")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Enhanced and selective delivery of enzyme therapy to 9L-glioma tumor via magnetic targeting of PEG-modified, ß-glucosidase- conjugated iron oxide nanoparticles
Authors Zhou J, Zhang J, Gao W
Received 21 December 2013
Accepted for publication 17 February 2014
Published 10 June 2014 Volume 2014:9(1) Pages 2905—2917
DOI https://doi.org/10.2147/IJN.S59556
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Jie Zhou,1,* Jian Zhang,2,* Wenxi Gao1
1Department of Urology, Hubei Hospital of Traditional Chinese Medicine, Hubei University of Chinese Medicine, Wuhan, People’s Republic of China; 2Department of Pharmaceutical Sciences, College of Pharmacy, University of Michigan, Ann Arbor, MI, USA
*These authors contributed equally to this work
Abstract: The stability of enzyme-conjugated magnetic iron oxide nanoparticles in plasma is of great importance for in vivo delivery of the conjugated enzyme. In this study, ß-glucosidase was conjugated on aminated magnetic iron oxide nanoparticles using the glutaraldehyde method (ß-Glu-MNP), and further PEGylated via N-hydroxysuccinimide chemistry. The PEG-modified, ß-glucosidase-immobilized magnetic iron oxide nanoparticles (PEG-ß-Glu-MNPs) were characterized by hydrodynamic diameter distribution, zeta potential, Fourier transform infrared spectroscopy, transmission electron microscopy, and a superconducting quantum interference device. The results showed that the multidomain structure and magnetization properties of these nanoparticles were conserved well throughout the synthesis steps, with an expected diameter increase and zeta potential shifts. The Michaelis constant was calculated to evaluate the activity of conjugated ß-glucosidase on the magnetic iron oxide nanoparticles, indicating 73.0% and 65.4% of enzyme activity remaining for ß-Glu-MNP and PEG-ß-Glu-MNP, respectively. Both magnetophoretic mobility analysis and pharmacokinetics showed improved in vitro/in vivo stability of PEG-ß-Glu-MNP compared with ß-Glu-MNP. In vivo magnetic targeting of PEG-ß-Glu-MNP was confirmed by magnetic resonance imaging and electron spin resonance analysis in a mouse model of subcutaneous 9L-glioma. Satisfactory accumulation of PEG-ß-Glu-MNP in tumor tissue was successfully achieved, with an iron content of 627±45 nmol Fe/g tissue and ß-glucosidase activity of 32.2±8.0 mU/g tissue.
Keywords: ß-glucosidase, enzyme/prodrug therapy, magnetic nanoparticles, magnetic targeting, 9L-glioma
Introduction
Conventional chemotherapy raises concerns about the severe side effects (such as immunosuppression and gastrointestinal disorders) due to the major disadvantages of the chemotherapeutics, ie, narrow therapeutic indices and low selectivity for neoplastic cells. Most currently available chemotherapeutics can nonspecifically affect all types of rapidly dividing cells, including normal cells, such as bone marrow cells and intestinal epithelial cells.1
A promising strategy to overcome these limitations is the use of directed enzyme prodrug therapy (DEPT), which has been investigated as a means to improve the tumor selectivity of therapeutics.2 This strategy comprises the targeted delivery of a prodrug-activating enzyme or its encoding gene to the tumor before administering a prodrug. After targeting and clearance of the enzyme from the circulation, the prodrug is administered and then converted to an active anticancer drug only in the tumor lesion, achieving enhanced anticancer efficacy and decreased systemic toxicity. To date, several DEPT strategies have been investigated, including gene DEPT, antibody DEPT, Clostridium DEPT, and, more recently, magnetic DEPT. Magnetic DEPT, which is attracting increasing attention, involves coupling the bioactive prodrug-activating enzyme to magnetic nanoparticles (MNP)3 that are then selectively delivered to the tumor by applying an external magnetic field. Of all the DEPT strategies, the β-glucosidase/amygdalin system,4 in which amygdalin is converted to hydrogen cyanide to kill tumor cells, is the most widely used. The nonspecific toxicity of hydrogen cyanide in normal cells/tissues can be greatly minimized by administering amygdalin with the maximum concentration ratio of β-glucosidase-conjugated MNP in tumor tissue and the blood circulation. Increasing accumulation of β-glucosidase in tumor tissue is extremely important for this targeted enzyme/prodrug (β-glucosidase/amygdalin) strategy to be successful.
Compared with other targeting carriers, MNP have unique advantages for targeted delivery of prodrug-activating enzymes.5–8 The conjugated prodrug-activating enzymes can be directed to and retained in solid tumor tissue by applying an external magnetic field to the magnetic cores.9 The enhanced permeability and retention effect in the tumor region also helps their local accumulation of MNP in tumor tissue.10,11 The favorable biocompatibility of MNP effectively avoids the immunogenicity issues facing other heterogeneous carriers, eg, antibodies and ligands.12 Further, as a specific and selective contrast agent for magnetic resonance imaging (MRI), the dynamic behavior of MNP can be monitored easily in vivo using real-time MRI.13 Further, the relatively large surface area of MNP enables multiple enzyme molecules to be immobilized on a single nanoparticle. Considering all these advantages, we conjugated β-glucosidase to MNP (β-Glu-MNP) using a glutaraldehyde crosslinking method. Our in vitro studies showed that the activity of β-glucosidase was conserved well throughout the synthesis process used for β-Glu-MNP, and the enzyme-conjugated MNP showed a slightly better magnetic response than that of the precursor nanoparticles.
However, the circulation time of β-Glu-MNP was noted to be very short (only about 30% of the initial dose was left in the circulation 20 minutes following administration) in our previous study.14 Improved stability in plasma benefits accumulation of MNP in tumor lesions via magnetic targeting and the enhanced permeability and retention effect. Polyethylene glycol (PEG), with the advantage of low recognition by the reticuloendothelial system, has been deemed to be the answer for delivery of drugs with a poor plasma pharmacokinetic profile. The stability of MNP in plasma can be greatly increased when modified with PEG.15 As reported previously, the plasma half-life and accumulation of PEGylated MNP at the tumor site was about 100-fold longer and 40.5-fold higher, respectively, than that of the parent fluid, MAG-D magnetic nanoparticles (D-MNP).16 Accumulation of PEGylated MNP in tumor tissue was further increased by applying magnetic targeting, and was found to be 5.6-fold greater than that using the enhanced permeability and retention effect only.17
In the present study, β-glucosidase was conjugated to aminated MNP using glutaraldehyde as the crosslinker. The β-Glu-MNP was then PEGylated (PEG-β-Glu-MNP) using simple N-hydroxysuccinimide chemistry. Enhanced and selective delivery of MNP and the conjugated enzyme was achieved by applying an external magnetic field and confirmed by MRI and electron spin resonance in a mouse model of subcutaneous 9L-glioma.
