Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Enhanced detection of single-cell-secreted proteins using a fluorescent immunoassay on the protein-G-terminated glass substrate
Authors Jeong Y, Lee KH, Park H, Choi J ![]()
Received 17 July 2015
Accepted for publication 29 October 2015
Published 24 November 2015 Volume 2015:10(1) Pages 7197—7205
DOI https://doi.org/10.2147/IJN.S92596
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Yoon Jeong,1,2 Kwan Hong Lee,1,2 Hansoo Park,3 Jonghoon Choi1,2
1Department of Bionano Technology, Graduate School, Hanyang University, Seoul, 2Department of Bionano Engineering, Hanyang University ERICA, Ansan, 3School of Integrative Engineering, Chung-Ang University, Seoul, South Korea
Abstract: We present an evaluation of protein-G-terminated glass slides that may contain a suitable substrate for aligning the orientation of antibodies to obtain better binding moiety to the target antigen. The results of the protein-G-terminated slides were compared with those obtained with epoxy-based slides to evaluate signal enhancement for human immunoglobulin G (IgG) targets, and an increase in the average fluorescence intensity was observed for the lowest measurable amount of IgG target in the assay using protein-G-terminated slides. Applying this strategy for signal amplification to single-cell assays improves the limits of detection for human IgG protein and cytokines (interleukin-2 and interferon-γ) captured from hybridomas. Our data indicate that protein-G-terminated slides have a higher binding capacity for antigens and have better spot-to-spot consistency than that of traditional epoxy-based slides. These properties would be beneficial in the detection of fine amounts of single-cell-secreted proteins, which may provide key insights into cell–cell communication and immune responses.
Keywords: microwell array, antibody’s orientation, single cell analysis, secreted cytokine, protein-G-terminated surface
Corrigendum for this paper has been published
Introduction
The communication between cells, essential for the maintenance of homeostasis, is a back-and-forth interaction among different proteins.1,2 Often, a breakdown in this intricate communication leads to a wide variety of diseases, thereby emphasizing the importance of protein–protein interactions. In particular, understanding the types and rates of proteins secreted by single cells is important for evaluating cell-to-cell communications. However, the limited protein detection range and the lack of detection sensitivity of current techniques are obstacles in the monitoring and study of secreted proteins from single cells.3,4 One of the ways to circumvent such concerns is the use of protein microarrays that are commonly utilized for the high-throughput screening and analysis of protein interactions.5–7 Protein microarrays rely on the capture of secreted proteins from cells on a support surface, for example, a nitrocellulose membrane, glass slide, or microtiter plate, and detection by the fluorescent dye-labeled probe molecules added to the array. Existing methods of detecting the capture of secreted proteins from single cells in microarrays are still impeded by their lack of detection sensitivity.
Traditional approaches to the capturing of cell-secreted proteins are based on the immunohistochemistry of the cells that rest on top of analyte-specific antibodies.8–10 This method, however, typically requires long incubation times (12–24 hours) in order to capture sufficient amounts of analytes for detection. Assays isolating individual cells in microwells for the detection of single-cell-secreted analytes also require this approach in order to probe the fluorescent signal from antibodies bound to the target proteins.11
A protein microarray utilizes microtechnology to probe interactions and functions of proteins in a high-throughput manner.12,13 Since the technology can analyze many proteins simultaneously, it has been widely applied in current biotechnology. The protein microarray consists of any reaction between the probe and the immobilized protein that emits a fluorescent signal that can be read by a laser scanner. Other benefits of protein microarray systems include their automated processes, fast analysis, high sensitivity, and requirement of only small quantities of samples and reagents.14 Protein microarrays were first introduced during the development of antibody microarrays in the 1980s.15 Subsequently, fast, high-throughput analysis was readily developed in protein assays since they are based on DNA microarrays.16,17
