")
Back to Journals » International Journal of Nanomedicine » Volume 11
Enhanced dissolution and oral absorption of tacrolimus by supersaturable self-emulsifying drug delivery system
Authors Lee DR, Ho MJ, Jung HJ, Cho HR, Park JS, Yoon S, Choi YS, Choi YW , Oh C, Kang MJ
Received 22 December 2015
Accepted for publication 29 January 2016
Published 18 March 2016 Volume 2016:11 Pages 1109—1117
DOI https://doi.org/10.2147/IJN.S102991
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Dae Ro Lee,1 Myoung Jin Ho,1 Hyuck Jun Jung,1 Ha Ra Cho,1 Jun Seo Park,1 Suk-Hyun Yoon,2 Yong Seok Choi,1 Young Wook Choi,3 Chung-Hun Oh,2,4,5 Myung Joo Kang1
1College of Pharmacy, 2Department of Medical Laser, Graduate School, Dankook University, Dongnam-gu, Choenan, Chungnam, 3College of Pharmacy, Chung-Ang University, Dongjak-gu, Seoul, 4Department of Oral Physiology, College of Dentistry, Dankook University, 5Abel Medi-Tech Inc., Dongnam-gu, Cheonan, Chungnam, Korea
Abstract: A new Soluplus (polyvinyl caprolactam–polyvinyl acetate–polyethylene glycol graft copolymer)-based supersaturable self-emulsifying drug delivery system (S-SEDDS) was formulated to enhance oral absorption of tacrolimus (FK506) with minimal use of oil, surfactant, and cosurfactant. A high payload supersaturable system (S-SEDDS) was prepared by incorporating Soluplus, as a precipitation inhibitor, to SEDDS consisting of Capmul MCM, Cremophor EL, and Transcutol (FK506:vehicle:Soluplus =1:15:1). In vitro dissolution profile and in vitro pharmacokinetic aspect of S-SEDDS in rats were comparatively evaluated with those of conventional SEDDS formulas containing four times greater content of vehicle components (FK506:vehicle =1:60). Both formulations formed spherical drug-loaded microemulsion <70 nm in size when in contact with aqueous medium. In an in vitro dissolution test in a nonsink condition, the amphiphilic polymer noticeably retarded drug precipitation and maintained >80% of accumulated dissolution rate for 24 hours, analogous to that from conventional SEDDS. Moreover, pharmacokinetic parameters of the maximum blood concentration and area under the curve from S-SEDDS formula in rats were not statistically different (P>0.05) than those of conventional SEDDS. The results suggest that the Soluplus-based supersaturable system can be an alternative to achieve a comparable in vitro dissolution profile and in vivo oral absorption with conventional SEDDS, with minimal use of vehicle ingredients.
Keywords: tacrolimus, supersaturation, precipitation inhibitor, Soluplus, microemulsion, oral bioavailability
Introduction
Tacrolimus (FK506), a macrolide immunosuppressant, has been prescribed as a first-line therapeutic agent to prevent organ rejection after liver, heart, kidney, and bone marrow transplantation.1,2 The calcineurin inhibitor exhibits superior immunosuppressive activity to cyclosporine A, providing long-term survival in patients after transplantation.3,4 However, oral FK506 therapy has been deterred by poor and variable intestinal absorption, predominantly caused by its low aqueous solubility (1–2 μg/mL) and extensive metabolism in the gastrointestinal (GI) tract.5,6 The oral bioavailability (BA) of the marketed product (Prograf; Astellas Pharma, Inc., Tokyo, Japan), a solid dispersion system with cellulose derivatives, was only 21% in patients with large intra- and interindividual variation (4%–89%).7
Several pharmaceutical technologies have been explored to boost dissolution and/or oral absorption of potent calcineurin inhibitors, including oily solution,8 cyclodextrin complex,9 polymeric micelle,10 and a self-emulsifying drug delivery system (SEDDS).11 SEDDS is an isotropic blend of oils, surfactants, and/or cosolvents, which rapidly forms oil-in-water emulsions upon contact with aqueous medium under gentle gastric motility.12,13 Hydrophobic compounds dissolved in the preconcentrates can be placed in an oil droplet in an aqueous environment with no precipitation, providing a large interfacial area for drug release and absorption. Borhade et al11 reported that the FK506-loaded SEDDS formulation composed of Capmul MCM (glyceryl caprylate), Cremophor EL (polyoxyl 35 hydrogenated castor oil), and Transcutol (diethylene glycol monoethyl ether) has a higher dissolution rate and in vivo immunosuppressant activity compared to the marketed product. However, the self-emulsifying system included considerable quantities of oil and (co)surfactants (~300 mg) to deliver 5 mg of active compound, which could cause severe GI side effects after long-term oral administration, such as intestinal mucosal irritation and/perturbation. In this regard, the use of (co)surfactants should be limited and/or minimized below their tolerable intake levels.