Materials and methods
Materials
All materials were obtained from commercial suppliers and used without further purification, unless otherwise noted. Starch-coated D-MNPs (50 mg/mL) were purchased from Chemicell GmbH (Berlin, Germany). β-glucosidase was sourced from Fisher Scientific (MP Biomedicals, Solon, OH, USA). Lacey carbon film-coated copper grids were obtained from Ted Pella (Redding, CA, USA). Iron standard (1,000 mg Fe/L) and yttrium internal standard (1,000 mg Fe/L) were purchased from GFS Chemicals (Powell, OH, USA). Methoxy PEG succinimidyl carboxymethyl ester (SC-NHS-PEG, 20 kDa) was obtained from Jenkem Technology (Beijing, People’s Republic of China). Glutaraldehyde solution (50%, w/v), 4-nitrophenyl β-D-glucopyranoside (GlcβNp), sodium phosphate (mono and dibasic), epichlorohydrin, concentrated ammonium hydroxide (containing 30% ammonia), and sodium hydroxide pellets were all obtained from Sigma-Aldrich (St Louis, MO, USA). All deionized water used in this study was produced using a Milli-Q A10 Biocel water purification system (Millipore, Billerica, MA, USA).
Preparation of PEG-β-Glu-MNP
Cross-linked, aminated D-MNP
Before enzyme conjugation and PEG modification, D-MNP was cross-linked and aminated by modifying the coating starch moieties using epichlorohydrin and concentrated ammonium hydroxide (30%, w/v), respectively (Figure 1).16 Briefly, 1.5 mL of D-MNP suspension (50 mg Fe/mL) was mixed with 2.0 mL of 6 M sodium hydroxide and digested for 15 minutes, followed by dropwise addition of 1.0 mL of epichlorohydrin with intense vortexing. The mixture was then incubated with shaking using a 3520 orbit shaker (Lab Line Instruments, Inc., Maharashtra, India) for 24 hours at 25°C. These cross-linked MNP were purified by dialysis and then incubated with 2.5 mL of concentrated ammonia (containing 30% ammonium hydroxide, w/v) for another 24 hours at 25°C. The cross-linked aminated product (DN-MNP) was also purified by dialysis and then concentrated using a Dynal magnetic separator (Invitrogen, Carlsbad, CA, USA) to 30–40 mg Fe/mL for further research.
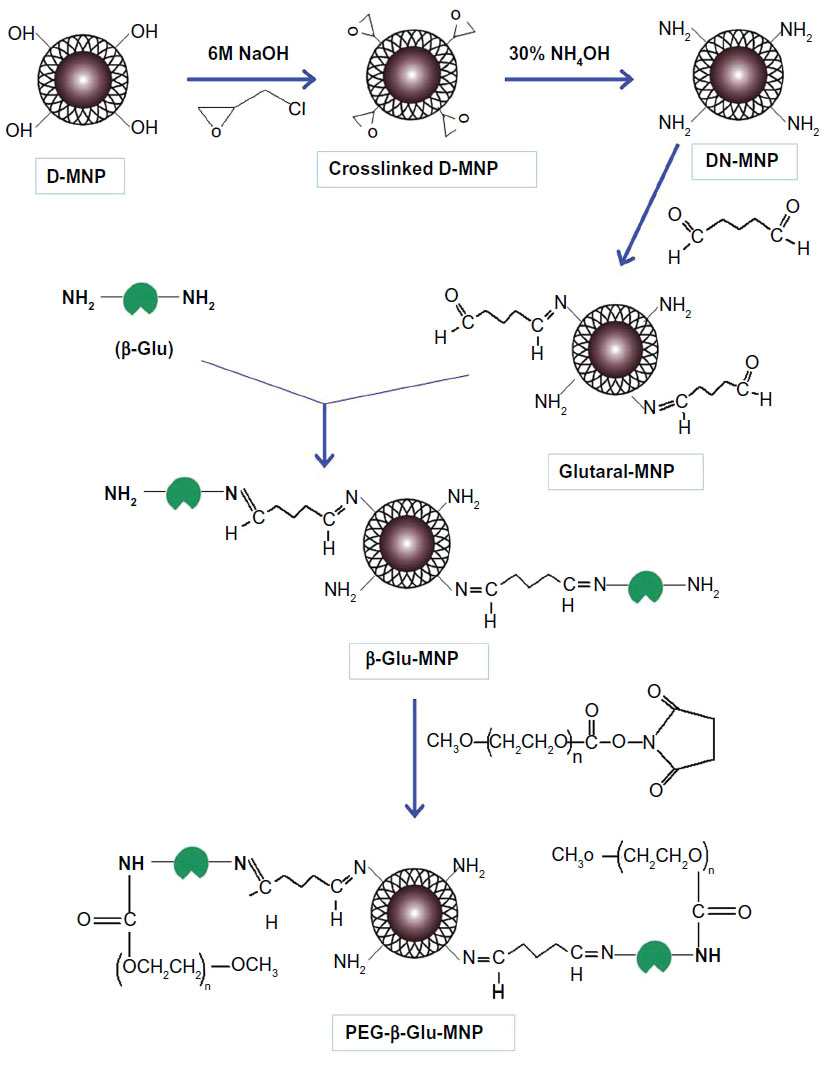 | Figure 1 Schematic synthesis of PEG-β-Glu-MNP. |
Conjugation of β-glucosidase on DN-MNP
In the current study, a simple glutaraldehyde method was used with modification for the conjugation of β-glucosidase onto DN-MNP.18 Briefly, 160 μL of aqueous glutaraldehyde solution (50%, w/v) were added to 8.0 mL of phosphate-buffered saline (0.15 M, pH 8.0, hereinafter the same unless otherwise noted). Next, 4.0 mL of DN-MNP phosphate-buffered saline suspension (10 mg Fe/mL) was added dropwise to the above-prepared glutaraldehyde solution with intense vortexing. This reaction was incubated at 25°C with mild shaking for 3 hours. The glutaraldehyde-conjugated nanoparticles (Glutaral-MNP) were concentrated by magnetic separation and washed in deionized water five times to remove the excess glutaraldehyde. The purified Glutaral-MNP was resuspended in phosphate-buffered saline (10 mg/mL) and incubated with β-glucosidase with a feed ratio of 1.45 (β-Glu/MNP, mg/mg Fe)14 and incubated for 3 hours at 25°C with gentle shaking. The β-glucosidase-conjugated product (β-Glu-MNP) was then purified by dialysis and concentrated by magnetic separation. The purified and concentrated nanoparticles were redispersed in deionized water using a sonicator operated at 10% amplitude for 2 minutes (Sonifier®, Branson Ultrasonics Corporation, CT, USA). The amount of conjugated enzyme on MNP was quantified using the bicinchoninic acid protein assay (Thermo Fisher Scientific, Lake Barrington, IL, USA).19
PEG modification of β-Glu-MNP
After enzyme conjugation, β-Glu-MNP was further modified with PEG (SC-NHS-PEG, 20 kDa) using N-hydroxysuccinimide chemistry. In detail, β-Glu-MNP were resuspended in 2.0 mL of phosphate-buffered saline (10 mg Fe/mL) and incubated with 1.0 mL of phosphate-buffered saline containing 20 mg of well dissolved SC-NHS-PEG for one hour at 25°C with mild shaking. After completion of synthesis, the PEGylated β-Glu-MNP (PEG-β-Glu-MNP) was diluted with deionized water, brought to a total volume of 7.0 mL, and then dispersed via sonication for 2 minutes. PEG-β-Glu-MNP was also purified by dialysis and concentrated by magnetic separation to a final concentration of about 20 mg Fe/mL and kept at 4°C for further study.