The performance of microarray functionalized slides (eg, in terms of signal intensity, signal-to-noise ratio, and spot homogeneity) is dependent on various factors, one of which is the surface moiety for the efficient immobilization of either protein or antibody samples. To improve the detection sensitivity of microarrays, current microarray slides are conferred with a diverse set of surface functionalities by coating them with various units, including, but not limited to, functional groups such as aldehyde, epoxy, and amino groups, and nucleic acids such as DNA.18,19 Antibodies are commonly conjugated onto such functionalized slides in an attempt to control and align their directionality. Yet, the nonspecific nature of antibody conjugation on glass slides results in a reduction of the assay efficiency. The need to detect low levels of target analyte further compounds the issue.20,21
Such glass slides with functionalized surface groups (eg, amine, aldehyde, and N-hydroxysuccinimide [NHS] ester groups) bind proteins and antibodies either through the formation of covalent bonds or by their adsorption or electrostatic interactions.18 The demand for a higher binding capacity for analytes and the need for decreased sample consumption while performing precise analyte quantification are leading to the application of these functionalized glass slides in single-cell microarrays.22,23
In this study, we evaluated the feasibility of protein-G-coated glass surfaces in terms of their antibody-binding capacity and resulting sensitivity by employing these surfaces to obtain single-cell secretory profiles of cytokines and by comparing these results with those obtained from experiments with epoxy-coated glass slides. We hypothesized that the use of protein-G-coated slides would be advantageous because of their affinity for the fragment crystallizable (Fc) portion of antibodies. By conferring the directionality of the antibodies to the functionalized slides, we expected that the sensitivity for target analytes secreted from single cells would subsequently be increased. Furthermore, the binding capacity was investigated using a microwell array of single cells, which would be of benefit to the functional analysis of cellular activity and regulation at the single-cell level.
Experimental methods
Reagents
Recombinant human immunoglobulin G1 (IgG1), interleukin-2 (IL-2), and interferon-γ (IFN-γ) were all purchased from R&D Systems (Minneapolis, MN, USA). All human blood cell samples were obtained from Research Blood Components (RBC), LLC which follows the American Association of Blood Banks guidelines for drawing donors. RBC, LLC’s regular population consists of healthy males and females between the ages of 18 and 65. All donors complete a uniform blood donor history questionnaire. An IRB approved consent form is obtained from each donor giving us permission to collect their blood and use or sell it at our discretion, for research purposes. Monoclonal antibodies were used for the capture and detection of human IgG, IL-2, and IFN-γ (Mabtech, Nacka Strand, Sweden). For detection, antibodies for each of the aforementioned proteins were labeled by dye conjugation using NHS ester-activated fluorescent dyes (Alexa Fluor 532, Alexa Fluor 647; Invitrogen, Carlsbad, CA, USA) and purified by a desalting column (Pierce, Rockford, IL, USA). The average degree of dye labeling was determined to be four–five dye molecules per antibody. Commercial protein-G-coated and epoxy-terminated glass slides (SuperEpoxy and SuperEpoxy 2 Substrate Slides [Protein] and Protein A/G Substrate Slides) were obtained from Arrayit Ltd, Sunnyvale, CA, USA.
Preparation of antibody-coated slides
Capture antibody was prepared at a concentration of 10 μg/mL and was added to the epoxy or protein G slides. Upon incubation in a humidity chamber at 37°C for 1 hour, the slides were subsequently blocked with blocking buffer (3% skim milk in deionized water) on a bench-top shaker for 30 minutes. The slides were then washed with phosphate-buffered saline–Tween 20 (PBS-T; 0.05% Tween 20 in PBS; Sigma-Aldrich, St Louis, MO, USA) for 15 minutes, followed by washing twice with PBS for 5 minutes each time, and then washing with deionized water for 20–30 seconds. Finally, the slides were dried by centrifugation in a microarray high-speed centrifuge (Arrayit Ltd) for 30 seconds. Various concentrations of IgG, IL-2, and IFN-γ were spotted onto both the epoxy and protein G slides. The slides were incubated in a humidity chamber at 37°C for 1 hour. Any remaining IgG was aspirated, and the functionalized slides were washed and dried as with the capture antibodies. The detection antibody was prepared at the concentration of 1 μg/mL, and the entire surface of epoxy slide and protein G slide were covered by using a slide cover. The slides were then incubated in a humidity chamber at 37°C for 1 hour, and then washed with PBS-T for 15 minutes, with PBS for 5 minutes, and with deionized water for 30 seconds. The arrays were imaged by using a GenePix 4000B microarray scanner (Molecular Devices LLC, Sunnyvale, CA, USA). The resulting fluorescence images were analyzed with GenePix ProMicroarray Image Analysis Software (Molecular Devices LLC).