In the last decade, supersaturable SEDDS (S-SEDDS) containing polymeric materials additionally, as a precipitation inhibitor, along with vehicle ingredients, has received much attention as an alternative to reduce the use of (co)surfactants, while preserving high dissolution rate and oral BA of conventional SEDDS formula.14–16 Decreased use of vehicle ingredients might lessen drug solubilization capacity and accelerate drug precipitation after dilution into the aqueous medium. However, precipitation inhibitors inhibit and/or retard drug crystallization, by delaying the rate of drug nucleation and crystal growth and/or by stabilizing the drug-loaded colloidal dispersion after adsorption onto colloidal surfaces.16–18 In fact, several previous studies have shown that the S-SEDDS formulations using hydroxypropyl methylcellulose (HPMC), polyvinyl pyrrolidone (PVP), or Soluplus (polyvinyl caprolactam–polyvinyl acetate–polyethylene glycol graft copolymer) as a precipitation inhibitor remarkably protracted the supersaturated state of the hydrophobic compounds, providing higher apparent concentration–time profile compared to that of the conventional SEDDS formulations.16,19,20
Herein, the aim of this study was to build a new FK506 S-SEDDS to achieve a comparable in vitro dissolution profile and in vivo oral absorption with conventional SEDDS, with the minimal use of oil, surfactant, and cosurfactant. Supersaturable systems of FK506 were formulated with HPMC, PVP, or Soluplus, and their physical characteristics, such as morphology, droplet size, and in vitro dissolution profile, were assessed. Moreover, the pharmacokinetic evaluation of FK506 from S-SEDDS formulation was carried out in rats with comparison to that of conventional SEDDS.
Materials and methods
Materials
FK506 monohydrate (purity >99.0 w/w%) was kindly supplied from Chong Kun Dang Pharm (Seoul, Korea). Ascomycin (purity >98% w/w%) used as an internal standard for FK506 Liquid Chromatography-Tandem Mass Spectrometry (LC–MS/MS) analysis was obtained from Sigma-Aldrich Co. (St Louis, MO, USA). Cremophor EL and Soluplus were kindly provided by BASF (Ludwigshafen, Germany). Capmul MCM C8 and Transcutol P were provided by Abitec Co. (Janesville, WI, USA) and Gattefosse (Saint Priest, France), respectively. PVP (Plasdone K17) and HPMC (Hypromellose 2910) were supplied by ISP Chemicals Co. (Shanghai, People’s Republic of China) and Shin-Etsu Chemical Co. (Tokyo, Japan), respectively. All other chemicals were of reagent and/or analytical grade and were used without further purification.
Preparation of FK506-loaded SEDDS and S-SEDDS formulations
The compositions of each formulation tested are listed in Table 1 and were prepared on a ten times larger scale for further experiments. The composition of F1 mimicked a previously described composition.11 FK506-loaded conventional SEDDS (F1–F3) were prepared by adding the active compound (50 mg) into an isotropic mixture of Capmul MCM/Cremophor EL/Transcutol P. The mixture was vortexed using a magnetic stirrer at 40°C until a transparent solution developed. For the preparation of S-SEDDS formulations (F4–F10) containing HPMC, PVP, or Soluplus as precipitation inhibitors, each polymer powder was added into the transparent SEDDS solution (F3). The mixture was then vortexed using a magnetic stirrer at room temperature to obtain a uniform suspension. The prepared self-emulsifying systems were stored at room temperature until tested.
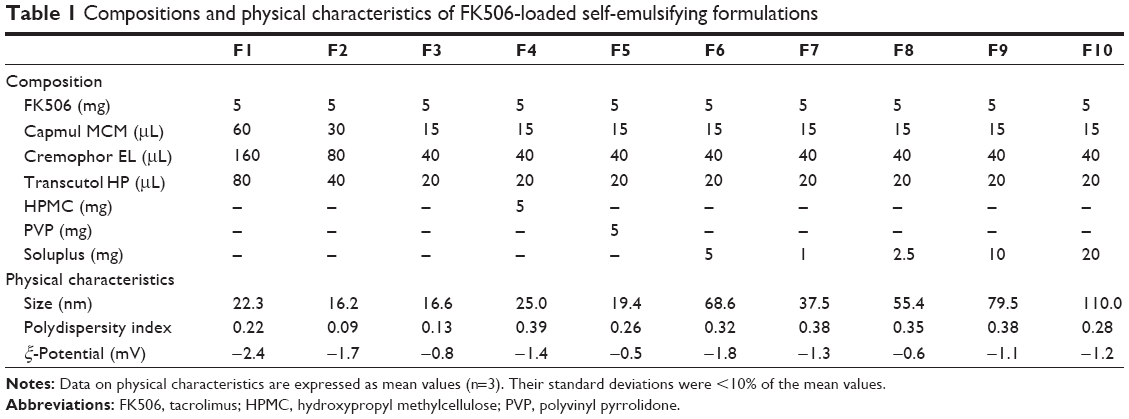 | Table 1 Compositions and physical characteristics of FK506-loaded self-emulsifying formulations |
Morphology observation by transmission electron microscopy
Each FK506-loaded preconcentrate (100 μL) was diluted with 1 mL of distilled water, and a drop of sample was deposited on a film-coated copper grid. The sample was immersed into uranyl acetate solution for 10 seconds to strain oil droplets. The dried sample was observed by transmission electron microscopy (TEM) (JEM 1010; JEOL, Tokyo, Japan) operating at an acceleration voltage of 80 kV.
Droplet size and ξ-potential
Each FK506-loaded preconcentrate (100 μL) was diluted with an appropriate volume of simulated gastric juice (10 mL) and then loaded into a cuvette placed in a thermostatic chamber at 25°C. The mean droplet size and ξ-potential of each aliquot was determined by dynamic light scattering using a Zetasizer Nano ZS apparatus (Malvern Instruments, Malvern, UK) at a scattering angle of 90°. All measurements were carried out in triplicate.
