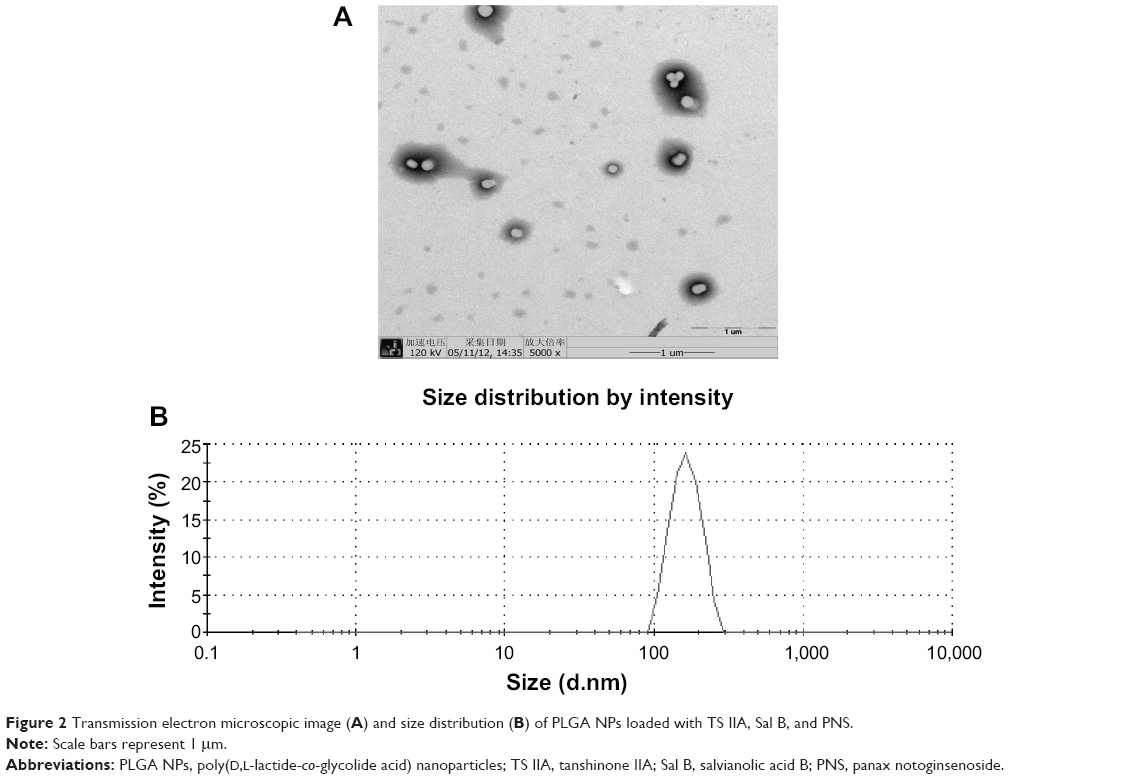 | Figure 2 Transmission electron microscopic image (A) and size distribution (B) of PLGA NPs loaded with TS IIA, Sal B, and PNS. |
According to the results of the RP-HPLC analysis, the entrapment efficiency of the NPs was 91.81% for TS IIA, 92.88% for Sal B, 37.87% for R1, 44.98% for Rg1, and 93.56% for Rb1. To prepare PLGA NPs loaded with TS IIA, Sal B, and PNS, the double emulsion/solvent evaporation method, the best technique for encapsulating hydrophilic drugs,32 was used with PLGA polymers with an L:G ratio of 50:50, which can improve the amount of the encapsulated water-soluble drugs. Compared to PNS, the stronger interaction of Sal B with the PLGA polymer may contribute to the relatively higher encapsulation efficiency of Sal B into PLGA NPs.
In vitro drug release of PLGA NPs
The in vitro cumulative release of coumarin-6 from the NPs was <2% in PBS (pH 7.4) for 72 hours. The release of coumarin-6 from the NPs in this study was slightly faster than in the previous report.33 The relative inertia of the coumarin-6 encapsulated in PLGA NPs guaranteed that the fluorescent probe was not released under physiological conditions. Consequently, coumarin-6 can be used as a fluorescence marker for in vivo tracing and targeting study of NPs. The release of free drugs (TS IIA, Sal B, and PNS) was used as the control.
