")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Enhanced antitumor activity of doxorubicin in breast cancer through the use of poly(butylcyanoacrylate) nanoparticles
Authors Cabeza L, Ortiz R, Arias JL , Prados J , Martínez MAR, Entrena JM, Luque R, Melguizo C
Received 16 September 2014
Accepted for publication 11 November 2014
Published 13 February 2015 Volume 2015:10(1) Pages 1291—1306
DOI https://doi.org/10.2147/IJN.S74378
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Laura Cabeza,1,* Raúl Ortiz,1,2,* José L Arias,3 Jose Prados,1,4 Maria Adolfina Ruiz Martínez,3 José M Entrena,5,6 Raquel Luque,7 Consolación Melguizo1,4
1Institute of Biopathology and Regenerative Medicine (IBIMER), Granada, Spain; 2Department of Health Science, University of Jaén, Jaén, Spain; 3Department of Pharmacy and Pharmaceutical Technology, University of Granada, Granada, Spain; 4Biosanitary Institute of Granada (ibs GRANADA), SAS-Universidad de Granada, Granada, Spain; 5Institute of Neuroscience, Biomedical Research Center, 6Animal Behavior Research Unit, Scientific Instrumentation Center, University of Granada, Armilla, Granada, Spain; 7Service of Medical Oncology, Virgen de las Nieves Hospital, Granada, Spain
*These authors contributed equally to this work
Abstract: The use of doxorubicin (DOX), one of the most effective antitumor molecules in the treatment of metastatic breast cancer, is limited by its low tumor selectivity and its severe side effects. Colloidal carriers based on biodegradable poly(butylcyanoacrylate) nanoparticles (PBCA NPs) may enhance DOX antitumor activity against breast cancer cells, thus allowing a reduction of the effective dose required for antitumor activity and consequently the level of associated toxicity. DOX loading onto PBCA NPs was investigated in this work via both drug entrapment and surface adsorption. Cytotoxicity assays with DOX-loaded NPs were performed in vitro using breast tumor cell lines (MCF-7 human and E0771 mouse cancer cells), and in vivo evaluating antitumor activity in immunocompetent C57BL/6 mice. The entrapment method yielded greater drug loading values and a controlled drug release profile. Neither in vitro nor in vivo cytotoxicity was observed for blank NPs. The 50% inhibitory concentration (IC50) of DOX-loaded PBCA NPs was significantly lower for MCF-7 and E0771 cancer cells (4 and 15 times, respectively) compared with free DOX. Furthermore, DOX-loaded PBCA NPs produced a tumor growth inhibition that was 40% greater than that observed with free DOX, thus reducing DOX toxicity during treatment. These results suggest that DOX-loaded PBCA NPs have great potential for improving the efficacy of DOX therapy against advanced breast cancers.
Keywords: biodegradable polymer, carcinoma, cytotoxicity, chemotherapeutic drug, drug delivery, nanopolymer
Introduction
Breast cancer is the most common malignancy affecting women in the world. Thirty percent of patients eventually develop an untreatable metastatic disease, their average life expectancy being no more than 3 years.1 At present, systemic chemotherapy is considered the standard strategy against metastatic breast cancer. Despite the important advances in breast cancer therapy, including the development of new molecules (eg, trastuzumab, fulvestrant, eribulinmesylate), combinations of well-established drugs continue to be the first-line therapy against the malignant disease.2 Doxorubicin (DOX)-based regimens, due to the molecule’s high antitumor efficacy, are one of the most interesting approaches toward improving the level of clinical success in treating metastatic breast cancers. However, DOX-based chemotherapy is characterized by poor tumor selectivity plus severe (dose-limiting) side effects in healthy tissues and cells.3,4 Furthermore, DOX can lead to drug resistance in breast cancer cells, which may be another pertinent cause of chemotherapy failure.5 Therefore, new research efforts are needed to reduce the effective dose required for antitumor activity, the toxicity, and the drug resistance associated with DOX in such chemotherapy programs.6
In this context, the use of nanotechnology may be a promising strategy to improve DOX efficacy and safety. Drug-delivery nanoplatforms have been formulated to protect the antitumor agents that are loaded onto them from in vivo metabolization and elimination (thus optimizing the pharmacokinetic profile of the antitumor agent), and to increase drug accumulation at the site of the tumor, thereby reducing the drug dose needed to obtain a greater antitumor effect and minimizing toxicity.7 Poly(alkylcyanoacrylates) have been used as promising nanoplatforms in targeted tissue/cell drug delivery, because of their well-known biodegradability and low toxicity in chronic treatments (multiple dosing), good tolerance, and biocompatibility.8–11 Furthermore, cell recovery after the metabolization of poly(alkylcyanoacrylate)-based nanoparticles (NPs) occurs easily in vivo, thanks to the very low contact time between healthy tissues and the NP biodegradation products that are carried away from the degradation site by the blood flow.12 These nanocarriers have been engineered for the efficient delivery of numerous anticancer drugs to the tumor site (targeted drug delivery), thus increasing the patient survival rate.13 In this regard, they have demonstrated a promising capacity to reverse the multidrug resistance mechanisms developed by malignant cells. For instance, the adsorption of drug-loaded poly(alkylcyanoacrylate) NPs onto the surface of cancer cells, along with the formation of drug–poly(cyanoacrylic acid) ion pairs (as polymer degradation occurs), has been described as preventing drug recognition by the P-glycoprotein.14 Some authors have produced a large volume of particularly interesting work with respect to the development of DOX-loaded poly(alkylcyanoacrylate) NP. However, their work is entirely focused on the application of such nanoplatforms to glioblastoma models.8,15–17 To the best of our knowledge, the possibilities of these DOX-loaded polymeric nanoplatforms have not been fully investigated in other cancer models, eg, that of breast cancers.
The present work, therefore, is devoted to the engineering of DOX-loaded poly(butylcyanoacrylate) (PBCA) NPs with the aim of optimizing DOX therapies used against breast cancers. To that end, the best formulation conditions have been investigated. Compared with the free drug, our nanoparticulate DOX delivery system gave rise to a significant enhancement of the in vitro antiproliferative efficacy of DOX molecules. In addition, in vivo experiments demonstrated that the DOX-loaded PBCA nanoparticulate formulations clearly reduced tumor growth without evidence of an associated systemic toxicity. Therefore, this (nano-)formulation may be a promising candidate for improving the efficacy of DOX therapies against advanced breast cancers, while also minimizing the toxicity associated with doxorubicin’s activity.
Materials and methods
Materials
All chemicals used were of analytical grade from Sigma-Aldrich Chemical Co. (Spain), except for DOX hydrochloride (purchased from Dr M Damas, San Cecilio Hospital, Granada, Spain), and butylcyanoacrylate (generous gift from Henkel Loctite, Ireland).
Preparation of poly(butylcyanoacrylate) nanoparticles
PBCA NPs were synthesized following the well-known emulsion/polymerization procedure for butylcyanoacrylate monomers in an aqueous solution.18,19 Briefly, a 1% (w/v) acetonic solution of the monomer was added dropwise, under stirring at 1,200 rpm, to 10 mL of an aqueous polymerization medium containing 10−4 N HNO3 and the stabilizing agent Pluronic® F-68 (Sigma-Aldrich, St Louis, MO, USA) (1%, w/v). The mixture was maintained under polymerization conditions for 3 hours, after which the medium was finally neutralized with 10 μL of an aqueous NaOH (10−1 M) solution to ensure total consumption of the monomer. The remaining acetone was then fully evaporated using a BuchiRotavapor® (BÜCHI Labortechnik AG, Flawil, Switzerland) rotary evaporator to obtain an aqueous suspension of PBCA NPs. Finally, the nanoparticulate system was cleaned by subjecting it to repeated cycles of centrifugation (60 min at 10,700 rpm using a Centrikon T-124 high-speed centrifuge, Kontron, France) and redispersion in water, until the conductivity of the supernatant was ≤10 μS/cm.
Loading to the polymeric nanoplatform with doxorubicin
The polymeric NPs were loaded with antitumor drug by: 1) DOX entrapment within the (nano-)matrix; and 2) DOX surface adsorption onto previously prepared NPs. DOX entrapment/adsorption within/onto the NPs was assessed through UV–Vis spectroscopy by determining the amount of drug remaining in the supernatant solutions (see below), which were obtained by filtering the nanoparticulate suspensions through a 0.1 μm membrane filter, after the entrapment/adsorption process. No DOX adsorption on the filter membrane was determined spectrophotometrically.
DOX entrapment involved the preparation of an aqueous polymerization medium, containing 10−4 N HNO3 and Pluronic® F-68 (1%, w/v) (Sigma-Aldrich), and the subsequent dropwise addition of an acetonic solution of the butylcyanoacrylate monomer (1%, w/v) containing increasing concentrations of DOX (10−4, 5×10−4, 10−3, 5×10−3, and 10−2 M). All other steps previously described to prepare the PBCA NPs remained unchanged. Finally, DOX surface adsorption was performed at 25.0°C±0.5°C by combining suspensions of PBCA NPs (≈2%, w/v) with aqueous DOX solutions of known concentrations (10−4, 5×10−4, 10−3, 5×10−3, and 10−2 M). The suspensions were then stirred (50 rpm) for 24 hours. All experiments were performed in triplicate.
Characterization methods
The geometry of the NPs was deduced from scanning electron microscopy images (Zeiss DSM 950 scanning electron microscope, Germany). Before taking images, a dilute aqueous NP suspension (≈0.1%, w/v) was sonicated for 5 minutes, and then drops of the suspension were placed on copper grids with Formvar film. The grids were then dried in a convection oven at 35.0°C±0.5°C.