X-ray powder diffraction
The drug crystallinity in the SEDDS and S-SEDDS formulations was determined using an X-ray diffractometer (Ultima IV; Rigaku Corporation, Tokyo, Japan) at room temperature. Each sample was added to the grid, and the diffraction pattern was measured using CuKα radiation source operating at 40 mA and 40 kV. Samples were scanned over a 2θ range of 5°–40° at a scanning rate of 2 seconds/step.
In vitro release test
In vitro dissolution testing was carried out using a shaking incubator. Each preparation containing 20 mg of FK506 was immersed in 100 mL of dissolution medium (pH 1.2, pH 6.8, and distilled water) maintained at 37°C±0.5°C. The shaking speed was 50 rpm. Approximately 1 mL of aliquots were removed at 0.5 hours, 1 hour, 2 hours, 4 hours, 6 hours, and 24 hours and were centrifuged at 13,000 rpm for 5 minutes. The supernatant was then appropriately diluted with the mobile phase, and the drug concentration was determined by high performance liquid chromatography (HPLC) analysis.
HPLC analyses were performed using a Waters HPLC system comprising a model 515 pump, model 717 plus auto sampler, and model 486 UV detector furnished with an ODS column (4.6 mm ×150 mm, TSK-Gel ODS 80™; Tosoh Bioscience, Tokyo, Japan). The mobile phase consisting of distilled water, isopropyl alcohol, and tetrahydrofuran at a volume ratio of 5:2:2 was eluted at a flow rate of 1.0 mL/min. The eluent was monitored at 220 nm, and the peak drug elution occurred at a retention time of 7.5 minutes. The calibration curve was linear in the drug concentration range of 5–100 μg/mL (y =36,221x +4,771, r2=0.9999). Intraday and interday precision ranged from 0.24% to 0.82% and from 2.57% to 3.31%, respectively.
In vivo oral absorption study
The study protocol and all animal experiments were approved by the Institutional Animal Care and Use Committee of Dankook University in Chungnam, Korea. Animal care and experimental methods were conducted according to the National Institute of Health Principles of Laboratory Animal Care guidelines. Sprague-Dawley male rats (200–250 g, 7–9 weeks of age) were procured from Orient Bio (Kyungki-do, Korea) and housed in a specific pathogen-free facility with food and water ad libitum. After a 7-day acclimatization period, the rats were randomly divided into three groups (n=5 per group) and were fasted for 16 hours prior to the experiment. Rats were orally administered conventional SEDDS formula (F1 or F3) or the optimized S-SEDDS (F6) through a syringe fitted with a flexible oral zoned needle at a dose of 5 mg/kg. Each preconcentrate was dispersed in distilled water at a drug concentration of 1 mg/mL before drug administration. Blood samples were obtained from the retro-orbital plexus using EDTA-treated syringes 0 hour, 0.25 hours, 0.5 hours, 1 hour, 2 hours, 4 hours, 6 hours, 12 hours, and 24 hours after oral administration. The blood samples were stored below −70°C, and the concentrations of FK506 in blood samples were analyzed using an LC–MS/MS procedure that was validated according to US Food and Drug Administration guidelines.21 The area under the curve (AUC) from 0 hours to 24 hours (AUC0–24 hours) was calculated using a BA Calc 2007 pharmacokinetic analysis program (Korea Food and Drug Administration, Cheongju, Korea). The maximum FK506 concentration in blood (Cmax) and the time taken to reach the maximum FK506 concentration (Tmax) were obtained from blood concentration–time profile. The level of statistical significance (P<0.05) was determined using the Student’s t-test.
Results and discussion
Morphological and physical characteristics of S-SEDDS formulations
Different FK506-loaded SEDDS (F1–F3) and S-SEDDS (F4–F10) formulations with several kinds of polymeric materials as precipitation inhibitors were fabricated and were characterized in terms of morphology, droplet size, surface charge, and drug crystallinity. The shape and surface of the microemulsion produced from either conventional SEDDS (F3) or S-SEDDS (F6) were scrutinized by TEM. Both SEDDS and S-SEDDS preconcentrates formed uniform spherical droplets ranging in diameter <100 nm (Figure 1). Little aggregation between oil droplets was observed in both SEDDS and S-SEDDS formulations.
 | Figure 1 Transmission electron microscopy micrographs of (A) FK506-loaded conventional SEDDS (F1) and (B) Soluplus-incorporated S-SEDDS (F6). |
The droplet size of the colloidal dispersion is an important factor affecting the rate and extent of drug release, as well as intestinal absorption of active compounds.22,23 The globular size of conventional SEDDS formulations (F1–F3) measured by dynamic light scattering was ~20 nm (Table 1), regardless of the ratio of the drug to SEDDS vehicle. Although the droplet size generally became large as the drug-to-vehicle ratio increased, there was no obvious trend in SEDDS F1–F3 formulations because the drug-to-vehicle ratio was extremely low in every formulation (1.7%–6.7%). The droplet size of HPMC- (F4) or PVP-incorporated S-SEDDS (F5) was analogous to that of the conventional SEDDS. On the other hand, the droplet size of the Soluplus-based formulations (F6–F10) was gradually increased as the extent of the polymer was increased, suggesting the incorporation and/or adsorption of the amphiphilic polymer onto the colloidal surface. Nevertheless, the droplet size of S-SEDDS formulations was still suitable for oral delivery of the calcinurin inhibitor. Bagwe et al24 reported that the microemulsions <100 nm in size are thermodynamically stable and provide a large surface area, preserving the drug in a solubilized state. The low polydispersity index of <0.4 in all S-SEDDS formulations implied a uniform size distribution. The surface charge of all formulations was neutral or slightly negative, probably due to the free fatty acid originated from the Capmul MCM oil ingredient.