Significantly different in vitro release profiles were observed for encapsulated TS IIA, Sal B, and PNS released from the PLGA NPs and for free drugs released from the saline containing 0.2% SDS (Figure 3). The variation in the release profiles of the drugs from NPs may be very likely due to the different solubilities of the drugs in the medium. Generally, compared to hydrophobic drugs, hydrophilic drugs have a faster diffusion rate and thus faster release from the polymer device. Because of the hydrophobicity of TS IIA, 0.2% SDS was applied as a surfactant to the release medium to promote the solubility of TS IIA and maintain sink condition. Free drugs were released rapidly; during the first 6 hours, >79% of the free TS IIA, Sal B, R1, Rg1, and Rb1 were released, which suggested that the free drugs could diffuse through the dialysis membrane freely. The cumulative release of free Sal B and Rg1 decreased at 6 hours and 8 hours, respectively, which may be due to their own degradation.34,35 Nanoparticle formulations exhibited a biphasic pattern of drug release for Sal B, R1, Rg1, and Rb1: burst release and sustained release. An initial burst release may be attributed to the immediate release of the surface-associated drug, and a prolonged release in the later stage is due to the slow diffusion of drug from the matrix. In vitro drug release behavior showed that NPs produced a sustained drug release. While 76.2% of Rg1, 76.0% of Rb1, 93.0% of Sal B, and 23.6% of TS IIA were released from NPs within 72 hours, R1 was completely released. We took 36 hours to complete the sample acquisition for the in vivo study. Thus, in vitro drug release was conducted for only 72 hours. A relatively slow release of TS IIA from the NPs was observed, which was consistent with the result obtained in a previous study.36 The release rate of TS IIA from the NPs and its concentration in the dissolution medium depend on the diffusion of the drug across the membrane and the partition coefficient of the drug between two immiscible phases, namely the polymeric phase and the aqueous environment in the dialysis bag.