Characterization of PEG-β-Glu-MNP
Particle size and zeta potential of MNP
The hydrodynamic diameter distribution (Z-average, intensity-weighted) and zeta potential of all the types of MNP suspension were measured by dynamic light scattering using a Nano ZS90 particle sizer (Malvern Instruments, Malvern, UK). Dilute suspensions of D-MNP, β-Glu-MNP, and PEG-β-Glu-MNP (about 0.1 mg Fe/mL) in deionized water were measured in triplicate.
Determination of PEG content
A modified barium iodide assay16 was used to quantify the amount of conjugated PEG. Briefly, 50 μL of MNP dispersion (20–30 mg Fe/mL) was digested in 100 μL of concentrated hydrochloride (HCl) and then diluted with 100 μL of deionized water. Next, 50 μL aliquots were added to 100 μL of 1% (w/v) barium iodide in a 96-well plate and mixed by gentle pipetting. Next, 100 μL of 0.001 N iodine solution was added to the magnetic nanoparticle/barium iodide mixture immediately prior to the spectrophotometric measurements. The samples were allowed to react for 30 minutes and were then measured at 535 nm using a Power Wave X340 spectrophotometer (Bio-Tek Instruments Inc., Winooski, VT, USA). Free SC-NHS-PEG was used for construction of the linear regression curve and all spectrophotometric measurements were performed in triplicate.
Fourier transform infrared analysis of PEG-β-Glu-MNP
All MNP sample suspensions (D-MNP, PEG-β-Glu-MNP, and β-Glu-MNP) were lyophilized prior to collection of infrared spectra using a Spectrum BX Fourier transform infrared spectrometer (Perkin Elmer, Waltham, MA, USA). The powdered MNP sample was mixed and ground with potassium bromide, then pressed into approximately 1 mm thick discs containing 1%–2% of MNP (w/w).
TEM images of MNP
Dilute particle suspensions (about 0.1 mg Fe/mL) were loaded onto lacey carbon film-coated copper grids from which static electricity had been removed, followed by ambient drying for preparation of transmission electron microscopy (TEM) samples. Representative TEM images of samples were acquired in the scanning TEM-bright field mode with an acceleration voltage of 200 kV using a 2010F analytical electron microscope (JEOL Ltd, Tokyo, Japan).
Measurement of magnetization
To assess the suitability of MNP for further magnetic targeting in vivo, the magnetization properties of MNP were measured using an MPMS-XL superconducting quantum interference device (SQUID) magnetometer (Quantum Design Inc., San Diego, CA, USA). Fluid samples were mounted in capsules and analyzed under varying DC magnetic fields (0–20,000 G) at 25°C.
Measurement of iron content
Inductively coupled plasma optical emission spectroscopy was carried out during preparation of the MNP to measure iron content of all samples using an Optima DV 2000 spectrometer (Perkin Elmer).20 In brief, a sample of MNP (about 10 μL) was digested in concentrated HCl (about 2 mL) and diluted with deionized water to a final concentration of 1–5 mg Fe/L. Samples were analyzed in triplicate at 238.204 nm. Dilutions of an iron standard (0–10 mg Fe/L) and an yttrium internal standard were used to construct the calibration curve.
Magnetophoretic mobility
Magnetophoretic mobility of the MNP suspensions was assessed by monitoring the decrease in iron concentration within the suspensions with duration of exposure to an external magnetic field.21 Briefly, samples (about 0.5 mg Fe/mL) were well dispersed in deionized water and placed into a 1 cm × 1 cm quartz cuvette fixed on the inner plate of a Dynal magnetic separator. Next, 10 μL aliquots were withdrawn at preset time points for measurement of iron content using inductively coupled plasma optical emission spectroscopy.
Characterization of immobilized β-glucosidase
Enzyme activity assay
GlcβNp was used as the substrate to measure both free and conjugated β-glucosidase activity throughout the study.22 Briefly, 0.1 mL of enzyme solution was added to 4.9 mL of citrate-phosphate buffer (0.1 mol/L, pH 6.0) containing GlcβNp and brought to a final enzyme concentration of 5.0 mmol/L, followed by incubation at 37°C. Next, 0.5 mL aliquots were sampled at preset intervals and mixed with 1.0 mL of carbonate buffer (stopper solution, 0.2 mol/L, pH 10.2). After centrifugation at 15,000 rpm for 20 minutes, the liberated p-nitrophenol, with a molar extinction coefficient of 18,300 mol/cm, was measured in the supernatant for absorbance at 405 nm. One β-glucosidase activity unit (1 U) was defined as the amount of enzyme required to hydrolyze 1 μmol per minute of the substrate (GlcβNp) under the test conditions.
Kinetic study of β-glucosidase
In order to measure the activity of the enzyme conjugated onto the MNP, the kinetics of free and conjugated β-glucosidase were determined using GlcβNp (0–4 nM) as the substrate. Michaelis-Menten parameters for free β-Glu, β-Glu-MNP, and PEG-β-Glu-MNP were obtained by fitting of the enzymatic hydrolysis ratio of β-glucosidase versus substrate concentration data on a Lineweaver-Burk plot. The apparent Michaelis constant (Km), indicating the binding affinity of β-glucosidase for GlcβNp, was calculated by applying Lineweaver-Burk plot analysis based on the following equation:
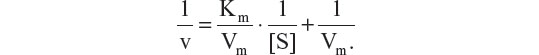
In vivo studies
All animal experiments were conducted according to protocols reviewed and approved by the University of Michigan Committee on Use and Care of Animals.
Pharmacokinetic study of PEG-β-Glu-MNP
Black C57BL6 mice (weight 20–25 g, from Harlan Laboratories, Indianapolis, IN, USA) were anesthetized by intraperitoneal injection of a ketamine/xylazine mixture (87/13 mg/kg body weight). D-MNP, β-Glu-MNP, or PEG-β-Glu-MNP suspension (12 mg Fe/kg body weight) was then administered through the lateral tail vein, consistent with previous studies.16 Next, 100 μL blood samples were collected by cardiac puncture at preset time intervals (n=3 per set) into 0.5 mL Eppendorf tubes spiked with 10 μL of heparin solution (5,000 U/mL). Immediately after collection of blood, the samples were centrifuged at 5,000 rpm for 5 minutes to separate plasma from whole blood. Next, 30 μL of plasma samples were added to electron spin resonance tubes using a microsyringe attached to a 12-inch long Teflon syringe needle and stored at −80°C for measurement of MNP concentration by electron spin resonance analysis (see section on quantitative iron content analysis using electron spin resonance spectroscopy). The area under the plasma concentration versus time profile was estimated numerically by a linear trapezoidal integration method.