Mean particle diameters were also determined in triplicate at 25.0°C±0.5°C by photon correlation spectroscopy (PCS) (Malvern Autosizer® 4700, Malvern Instruments Ltd., Malvern, UK), with the scattering angle set at 60°. Measurements were made after suitably diluting the aqueous NP dispersions (≈0.1%, w/v). The stability of the DOX-loaded PBCA NPs was assessed by measuring both their size and DOX loading values after 1 month of storage in water at 4.0°C±0.5°C. The surface electrical properties of the PBCA NPs (in a ≈0.1% w/v aqueous suspension) were examined by electrophoresis (Malvern Zetasizer 2000 electrophoresis device, Malvern Instruments Ltd.) at pH 4 (adjusted with the addition of 10−4 N HNO3) and in a 10−3 M KNO3 solution. Measurements were performed at 25.0°C±0.5°C, after the DOX solution and PCBA NP suspension had been mixed under mechanical stirring (200 rpm) for 24 hours at the same temperature. The experimental uncertainty of the measurements was less than 5%. The O’Brien and White theory was used to convert the electrophoretic mobility (ue) data into zeta potential (ζ) values.20
UV–Vis absorption measurements (8500 UV–Vis Dinko spectrophotometer, Dinko, Spain) to determine DOX concentration in all the systems investigated were conducted at the maximum absorbance wavelength (481 nm), using quartz cells of 1 cm path length. Good linearity was observed at this wavelength, and the method was validated and verified for accuracy, precision, and linearity in all conditions tested. The reproducibility of the drug solutions’ UV–Vis absorbance spectra was assured throughout the time period required for the experiments performed in this work by wrapping all glassware with aluminum foil to protect them against ambient light. DOX entrapment/adsorption measurements were carried out in triplicate by applying Beer’s law to the UV–Vis absorbance of the supernatant solutions obtained by filtration (see above). For the method to be accurate, we considered the contribution to the absorbance of sources other than variations in drug concentration (mainly the presence of Pluronic® F-68 [Sigma-Aldrich] and PBCA degradation products in the supernatant solutions) by subtracting the absorbance of the supernatant produced under the same conditions but without DOX.21–23
DOX incorporation to the PBCA NPs was expressed in terms of drug entrapment efficiency (%) and drug loading (%). These parameters were calculated as follows:
|
|

|
|
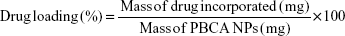
In vitro release of doxorubicin from poly(butylcyanoacrylate) nanoparticles
The dialysis bag method was used in triplicate to subsequently determine the amount of DOX released from PBCA NPs after its incorporation. Concretely, this characterization was performed on the PBCA NPs with the highest drug entrapment efficiencies (Figure 1B), ie, ≈22.3% for the adsorption method and ≈49.3% for the entrapment technique. Phosphate buffered saline (PBS) (pH =7.4±0.1) maintained at 37.0°C±0.5°C was used as the release medium for all of the experiments. The dialysis bag (2,000 Da cutoff; Spectrum® Spectra/Por® 6 dialysis membrane tubing, Spectrum Laboratories, Inc., Rancho Dominguez, CA, USA) retained the NPs and allowed the free DOX molecules to diffuse into the dissolution medium. About 2 mL of a drug-loaded PBCA NP suspension (containing 2 mg/mL of DOX) was poured into the bags (previously soaked in water for 12 h) with the two ends held by clamps. The bags were then placed in a conical flask containing 50 mL of the receiving phase (PBS, pH 7.4), which was stirred at 250 rpm. At predetermined times (0.08, 0.25, 0.50, 0.75, 1, 2, 3, 6, 12, and 24 hours), 3 mL samples of the receiving phase were withdrawn for UV–Vis analysis at 481 nm. An equal volume of PBS, maintained at the same temperature, was added to the release medium after sampling to ensure sink conditions.
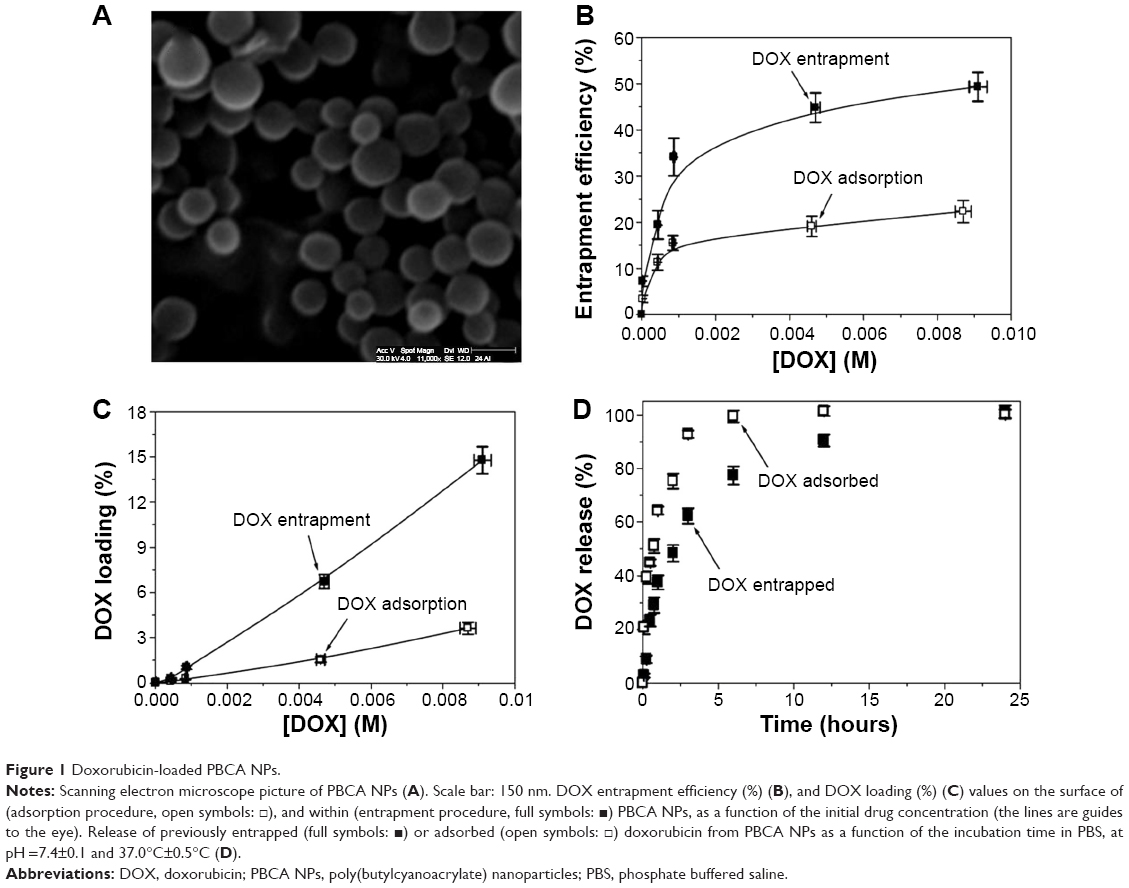 | Figure 1 Doxorubicin-loaded PBCA NPs. |
Cell culture
The MCF-7 human breast cancer cell line (European Collection of Cell Culture) and E0771 mouse breast cancer cell line (isolated from an immunocompetent C57BL/6 mouse and provided by Robin Anderson from Peter MacCallum Cancer Center, East Melbourne, Australia) were used. Both estrogen receptor-positive cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Sigma-Aldrich) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and 10 mM HEPES (4-(2-Hydroxyethyl)piperazine-1-ethanesulfonic acid) buffer, then maintained in a monolayer culture at 37.0°C±0.5°C under a humidified atmosphere of 5% CO2 in air.