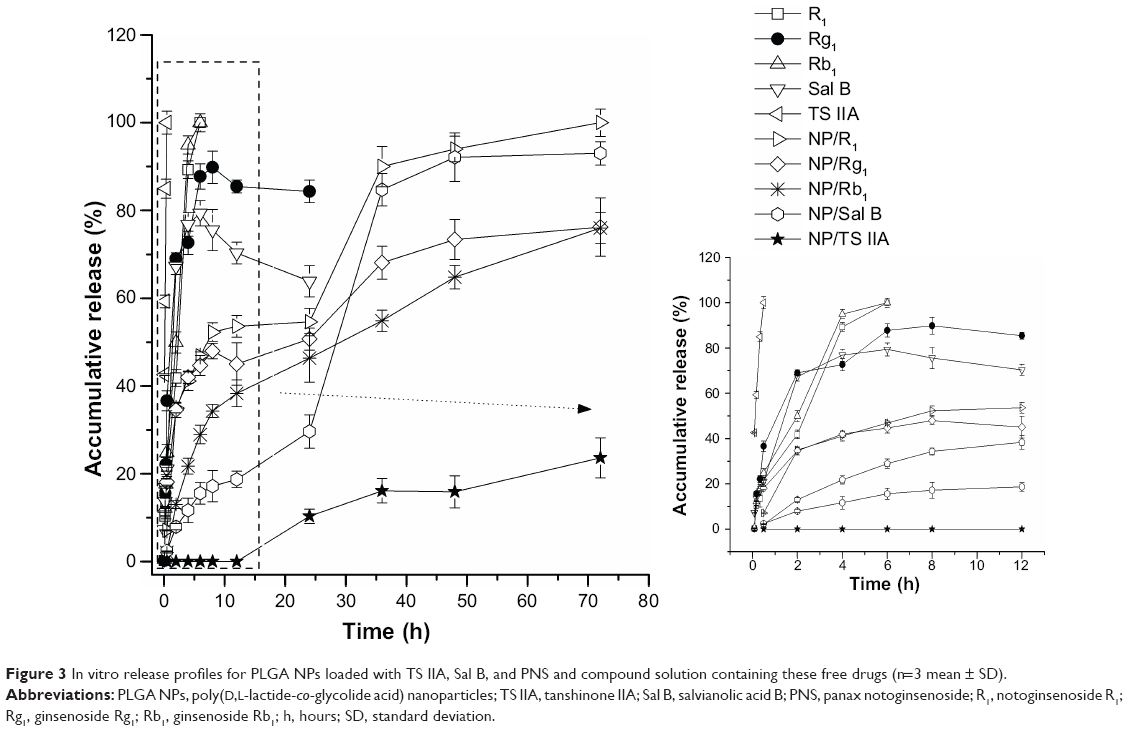 | Figure 3 In vitro release profiles for PLGA NPs loaded with TS IIA, Sal B, and PNS and compound solution containing these free drugs (n=3 mean ± SD). |
In vivo pharmacokinetic study
The pharmacokinetic parameters were determined using the non-compartmental method by the DAS (Drug and Statistic for Windows) software (version 3.1.1, Shanghai BioGuider Medicinal Technology Co, Ltd, People’s Republic of China). The pharmacokinetic parameters of coumarin-6 are shown in Table 1. The concentration versus time profiles after the RW administration of coumarin-6 NPs and its solution in the PL are presented in Figure 4.
 | Table 1 Pharmacokinetic parameters of coumarin-6 after RW administration of coumarin-6 nanoparticles and solution in PL |
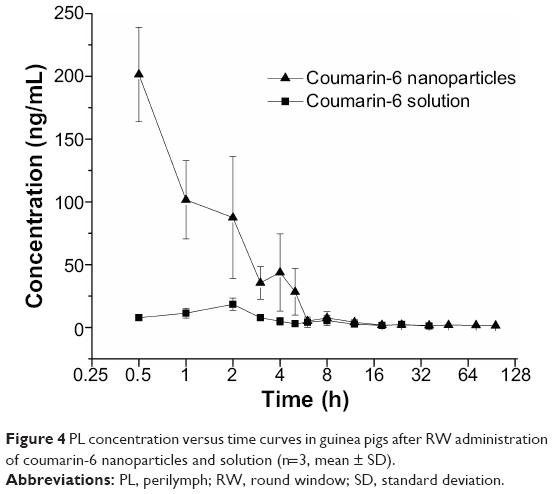 | Figure 4 PL concentration versus time curves in guinea pigs after RW administration of coumarin-6 nanoparticles and solution (n=3, mean ± SD). |
No coumarin-6 was detected after 36 hours in PL after the RW administration of the coumarin-6 solution. However, coumarin-6 was found in the PL up to 96 hours following RW administration of coumarin-6 NPs. An infinitesimal amount of coumarin-6 was detected in the plasma of several guinea pigs after the RW administration of coumarin-6 NPs or solution. The AUC0–t (area under the curve) of coumarin-6 in the PL after the RW administration of NPs was 4.7-fold higher and the Cmax (maximum concentration) was 10.9-fold higher than after the RW administration of a coumarin-6 solution. Due to the unique nanoscale size of the particles, the PLGA NPs readily carried substances across the RWM to the PL.37 These results indicated that the application of the PLGA NPs to the RWM significantly improved the drug distribution within the inner ear. PLGA is a biodegradable carrier with controllable degradation rates, which protects the encapsulated drugs, so coumarin-6 had a sustained release from NPs in the inner ear. Therefore, the concentration of coumarin-6 in inner ear PL after the local application of NPs is apparently higher than that of the solution. All these results demonstrated that the topical application of PLGA NPs to the RWM might be an effective strategy for the treatment of inner ear diseases.