X-ray powder diffractograms of the drug powder, SEDDS (F1), and S-SEDDS (F4–F6) are depicted in Figure 2. The characteristic peaks of FK506 powder (Figure 2a) were not observed in the diffractograms of SEDDS F1 (Figure 2b) and S-SEDDS compositions (Figure 2c–e). This signifies that the calcineurin inhibitor preserves amorphous, solubilized state in all supersaturable formulations, with no drug precipitation.
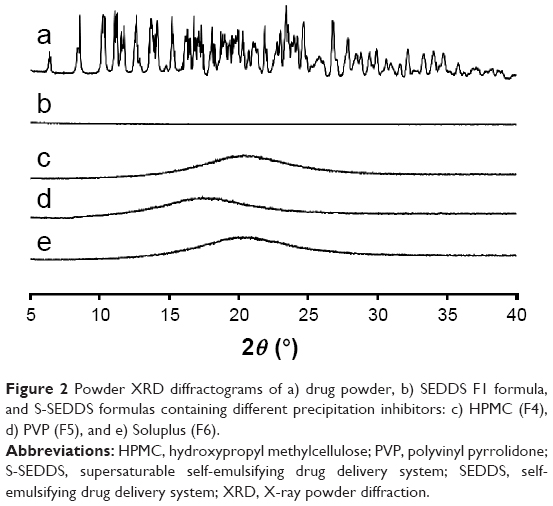 | Figure 2 Powder XRD diffractograms of a) drug powder, b) SEDDS F1 formula, and S-SEDDS formulas containing different precipitation inhibitors: c) HPMC (F4), d) PVP (F5), and e) Soluplus (F6). |
In vitro dissolution study
To distinctively evaluate the influence of formulation variables (eg, ratio of the drug to SEDDS vehicle and kind/amount of precipitation inhibitor) on the degree of supersaturation of FK506 in aqueous medium, an in vitro dissolution test was carried out under nonsink condition. The primary test medium was simulated gastric fluid (pH 1.2), because the active compound exhibits rapid and profound absorption behavior in the upper GI tract.5,6
The influence of the amount of drug loaded in conventional self-emulsifying formulations (F1–F3) on the drug release profile after dilution with aqueous medium (pH 1.2) was evaluated (Figure 3). In all formulations tested (F1–F3), the drug concentration in acidic medium peaked at the first sampling time with a high dissolution rate (>90%), suggesting that these preconcentrates rapidly formed drug-loaded fine droplets upon contact with aqueous medium, regardless of the quantity of drug. The F1 (FK506:vehicle =1:60) and F2 formulations (FK506:vehicle =1:30) permitted a high drug concentration for an extended period, displaying >90% accumulative release rate at 24 hours. However, in the case of F3, a fourfold high payload formula (FK506:vehicle =1:15) compared to F1, the drug concentration in the medium fell precipitously with time, because the water-insoluble compound was thermodynamically unstable at elevated concentration exceeding its equilibrated solubility. The amount of drug dissolved at 24 hours was markedly decreased to one-fifth of its original amount at 30 minutes. The solubilization capacity of lipid formulations drops logarithmically when diluted into an aqueous medium.25
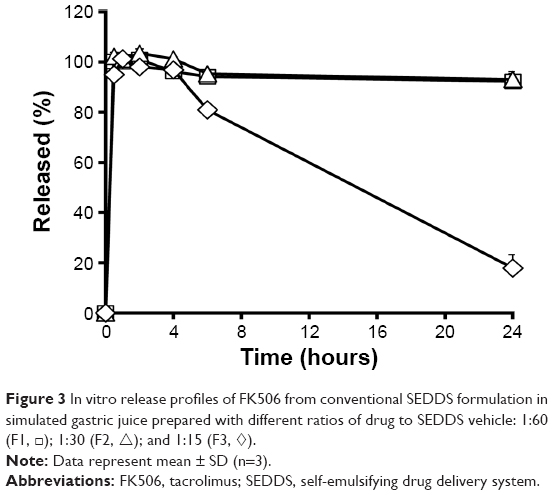 | Figure 3 In vitro release profiles of FK506 from conventional SEDDS formulation in simulated gastric juice prepared with different ratios of drug to SEDDS vehicle: 1:60 (F1, □); 1:30 (F2, △); and 1:15 (F3, ◊). |
Next, prototype S-SEDDS formulations were constructed by adding several polymeric materials (HPMC, PVP, and Soluplus) to SEDDS F3 formula to achieve the analogous dissolution profile with F1, with only one-quarter the quantity of SEDDS vehicle compared to F1. Cellulose derivatives, such as HPMC, HPMC acetate succinate, and hydroxypropyl cellulose, and vinyl polymers, including PVP, polyvinyl alcohol, and polyacrylic acid, are effective inhibitors of drug precipitation in solid dispersion or lipid-based formulations. These hydrophilic polymers have been reported to kinetically retard or inhibit drug crystallization by directly interfering with drug nucleation or crystal growth by intramolecular interaction.18 Yamashita et al26 reported that HPMC-based solid dispersion maintained the highest concentration of FK506 in an aqueous medium compared to polyethylene glycol or PVP-based compositions, by enhancing wettability and stabilizing the metastable state of active compound. On the other hand, the Soluplus amphiphilic polymer was reported to hinder aggregation and/or destruction of the drug-loaded