Cytotoxicity assay
Cells were seeded into 24-well plates (104 cells/well for MCF-7, 1.5×104 cells/well for E0771) and incubated with increasing concentrations of free DOX (0.05–5.00 μM), DOX-loaded PBCA NPs (equal to 0.05–5.00 μM of free DOX) and blank PBCA NPs (ie, with no DOX loading, and at similar dilutions to drug-loaded NPs). Cytotoxic activity was assessed by determining the 50% inhibitory concentration (IC50) values obtained from the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide-based colorimetric assay (MTT assay). After 8, 24, and 48 hours of incubation, 20 μL of an MTT solution (5 mg/mL of cell culture medium) was added to each well. After incubation for 4 hours at 37°C±0.5°C, the culture medium was removed, and the resultant formazan crystals were dissolved in 200 μL of dimethyl sulfoxide (DMSO). The absorbance of the converted dye, which is proportional to the number of viable cells, was measured at 570 and 690 nm using a Titertek Multiskan™ colorimeter (Flow Laboratories, Irvine, UK). The percentage of surviving cells was expressed as relative growth rates (RGR, %) and was calculated as follows:
|
|
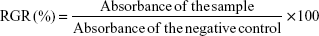
Intracellular uptake of doxorubicin
The intracellular distribution of free DOX and DOX derived from drug-loaded PBCA NPs was visualized by fluorescence microscopy. MCF-7 and E0771 cells were seeded into 8-well chamber slides (BD Biosciences, Erembodegem, Belgium) (5×103 cells/well) in 300 μL of DMEM. According to Li et al25 the cells were incubated with high doses of free DOX (10.0 and 43.1 μM) and DOX-loaded PBCA NPs (equal to 10.0 and 43.1 μM of free DOX) for different periods (from 1 to 24 h). A 100 nM solution of 4′,6-diamidino-2-phenylindole, DAPI, (Invitrogen™, Thermo Fisher Scientific, Waltham, MA, USA) was used for nuclear staining. The cells were then rinsed with PBS, mounted, and examined with fluorescent microscopy (Nikon Eclipse 50i microscope, Nikon Instruments Inc., Melville, NY, USA). DOX was excited at 570 nm and DAPI nuclear stains at 358 nm. In addition, FACScan (Becton Dickinson, San Jose, CA, USA) was used for the quantification of DOX incorporation by cells. Cells were seeded in 6-well plates (1.5×105 cells/well) in 2 mL of DMEM. After short incubations with DOX and DOX-loaded PBCA NPs (for 0.5, 1, 1.5, 2, and 4 h), the cells were washed with PBS. They were then harvested with a solution of PBS-ethylene-diamine-tetraacetic acid (0.02%) (EDTA), transferred to universal screw cap tubes, centrifuged (600× g for 5 min), and washed twice with PBS. FACScan analysis was used to obtain the mean value of fluorescence. The increase in mean fluorescence (IMF, %) was calculated as follows:
|
|

Transmission electron microscopy
Parental MCF-7 cells (control) and MCF-7 cells treated with DOX-loaded PBCA NPs (25 μg/mL) were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.2) at 25.0°C±0.5°C for 1 hour. Postfixation was performed at 25.0°C±0.5°C for 2 hours with 1% osmium tetroxide (OsO4) in cacodylate buffer (pH 7.2), and sections were then dehydrated in 100% ethanol before a final dehydration in propylene oxide. Samples were then embedded in Epon 812 resin. Ultrathin sections were stained with uranyl acetate and lead citrate, and then analyzed by optical and transmission electronic microscopy (Hitachi H7000 transmission electron microscope, Tokyo, Japan).24
Tumor induction and treatment
Female C57BL/6 mice (Scientific Instrumentation Center, University of Granada) were used in the in vivo study. All mice (body weight: 25–30 g) were maintained in a laminar airflow cabinet placed in a room maintained at 37.0°C±0.5°C and 40%–70% relative humidity, with a 12-hour light/dark cycle under specific pathogen-free conditions. The study was approved by the Ethics Committee of the School of Medicine (University of Granada) and performed according to its guidelines. Pilot experiments were previously conducted to determine the number of malignant cells for subcutaneous injection. After that, tumors were induced by subcutaneous injection of 5×105 E0771 cells into the left flanks of C57BL/6 mice. When the tumor was palpable, animals were randomly divided into four groups (n=10) corresponding to the treatments with saline solution (group I), free DOX solution (group II), blank PBCA NPs (with no DOX loading) (group III), and DOX-loaded PBCA NPs (group IV). The doses of DOX administered were 10 mg/kg of body mass in all of the formulations containing the antitumor drug. Each mouse was intravenously administered the formulation every 3 days up to a total number of 5 times. Weights and deaths were carefully recorded throughout the period, and the tumors’ largest diameter a plus the second largest diameter b perpendicular to a was measured using a digital calliper. The tumor volume (V, mm3) was calculated as follows:
|
|
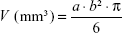
Statistical analysis
Statistical analysis was performed by using the Student’s t-test (SPSS version 7.5, SPSS, Chicago, IL, USA). Data with P<0.05 and P<0.001 were considered significant and very significant, respectively. A Kaplan–Meier method was used to determine the probability of mice survival, and the log-rank test was used to compare the fraction of surviving mice between groups (α=0.05).
Results
Particle geometry and surface electrical properties
The synthesis of PBCA NPs following an emulsion/polymerization method allowed, in agreement with previous studies, the formation of highly stabile spherical particles with a narrow colloidal size distribution (average diameter of 135±20 nm with a polydispersity index of 0.071, Figure 1A); as such, they are well suited to parenteral administration.22,25 DOX loading did not influence either the NP geometry or the quality of the nanoparticulate suspensions. Finally, PCS measurements after 1 month of storage at 4.0°C±0.5°C in water confirmed that the size of the particles did not change. In addition, there was no observation of the formation of bulky sediments or aggregates, NP aggregation or DOX precipitation, and/or release during this time period. Therefore, if the (nano-)preparations were not used directly once prepared, they were kept under these storage conditions until used. Finally, the electrokinetic characterization of the PBCA NPs (≈0.1%, w/v aqueous suspension) (ζ=−3.6±0.2 mV, at pH 4, 25.0°C±0.5°C and 10−3 M KNO3) was in agreement with previous electrophoretic studies on PBCA NPs in which a negative surface charge was observed at pHs ≥3.26
Doxorubicin loading to poly(butylcyanoacrylate) nanoparticles
We first investigated the influence of the drug-loading procedure on the quantity of DOX incorporated by the PBCA NPs: drug entrapment within the PBCA (nano-)matrix, and DOX surface adsorption onto previously synthesized PBCA NPs (see above). Interestingly, compared with DOX adsorption onto the NP surfaces, both the entrapment efficiency (%) and the drug loading (%) were significantly enhanced when DOX was entrapped within the NP matrices, independent of the initial DOX concentration. For instance, when the initial drug concentration was 0.01 M, these parameters, respectively, increased from 22.3%±2.4% and 3.6%±0.4%, when DOX was adsorbed onto the NP surface, to 49.3%±3.1% and 14.8%±0.9%, when the drug was entrapped within the NP network (Figure 1B and C). Finally, drug concentration positively influenced DOX incorporation to the PBCA NPs, for both of the drug-loading procedures employed (entrapment and surface adsorption). In the case of the adsorption method, over the range of initial DOX concentrations from 10−5 to 10−2 M, the entrapment efficiency and drug loading values increased from 3.4%±0.8% and 0.006%±0.001% up to 22.3%±2.4% and 3.6%±0.4%, respectively. For the entrapment technique, the corresponding values rose from 7.2%±1.1% and 0.022%±0.001% to 49.3%±3.1% and 14.8%±0.9%, respectively, over the same initial DOX concentration range (10–5 to 10–2 M). In any case, a tendency toward saturation at higher concentrations was apparent (Figure 1B).
Doxorubicin release from poly(butylcyanoacrylate) nanoparticles
DOX release from PBCA NPs at pH 7.4 and 37.0°C showed a biphasic, sustained process that was characterized by an initial rapid, burst phase (up to ≈37% and ≈64% drug released in 1 hour for the cases of DOX entrapment within NPs and surface adsorption onto NPs, respectively), followed by a more sustained release of the remaining DOX molecules over a period of 23 hours for DOX entrapment and 5 hours for surface adsorption (Figure 1D). Taking our results into consideration, we decided to perform all in vitro and in vivo antitumor studies using the PBCA NPs that had the highest DOX entrapment efficiency (ie, ≈49%, Figure 1B) and that were characterized by a sustained drug release profile (Figure 1D): therefore, we used DOX-loaded NPs that were synthesized by the entrapment procedure using a fixed initial drug concentration of 0.01 M.
Cytotoxicity of doxorubicin-loaded poly(butylcyanoacrylate) nanoparticles
Before the antitumor tests, the toxicity of blank PBCA NPs (with no DOX loading) was investigated in MCF-7 and E0771 cells. No significant differences were observed between the negative control and the cells exposed to increasing concentrations of PBCA NPs (Figure 2). Thus, the lack of cytotoxicity of the PBCA NPs alone meant they could safely be used as drug carriers. On the other hand, the cytotoxicity of DOX-loaded PBCA NPs against MCF-7 breast carcinoma cells (IC50: 0.5 μM) was significantly greater than that of free DOX (IC50: 2 μM) (P<0.001) after 8 hours of incubation. This difference between the IC50 of DOX-loaded PBCA NPs and free DOX could still be observed after 48 hours of incubation (Figure 3A). In addition, the IC50 of DOX-loaded PBCA NPs in E0771 cells was significantly lower than the IC50 of the free antitumor drug (P<0.05). DOX loaded within PBCA NPs yielded an IC50 (0.2 μM) 15 times lower than that of free DOX (IC50: 3 μM) after 8 hours of exposure and 21.7 times lower (0.06 μM) after 48 hours of exposure (free DOX IC50: 1.3 μM) (Figure 3B).