Compound Danshen Tablets containing Danshen and Sanqi in Chinese Pharmacopoeia require that the doses of TS IIA and Sal B given at a single time are not <0.6 mg and <15 mg, respectively. Each dosage of PNS intravenous or intramuscular injections “Xueshuantong” for an adult is 150 mg. Referring to Chinese Pharmacopoeia for the dosage of compound Danshen tablets, the human dose was converted to the guinea pig equivalent dose based on body surface area conversion. The dosages given to the guinea pigs were as follows: Sal B was 1.796–2.026 mg, TS IIA was 0.0718–0.0810 mg, and PNS was 2.900–3.500 mg. Because tympanic capacity is limited, the dosing volume should not exceed 0.1 mL. Thus, the formulation concentrations of Sal B, TS IIA, and PNS were 20.0 mg/mL, 0.80 mg/mL, and 30 mg/mL, respectively. However, TS IIA could not be detected at this dose, so we used its maximum solubility in mixed solvents (ethanol/glycerol/PEG 400 =2:1:2) of 1.83 mg/mL as its dosage. The contents of R1, Rg1, and Rb1 were determined to be 26.1%, 36.5%, and 31.8%, respectively, and the guinea pigs weighed 250 g. Therefore, the doses of the compound solution were equivalent to 8 mg/kg Sal B, 0.73 mg/kg TS IIA, 3.13 mg/kg R1, 4.38 mg/kg Rg1, and 3.82 mg/kg Rb1. The same volume of nanosuspension containing compound drugs administered to the inner ear could not reach the same doses as the compound solution because of the limited capacity of the NPs to encapsulate drugs in a finite administration volume. To detect these agents in the animal study, we used the optimized NPs to calculate the dose.
The pharmacokinetic parameters of the PLGA NPs loaded with TS IIA, Sal B, and PNS are shown in Table 2. The concentration versus time profiles in different tissues after the RW administration of NPs or solution are presented in Figures 5–8. The administered dosages of R1, Rg1, and Rb1 in the compound solution were 3.0, 2.1, and 1.7 times as large as those in the NP formulations administered via RM, respectively. However, the Cmax values of R1, Rg1, and Rb1 in the PL after the delivery of the NPs were respectively 14.4-, 10.0-, and 16.7-fold higher compared with those of the compound solution, and the AUC0–t of R1, Rg1, and Rb1 was 4.0-, 3.1-, and 7.1-fold greater, respectively. The administration doses of Sal B and TS IIA in the compound solution were much larger than that in the NPs; however, both the Cmax and the AUC0–t of Sal B and TS IIA in the PL were not significantly higher. Although 10.4- and 58.7-fold