Induction of subcutaneous 9L-glioma model
The magnetic targeting of PEG-β-Glu-MNP was evaluated in a mouse model of subcutaneous 9L-glioma. Briefly, 9L-glioma cells (Brain Tumor Research Center, University of California, San Francisco, CA, USA) were cultured in 100 mm culture flasks to confluence in Dulbecco’s Modified Eagle’s Medium supplemented with 10% heat-inactivated fetal bovine serum, 100 IU/mL of penicillin, and 100 mg/mL of streptomycin at 37°C in a humidified atmosphere of 5% CO2. Prior to implantation, the cells were harvested and resuspended in serum-free Dulbecco’s Modified Eagle’s Medium at a concentration of 4×107 cells/mL. The cell suspension (50 μL) was implanted in the right flank of the anesthetized mouse. Magnetic targeting experiments were conducted when tumor volumes reached about 300–400 mm3.
Magnetic targeting of PEG-β-Glu-MNP in mouse model of 9L-glioma monitored by MRI
Briefly, a magnetic field strength of about 320 mT was achieved by mounting a small NdFeB cylindrical ferromagnet (9 mm in diameter) on three tandem DYOYO-52 permanent magnets (K&J Magnetics, Plumsteadville, PA, USA). The tumor-bearing mouse was anesthetized using the same method mentioned in an earlier section and then placed on a platform with the tumor positioned directly on the pole face of the small magnet. The mouse was then injected with the MNP suspension (12 mg Fe/kg) through the lateral tail vein and kept in the magnetic field for one hour.
MRI experiments were carried out with using a protocol described previously with modification19 using a 30 cm horizontal-bore, 7T Direct Drive small animal imaging system (Varian, Walnut Creek, CA, USA). A thermostated circulating water bath was used to keep the temperature inside the magnet at 37°C throughout the MRI scanning process. T2-weighted images were acquired prior to and at one hour postinjection for animals with or without magnetic targeting. After the MRI scans, the mice were euthanized immediately by an overdose of CO2 inhalation and tissue samples were taken and kept at −80°C until further analysis.
Ex vivo analysis of MNP in excised tissue samples
Quantitative iron content analysis using electron spin resonance spectroscopy
Quantitative analysis of iron content in the plasma (see Pharmacokinetics section) and tumor tissue samples was done using electron spin resonance as described previously.14 Briefly, electron spin resonance spectra were acquired using an EMX electron spin resonance spectrometer (Bruker Corporation, Billerica, MA, USA) equipped with a liquid nitrogen cryostat. The acquisition parameters were set as follows: −128°C for temperature, 9.2 GHz for resonant frequency, and 20 mW for microwave power. The combination of receiver gain (1×104 or 1×105) and modulation amplitude (1 G or 5 G) was dependent on the type of sample as well as the intensity of the observed spectra. A WinEPR workstation (Bruker Corporation) was used to obtain the double integral values of the spectra. Neither the control tumor tissue samples (without exposure to MNP) nor the plasma samples showed visible double integral values when compared with the tested samples, so background correction was not necessary. Calibration standards were constructed by preparing electron spin resonance samples of dilutions (0–50 μg Fe/mL) of MNP stock with known iron concentration.
Analysis of β-glucosidase activity in tumor tissue samples
To minimize the effect of endogenous β-glucosidase activity, the β-glucosidase activity in the excised tissue samples was assayed using a spectrophotometric method as described previously with modification.19 Tissue samples (~100 mg) were homogenized at 0°C in 50 μL of lysis buffer (Tropix Inc., Bedford, MA, USA) using a pestle tissue grinder. The homogenates were diluted with 950 μL of 100 mmol/L pH 7.0 HEPES buffer containing 100 mmol/L KCL and 1 mmol/L MgSO4. The samples were dispersed in HEPES buffer by sonication at 30% amplitude for 5 seconds. The enzyme activity of the homogenates was determined at 37°C using the method described in an earlier section (Enzyme activity assay). To determine the endogenous enzyme activity, the activity of β-glucosidase in tissue samples from the control group was also measured and subtracted from the enzyme activity of the tissue samples from the experimental groups to determine the net activity caused by the targeted β-glucosidase.
Statistical analysis
The data are presented as the mean ± standard deviation unless otherwise indicated. Statistical Package for the Social Sciences version 16.0 software (SPSS Inc., Chicago, IL, USA) was used for the data analysis. β-glucosidase activity in the excised tumor tissues was compared using one-way analysis of variance, followed by Tukey’s multiple comparison test. A P-value of <0.05 was considered to indicate a statistically significant difference.
Results
Size distribution and zeta potential
D-MNP have shown their potential application in magnetic targeting in previous studies.16 The hydroxyl groups on the coating starch of the parent D-MNP enabled successful cross-linking via epichlorohydrin and amination with concentrated ammonia. Glutaraldehyde was used to crosslink β-glucosidase and DN-MNP via Schiff’s base formation. β-Glu-MNP was then PEGylated via conjugation of the amine group on β-glucosidase and the amine-reactive N-hydroxysuccinimide ester of SC-NHS-PEH (Figure 1).
Crosslinking and amination the D-MNP resulted in an expected increase in particle size from 102.7±2.2 nm to 138.3±3.1 nm (DN-MNP). Satisfactory amination of D-MNP to DN-MNP was confirmed by observation of a positive shift in zeta potential from –1.6±0.7 mV to +41.6±1.5 mV. After modification of DN-MNP with glutaraldehyde, the average particle size of Glutaral-MNP increased to 231.1±4.3 nm.The zeta potential of Glutaral-MNP varied from positive to slightly negative (–7.9±2.2 mV). The zeta potential, however, shifted back to positive (+17.5±2.3 mV) after conjugation of β-Glu, confirming another favorable Schiff’s base formation between the amine group on the enzyme and the end aldehyde group on Glutaral-MNP. Immobilization of β-glucosidase and PEGylation also resulted in increases in the average hydrodynamic diameter of MNP from 231.1±4.3 nm to 253.3±8.4 nm (β-Glu-MNP) and 267.3±0.1 nm (PEG-β-Glu-MNP), respectively (Table 1). Of note, the zeta potential of PEG-β-Glu-MNP showed a shift toward neutrality (–4.3±1.1 mV) due to coverage of part of the particle surface by the neutral long chain of PEG.
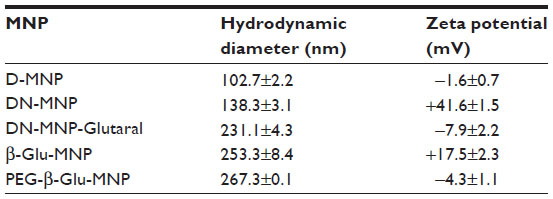 | Table 1 Hydrodynamic diameter and zeta potential characterization of MNP throughout the synthesis steps |
All the expected increases in hydrodynamic diameter and variations in zeta potential of the MNP confirmed correct synthesis of PEG-β-Glu-MNP from the precursor D-MNP.