oil droplet in gastric fluid.16,27 The release profiles of FK506 from S-SEDDS formulations containing different kinds of precipitation inhibitors (HPMC, PVP, or Soluplus) (F4–F6) in acidic medium (pH 1.2) are shown in Figure 4. In the case of hydrophilic polymers (HPMC and PVP), there were no differences in drug release compared to conventional SEDDS F3. The drug concentration was promptly boosted >90% for an initial period of 30 minutes but underwent rapid drug precipitation to <20% within 24 hours, analogous to the pattern from the conventional SEDDS F3 formula. The results indicated that these hydrophilic polymers could not effectively delay nucleation and/or crystal growth of drug molecules under nonsink condition, leading to substantial drug precipitation. Although HPMC is rich in hydroxyl groups capable of forming hydrogen bonding with FK506, the suspended polymer in the oily vehicle would not readily approach free drug molecules and/or crystals liberated from the droplets. Thus, the hydrophilic polymer could not delay the drug nucleation or crystal growth via their intramolecular interactions in the aqueous medium. In contrast, the addition of a small amount of Soluplus (5 mg) into the conventional SEDDS (F3) formulation dramatically prolonged the supersaturated state of FK506 during the experimental period. The amphiphilic polymer effectively inhibited drug precipitation, maintaining dissolution rate ~85% after 24 hours. This observation is consistent with the previous report that the incorporation of the amphiphilic polymer into the self-emulsifying formulation resulted in a slower precipitation of dutasteride by virtue of a precipitation inhibitor, maintaining three times greater dissolution rate after 2 hours compared to conventional lipid formulation.16 The polycaprolactam moiety, a hydrophobic compartment of the amphiphilic polymer, would be preferentially adsorbed and/or incorporated into the drug-loaded globules, forming a more condensed structure. Moreover, the hydrophilic polyethylene glycol groups would sterically stabilize the drug-loaded colloidal dispersion after dilution into the aqueous medium.
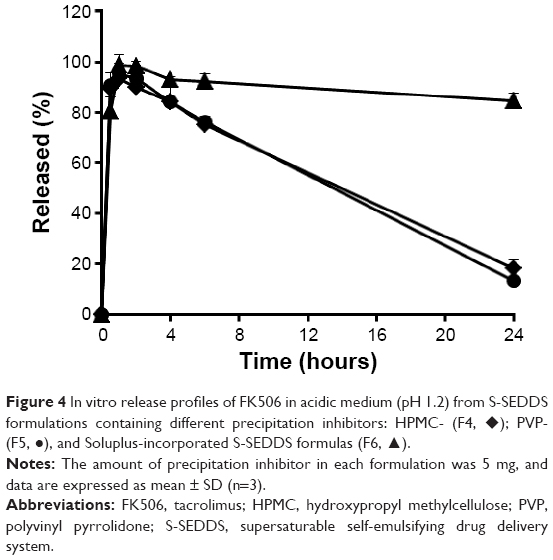 | Figure 4 In vitro release profiles of FK506 in acidic medium (pH 1.2) from S-SEDDS formulations containing different precipitation inhibitors: HPMC- (F4, ♦); PVP- (F5, •), and Soluplus-incorporated S-SEDDS formulas (F6, ▴). |
A series of S-SEDDS formulations containing different amounts of Soluplus (6.7%, 1.3%, 3.3%, 13.3%, and 26.6% to SEDDS vehicle; F6–F10) were prepared to evaluate the influence of the amount of Soluplus on the degree of the supersaturated state. As shown in Figure 5, F6 and F8 formulations with either 5 mg or 2.5 mg of the amphiphilic polymer, respectively, provided a higher apparent drug concentration profile compared to the other formulations; >80% dissolution rate at 24 hours. Conversely, the presence of >5 mg polymer markedly decreased the rate and extent of drug released, with the dissolution rate of F9 and F10 at 24 hours being only 70% and 37%, respectively. It may be that when the amount of Soluplus exceeds the critical level, the polymer drives loose packing of interfacial layers, finally destabilizing drug-loaded colloides.27 In fact, there was a tendency for increased globular size of Soluplus-based S-SEDDS formulas as the proportion of the amphiphilic polymer was increased (Table 1). It is also probable that, when the amount of the amphiphiles exceeds the critical micellar concentration in the medium, the polymer might aggregate to form its own micelle structure, neither being primarily incorporated nor stabilizing emulsions.