Preparation of cells
A hybridoma cell line (NI 4 426/1A6-10; obtained from Public Health England, London, UK) secreting human anti-IL-4 IgG was cultured in Dulbecco’s Modified Eagle’s Medium (Sigma-Aldrich) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 mg/mL streptomycin.
Peripheral blood mononuclear cells (PBMCs) were obtained by Percoll (Sigma-Aldrich) isolation from heparinized whole blood (Research Blood Components, LLC, Boston, MA, USA). Frozen PBMCs were thawed and cultured in a HL-1 medium (Mediatech, Herndon, VA, USA) supplemented with 10% fetal bovine serum (PAA Laboratories, Cölbe, Germany), 100 U/mL penicillin, 100 mg/mL streptomycin, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 50 mM 2-mercaptoethanol, 1 mM sodium pyruvate, and 0.1 mM nonessential amino acids. In order to stimulate PBMCs, the cells were treated with a mixture of lipopolysaccharide (0.5 μg/mL), cytosine-phosphate-guanosine (CpG; 5 μg/mL), and tetanus toxoid (1.5 μg/mL) and were incubated at 37°C with 5% CO2 for 24 hours.
Preparation of microwell devices
The microwell arrays were prepared as previously described.24 Briefly, microwells were fabricated by preparing a polydimethylsiloxane mixture (Dow Corning, Midland, MI, USA) into a custom-built silicon wafer mold and cured at 80°C for 2 hours. The sizes of microwells were fixed at 50 μm in all dimensions. The microwell devices were sterilized in a plasma chamber (Femto Science, Suwon, South Korea) for 1 minute.
On-chip cell imaging and single-cell protein array
After washing, the stimulated cells were incubated with 1 mM of calcein violet AM stain (Invitrogen) for 30 minutes to evaluate their viability. Next, a suspension of washed cells was loaded into the microwells and adjusted to obtain a cell density ~1 cell/well, as described previously.24,25 Images of the cells in the microwells were obtained by using an automated inverted epifluorescence microscope (Zeiss, Oberkochen, Germany) equipped for live-cell imaging (temperature and CO2 control). Protein G and epoxy glass slides were coated with a mixture of three capture antibodies (10 μg/mL; human IgG1, IL-2, and IFN-γ). The functionalized slide was then used for microarray printing with cell-loaded arrays of microwells for 1 hour, as reported previously.24 Following the printing processes, the functionalized slides were blocked with 3% skim milk in PBS and washed with PBS-T and then PBS.
Detection of signal in the protein array
For the detection of secreted proteins and spotted recombinant IgG1, the slides were incubated with fluorescent dye-conjugated detection antibodies (1 μg/mL) for 1 hour at room temperature. Following incubation, the slides were briefly washed with PBS-T and PBS and then dried.
The arrays were imaged using a GenePix 4000B microarray scanner (Molecular Devices LLC). These data were filtered to remove features that contained saturated pixels (% Sat >1) or that exhibited a high degree of covariance >60.
Statistical analysis
The limit of detection was calculated from the individual calibration curves as 3× σ/s (σ, the standard deviation of the lowest concentration of target; s, the slope of the calibration curve in the linear range). Fisher’s test and Student’s t-tests were performed using GraphPad Prism (Version 5.0b for Mac OS X, GraphPad Software, San Diego, CA, USA).
Results and discussion
Antibodies (Ig) are relatively large proteins (~150 kDa) consisting of two light and two heavy chains. The typical structure of an antibody consists of a roughly asymmetric Y-shape containing many molecules, one type of which are amino acids. While the amino acids in the Fc region share the same sequence, the ones in the fragment antigen-binding (Fab) region have different sequences. This, therefore, allows the specific binding of antibodies to various antigens.26 In Figure 1A, schemes of three possible orientations of antibody binding with a substrate are illustrated, excluding the possible steric hindrance. Single-site attachment or multiple-site attachment occurs when the affinity ligand of an antibody is attached to the substrates by one or more functional groups, respectively. The proper orientation for alignment of the antibodies can only be obtained in the case where the Fc regions bind to a single ligand molecule on the surface. Therefore, any improperly orientated positions could block the Fab regions. For an effective binding moiety, two binding sites at two Fab regions should be exposed to the target analytes. As shown in Figure 1B, both types of slides were treated with human IgG antibodies (with decreasing concentrations from 34 to 4.3 μg/mL) and then compared in terms of their limits of detection. The protein G slides showed higher fluorescence intensities than those of the epoxy-coated slides and also displayed an extended detection limit. This result indicates that protein-G-coated substrate could be used as a suitable surface for the preparation of antibodies and detection of analytes at low concentrations.