Fourier transform infrared, TEM, SQUID, and magnetic mobility of PEG-β-Glu-MNP
Figure 2 shows the Fourier transform infrared spectra for PEG, β-glucosidase, D-MNP, PEG-β-Glu-MNP, and β-Glu-MNP. The infrared spectra for PEG and PEG-β-Glu-MNP showed the hallmark absorption peak of C-O-C ether bond stretching vibrations (at 1,100 cm−1), indicating correct attachment of the PEG moieties of PEG-β-Glu-MNP. Further, the peaks at 2,934.8 cm−1 and 1,543.1 cm−1 shown in the infrared spectra of free β-glucosidase, PEG-β-Glu-MNP, and β-Glu-MNP are assigned to the amide bonds of β-glucosidase, confirming successful conjugation of β-glucosidase on both types of MNP. Compared with the infrared spectrum of D-MNP, the presence of new absorption peaks in the spectra of PEG-β-Glu-MNP (1,100 cm−1, 2,934.8 cm−1, and 1,543.1 cm−1) and β-Glu-MNP (2,934.8 cm−1 and 1,543.1 cm−1) indicated correct synthesis of β-Glu-MNP and PEG-β-Glu-MNP.
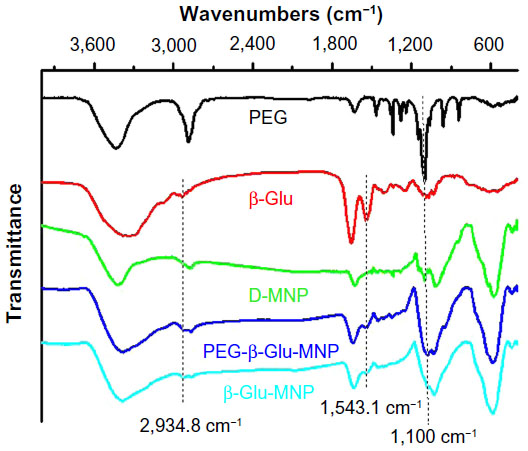 | Figure 2 Fourier transform infrared spectra of PEG (black), β-Glu (red), D-MNP (green), β-Glu-MNP (azure), and PEG-β-Glu-MNP (blue). |
The morphology of the larger coated D-MNP cores comprised of multiple smaller iron oxide domains was conserved well throughout the synthesis steps, and is clearly demonstrated by the TEM images of MNP shown in Figure 3. These TEM images also confirmed the size increases of MNP measured by dynamic light scattering, especially the increase in size of the MNP during glutaraldehyde crosslinking.
 | Figure 3 Scanning transmission electron microscopy-bright field images of (A) D-MNP and (B) PEG-β-Glu-MNP. |
The superparamagnetic properties of D-MNP were also conserved well during synthesis of PEG-β-Glu-MNP (Figure 4A). None of the magnetization curves for D-MNP, β-Glu-MNP, and PEG-β-Glu-MNP showed hysteresis or remanent magnetization. The high saturation magnetization of PEG-β-Glu-MNP (about 60 emu/g Fe) suggests the feasibility of magnetic targeting of the enzyme-conjugated nanoparticles.
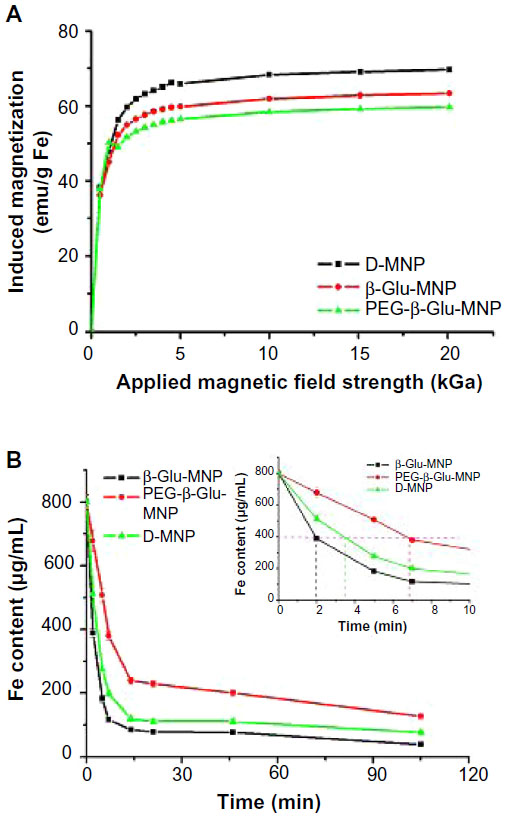 | Figure 4 (A) Magnetization properties of D-MNP, β-Glu-MNP, and PEG-β-Glu-MNP via superconducting quantum interference device analysis. (B) Magnetophoretic mobility of the magnetic nanoparticles. The iron concentrations of nanoparticle suspensions represent the amount of the nanoparticles not captured by the magnetic field. Inset image shows the data of time (1–10 minutes) with an expanded scale. |
Measurements of magnetophoretic mobility for all three types of MNP demonstrated the moving speeds of MNP (VMNP) in the applied magnetic field: Vβ-Glu-MNP > VD-MNP > VPEG-β-Glu-MNP (Figure 4B). The migration half-time (t1/2), defined as the time required to achieve a 50% decrease in iron concentration, was about 2 minutes, 3.5 minutes, and 7 minutes for β-Glu-MNP, D-MNP, and PEG-β-Glu-MNP, respectively.
Enzyme activity of β-glucosidase conjugated on MNP
Corresponding Km values, indicating the affinity of the enzyme for the substrate and the maximum reaction rate (Vmax) of β-glucosidase are shown in Table 2 and Figure 5. The apparent Km value of the free enzyme was determined to be 2.23 mmol/L, and this was retained with β-Glu-MNP and PEG-β-Glu-MNP, which showed Km values of 2.27 mmol/L and 2.25 mmol/L, respectively. After conjugation, β-Glu-MNP and PEG-β-Glu-MNP showed maximum reaction rates of 23.7 μmol per minute per mg and 21.2 μmol per minute per mg, respectively, which were, respectively, about 73.0% and 65.4% that of the free enzyme (32.4 μmol per minute per mg). In our previous study,14 conjugated β-glucosidase showed maximum enzyme activity of 1.17 U/mg iron (44.1 μg enzyme/mg Fe) with an optimized feed ratio of β-glucosidase and MNP (1.45 mg protein/mg Fe). After PEGylation of β-Glu-MNP, the enzyme activity of the immobilized β-glucosidase decreased slightly to 1.05 U/mg Fe.