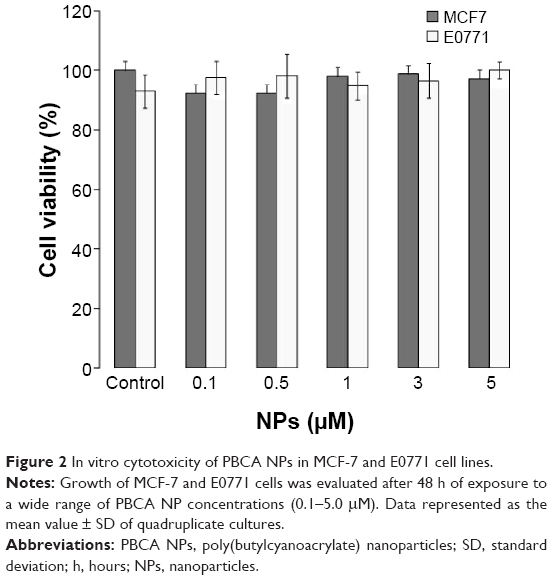 | Figure 2 In vitro cytotoxicity of PBCA NPs in MCF-7 and E0771 cell lines. |
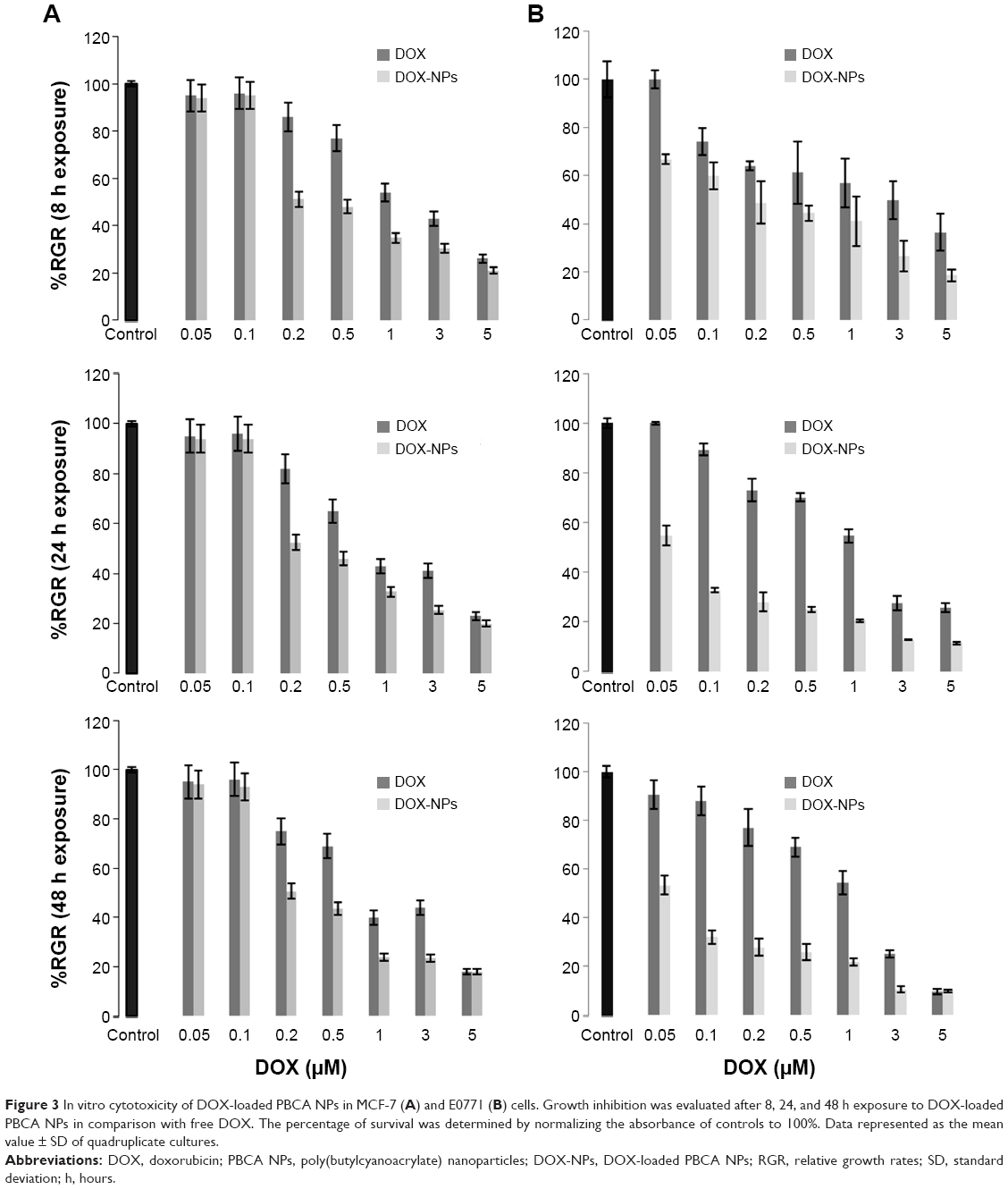 | Figure 3 In vitro cytotoxicity of DOX-loaded PBCA NPs in MCF-7 (A) and E0771 (B) cells. Growth inhibition was evaluated after 8, 24, and 48 h exposure to DOX-loaded PBCA NPs in comparison with free DOX. The percentage of survival was determined by normalizing the absorbance of controls to 100%. Data represented as the mean value ± SD of quadruplicate cultures. |
Distribution of doxorubicin-loaded poly(butylcyanoacrylate) nanoparticles
MCF-7 and E0771 cell lines treated with DOX-loaded PBCA NPs and free DOX at different times and concentrations were visualized by fluorescence microscopy. At any given moment, DOX-loaded PBCA NPs induced a more intense fluorescence (in both cytoplasm and nucleus) than free DOX, suggesting a greater DOX uptake by malignant cells (incorporation into the nucleus) when drug molecules were loaded within/onto NPs (Figure 4). Both cell lines were characterized by a similar drug incorporation and intracellular drug localization.
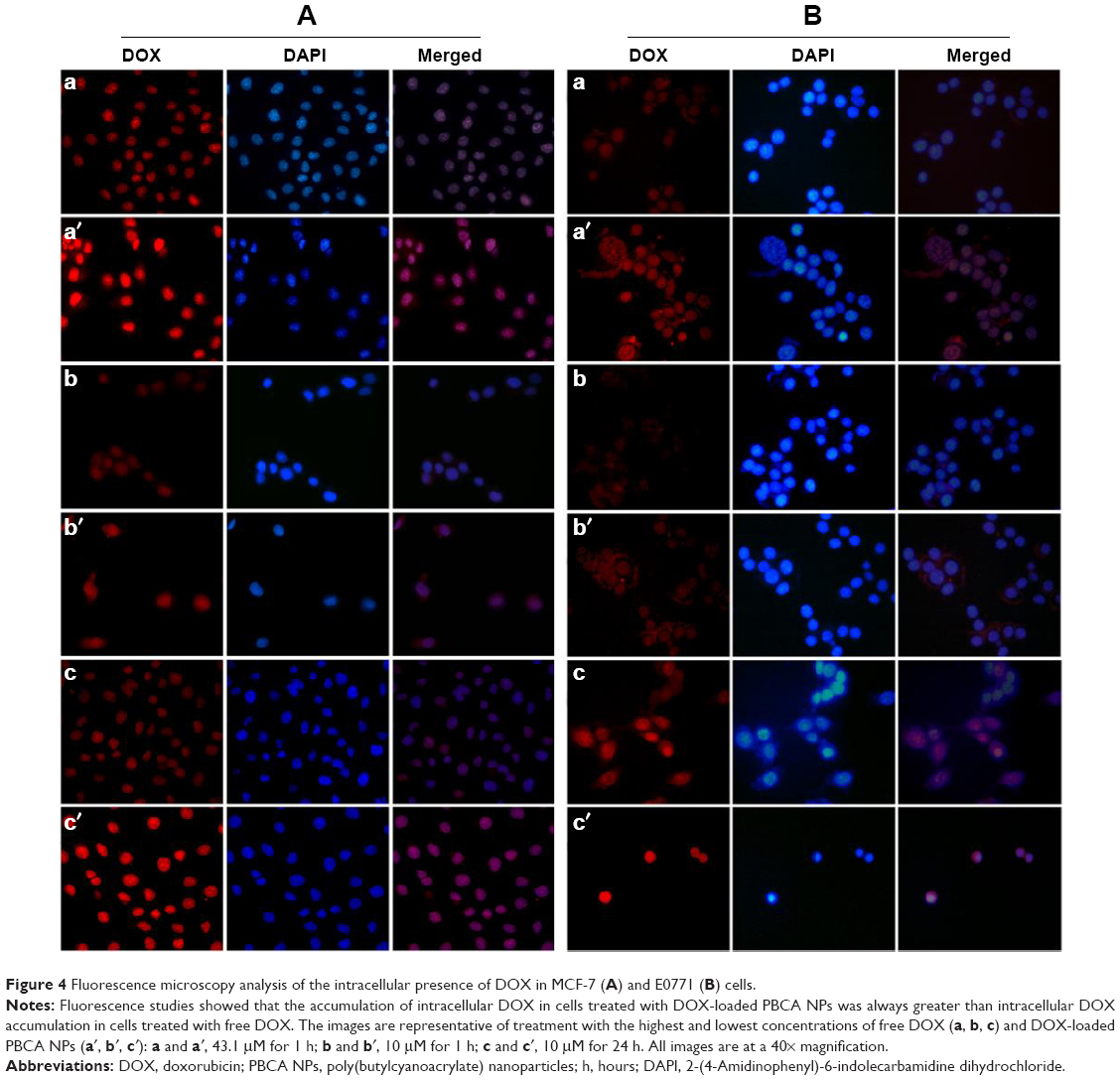 | Figure 4 Fluorescence microscopy analysis of the intracellular presence of DOX in MCF-7 (A) and E0771 (B) cells. |
FACScan analysis of doxorubicin location inside cells
To evaluate drug accumulation inside the malignant cells, we conducted a fluorocytometric analysis during the first hours of the treatment (Figure 5). When MCF-7 cells were incubated with high doses of DOX-loaded NPs for 0.5, 1, 1.5, 2, and 4 hours, relative increases in fluorescence were observed (51.9%, 42.7%, 50.6%, 33.1%, and 34.2%, respectively) in comparison with tumor cells treated with free DOX (Figure 5A). Qualitatively, similar results were obtained for E0771 cells when comparing DOX-loaded NPs and free DOX, where the relative increases in fluorescence after the same periods of exposure were 53.0%, 29.4%, 26.7%, 35.8%, and 15.5%, respectively (Figure 5B). This finding could be the consequence of a greater and more rapid DOX uptake when the drug is encapsulated within the PBCA NPs. As a result, a higher DOX concentration within the cell nucleus was maintained at all times.