 | Figure 1 Detection of human IgG protein in an immunochemistry assay on protein G or epoxy slides. |
Antibodies mainly adsorb onto epoxy slides, but a few free amines present on an antibody can directly react with the epoxy groups on the substrate, resulting in secondary amine bonds.27,28 Thus, the random immobilization of antibodies would occur on the epoxy-coated slides, and also, a small portion of antibodies may bind with the surface in the proper orientation for exposing the Fab regions. The random adsorption of antibodies on the epoxy-coated surface results from the reactions of random primary amino groups on the antibody, and thus would lead to less region-specific immobilization. The major disadvantage of this is not only that all of the antibody-binding sites would become occupied, but also that the maximal antibody-binding efficacy would not be reached.29 In contrast, protein G would expose the binding sites of antibodies by the site-specific attachment of the affinity ligand to the antibodies’ Fc regions.29,30 Antibodies can be directionally adsorbed onto protein G glass slide because a repeating 55-residue domain of protein G binds strongly to the Fc region of IgG, which is the tail region of an antibody.31 Although the constant domains of Fab and Fc are structurally related, protein G employs two different regions of its surface to bind to the Fab and Fc regions of IgG, displaying approximately tenfold higher affinity for the Fc region.31–33 It is desirable to control antibody orientation on the substrates in immunoassays in order to utilize them as useful tools for the binding and detection of monoclonal and polyclonal antibodies via affinity interactions. To this end, protein G slides would have broader specificity than epoxy-based slides.
Recombinant human IL-2 or IFN-γ was spotted on the epoxy and protein G slides coated with capture antibodies, and these cytokines were each detected using the respective antibodies labeled with fluorescent molecules (Figure 2). The results for the epoxy-slide spots are not shown in this figure due to negligible affinity and fluorescence signal observed for IL-2 and IFN-γ. A quantitative evaluation and comparison of the spotting results is shown in Figure 2C and D, respectively. On the protein-G-terminated slides, it was possible to detect the target antigens in the low-concentration sample and also to obtain higher fluorescent signals than those obtained on the epoxy-coated slides. More Fab and Fc regions were expected to bind onto the epoxy surfaces in a random fashion, and subsequently, decreased binding signal was observed. While it was expected that the protein-G-modified surfaces prepared with antibodies immobilized in an oriented manner would yield greater binding signal. Our results clearly show that the fluorescence intensities observed for the cytokines in the concentration range of ng/mL to pg/mL on the protein-G-modified slides were greater than those observed on the epoxy slides. These results indicate that using the protein G slides would provide the sensitivity needed to detect cytokine concentrations of less than 1 pg/mL, and that the antigen-binding sites (Fab regions) on the surface of protein G slides were more accessible to the analytes, whereas many antigen-binding sites might have been blocked on the epoxy-coated slides. Hence, site-oriented immobilization plays an important role in enhancing the detection signals for the protein-G-modified slides.