 | Table 2 Enzyme activity measurements of free and immobilized β-glucosidase |
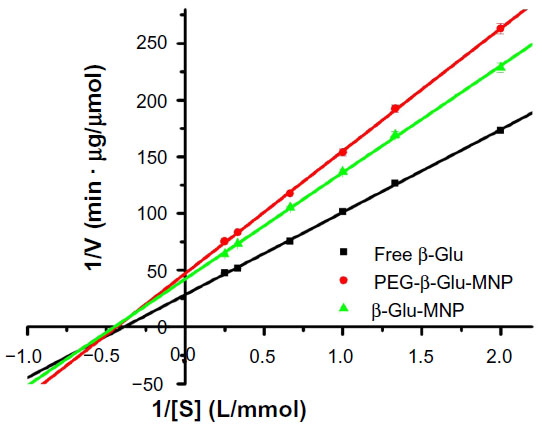 | Figure 5 Lineweaver-Burk plot analysis of the enzymatic kinetics of free β-Glu, β-Glu-MNP, and PEG-β-Glu-MNP. |
In vivo pharmacokinetics and tumor accumulation of PEG-β-Glu-MNP
The promising in vitro performance of PEG-β-Glu-MNP encouraged us to evaluate its potential for in vivo application. Electron spin resonance analysis of MNP concentrations in plasma samples following intravenous administration indicated that D-MNP had a short blood circulation time. Less than one fifth (14.4%) of the initial dose (14.78 μg Fe/mL) remained in the systemic circulation at 20 minutes post-administration, and similar results have been reported elsewhere.23 PEG-β-Glu-MNP and β-Glu-MNP showed markedly increased stability in blood, with more than 62% and 34% of the initial blood MNP concentrations, respectively, still remaining in the circulation at 20 minutes post-administration. The area under the curve, equal to the total plasma exposure of MNP for PEG-β-Glu-MNP (8,579±568 μg Fe/mL · minute) and β-Glu-MNP (2,791±234 μg Fe/mL · minute) was approximately 8.3-fold and 2.7-fold (P<0.001 for both) greater than that of D-MNP (1,031±123 μg Fe/mL · minute), (Figure 6).
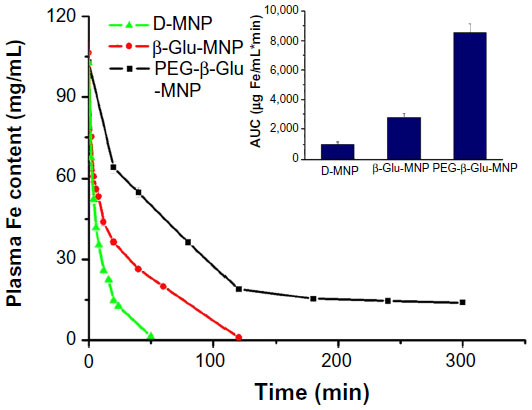 | Figure 6 Plasma concentration-time profiles of D-MNP, β-Glu-MNP, and PEG-β-Glu-MNP. Inset: corresponding area under the concentration-time curves (AUC) for D-MNP, β-Glu-MNP, and PEG-β-Glu-MNP. |
MRI monitoring of MNP showed accumulation of D-MNP, β-Glu-MNP, and PEG-β-Glu-MNP in tumor lesions at one hour post-administration with/without magnetic targeting (Figure 7). In baseline T2-weighted scans (preinjection or pretargeting images shown in Figure 7), the subcutaneous tumors are clearly visible as hyperintense regions located in the mouse flank. Encouragingly, the regions of MNP accumulation were spatially consistent with the tumor lesions (hyperintense regions) on the preinjection T2-weighted images taken prior to MNP administration), confirming considerably increased accumulation of MNP in the tumor lesions. Due to the poor stability of D-MNP in plasma, almost no accumulation of D-MNP in tumor tissue was observed in the mice even with magnetic targeting. Compared with D-MNP-injected mice, the MRI images of animals administered β-Glu-MNP or PEG-β-Glu-MNP showed much better nanoparticle accumulation in tumor tissue, displaying obvious negative contrasts. For mice treated with PEG-β-Glu-MNP in particular, part of the negative contrast area in the tumor was almost as dark as the background. The representative MRI images also showed that accumulation of MNP at the tumor site via the enhanced permeability and retention effect only was not enough. Without magnetic targeting, almost no negative contrast was seen in the tumor tissue from PEG-β-Glu-MNP-treated mice at one hour postinjection.
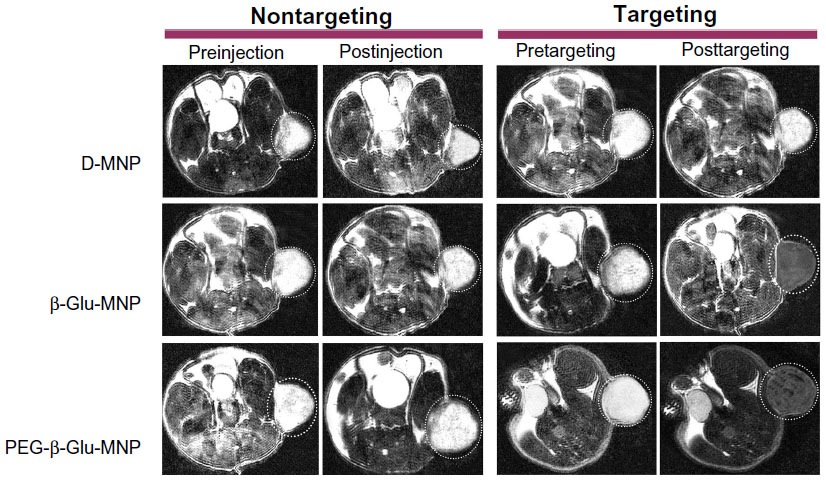 | Figure 7 Representative magnetic resonance images of mice bearing subcutaneous (flank) 9L-glioma tumors injected with D-MNP, β-Glu-MNP, or PEG-β-Glu-MNP (12 mg Fe/kg). For each MNP type, magnetic resonance images were taken prior to and at 60 minutes post-administration of MNP with/without magnetic targeting. |
All MRI results were further quantified by ex vivo analysis of MNP concentrations in excised tumor tissues taken one hour post-MNP administration. As shown in Figure 8, higher MNP concentrations were detected in tumor lesions from mice administered PEG-β-Glu-MNP (627±45 nmol Fe/g tissue, 8.5-fold higher) or β-Glu-MNP (289±58 nmol Fe/g tissue, 3.9-fold higher), compared with those injected with D-MNP (74±30 nmol Fe/g tissue). Moreover, magnetic targeting resulted in significantly greater accumulation of all three types of MNP in tumors than those of their nontargeted counterparts (8.7-fold lower for PEG-β-Glu-MNP at 71.8±5.1 nmol Fe/g tissue; 14.5-fold lower for β-Glu-MNP at 20.0±5.0 nmol Fe/g tissue; and 8.0-fold lower for D-MNP at 9.2±3.2 nmol Fe/g tissue).