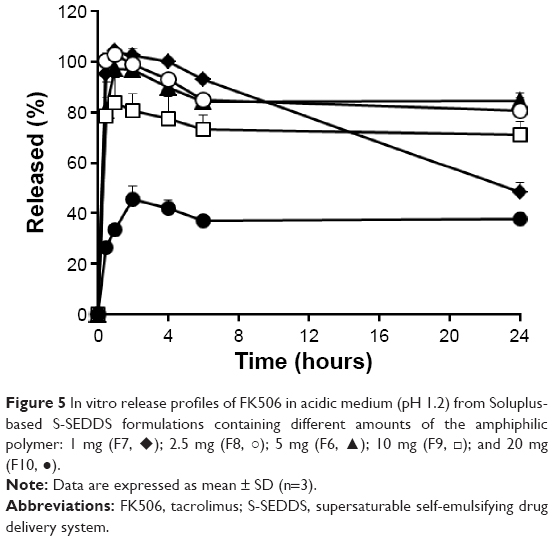 | Figure 5 In vitro release profiles of FK506 in acidic medium (pH 1.2) from Soluplus-based S-SEDDS formulations containing different amounts of the amphiphilic polymer: 1 mg (F7, ♦); 2.5 mg (F8, ○); 5 mg (F6, ▴); 10 mg (F9, □); and 20 mg (F10, •). |
In order to investigate the potential interaction between the self-emulsifying system and the amphiphilic polymer, the dissolution profile of FK506 from the physical mixtures of drug and Soluplus (weight ratio of 1:1, 1:2.5, 1:5, and 1:10) is further evaluated (Figure 6). In contrast to S-SEDDS formulations, the extent of drug released was linearly increased as the proportion of the polymer was increased, by micellar solubilization. After 24 hours, ~4.8%, 6.5%, 8.5%, and 16.4% of the drug was released from the physical mixtures in weight ratio of 1:1, 1:2.5, 1:5, and 1:10, respectively. A previous study has shown that the micelles formed by the amphiphilic polymer efficiently entrapped itraconazole, a hydrophobic compound inner compartment, providing the high apparent solubility in the aqueous medium.28 However, the amount of FK506 solubilized by Soluplus, in this study, was much lower than conventional SEDDS or S-SEDDS formulas. It is, thus, noteworthy that the dissolution rate obtained from the S-SEDDS F6 at 24 hours (85%) was remarkably higher than the sum of those from SEDDS F3 (17%) and the corresponding physical mixture (4.8%), revealing a synergistic interaction between SEDDS and the amphiphilic polymer on the degree of supersaturation of FK506 in the aqueous medium.
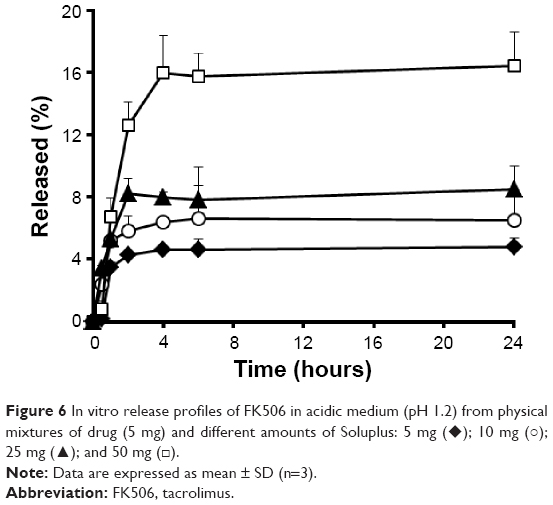 | Figure 6 In vitro release profiles of FK506 in acidic medium (pH 1.2) from physical mixtures of drug (5 mg) and different amounts of Soluplus: 5 mg (♦); 10 mg (○); 25 mg (▴); and 50 mg (□). |
On the other hand, the release profile of FK506 from the F6 formulation containing 5 mg of Soluplus was pH independent in both aspects of rate and extent of drug released, exhibiting >80% of accumulated release rate at 24 hours in pH 1.2, pH 6.8, and distilled water (Figure 7). Taken together, S-SEDDS F6, which displayed a comparable dissolution profile to F1, was chosen for further in vivo absorption study.
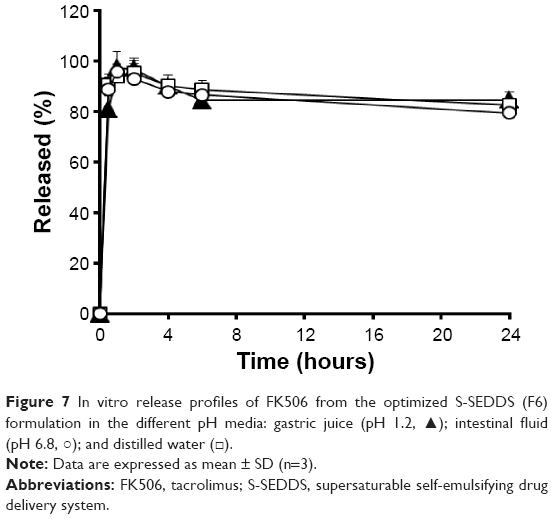 | Figure 7 In vitro release profiles of FK506 from the optimized S-SEDDS (F6) formulation in the different pH media: gastric juice (pH 1.2, ▴); intestinal fluid (pH 6.8, ○); and distilled water (□). |
In vivo absorption study in rats
The mean blood concentration versus time profiles of FK506 after oral administration to rats of two conventional SEDDS (F1 and F3) or the optimized S-SEDDS (F6) at a dose of 5 mg/kg are depicted in Figure 8. The corresponding pharmacokinetic parameters of FK506 – AUC0–24 hours, Cmax, and Tmax – are listed in Table 2. Upon oral administration, the blood concentration of FK506 was sharply elevated in all formulations, exhibiting a maximum drug concentration (Cmax) at 30 minutes after administration. These self-emulsifying formulations administered, spontaneously formed fine drug-loaded oil droplets, presenting the poorly water-soluble compound in a dissolved form. Of the two conventional SEDDS (F1 and F3), F1 showed significantly higher AUC0–24 hours (P<0.05) and Cmax (P<0.05) values compared to F3, with higher solubilizing capacity. These data were consistent with the in vitro dissolution data, showing that the larger amount of vehicle component provides a greater colloidal stability and solubilizing capacity in aqueous medium.