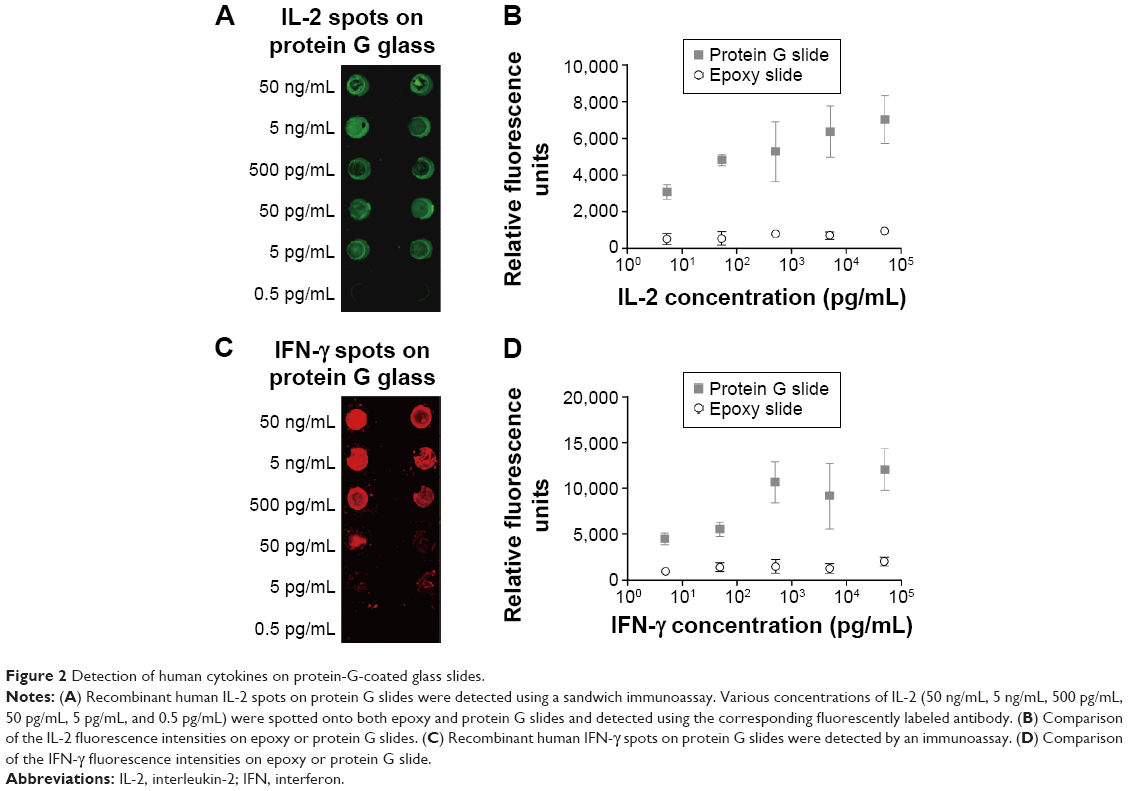 | Figure 2 Detection of human cytokines on protein-G-coated glass slides. |
In order to confirm the increased sensitivity of the protein assay, we evaluated the protein-G-modified glass slide for the capturing of single-cell-secreted proteins using an array of microfabricated wells. Because the cells in microwells can be retained after printing to utilize their cytokine release, the system would be particularly useful for the monitoring of nonsacrificed, viable cell responses and also for the repeated capturing of cytokines from the same cells.25 In Figure 3A, a cross-sectional schematic illustration of the microwell system is shown for the printing of cell-secreted proteins. After the suspension of cells was placed onto the array of microwells, the cells were settled down into the wells by gravity at an average density of 1–2 cells/well (Figure 3B). Next, the microwells were covered with two different types of glass slides that were precoated with specific IgG capture antibodies. After incubation (~1 hour), fluorescently labeled reagents were added to the glass slides for analyte detection. After the detachment of the glass slides, hybridomas were retained in the microwells for a continuous incubation period. We first hypothesized that the protein-G-terminated glass substrate could adsorb capture antibodies in the proper orientation. The comparative fluorescent microscope images (Figure 3C and D) for the IgG detection clearly showed that the protein-G-terminated glass slides had higher signal intensities than those on the epoxy-based slides. A greater detection range would be beneficial for capturing the small amounts of single cell-secreted proteins, and proper immobilization significantly improves the antibody–antigen binding ratio in a sandwich immunoassay. Previous studies11,25 used epoxy-based slides to capture the cytokines or immunoglobulins secreted from isolated individual cells, while our results show that, first, the application of protein-G-terminated substrates for the immobilization of antibodies in an immunoassay increases its sensitivity for the detection of cytokines from hybridomas. This is due to the correct orientation of antibodies that occurs on a surface coated with such substrates.