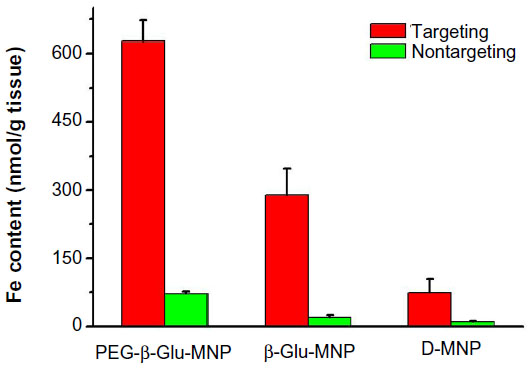 | Figure 8 Measurement of iron content in targeted or nontargeted tumor tissue of mice administered with D-MNP, β-Glu-MNP, or PEG-β-Glu-MNP. |
While the greatly enhanced delivery of MNP to tumors was encouraging, a more important index for evaluating the therapeutic potential of this MDEPT system was the activity of the delivered β-glucosidase in the excised tumors. Tissue samples from control mice, which were not treated with β-glucosidase, showed endogenous background β-glucosidase activity of 17.6 mU/g tissue, which was subtracted for correction of the delivery enzyme activity in tumor lesions. We found 6.07-fold and 3.84-fold higher (P<0.001 for both) β-glucosidase activity in tumor samples excised from the groups targeted with PEG-β-Glu-MNP (32.2±8.0 mU/g tissue) and β-Glu-MNP (20.1±5.0 mU/g tissue), respectively, compared with their nontargeted counterparts (5.3±1.6 mU/g tissue and 5.2±1.5 mU/g tissue, respectively, Figure 9). Enzyme activity in tumor tissues excised from mice injected with free β-glucosidase was also analyzed for comparison. Without the help of MNP and magnetic targeting, only 6.2±2.8 mU/g tissue of enzyme was delivered to these tumor lesions. Conjugation of the enzyme on MNP, PEGylation for better plasma stability, and magnetic targeting together ensured selective delivery of β-glucosidase to the subcutaneous tumor site.
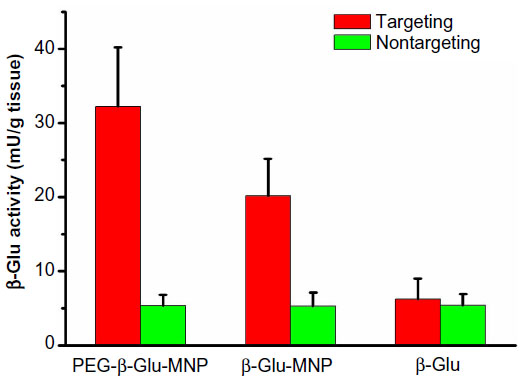 | Figure 9 Activity of β-glucosidase in the tumor lesion of the mice administered β-glucosidase (control), β-Glu-MNP, or PEG-β-Glu-MNP. Tumors of magnetically-targeted mice administered PEG-β-Glu-MNP exhibited significantly higher β-glucosidase activity than any other analyzed samples (P<0.01), confirming the selective tumor targeting of PEG-β-Glu-MNP. |
Discussion
In this study, β-glucosidase was successfully conjugated on aminated MNP (DN-MNP), followed by PEGylation of β-Glu-MNP using N-hydroxysuccinimide chemistry for better stability in plasma. Due to the longer blood circulation time, PEG-β-Glu-MNP showed better accumulation of both MNP concentration and β-glucosidase activity in subcutaneous 9L-glioma tumors. To achieve maximum conjugation of the enzyme to MNP, amine groups on the enzyme, rather than the nanoparticles, were used for PEGylation. If PEGylation had been carried out by using the amine groups on DN-MNP, more of the particle surface would be shielded by the PEG long chains, and much less particle surface area would be available for enzyme conjugation. It is well known that hydrolysis of the N-hydroxysuccinimide ester is the major competing reaction for N-hydroxysuccinimide chemistry in a water system, the rate of which increases rapidly when the pH value is above 7.0. As a result, a high feed ratio (PEG to particles, 1:1, mg/mg Fe), mild pH conditions (7.4), and a short reaction time were chosen for this PEGylation process. The results of a barium iodide assay showed conjugation of 0.23 nmol PEG/mg of iron, indicating an average of 80 PEG molecules per particle.
Except for size and surface chemistry, the response of nanoparticles to the applied magnetic field is also very crucial for their application in vivo. Based on the results of the magnetization measurements, the saturation magnetization of MNP decreased slightly from 69.7 emu/g Fe (D-MNP) to 63.3 emu/g Fe (β-Glu-MNP) and 59.7 emu/g Fe (PEG-β-Glu-MNP). The modifications made to the MNP (amination, enzyme conjugation, and PEGylation) improved the physicochemical properties of the parent D-MNP and decreased the response of MNP to the magnetic field at the same time. As a result, the magnetophoretic mobility rate of PEG-β-Glu-MNP was slower than that of β-Glu-MNP due to improved stability and the decreased magnetic response of PEG-β-Glu-MNP during the PEGylation process.
According to some previous studies,24,25 PEGylation could have both positive and negative effects on the activity of the immobilized enzyme. In the current study, the immobilized β-Glu, with or without PEG modification, maintained a similar affinity for the GlcβNp substrate. However, the Km of β-glucosidase immobilized on MNP was about 4-fold lower than the value reported in the literature (9.4±1 mmol/L) when β-glucosidase was immobilized on microgels.26 The Vmax of the immobilized β-glucosidase was also lower than that of free β-glucosidase due to inactivation of the enzyme, the change in enzyme structure, and/or cross-linking of the enzyme caused by glutaraldehyde. These findings underscore the value of MNP as carriers for enzymes.26,27
The pharmacokinetic results showed improved stability of β-Glu-MNP in plasma when compared with the precursor D-MNP. However, the blood circulation time of β-Glu-MNP is still not long enough, with only 20% of the initial dose remaining in the plasma at one hour post-administration, indicating fast recognition and clearance of β-Glu-MNP by the reticuloendothelial system. The blood circulation time of β-Glu-MNP was extended by PEG modification. About 20% of the initial dose remained in the blood circulation even at 2 hours postinjection, confirming the protective effect of PEG on MNP. The long PEG chain on the nanoparticle surface can help the MNP to escape recognition and clearance by the reticuloendothelial system. The blood circulation time of PEG-β-Glu-MNP is long enough for its selective accumulation in tumor tissue, according to previous studies.13,16 Owing to the limited surface area of MNP and the relatively long circulation time of PEG-β-Glu-MNP, PEG moieties were conjugated to enzyme, instead of MNP surface, for the purpose of maximizing enzyme activity in the targeted tumor tissue (Figure 1).16,28 Instead, enzyme conjugation was carried out with a preference for maximum enzyme activity in the targeted tumor tissue.
One of the conundrums of the enzyme/prodrug strategy is determining the optimal timing of prodrug administration due to the difficulty of real-time monitoring of enzyme concentrations in the circulation and at the tumor site. Earlier administration of the prodrug will result in activation of the prodrug by the enzyme left in the circulation, causing severe systemic toxicity. On the other hand, later administration of the prodrug will miss the maximum enzyme activity in the tumor lesion, leading to a poor therapeutic effect. After conjugation and magnetic targeting, accumulation of the enzyme at the tumor site can be monitored indirectly by real-time MRI analysis, allowing the optimal timing for prodrug administration to be determined. With PEGylation and magnetic targeting, accumulation of MNP at the tumor site was greatly increased (from 20.0±4.9 nmol Fe/g tissue to 627.2±45.2 nmol Fe/g tissue). Meanwhile, the enzyme activity at the tumor site increased from 5.2±1.8 mU/g tissue to 32.2±7.9 mU/g tissue. Maximum enzyme activity at the tumor site was achieved immediately after magnetic targeting, and decreased gradually thereafter. Therefore, the prodrug could be administered when the rate of prodrug activation by blood enzyme is negligible or acceptable according to the pharmacokinetic results.