lower doses of Sal B and TS IIA were, respectively, administered with the NPs compared to the compound solution, the AUC0–t values of Sal B and TS IIA in the PL were only 4.1- and 1.9-fold, respectively, lower compared to the compound solution, and the Cmax values of Sal B and TS IIA were 1.9- and 3.8-fold lower, respectively. The pharmaceutical concentrations in the PL were much higher than those in the plasma through the local administration of PLGA nanoparticles or drug solution. The maximum PL plasma concentration ratios of R1, Rg1, and Rb1 after the application of NPs were obviously higher than those of drug solution. In addition, the plasma concentrations of Sal B and TS IIA with administration of NPs were higher than those with the drug solution. We speculated that the existing surfactants (eg, PVA) in the NP colloidal suspension might enhance the transportation of Sal B and TS IIA from the PL to the plasma. Further studies would be required to confirm this speculation. On the whole, these results show that the NPs are promising vehicles to transport potential compound drugs to the inner ear and can result in increased absorption after RW administration. The enhanced bioavailability of the NPs in the inner ear may be the result of several factors as follows: 1) The permeability of the RWM. The RWM is a membranous opening in the bone within the scala tympani that acts as a barrier to separate the PL from the middle ear space and only allows select substances to cross from the middle ear to the inner ear because of its relative permeability.38,39 NPs readily cross the RWM.20,21 In addition, the bioadhesive properties of the polymer may have contributed to the increased rate of absorption.40 NPs deposited locally in the middle ear cavity could be transported by pinocytosis through the RWM, and then enter the perilymph through lymphatic and blood vessels.9 2) Middle ear clearance. The injected drug can be eliminated from the RW niche by the middle ear mucosa or through the Eustachian tube.10 Thus, the perilymph drug level will be greatly affected by the presence of middle ear clearance. The higher viscosity of the NP colloidal suspension would prolong contact with the RWM and allow greater access to the inner ear, which will further increase the efficiency of drug delivery across the RWM. 3) The protection of NPs. NPs diffusing into the inner ear fluids through the RWM protect drugs in the PLGA matrix against undesired clearance and their own degradation,41 which will further modulate their release. Moreover, PLGA nanoparticulate systems are able to convert poorly soluble, poorly absorbed, and labile biologically active substances into promising deliverable drugs.19