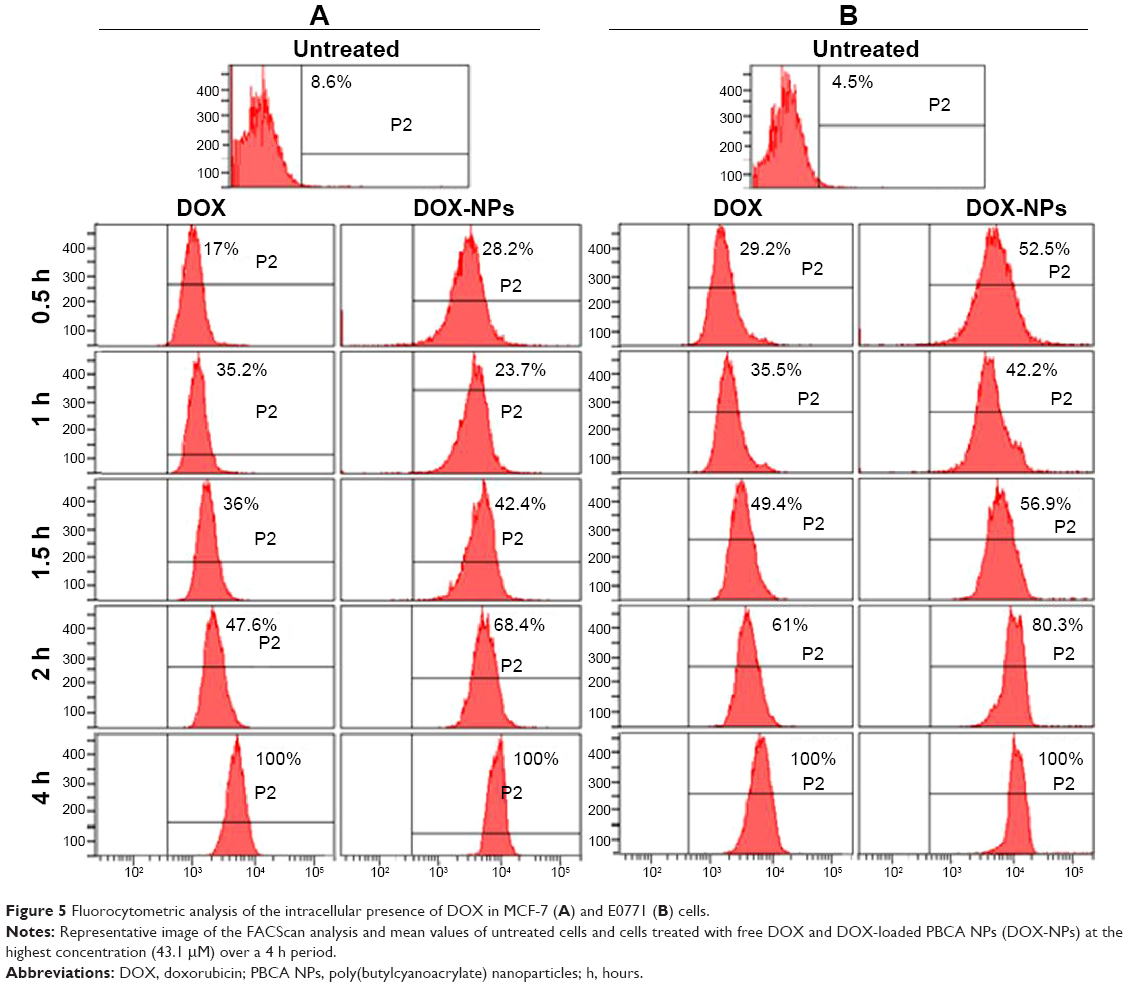 | Figure 5 Fluorocytometric analysis of the intracellular presence of DOX in MCF-7 (A) and E0771 (B) cells. |
Ultrastructural changes in breast cancer cells treated with doxorubicin-loaded poly(butylcyanoacrylate) nanoparticles
MCF-7 cells were assessed using transmission electron microscopy to determine the nature of any ultrastructural changes. As shown in Figure 6, treatment with both free DOX and DOX-loaded PBCA NPs caused dramatic changes in the cell nucleus after 4 hours of exposure to the drug. However, only DOX-loaded PBCA NPs induced significant nuclear alterations even after just 1 hour of exposure. In addition, MCF-7 cells treated with DOX-loaded NPs showed higher levels of mitochondrial damage compared with cells treated with free DOX for the same period and at the same concentration. Finally, neither of the treatments induced significant differences in other cytoplasm structures.
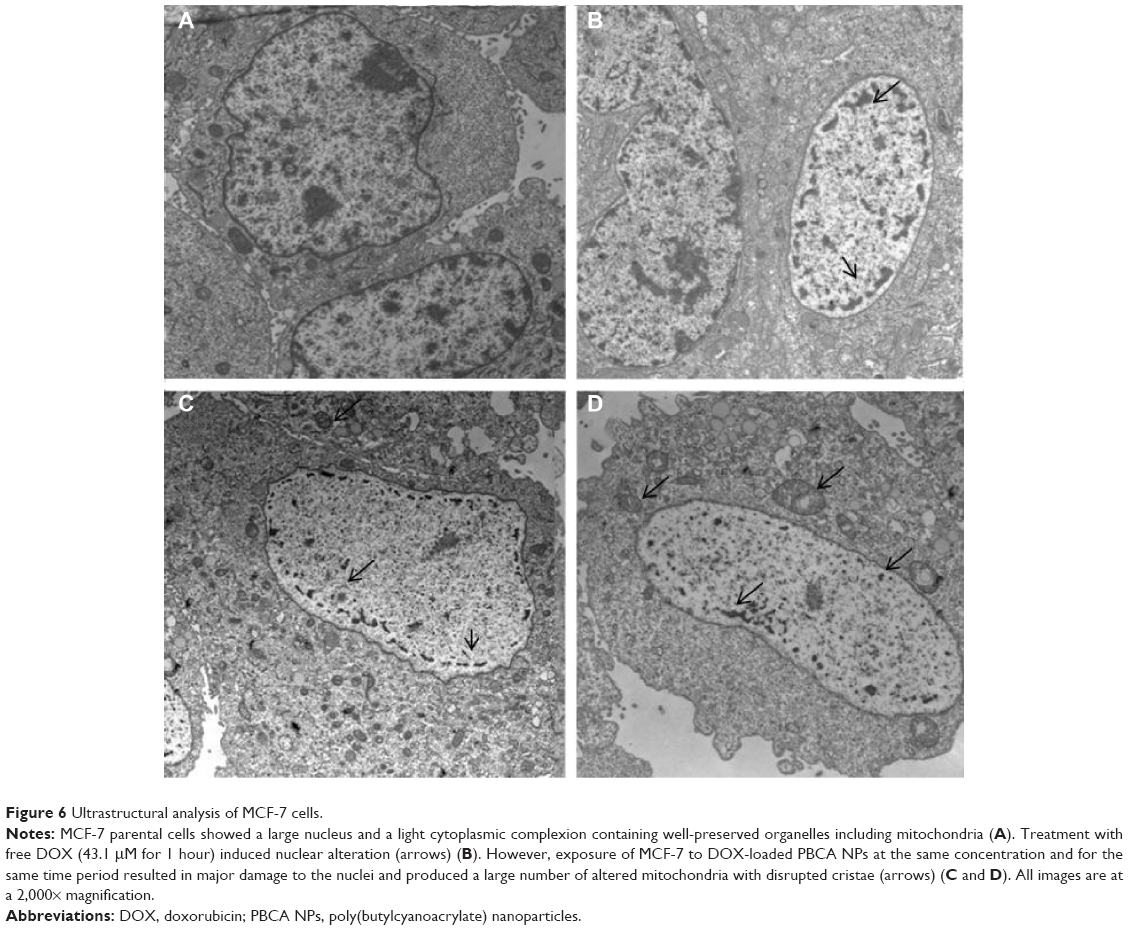 | Figure 6 Ultrastructural analysis of MCF-7 cells. |
In vivo breast cancer growth suppression and mice survival
The potential of DOX-loaded PBCA NPs to improve in vivo tumor cell death rates was evaluated using subcutaneous E0117 tumor-bearing immunocompetent C57BL/6 mice. The intravenous administration of DOX-loaded NPs was able to induce a significant inhibition of tumor growth (Figure 7). In fact, tumor volumes of mice treated with DOX-loaded NPs were significantly smaller than those of control mice (P<0.01). At the end of the in vivo study (day 33), tumor growth was inhibited by up to 40% in mice treated with DOX-loaded NPs compared with mice treated with free DOX. The intravenous injection of blank NPs did not influence the typical growth of untreated control tumors. On the other hand, both treatments (free DOX and DOX-loaded NPs) permitted a greater survival in comparison with the control group and the blank NPs group. The Kaplan–Meier curve also showed a 0.2-fold increase in the fraction of surviving mice when treated with DOX-loaded NPs in comparison with mice treated with free DOX (Figure 8). However, this increase was not significant according to the log-rank test.