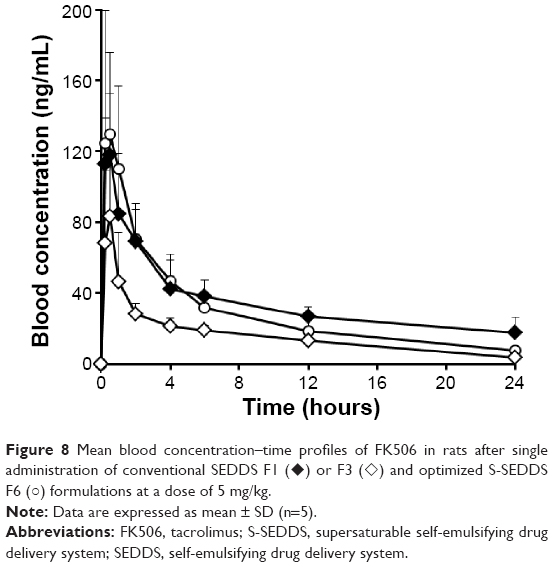 | Figure 8 Mean blood concentration–time profiles of FK506 in rats after single administration of conventional SEDDS F1 (♦) or F3 (◊) and optimized S-SEDDS F6 (○) formulations at a dose of 5 mg/kg. |
 | Table 2 Pharmacokinetic parameters of FK506 following oral administration of conventional SEDDS (F1 and F3) and S-SEDDS formulations (F6) in rats at a dose of 5 mg/kg |
However, surprisingly, the optimized S-SEDDS (F6) containing one-quarter the amount of SEDDS vehicle compared to F1 exhibited comparable blood concentration–time profile to that of F1, while providing higher concentration–time profile compared to F3. This observation indicated that the incorporation of the amphiphilic polymer into SEDDS drastically inhibited drug precipitation and extended the supersaturated state of the compound in aqueous medium, as demonstrated by in vitro release test. The Cmax values for F1 and F6 were 118.0±34.3 ng/mL and 129.4±46.2 ng/mL, respectively. The AUC0–24 hours values for these formulations were 791.8±157.8 ng/mL and 762.9±123.3 ng/mL, respectively. These parameters obtained from F1 and F6 were statistically similar (P>0.05). On the other hand, the Cmax and AUC0–24 hours values for the S-SEDDS formula were 1.5-fold (P<0.05) and 2.1-fold (P<0.05) greater than that of the conventional SEDDS F3. From these findings, we concluded that the oral absorption of FK506 can be markedly facilitated by incorporating a small quantity of the amphiphilic polymer (5 mg) as a precipitation inhibitor to the conventional SEDDS formulation of FK506.
Conclusion
To improve the oral absorption of FK506 with minimal use of oil, surfactant, and cosolvent, a new S-SEDDS formulation was fabricated by incorporating Soluplus (5 mg) as a precipitation inhibitor to the conventional SEDDS. Soluplus-based S-SEDDS exhibited a markedly higher dissolution rate compared to conventional SEDDS with an equal quantity of vehicle ingredients, with a release rate comparable to that of the conventional SEDDS prepared with four times more oil, surfactant, and cosolvent. A pharmacokinetic study carried out in rats revealed that the pharmacokinetic parameters of AUC0–24 hours, Cmax, and Tmax of S-SEDDS were not statistically different (P>0.05) than those of SEDDS prepared with four times more vehicle, a potentially harmful ingredient after long-term administration. Thus, the use of supersaturable formulation can be an effective approach for boosting dissolution and oral absorption of poorly water-soluble compounds, including FK506.
Acknowledgments
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT, and Future Planning (NRF-2013R1A1A1058935). This work (Grant no C0236963) was supported by Business for Cooperative R&D between Industry, Academy, and Research Institute funded by Korea’s Small and Medium Business Administration in 2014.
Disclosure
The authors report no conflicts of interest in this work.