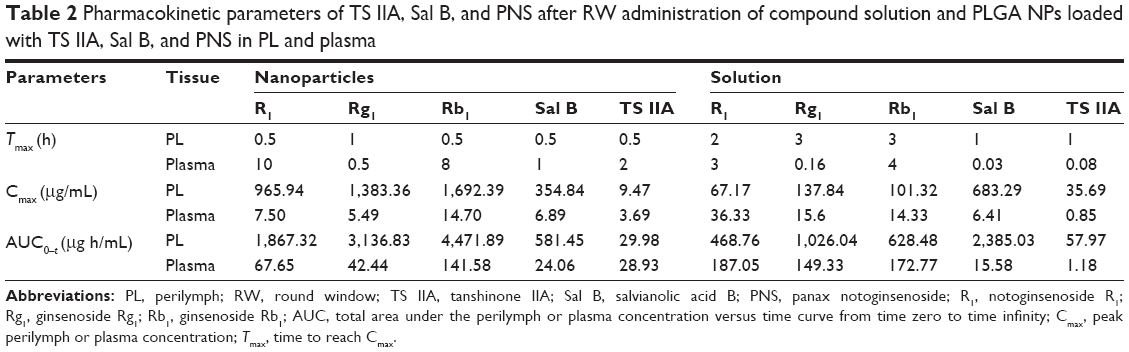 | Table 2 Pharmacokinetic parameters of TS IIA, Sal B, and PNS after RW administration of compound solution and PLGA NPs loaded with TS IIA, Sal B, and PNS in PL and plasma |
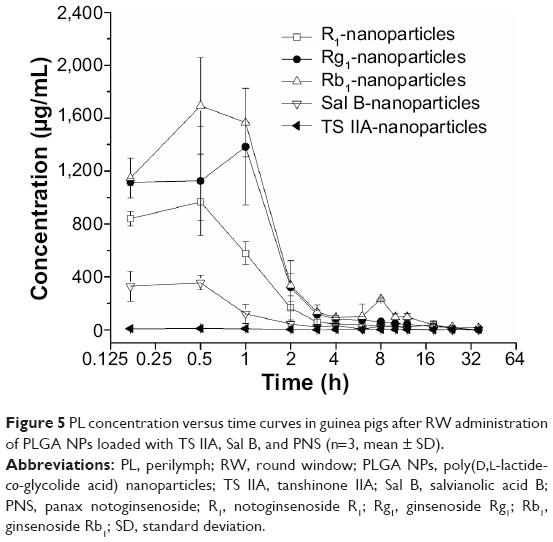 | Figure 5 PL concentration versus time curves in guinea pigs after RW administration of PLGA NPs loaded with TS IIA, Sal B, and PNS (n=3, mean ± SD). |
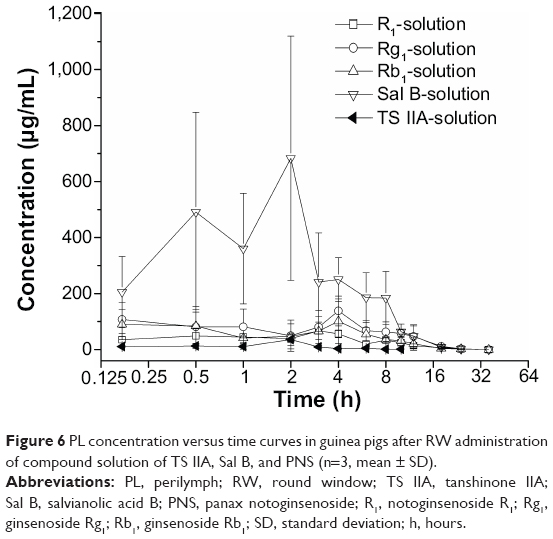 | Figure 6 PL concentration versus time curves in guinea pigs after RW administration of compound solution of TS IIA, Sal B, and PNS (n=3, mean ± SD). |
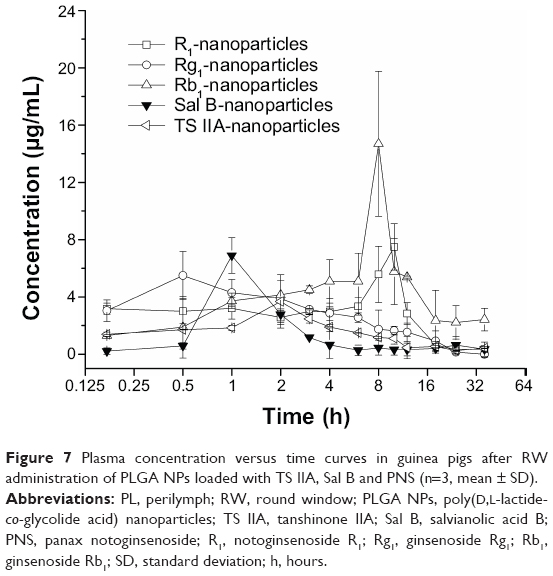 | Figure 7 Plasma concentration versus time curves in guinea pigs after RW administration of PLGA NPs loaded with TS IIA, Sal B and PNS (n=3, mean ± SD). |
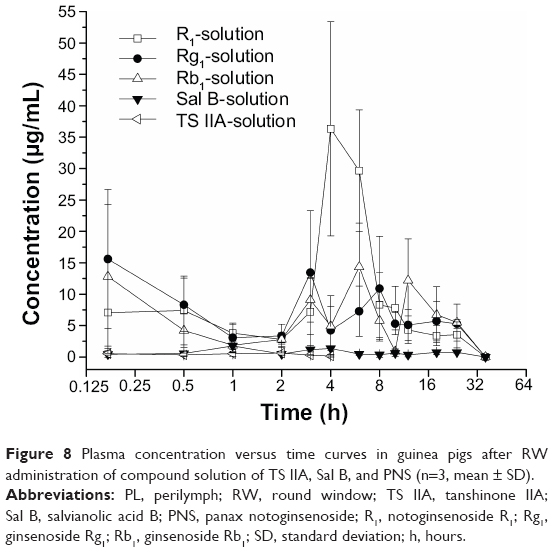 | Figure 8 Plasma concentration versus time curves in guinea pigs after RW administration of compound solution of TS IIA, Sal B, and PNS (n=3, mean ± SD). |
Conclusion
In the present study, we successfully prepared PLGA NPs loaded with coumarin-6 or compound drugs of Salvia miltiorrhiza and Panax notoginseng with satisfactory encapsulation efficiency. Coumarin-6 loaded PLGA NPs were identified in the inner ear fluids after RW application, suggesting that PLGA NPs can permeate through the RWM. PLGA NPs loaded with the compound components passing through the RWM to the inner ear was also confirmed. PLGA NPs with this powerful capability were able to carry single or multiple agents to traverse the RWM due to their unique properties at nanoscale dimensions, thus resulting in increased drug absorption in the inner ear. Local bioavailability in the inner ear was enhanced remarkably by the NP delivery system after RW application, suggesting that PLGA NPs provide a promising vehicle for effective inner ear drug delivery across the RWM.
Acknowledgments
This work was supported by the National Nature Science Foundation of China (Grant No 81274097), the Project for Zhujiang Science and Technology New Star of Guangzhou (Grant No 2013J2200059), the Cultivation Foundation for Distinguished Young Teachers in Higher Education of Guangdong (Grant No Yq2013099), and the Project of the Department of Education of Guangdong Province (Grant No 2012KJCX0069).