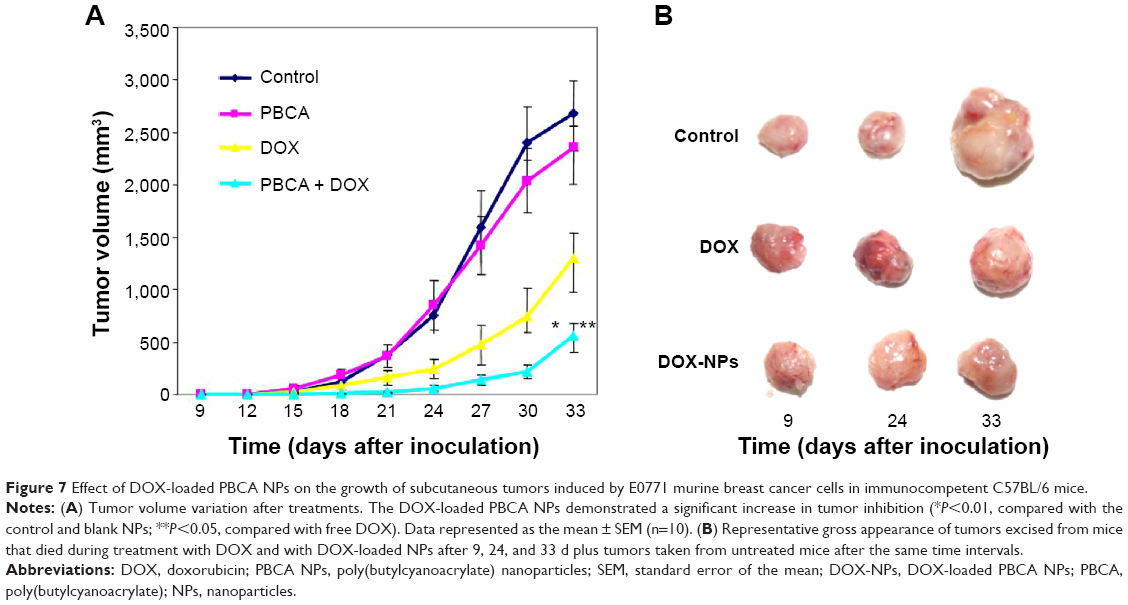 | Figure 7 Effect of DOX-loaded PBCA NPs on the growth of subcutaneous tumors induced by E0771 murine breast cancer cells in immunocompetent C57BL/6 mice. |
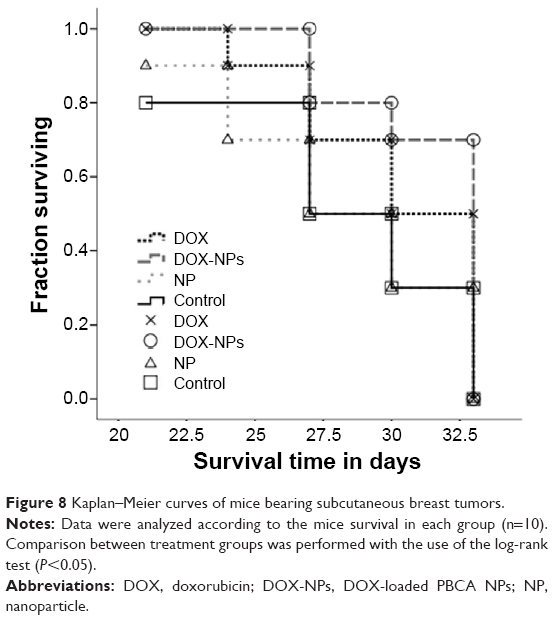 | Figure 8 Kaplan–Meier curves of mice bearing subcutaneous breast tumors. |
In vivo toxicity
Body weight of the mice was monitored as a clinical sign of the in vivo toxicity of the treatment. According to Figure 9, mice treated with blank NPs showed neither increased mortality nor noticeable losses of body weight compared with the controls. In contrast, mice treated with free DOX and DOX-loaded PBCA NPs showed a significant loss in body weight compared with the controls (P<0.001). Interestingly, during the treatment administration period (15 days), the weight loss was statistically lower (P<0.001) in mice treated with DOX-loaded PBCA NPs than in those treated with free DOX, suggesting that NPs decreased drug toxicity. After withdrawing the treatment, both groups recovered their body weight, resulting in similar values at the end of the experiment (33 days).
 | Figure 9 Evolution of body weight of immunocompetent C57BL/6 mice during and after treatment. |
Discussion
In the present study, we reported a DOX-loaded PBCA nanoparticulate system that demonstrated significantly more in vitro activity against breast cancer tumor cells than the free antitumor drug, doxorubicin, inducing a 40% relative decrease in in vivo tumor growth when administered intravenously. Furthermore, the polymeric nanocarrier was able to reduce systemic toxicity, at least throughout the duration of the treatment. These results suggest that PBCA NPs may improve the efficacy of DOX therapies against advanced breast cancers.
We have developed a reproducible synthesis procedure for the formulation of DOX-loaded PBCA NPs to target breast cancer cells. The synthesis methodology is based on the emulsion/polymerization of butylcyanoacrylate monomer in an aqueous solution.18,19 The procedure afforded the synthesis of spherical, stable DOX-loaded NPs with a number average diameter <160 nm (Figure 1A). These relatively small particles are expected to facilitate the uptake and accumulation of DOX by and within malignant cells. In fact, it has been stated that drug carriers ranging from 100 to 200 nm in size enter cancer cells by receptor-mediated endocytosis.27 The optimum synthesis conditions were taken as those that yielded the highest drug entrapment efficiencies and drug loading values (Figure 1B and C). In detail, the drug entrapment procedure yielded the best DOX loading values, as is generally the case when drug molecules are added to the cyanoacrylate monomer solution before NP formation. This is probably due to the difficulty experienced by the antitumor drug in breaking away from the growing polymeric (nano-)network once it is entrapped within it.19 In addition, a strong interaction between DOX molecules and the growing PBCA network has previously been described during emulsion/polymerization synthesis, a consequence of hydrogen bond formation between the drug’s ammonium groups and the PBCA’s cyano groups.28
Despite the fact that an attractive electrostatic interaction may exist between the positively charged drug molecules and the negatively charged polymeric surface, DOX loading values (%) for the surface adsorption method can be considered relatively low (<4%) (Figure 1C).29,30 In fact, the approximation of this hydrophilic chemotherapy agent from the aqueous phase to the hydrophobic NP surface is expected to be thermodynamically unfavorable (when considering the decimal logarithm of the partitioning coefficient at pH 5.8 in an n-octanol/water system [log10Doct/water] =−0.45).26,31
Regarding the initial drug concentration, a positive effect on drug vehiculization was observed upon increasing [DOX] in the aqueous solution, as has been reported previously with other drugs.19,22 We did not investigate the influence of either monomer or stabilizing agent concentrations on drug entrapment because no relevant effect had previously been reported.22,25,32,33
With respect to previously published investigations into the development of DOX-loaded poly(alkylcyanoacrylate) NPs, although it has been demonstrated that 80% of the drug used in DOX loading experiments can be associated with the NPs, the mean particle size of these nanoformulations (ie, 270±30 nm) is greater than the one obtained when following the synthesis described in this contribution (ie, 135±20 nm).8 Thus, a more favorable biological fate of the targeted drug-delivery system can be expected for the case of the nanoformulation developed by our research group.
DOX release from PBCA NPs at pH 7.4 and 37°C exhibited a biphasic, sustained profile (Figure 1D), characteristic of this polymer family, which could be a consequence of NP disintegration via surface erosion, drug diffusion through the polymeric matrix, or both.19,22,34,35 More specifically, the rapid drug release during the first phase could be due to the loss of DOX–molecules that are surface associated and/or poorly entrapped (only adsorbed to the surface pores). These molecules rapidly diffused into the incubation medium. On the contrary, DOX release during the slower second phase may correspond to the fraction of the drug that is embedded deeply within the PBCA (nano-)matrix and that must follow a longer diffusion path before release into the surrounding medium. Finally, the faster release of DOX that occurs when the drug is adsorbed onto the NP surface may well be due to the weak physisorption of the hydrophilic drug onto the hydrophobic PBCA surface.
In order to define the antitumor activity of DOX-loaded PBCA NPs, they were tested in human MCF-7 and mouse E0771 breast cancer cell lines. No toxicity has been reported for the PBCA NPs, thus indicating that they provide a safe method for transporting biomolecules within living systems, and that any cytotoxic effects were only due to the loaded drug molecules. Interestingly, DOX-loaded PBCA NPs induced a greater inhibition of cell proliferation than free DOX in both cell lines, and in only a very short exposure time (8 h). After 48 hours, DOX-loaded NPs induced a significant decrease of the IC50 in MCF-7 and E0771 cells in comparison with free DOX. To date, no significant increase in the antitumor DOX activity using poly(alkylcyanoacrylate) NPs has been described. Previously, DOX-loaded poly(isohexylcyanoacrylate) (PIHCA) NPs demonstrated some benefit in terms of cell cytotoxicity.36 Recently, Wohlfart et al37,38 reported a significant but lesser increase in DOX activity in 101/8 rat glioblastoma cells using DOX-loaded PIHCA NPs. Duan et al39 also demonstrated a significant decrease in the IC50 of DOX-loaded PBCA NPs in relation to free DOX, but only when the NPs were surface modified with folic acid. The same authors demonstrated a reduction in the viability of MCF-7 cells using DOX-cyanoacrylate, thanks to the down-regulation of P-glycoprotein and to the coadministration of curcumin.40 Although the cytotoxicity of anthracyclines has been observed to increase when entrapped within PBCA NPs, the mechanism of modulation of this drug activity has not yet been determined. One possibility is the use of surfactants that can act upon resistant mechanisms or enhance the plasma membrane permeability.41,42 In fact, it has been proposed that the presence of Pluronic® F-68 (Sigma-Aldrich) (a nonionic surfactant), which was used to synthesize the DOX-loaded PBCA NPs in the present study, may lead to an increased cytotoxicity.43,44
Fluorescence microscopy and flow cytometry analysis were employed in an attempt to explain the enhancement of DOX cytotoxicity when the drug was incorporated within/on the surface of PBCA NPs. A greater DOX concentration was observed within malignant cells compared with the use of free DOX, thus suggesting that the NPs can improve drug incorporation into the cells. In fact, different intracellular concentrations and locations (nucleus or cytoplasm) have been reported when DOX is loaded within/on the surface of the NPs compared with the concentrations attained when using free DOX–this has been attributed to a simple diffusion or endocytosis process.45,46 Free DOX and DOX-loaded NPs predominantly situated themselves within the nucleus of both MCF7 and E0771 cells. However, when the antitumor drug was loaded in NPs, DOX also accumulated in the cytoplasm after a short period of exposure. Reasons justifying the different DOX accumulation profiles inside malignant cells are lacking. Some authors have hypothesized that the presence of surfactants (eg, Pluronic® F-68, Sigma-Aldrich) could alter the membrane transport proteins, thus enhancing drug entry into tumor cells.43,44 Whatever the reason, the higher DOX concentration inside MCF-7 cells (when it is delivered via loaded NPs) could explain the significant mitochondrial damage and nuclear alterations observed through transmission electron microscopy. In fact, morphological and ultrastructural changes caused by DOX in H9C2 rat myoblast cells (chromatin clumping, swollen mitochondria, disruption of the nuclear membrane structure, and cytoplasm vacuolization) were directly associated with the drug concentration.47 The actual mechanism(s) that results in the significantly higher cytotoxicity of DOX-loaded PBCA NPs against MCF-7 and E0771 cells compared with the cytotoxicity of free DOX is not clear. It is known that, whereas the free drug enters cells by simple diffusion, drug-loaded NPs may provide a different cell entry mechanism such as endocytosis, pinocytosis, or phagocytosis, which is still under discussion.48,49 It has been suggested that drug-loaded NPs are too large to pass through both the plasma and nuclear membranes. Thus, the presence of the drug within the cell nucleus must occur only once it has been released from inside the NPs.50,51 The significant decrease in DOX IC50 values in our cell lines after treatment with DOX-loaded PBCA NPs could be explained by the greater degree of drug incorporation into the cell when the NP biodegrades in the endosome and releases the DOX before finally reaching the nucleus.51,52
On the other hand, when DOX-loaded PBCA NPs were administered to tumor-bearing mice, a significantly greater inhibition of tumor growth (40%) was obtained in comparison with free DOX. This observation represents a substantial improvement in drug activity against breast cancer tumors. Our results demonstrated a similar or greater breast cancer growth inhibition when compared with the literature.53–55 In the latter case, for example, a triblock N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer loaded with DOX molecules has demonstrated a 26-fold increase in the IC50 of the drug against 4T1 mammary carcinoma cells, but a mere 21.4% tumor growth inhibition compared with the free drug.56 The in vivo results reported here for DOX-loaded PBCA NPs can be directly related to the greater in vitro effect of DOX-loaded NPs. However, the use of NPs by itself could increase the concentration of drug inside the tumor tissue, where an enhanced permeability and retention (EPR) effect and a low lymphatic macromolecular clearance exist.57 Furthermore, one of our main findings was the lower toxicity induced by the DOX-loaded PBCA NP treatment in relation to treatment with free DOX. Following the work of Yang et al56 who reduced DOX toxicity using a block copolymer DOX conjugate, we assessed the fluctuation of mice body weight to determine the toxicity of the in vivo treatment. Our results showed a similar weight evolution for both untreated mice and mice treated with blank NPs. In contrast, a 15-day treatment with DOX-loaded PBCA NPs resulted in the DOX causing a significant body weight loss in relation to the controls. Interestingly, this weight loss of body mass was statistically lower (P<0.01) with DOX-loaded PBCA NPs than free DOX, suggesting that loading DOX within/onto NPs decreased its toxicity. After withdrawing the treatment, body weight was recovered by both groups, being similar at the end of the experiment (33 days). In contrast, others’ studies showed no significant differences in mice body weight between treatments with DOX-loaded PBCA NPs and those employing free DOX, although for the case in reference both treatments were assayed in intracranial gliomas.58 However, despite the positive results obtained in terms of reducing tumor growth and drug toxicity, the treatment schedule described here was not enough to ensure a clear and significant modulation in mouse survival – probably related to the relatively short treatment period. Nevertheless, the increased antitumor activity of DOX against breast cancer cells when loaded within/onto PBCA NPs could be used to reduce the therapeutic dose of the drug as part of a strategy to limit its side effects.