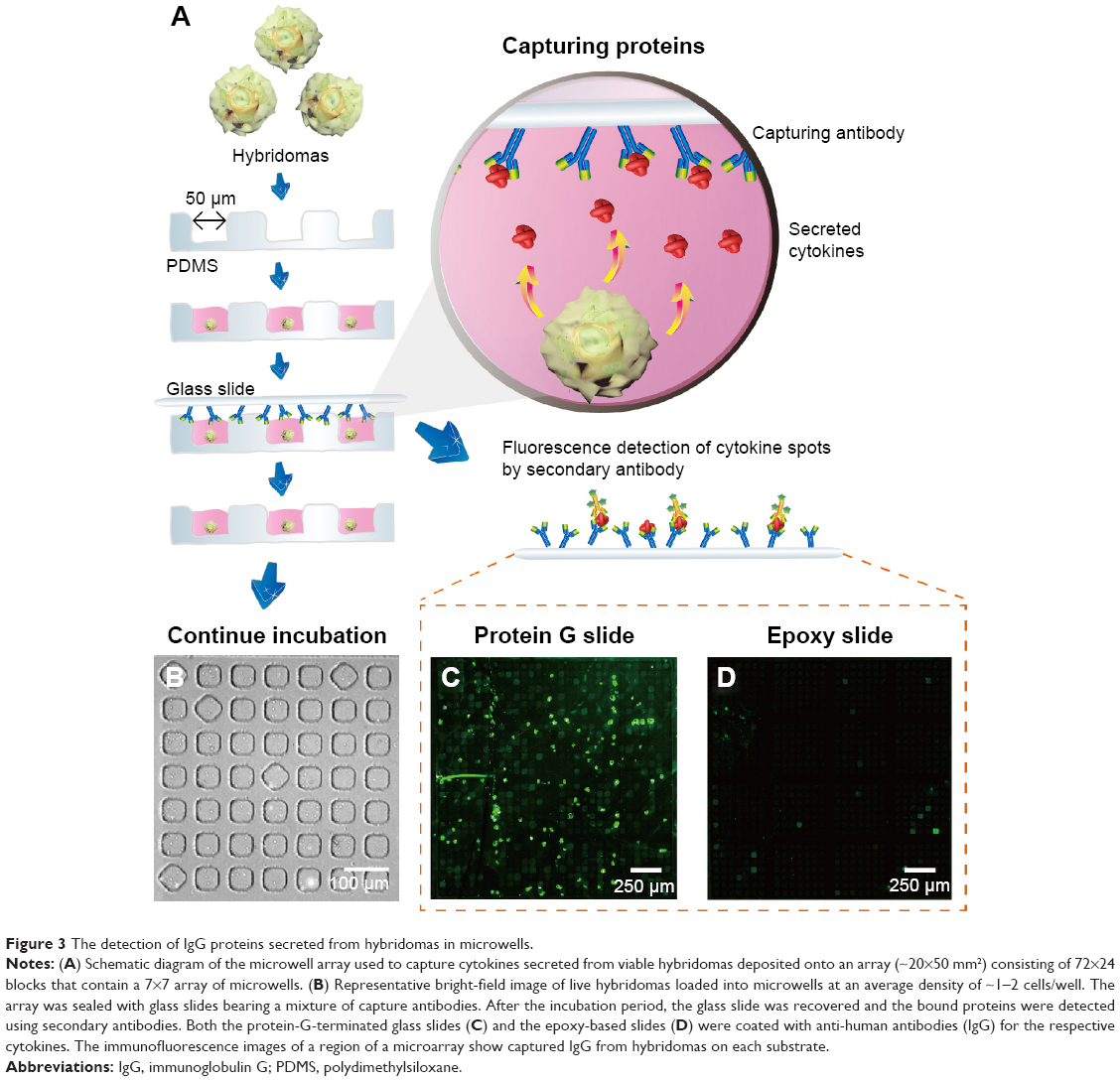 | Figure 3 The detection of IgG proteins secreted from hybridomas in microwells. |
In addition, we studied the feasibility of obtaining high sensitivity in capturing primary cell-secreted cytokines using protein-G-terminated glass slides in a microwell assay. The protein-G-coated glass slides were functionalized with antibodies for human IL-2 and IFN-γ, and were then sandwiched with a microwell assay isolating human PBMCs. The approach we describe in Figure 4 reveals that it was possible to obtain results for the repetitive detection of different cytokines released from the same subset of PBMCs in the microwells. The combined fluorescence images (Figure 4A) show that the live PBMCs visualized in the microwells had different surface cluster-of-differentiation markers for immune cells. For the evaluation of the detection sensitivity, protein-G-terminated glass slides were compared with the epoxy-coated slides in terms of the number of positive spots and their fluorescence intensities, representing the capture of IL-2 and IFN-γ (Figure 4B). Figure 4C correlates the number of positive spots for each cytokine with the data collected from each microwell by image analysis. The number of secretion events detected in the protein G slides was two- to threefold higher than that in the epoxy slides. As shown in Figure 4D, the relative fluorescence intensity was also statistically significantly higher in the protein G slides than that in the epoxy slides for both IL-2 and IFN-γ detection.