References
Kino T, Hatanaka H, Miyata S, et al. FK-506, a novel immunosuppressant isolated from a Streptomyces. II. Immunosuppressive effect of FK-506 in vitro. J Antibiot. 1987;40:1256–1265. | ||
Spencer CM, Goa KL, Gillis JC. Tacrolimus. An update of its pharmacology and clinical efficacy in the management of organ transplantation. Drugs. 1997;54:925–975. | ||
Geissler EK, Schlitt HJ. Immunosuppression for liver transplantation. Gut. 2009;58:452–463. | ||
Cardenas ME, Zhu D, Heitman J. Molecular mechanisms of immunosuppression by cyclosporine, FK506, and rapamycin. Curr Opin Nephrol Hypertens. 1995;4:472–477. | ||
Venkataramanan R, Swaminathan A, Prasad T, et al. Clinical pharmacokinetics of tacrolimus. Clin Pharmacokinet. 1995;29:404–430. | ||
Tamura S, Ohike A, Ibuki R, Amidon GL, Yamashita S. Tacrolimus is a class II low-solubility high-permeability drug: the effect of P-glycoprotein efflux on regional permeability of tacrolimus in rats. J Pharm Sci. 2002;91:719–729. | ||
Christine E, Susan E. Clinical pharmacokinetics and pharmacodynamics of tacrolimus in solid organ transplantation. Clin Pharmacokinet. 2004;43:623–653. | ||
Honbo T, Kobayashi M, Hane K, Hata T, Ueda Y. The oral dosage form of FK-506. Transplant Proc. 1987;19:17–22. | ||
Arima H, Yunomae K, Miyake K, Irie T, Hirayama F, Uekama K. Comparative studies of the enhancing effects of cyclodextrins on the solubility and oral bioavailability of tacrolimus in rats. J Pharm Sci. 2001;90:690–701. | ||
Wang Y, Wang C, Fu S, et al. Preparation of tacrolimus loaded micelles based on poly(ε-caprolactone)-poly(ethylene glycol)-poly(ε-caprolactone). Int J Pharm. 2011;407:184–189. | ||
Borhade V, Nair H, Hegde D. Design and evaluation of self-microemulsifying drug delivery system (SMEDDS) of tacrolimus. AAPS PharmSciTech. 2008;9:13–21. | ||
Constantinides PP. Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharm Res. 1995;12:1561–1572. | ||
Constantinides PP, Scalart JP. Formulation and physical characterization of water-in-oil microemulsions containing long- versus medium-chain glycerides. Int J Pharm. 1997;158:57–68. | ||
Gao P, Rush BD, Pfund WP, et al. Development of a supersaturable SEDDS (S-SEDDS) formulation of paclitaxel with improved oral bioavailability. J Pharm Sci. 2003;92:2386–2398. | ||
Gao P, Akrami A, Alvarez F, et al. Characterization and optimization of AMG 517 supersaturatable self-emulsifying drug delivery system (S-SEDDS) for improved oral absorption. J Pharm Sci. 2009;98:516–528. | ||
Lee DH, Yeom DW, Song YS, et al. Improved oral absorption of dutasteride via Soluplus®-based supersaturable self-emulsifying drug delivery system (S-SEDDS). Int J Pharm. 2015;478:341–347. | ||
Usui F, Maeda K, Kusai A, Nishimura K, Keiji Y. Inhibitory effects of water-soluble polymers on precipitation of RS-8359. Int J Pharm. 1997;154:59–66. | ||
Xu S, Dai WG. Drug precipitation inhibitors in supersaturable formulations. Int J Pharm. 2013;453:36–43. | ||
Wei Y, Ye X, Shang X, et al. Enhanced oral bioavailability of silybin by a supersaturatable self-emulsifying drug delivery system (S-SEDDS). Colloids Surf A Physicochem Eng Asp. 2012;396:22–28. | ||
Chen ZQ, Liu Y, Zhao JH, Wang L, Feng NP. Improved oral bioavailability of poorly water-soluble indirubin by a supersaturatable self-microemulsifying drug delivery system. Int J Nanomedicine. 2012;7:1115–1125. | ||
Park JS, Cho HR, Kang MJ, Choi YS. A rapid and sensitive method to determine tacrolimus in rat whole blood using liquid–liquid extraction with mild temperature ultrasonication and LC–MS/MS. Arch Pharm Res. 2016;39(1):73–82. | ||
Shah NH, Carvajal MT, Patel CI, Infeld MH, Malick AW. Self-emulsifying drug delivery systems (SEDDS) with polyglycolyzed glycerides for improving in vitro dissolution and oral absorption of lipophilic drugs. Int J Pharm. 1994;106:15–23. | ||
Tarr BD, Yalkowsky SH. Enhanced intestinal absorption of cyclosporine in rats through the reduction of emulsion droplet size. Pharm Res. 1989;6:40–43. | ||
Bagwe RP, Kanicky JR, Palla BJ, Patanjali PK, Shah DO. Improved drug delivery using microemulsions: rationale, recent progress, and new horizons. Crit Rev Ther Drug Carrier Syst. 2001;18:77–140. | ||
Pouton CW. Formulation of poorly water-soluble drugs for oral administration: physicochemical and physiological issues and the lipid formulation classification system. Eur J Pharm Sci. 2006;29:278–287. | ||
Yamashita K, Nakate T, Okimoto K, et al. Establishment of new preparation method for solid dispersion formulation of tacrolimus. Int J Pharm. 2003;267:79–91. | ||
Song WH, Park JH, Yeom DW, et al. Enhanced dissolution of celecoxib by supersaturating self-emulsifying drug delivery system (S-SEDDS) formulation. Arch Pharm Res. 2013;36:69–78. | ||
Zhong Y, Jing G, Tian B, et al. Supersaturation induced by Itraconazole/Soluplus® micelles provided high GI absorption in vivo. Asian J Pharm Sci. Epub 2015 Sep 2. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.