Disclosure
The authors report no conflicts of interest in this work.
References
Angelborg C, Hillerdal M, Hultcrantz E, Larsen HC. The microsphere method for studies of inner ear blood flow. ORL J Otorhinolaryngol Relat Spec. 1988;50(6):355–362. | ||
Juhn SK, Rybak LP. Labyrinthine barriers and cochlear homeostasis. Acta Otolaryngol. 1981;91(5–6):529–534. | ||
Inamura N, Salt AN. Permeability changes of the blood–labyrinth barrier measured in vivo during experimental treatments. Hear Res. 1992;61(1–2):12–18. | ||
Juhn SK, Hunter BA, Odland RM. Blood-labyrinth barrier and fluid dynamics of the inner ear. Int Tinnitus J. 2001;7(2):72–83. | ||
Mukherjea D, Rybak LP, Sheehan KE, et al. Current strategies for drug delivery to the inner ear. Expert Opin Drug Discov. 2011;6(5):491–505. | ||
Wagner N, Cayé-Thomasen P, Laurell G, Bagger-Sjöbäck D, Thomsen J. Cochlear hair cell loss in single-dose versus continuous round window administration of gentamicin. Acta Otolaryngol. 2005;125(4):340–345. | ||
Salt AN, Plontke SK. Principles of local drug delivery to the inner ear. Audiol Neurootol. 2009;14(6):350–360. | ||
Liu H, Feng L, Tolia G, Liddell MR, Hao J, Li SK. Evaluation of intratympanic formulations for inner ear delivery: methodology and sustained release formulation testing. Drug Dev Ind Pharm. 2014;40(7):896–903. | ||
Chen G, Zhang X, Yang F, Mu L. Disposition of nanoparticle-based delivery system via inner ear administration. Curr Drug Metab. 2010;11(10):886–897. | ||
Paulson DP, Abuzeid W, Jiang H, Oe T, O’Malley BW, Li D. A novel controlled local drug delivery system for inner ear disease. Laryngoscope. 2008;118(4):706–711. | ||
Minor LB, Schessel DA, Carey JP. Meniere’s disease. Curr Opin Neurol. 2004;17(1):9–16. | ||
Lefebvre PP, Staecker H. Steroid perfusion of the inner ear for sudden sensorineural hearing loss after failure of conventional therapy: a pilot study. Acta Otolaryngol. 2002;122(7):698–702. | ||
Goycoolea MV. The round window membrane under normal and pathological conditions. Acta Otolaryngol Suppl. 1992;493:43–55. | ||
Bjurstrom S, Slepecky N, Angelborg C. A histopathological study of the inner ear after administration of hyaluronan into the middle ear of the guinea pig. Acta Otolaryngol Suppl. 1987;442:62–65. | ||
Du X, Chen K, Kuriyavar S, et al. Magnetic targeted delivery of dexamethasone acetate across the round window membrane in guinea pigs. Otol Neurotol. 2013;34(1):41–47. | ||
Buckiová D, Ranjan S, Newman TA, et al. Minimally invasive drug delivery to the cochlea through application of nanoparticles to the round window membrane. Nanomedicine (Lond). 2012;7(9):1339–1354. | ||
Pritz CO, Dudás J, Rask-Andersen H, Schrott-Fischer A, Glueckert R. Nanomedicine strategies for drug delivery to the ear. Nanomedicine (Lond). 2013;8(7):1155–1172. | ||
Di Toro R, Betti V, Spampinato S. Biocompatibility and integrin mediated adhesion of human osteoblasts to PLGA copolymers. Eur J Pharm Sci. 2004;21(2–3):161–169. | ||
Acharya S, Sahoo SK. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv Drug Deliv Rev. 2011;63(3):170–183. | ||
Tamura T, Kita T, Nakagawa T, et al. Drug delivery to the cochlea using PLGA nanoparticles. Laryngoscope. 2005;115(11):2000–2005. | ||
Ge X, Jackson RL, Liu J, et al. Distribution of PLGA nanoparticles in chinchilla cochleae. Otolaryngol Head Neck Surg. 2007;137(4):619–623. | ||