 | Figure 4 Detection of secreted cytokines (IL-2 and IFN-γ) from stimulated PBMCs in a microwell assay using two types of substrates. |
These results confirm that increased detection sensitivity for cell-secreted cytokines can be obtained by minimizing the nonspecific antibody binding on the substrate and by increasing the possibility of correct orientations of the capture antibodies on the surface of protein-G-terminated glass.
Therefore, it would be important in a protein assay, particularly for single-cell analysis, to ensure that the affinity ligands are immobilized on the substrate and that the binding regions of the antibodies are exposed and free to interact with the target molecules. As we show in this study by the use of comparative results, the type of surface used in an assay can play an important role in the efficacy, number of detection events, and fluorescent signal intensity of protein assays.
Conclusion
Herein, we demonstrated the increased detection capacity of protein assays when employing protein-G-modified substrates for the preparation of capture antibodies. When the geometry of the antibodies is taken into account, it can be inferred that they were directionally conjugated onto the protein G slide, because only the Fc portion of the antibody can be conjugated on the substrate. We show that this approach allowed increased detection events and fluorescence intensity for the capture of proteins or cytokines both from hybridomas and human PBMCs in the microarrays. The protein G on the slides may serve as an effective substrate for capturing minute amounts of single-cell-secreted cytokines, which are important markers in the study of cell–cell signaling between immune cells.
Acknowledgment
This work was supported by the research fund of Hanyang University (HY-2014-2082).
Disclosure
The authors report no conflicts of interest in this work.
References
Di Carlo D, Lee LP. Dynamic single-cell analysis for quantitative biology. Anal Chem. 2006;78(23):7918–7925. | ||
Jeong Y, Choi J, Lee KH. Technology advancement for integrative stem cell analyses. Tissue Eng Part B Rev. 2014;20(6):669–682. | ||
Han Q, Bradshaw EM, Nilsson B, et al. Multidimensional analysis of the frequencies and rates of cytokine secretion from single cells by quantitative microengraving. Lab Chip. 2010;10(11):1391–1400. | ||
Raphael MP, Christodoulides JA, Delehanty JB, et al. Quantitative imaging of protein secretions from single cells in real time. Biophys J. 2013;105(3):602–608. | ||
MacBeath G, Schreiber SL. Printing proteins as microarrays for high-throughput function determination. Science. 2000;289(5485):1760–1763. | ||
Wacker R, Schroder H, Niemeyer CM. Performance of antibody microarrays fabricated by either DNA-directed immobilization, direct spotting, or streptavidin-biotin attachment: a comparative study. Anal Biochem. 2004;330(2):281–287. | ||
Sun H, Tan LP, Gao L, Yao SQ. High-throughput screening of catalytically inactive mutants of protein tyrosine phosphatases (PTPs) in a phosphopeptide microarray. Chem Commun (Camb). 2009;(6): 677–679. | ||
Hsu SM, Raine L, Fanger HA. Comparative study of the peroxidase-antiperoxidase method and an avidin-biotin complex method for studying polypeptide hormones with radioimmunoassay antibodies. Am J Clin Pathol. 1981;75(5):734–738. | ||
Hsu SM, Soban E. Color modification of diaminobenzidine (DAB) precipitation by metallic ions and its application for double immunohistochemistry. J Histochem Cytochem. 1982;30(10):1079–1082. | ||
de Matos LL, Trufelli DC, de Matos MG, et al. Immunohistochemistry as an important tool in biomarkers detection and clinical practice. Biomark Insights. 2010;5:9–20. | ||