Certainly, other nanosystems for DOX release such as lipid vesicles or liposomes with a high biodegradability and biocompatibility also offer advantages in breast cancer treatment.59 Strategies that include targeting ligands on DOX-loaded lipid vesicles, which are accessible by tumor acidification, have allowed the release of drugs after NP endocytosis. This system increased the percentage of in vitro cell death by almost threefold.60 In addition, in murine xenograft models, lipid vesicles with DOX-loaded pH-triggered anti-HER2/neu cells showed a greater reduction in tumor volume compared with FDA-approved DOX-loaded vesicles.61 However, the clinical application of lipid vesicles or liposomes requires optimization in terms of both size and surface to resolve some of their disadvantages, such as their rapid renal clearance, recognition by the reticuloendothelial system, or low penetration in tumors. By contrast, polymeric NPs similar to those used here have already demonstrated a homogeneous size, a greater ability to solubilize hydrophobic drugs, a sustained release of the drug plus highly stable and customizable physicochemical properties, which means they will be accepted for clinical use in the near future.62 Further assays involving both NP types will be necessary to obtain a quantitative improvement in the response of breast cancer patients.
Conclusion
We have reported the optimal preparation conditions required to synthesize DOX-loaded PBCA NPs suitable for parenteral administration. Compared with surface adsorption, DOX incorporation within the NP matrix during the polymerization process has led to greater drug loading values and a slower drug release profile. The preclinical in vitro results described here have demonstrated the efficacy of DOX-loaded NPs against human and mouse breast carcinoma cells, given the important reduction of the IC50. In vivo studies have shown that DOX-loaded NPs can induce a significant inhibition of tumor growth and that the use of PBCA NPs reduces DOX toxicity. These results suggest that PBCA NPs may be used to improve the efficacy of DOX therapies against advanced breast cancers. The greater antitumor activity of DOX-loaded PBCA NPs could be used to reduce the dose of DOX needed to obtain an adequate antitumor effect but with almost negligible toxicity.
Acknowledgments
This investigation was funded by FEDER, Plan Nacional de Investigación Científica, Desarrollo e Innovación Tecnológica (I + D + I), Instituto de Salud Carlos III (FIS) through projects Nos PI11/01862 and PI11/02571, and by the Consejería de Salud de la Junta de Andalucía through project No PI-0338. The authors wish to express their gratitude to G Ortiz Ferron (CIC, University of Granada, Spain) for his skillful assistance with cytometry experiments.
Disclosure
The authors report no conflicts of interest in this work.
References
Redig AJ, McAllister SS. Breast cancer as a systemic disease: a view of metastasis. J Intern Med. 2013;274(2):113–126. | ||
Morris PG, McArthur HL, Hudis CA. Therapeutic options for metastatic breast cancer. Expert Opin Pharmacother. 2009;10(6):967–981. | ||
Carvalho C, Santos RX, Cardoso S, et al. Doxorubicin: the good, the bad and the ugly effect. Curr Med Chem. 2009;16(25):3267–3285. | ||
Toldo S, Goehe RW, Lotrionte M, et al. Comparative cardiac toxicity of anthracyclines in vitro and in vivo in the mouse. PLoS One. 2013;8(3):1–8. | ||
Peetla C, Bhave R, Vijayaraghavalu S, Stine A, Kooijman E, Labhasetwar V. Drug resistance in breast cancer cells: biophysical characterization of and doxorubicin interactions with membrane lipids. Mol Pharm. 2010;7(6):2334–2348. | ||
Tang SC. Strategies to decrease taxanes toxicities in the adjuvant treatment of early breast cancer. Cancer Invest. 2009;27(2):206–214. | ||
Prados J, Melguizo C, Ortiz R, et al. Doxorubicin-loaded nanoparticles: new advances in breast cancer therapy. Anticancer Agents Med Chem. 2012;12(9):1058–1070. | ||
Gelperina SE, Khalansky AS, Skidan IN, et al. Toxicological studies of doxorubicin bound to polysorbate 80-coated poly(butyl cyanoacrylate) nanoparticles in healthy rats and rats with intracranial glioblastoma. Toxicol Lett. 2002;126(2):131–141. | ||
Vauthier C, Dubernet C, Fattal E, Pinto-Alphandary H, Couvreur P. Poly(alkylcyanoacrylates) as biodegradable materials for biomedical applications. Adv Drug Deliv Rev. 2003;55(4):519–548. | ||
Merle P, Si-Ahmed S, Habersetzer F, et al. Phase 1 study of intra-arterial hepatic (IAH) delivery of doxorubicin-transdrug® (DT) for patients with advanced hepatocellular carcinoma (HCC). J Clin Virol. 2006; 24(18):179. | ||
Mestieri LB, Saska S, Carrodeguas RG, Gaspar AM. Evaluation of n-butyl cyanoacrylate adhesive in rat subcutaneous tissue. Dermatol Surg. 2012;38(5):767–771. | ||
Vauthier C, Dubernet C, Chauvierre C, Brigger I, Couvreur P. Drug delivery to resistant tumors: the potential of poly(alkyl cyanoacrylate) nanoparticles. J Control Release. 2003;93(2):151–160. | ||
Zhou Q, Sun X, Zeng L, Liu J, Zhang Z. A randomized multicenter phase II clinical trial of mitoxantrone-loaded nanoparticles in the treatment of 108 patients with unresected hepatocellular carcinoma. Nanomedicine. 2009;5(4):419–423. | ||
Barraud L, Merle P, Soma E, et al. Increase of DOX sensitivity by doxorubicin-loading into NPs for hepatocellular carcinoma cells in vitro and in vivo. J Hepatol. 2005;42(5):736–743. | ||
Ambruosi A, Khalansky AS, Yamamoto H, Gelperina SE, Begley DJ, Kreuter J. Biodistribution of polysorbate 80-coated doxorubicin-loaded [14C]-poly(butyl cyanoacrylate) nanoparticles after intravenous administration to glioblastoma-bearing rats. J Drug Target. 2006;14(2): 97–105. | ||
De Juan BS, Von Briesen H, Gelperina SE, Kreuter J. Cytotoxicity of doxorubicin bound to poly(butyl cyanoacrylate) nanoparticles in rat glioma cell lines using different assays. J Drug Target. 2006;14(9): 614–622. | ||
Hekmatara T, Bernreuther C, Khalansky AS, et al. Efficient systemic therapy of rat glioblastoma by nanoparticle-bound doxorubicin is due to antiangiogenic effects. Clin Neuropathol. 2009;28(3):153–164. | ||
Couvreur P, Kante B, Roland M, Guiot P, Bauduin P, Speiser P. Polycyanoacrylate nanocapsules as potential lysosomotropic carriers: preparation, morphological and sorptive properties. J Pharm Pharmacol. 1979;31(5):331–332. | ||
Arias JL, Reddy LH, Couvreur P. Polymeric nanoparticulate system augmented the anticancer therapeutic efficacy of gemcitabine. J Drug Target. 2009;17(8):586–598. | ||
O’Brien RW, White LR. Electrophoretic mobility of a spherical colloidal particle. J Chem Soc Faraday Trans. 1978;2(74):1607–1626. | ||
Sullivan CO, Birkinshaw C. In vitro degradation of insulin-loaded poly(n-butylcyanoacrylate) nanoparticles. Biomaterials. 2004;25(18): 4375–4382. | ||
Arias JL, Ruiz MA, López-Viota M, Delgado AV. Poly(alkylcyanoacrylate) colloidal particles as vehicles for antitumour drug delivery: a comparative study. Colloids Surf B Biointerfaces. 2008;62(1):64–70. | ||
Li YL, Zhu L, Liu Z, et al. Reversibly stabilized multifunctional dextran nanoparticles efficiently deliver doxorubicin into the nuclei of cancer cells. Angew Chem Int Ed Engl. 2009;48(52):9914–9918. | ||
Prados J, Melguizo C, Rama AR, et al. Gef gene therapy enhances the therapeutic efficacy of doxorubicin to combat growth of MCF-7 breast cancer cells. Cancer Chemother Pharmacol. 2010;66(1):69–78. | ||