Patel NR, Damann K, Leonardi C, Sabliov CM. Size dependency of PLGA-nanoparticle uptake and antifungal activity against Aspergillus flavus. Nanomedicine (Lond). 2011;6(8):1381–1395. | ||
Chen YC, Hsieh WY, Lee WF, Zeng DT. Effects of surface modification of PLGA-PEG-PLGA nanoparticles on loperamide delivery efficiency across the blood–brain barrier. J Biomater Appl. 2013;27(7):909–922. | ||
Zhang X, Chen G, Mu L. Research progress of novel dosage forms and pharmaceutical technologies for effective components of Danshen and Sanqi and their combination. Zhong Cheng Yao. 2011;33:1568–1570. | ||
Guo G, Li B, Wang Y, et al. Effects of salvianolic acid B on proliferation, neurite outgrowth and differentiation of neural stem cells derived from the cerebral cortex of embryonic mice. Sci China Life Sci. 2010;53(6):653–662. | ||
Qiao Z, Ma J, Liu H. Evaluation of the antioxidant potential of Salvia miltiorrhiza ethanol extract in a rat model of ischemia-reperfusion injury. Molecules. 2011;16(12):10002–10012. | ||
Fujita K, Hakuba N, Hata R, et al. Ginsenoside Rb1 protects against damage to the spiral ganglion cells after cochlear ischemia. Neurosci Lett. 2007;415(2):113–117. | ||
Lin XQ. Curative effect survey of compound root of red rooted salvia injection liquid treating sudden deafness. Anhui Yi Yao. 2001;5:175–176. | ||
Huang ZP. Two cases of Danshen tablets and other drug for treatment of Ménière’s disease. Nei Moivgol Med J. 2005;3:227. | ||
Gao D, Tang S, Tong Q. Oleanolic acid liposomes with polyethylene glycol modification: promising antitumor drug delivery. Int J Nanomedicine. 2012;7:3517–3526. | ||
Fei X, Chen X, Liang G, et al. Preparation, characterization, and biodistribution of breviscapine proliposomes in heart. J Drug Target. 2009;17(5):408–414. | ||
Jain RA. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials. 2000;21(23):2475–2490. | ||
Lu W, Zhang Y, Tan YZ, Hu KL, Jiang XG, Fu SK. Cationic albumin-conjugated pegylated nanoparticles as novel drug carrier for brain delivery. J Control Release. 2005;107(3):428–448. | ||
Chen YF, Jaw I, Shiao MS. Determination and pharmacokinetic analysis of salvianolic acid B in rat blood and bile by microdialysis and liquid chromatography. J Chromatogr A. 2005;1088(1–2):140–145. | ||
Han M, Fang XL. Difference in oral absorption of ginsenoside Rg1 between in vitro and in vivo models. Acta Pharmacol Sin. 2006;27(4):499–505. | ||
Li Q, Wang Y, Feng N, Fan Z, Sun J, Nan Y. Novel polymeric nanoparticles containing tanshinone IIA for the treatment of hepatoma. J Drug Target. 2008;16(10):725–732. | ||
Goycoolea MV. Clinical aspects of round window membrane permeability under normal and pathological conditions. Acta Otolaryngol. 2001;121(4):437–447. | ||
Swan EE, Mescher MJ, Sewell WF, Tao SL, Borenstein JT. Inner ear drug delivery for auditory applications. Adv Drug Deliv Rev. 2008;60(15):1583–1599. | ||
Juhn SK, Hamaguchi Y, Goycoolea M. Review of round window membrane permeability. Acta Otolaryngol Suppl. 1989;457:43–48. | ||
He W, Horn SW, Hussain MD. Improved bioavailability of orally administered mifepristone from PLGA nanoparticles. Int J Pharm. 2007;334(1–2):173–178. | ||
Kumar A, Chen F, Mozhi A, et al. Innovative pharmaceutical development based on unique properties of nanoscale delivery formulation. Nanoscale. 2013;5(18):8307–8325. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.