Choi J, Hwang MP, Lee JW, Lee KH. A glimpse into the interactions of cells in a microenvironment: the modulation of T cells by mesenchymal stem cells. Int J Nanomedicine. 2014;9:127–139. | ||
Magnan CN, Zeller M, Kayala MA, et al. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics. 2010;26(23):2936–2943. | ||
Hospach I, Joseph Y, Mai M, et al. Fabrication of homogeneous high-density antibody microarrays for cytokine detection. Microarrays. 2014;3(4):282–301. | ||
Waentig L, Techritz S, Jakubowski N, et al. A multi-parametric microarray for protein profiling: simultaneous analysis of 8 different cytochromes via differentially element tagged antibodies and laser ablation ICP-MS. Analyst. 2013;138(21):6309–6315. | ||
Chang TW. Binding of cells to matrixes of distinct antibodies coated on solid surface. J Immunol Methods. 1983;65(1–2):217–223. | ||
Schena M, Shalon D, Davis RW, et al. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270(5235):467–470. | ||
Schaferling M, Nagl S. Optical technologies for the read out and quality control of DNA and protein microarrays. Anal Bioanal Chem. 2006;385(3):500–517. | ||
Kusnezow W, Jacob A, Walijew A, et al. Antibody microarrays: an evaluation of production parameters. Proteomics. 2003;3(3):254–264. | ||
Seurynck-Servoss SL, White AM, Baird CL, et al. Evaluation of surface chemistries for antibody microarrays. Anal Biochem. 2007;371(1):105–115. | ||
Kausaite-Minkstimiene A, Ramanaviciene A, Kirlyte J, et al. Comparative study of random and oriented antibody immobilization techniques on the binding capacity of immunosensor. Anal Chem. 2010;82(15):6401–6408. | ||
Song HY, Zhou X, Hobley J, et al. Comparative study of random and oriented antibody immobilization as measured by dual polarization interferometry and surface plasmon resonance spectroscopy. Langmuir. 2012;28(1):997–1004. | ||
Trilling AK, Beekwilder J, Zuilhof H. Antibody orientation on biosensor surfaces: a minireview. Analyst. 2013;138(6):1619–1627. | ||
Makaraviciute A, Ramanaviciene A. Site-directed antibody immobilization techniques for immunosensors. Biosens Bioelectron. 2013;50:460–471. | ||
Ogunniyi AO, Story CM, Papa E, et al. Screening individual hybridomas by microengraving to discover monoclonal antibodies. Nat Protoc. 2009;4(5):767–782. | ||
Love JC, Ronan JL, Grotenbreg GM, et al. A microengraving method for rapid selection of single cells producing antigen-specific antibodies. Nat Biotechnol. 2006;24(6):703–707. | ||
Wang W, Singh S, Zeng DL, et al. Antibody structure, instability, and formulation. J Pharm Sci. 2007;96(1):1–26. | ||
Leckband D, Langer R. An approach for the stable immobilization of proteins. Biotechnol Bioeng. 1991;37(3):227–237. | ||
Mateo C, Torres R, Fernandez-Lorente G, et al. Epoxy-amino groups: a new tool for improved immobilization of proteins by the epoxy method. Biomacromolecules. 2003;4(3):772–777. | ||
Beyer NH, Hansen MZ, Schou C, et al. Optimization of antibody immobilization for on-line or off-line immunoaffinity chromatography. J Sep Sci. 2009;32(10):1592–1604. | ||
Akerstrom B, Brodin T, Reis K, et al. Protein G: a powerful tool for binding and detection of monoclonal and polyclonal antibodies. J Immunol. 1985;135(4):2589–2592. | ||
Gronenborn AM, Filpula DR, Essig NZ, et al. A novel, highly stable fold of the immunoglobulin binding domain of streptococcal protein G. Science. 1991;253(5020):657–661. | ||
Kato K, Lian LY, Barsukov IL, et al. Model for the complex between protein G and an antibody Fc fragment in solution. Structure. 1995;3(1):79–85. | ||
Sinha S, Zhang L, Duan S, et al. Effect of protein structure on deamidation rate in the Fc fragment of an IgG1 monoclonal antibody. Protein Sci. 2009;18(8):1573–1584. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.