Arias JL, Gallardo V, Ruiz MA, Delgado AV. Ftorafur loading and controlled release from poly(ethyl-2-cyanoacrylate) and poly(butylcyanoacrylate) nanospheres. Int J Pharm. 2007;337(1–2): 282–290. | ||
Arias JL, Gallardo V, Linares-Molinero F, Delgado AV. Preparation and characterization of carbonyl iron/poly(butylcyanoacrylate) core/shell nanoparticles. J Colloid Interface Sci. 2006;299(2):599–607. | ||
Decuzzi P, Pasqualini R, Arap W, Ferrari M. Intravascular delivery of particulate systems: does geometry really matter? Pharm Res. 2009; 26(1):235–243. | ||
Poupaert JH, Couvreur P. A computationally derived structural model of doxorubicin interacting with oligomeric polyalkylcyanoacrylate in nanoparticles. J Control Release. 2003;92(1–2):19–26. | ||
Yu MK, Jeong YY, Park J, et al. Drug-loaded superparamagnetic iron oxide nanoparticles for combined cancer imaging and therapy in vivo. Angew Chem Int Ed Engl. 2008;47(29):5362–5365. | ||
Gaihre B, Khil MS, Lee DR, Kim HY. Gelatin-coated magnetic iron oxide nanoparticles as carrier system: drug loading and in vitro drug release study. Int J Pharm. 2009;365(1–2):180–189. | ||
Formariz TP, Sarmento VHV, Silva-Junior AA, Scarpa MV, Santilli CV, Oliveira AG. Doxorubicin biocompatible O/W microemulsion stabilized by mixed surfactant containing soya phosphatidylcholine. Colloids Surf B Biointerfaces. 2006;51(1):54–61. | ||
Llovet MI, Egea MA, Valero J, Alsina MA, García ML, Chauvet A. Methotrexate loaded nanoparticles: analysis of drug content and study of the matrix structure. Drug Dev Ind Pharm. 1995;21(15):1761–1771. | ||
Arias JL, Martínez-Soler GI, López-Viota M, Ruiz MA. Formulation of chitosan NPs loaded with metronidazole for the treatment of infectious diseases. Lett Drug Des Discov. 2010;7(2):70–78. | ||
Soma CE, Dubernet C, Bentolila D, Benita S, Couvreur P. Reversion of multidrug resistance by co-encapsulation of doxorubicin and cyclosporin A in polyalkylcyanoacrylate nanoparticles. Biomaterials. 2000;21(1):1–7. | ||
Graf A, McDowell A, Rades T. Poly(alkylcyanoacrylate) nanoparticles for enhanced delivery of therapeutics–is there real potential? Expert Opin Drug Deliv. 2009;6(4):371–387. | ||
Bennis S, Chapey C, Couvreur P, Robert J. Enhanced cytotoxicity of doxorubicin encapsulated in polyisohexylcyanoacrylatenanospheres against multidrug-resistant tumour cells in culture. Eur J Cancer. 1994;30(1):89–93. | ||
Wohlfart S, Khalansky AS, Bernreuther C, et al. Treatment of glioblastoma with poly(isohexyl cyanoacrylate) nanoparticles. Int J Pharm. 2011;415(1–2):244–251. | ||
Wohlfart S, Khalansky AS, Gelperina S, Begley D, Kreuter J. Kinetics of transport of doxorubicin bound to nanoparticles across the blood–brain barrier. J Control Release. 2011;154(1):103–107. | ||
Duan J, Liu M, Zhang Y, Zhao J, Pan Y, Yang X. Folate-decorated chitosan/doxorubicin poly(butyl)cyanoacrylate nanoparticles for tumor-targeted drug delivery. J Nanopart Res. 2012;14(4):1–9. | ||
Duan J, Mansour HM, Zhang Y, et al. Reversion of multidrug resistance by co-encapsulation of doxorubicin and curcumin in chitosan/poly(butyl cyanoacrylate) nanoparticles. Int J Pharm. 2012;426(1–2):193–201. | ||
Ambruosi A, Gelperina S, Khalansky A, Tanski S, Theisen A, Kreuter J. Influence of surfactants, polymer and doxorubicin loading on the anti-tumour effect of poly(butyl cyanoacrylate) nanoparticles in a rat glioma model. J Microencapsul. 2006;23(5):582–592. | ||
Li-Blatter X, Nervi P, Seelig A. Detergents as intrinsic P-glycoprotein substrates and inhibitors. Biochim Biophys Acta. 2009;1788(10): 2335–2344. | ||
Yordanov G, Skrobanska R, Evangelatov A. Entrapment of epirubicin in poly(butyl cyanoacrylate) colloidal nanospheres by nanoprecipitation: formulation development and in vitro studies on cancer cell lines. Colloids Surf B Biointerfaces. 2012;92:98–105. | ||
Yordanov G, Evangelatov A, Skrobanska R. Epirubicin loaded to pre-polymerized poly(butyl cyanoacrylate) nanoparticles: preparation and in vitro evaluation in human lung adenocarcinoma cells. Colloids Surf B Biointerfaces. 2013;107:115–123. | ||
Upadhyay KK, Bhatt AN, Mishra AK, et al. The intracellular drug delivery and anti tumor activity of doxorubicin loaded poly(gamma-benzyl L-glutamate)-b-hyaluronanpolymersomes. Biomaterials. 2010;31(10):2882–2892. | ||
Ren D, Kratz F, Wang SW. Protein nanocapsules containing doxorubicin as a pH-responsive delivery system. Small. 2011;7(8):1051–1060. | ||
Sardão VA, Oliveira PJ, Holy J, Oliveira CR, Wallace KB. Morphological alterations induced by doxorubicin on H9c2 myoblasts: nuclear, mitochondrial, and cytoskeletal targets. Cell Biol Toxicol. 2009;25(3):227–243. | ||
Hillaireau H, Couvreur P. Nanocarriers’ entry into the cell: relevance to drug delivery. Cell Mol Life Sci. 2009;66(17):2873–2896. | ||
Ma N, Ma C, Li C, et al. Influence of nanoparticle shape, size, and surface functionalization on cellular uptake. J Nanosci Nanotechnol. 2013;13(10):6485–6498. | ||
Sahay G, Alakhova DY, Kabanov AV. Endocytosis of nanomedicines. J Control Release. 2010;145(3):182–195. | ||
Iversena TG, Skotlanda T, Sandvig K. Endocytosis and intracellular transport of nanoparticles: present knowledge and need for future studies. Nano Today. 2011;6(2):176–185. | ||
Arora HC, Jensen MP, Yuan Y, et al. Nanocarriers enhance Doxorubicin uptake in drug-resistant ovarian cancer cells. Cancer Res. 2012;72(3): 769–778. | ||
Attia AB, Yang C, Tan JP, et al. The effect of kinetic stability on biodistribution and anti-tumor efficacy of drug-loaded biodegradable polymeric micelles. Biomaterials. 2013;34(12):3132–3140. | ||
Pu Y, Chang S, Yuan H, Wang G, He B, Gu Z. The anti-tumor efficiency of poly(L-glutamic acid) dendrimers with polyhedral oligomericsilsesquioxane cores. Biomaterials. 2013;34(14):3658–3666. | ||
Zou A, Chen Y, Huo M, et al. In vivo studies of octreotide-modified N-octyl-O, N-carboxymethyl chitosan micelles loaded with doxorubicin for tumor-targeted delivery. J Pharm Sci. 2013;102(1):126–135. | ||
Yang Y, Pan D, Luo K, Li L, Gu Z. Biodegradable and amphiphilic block copolymer-doxorubicin conjugate as polymeric nanoscale drug delivery vehicle for breast cancer therapy. Biomaterials. 2013;34(33): 8430–8443. | ||
Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev. 2013;65(1):71–79. | ||
Petri B, Bootz A, Khalansky A, et al. Chemotherapy of brain tumour using doxorubicin bound to surfactant-coated poly(butyl cyanoacrylate) nanoparticles: revisiting the role of surfactants. J Control Release. 2007; 117(1):51–58. | ||
Biswas S, Dodwadkar NS, Deshpande PP, Parab S, Torchilin VP. Surface functionalization of doxorubicin-loaded liposomes with octa-arginine for enhanced anticancer activity. Eur J Pharm Biopharm. 2013;84(3):517–525. | ||
Karve S, Bandekar A, Ali MR, Sofou S. The pH-dependent association with cancer cells of tunable functionalized lipid vesicles with encapsulated doxorubicin for high cell-kill selectivity. Biomaterials. 2010;31(15):4409–4416. | ||
Bandekar A, Karve S, Chang MY, Mu Q, Rotolo J, Sofou S. Antitumor efficacy following the intracellular and interstitial release of liposomal doxorubicin. Biomaterials. 2012;33(17):4345–4352. | ||
Hu CM, Aryal S, Zhang L. Nanoparticle-assisted combination therapy for effective cancer treatment. Ther Deliv. 2010;1(2):321–334. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.