Quantitative analysis of MNP concentration in the excised tumor tissue was done using electron spin resonance measurements, which were consistent with the MRI results. Although the saturation magnetization of β-Glu-MNP was slightly decreased after modification with PEG, a 2.2-fold higher nanoparticle concentration was detected in tumors from mice injected with PEG-β-Glu-MNP compared with those injected with β-Glu-MNP. Because of the better stability (and thus better enhanced permeability and retention effect) of PEG-β-Glu-MNP in plasma compared with that of β-Glu-MNP, accumulation of PEG-β-Glu-MNP at the tumor site was 3.6-fold higher than that of β-Glu-MNP when no magnetic field was applied. Compared with accumulation of MNP in tumor tissue, the activity of β-glucosidase in excised tumor tissue is a more important parameter for application of this PEGylated and enzyme-conjugated MDEPT system. According to optimal measurements, the average enzyme activity in tumors from PEG-β-Glu-MNP-treated mice was 1.6-fold and 5.2-fold higher than that in tumors from β-Glu-MNP-treated and free β-glucosidase-treated mice, respectively, which is consistent with the quantitative analysis of MNP in excised tumor lesions. The activity of β-glucosidase in the magnetically targeted tumor tissue was as high as 400 mU/g tissue, making PEG-β-Glu-MNP a suitable MDEPT candidate for use as an antitumoral agent.
Conclusion
β-glucosidase was successfully conjugated to MNP and further modified with PEG using the glutaraldehyde method and NHS chemistry. The immobilized β-glucosidase retained 65.4% residual enzyme activity when compared with that of the free enzyme. PEG modification of β-Glu-MNP improved the plasma stability and tumor targeting of β-Glu-MNP without compromising enzyme activity, rendering this MDEPT system suitable for in vivo application. Magnetic targeting of PEG-β-Glu-MNP was confirmed in mice bearing subcutaneous 9L-glioma tumors, and was demonstrated by MRI (in vivo, qualitative) as well as electron spin resonance analysis (ex vivo, quantitative). The encouraging results with regard to MNP concentration and enzyme activity in excised tumor tissue suggest that PEG-β-Glu-MNP warrant further study with a view to clinical application of magnetic directed enzyme/prodrug therapy.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant numbers 30700836 and 81160314) and China Postdoctoral Science Foundation projects (Grant numbers 20070420152 and 200801268). The authors would like to thank Professor Bart M Bartlett (Department of Chemistry, University of Michigan, Ann Arbor, MI, USA) for his help with the magnetization measurements and Dr Kai Sun (Department of Materials Science and Engineering, University of Michigan) for his help with the TEM analyses.
Disclosure
The authors report no conflicts of interest in this work.
References
Lindley C, McCune JS, Thomason TE, et al. Perception of chemotherapy side effects cancer versus noncancer patients. Cancer Pract. 1999;7:59–65. | |
Rooseboom M, Commandeur JN, Vermeulen NP. Enzyme-catalyzed activation of anticancer prodrugs. Pharmacol Rev. 2004;56:53–102. | |
Gwenin VV, Gwenin CD, Kalaji M. Colloidal gold modified with a genetically engineered nitroreductase: toward a novel enzyme delivery system for cancer prodrug therapy. Langmuir. 2011;27:14300–14307. | |
Syrigos KN, Rowlinson-Busza G, Epenetos AA. In vitro cytotoxicity following specific activation of amygdalin by beta-glucosidase conjugated to a bladder cancer-associated monoclonal antibody. Int J Cancer. 1998;78:712–719. | |
McBain SC, Yiu HH, Dobson J. Magnetic nanoparticles for gene and drug delivery. Int J Nanomedicine. 2008;3:169–180. | |
Cole AJ, Yang VC, David AE. Cancer theranostics: the rise of targeted magnetic nanoparticles. Trends Biotechnol. 2011;29:323–332. | |
Chorny M, Hood E, Levy RJ, Muzykantov VR. Endothelial delivery of antioxidant enzymes loaded into non-polymeric magnetic nanoparticles. J Control Release. 2010;146:144–151. | |
Kumar S, Jana AK, Dhamija I, Singla Y, Maiti M. Preparation, characterization and targeted delivery of serratiopeptidase immobilized on amino-functionalized magnetic nanoparticles. Eur J Pharm Biopharm. 2013;85(3 Pt A):413–426. | |
Tewes F, Ehrhardt C, Healy AM. Superparamagnetic iron oxide nanoparticles (SPIONs)-loaded Trojan microparticles for targeted aerosol delivery to the lung. Eur J Pharm Biopharm. 2014;86(1):98–104. | |
Oh KS, Lee H, Kim JY, et al. The multilayer nanoparticles formed by layer by layer approach for cancer-targeting therapy. J Control Release. 2013;165:9–15. | |
Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63:136–151. | |
Pasquetto MV, Vecchia L, Covini D, Digilio R, Scotti C. Targeted drug delivery using immunoconjugates: principles and applications. J Immunother. 2011;34:611–628. | |
Chertok B, Moffat BA, David AE, et al. Iron oxide nanoparticles as a drug delivery vehicle for MRI monitored magnetic targeting of brain tumors. Biomaterials. 2008;29:487–496. | |
Zhou J, Zhang J, David AE, Yang VC. Magnetic tumor targeting of β-glucosidase immobilized iron oxide nanoparticles. Nanotechnology. 2013;24:375102–375114. | |
Allard-Vannier E, Cohen-Jonathan S, Gautier J, et al. PEGylated magnetic nanocarriers for doxorubicin delivery: a quantitative determination of stealthiness in vitro and in vivo. Eur J Pharm Biopharm. 2012;81:498–505. | |
Cole AJ, David AE, Wang J, Galbán CJ, Hill HL, Yang VC. Polyethylene glycol modified, cross-linked starch-coated iron oxide nanoparticles for enhanced magnetic tumor targeting. Biomaterials. 2011;32:2183–2193. | |
Cole AJ, David AE, Wang J, Galbán CJ, Yang VC. Magnetic brain tumor targeting and biodistribution of long-circulating PEG-modified, cross-linked starch-coated iron oxide nanoparticles. Biomaterials. 2011;32:6291–6301. | |
Song C, Sheng L, Zhang X. Preparation and characterization of a thermostable enzyme (Mn-SOD) immobilized on supermagnetic nanoparticles. Appl Microbiol Biotechnol. 2012;96:123–132. | |
Chertok B, David AE, Yang VC. Magnetically-enabled and MR-monitored selective brain tumor protein delivery in rats via magnetic nanocarriers. Biomaterials. 2011;32:6245–6253. | |
Chertok B, Cole AJ, David AE, Yang VC. Comparison of electron spin resonance spectroscopy and inductively-coupled plasma optical emission spectroscopy for biodistribution analysis of iron-oxide nanoparticles. Mol Pharm. 2010;7:375–385. | |
Yu F, Zhang L, Huang Y, Sun K, David AE, Yang VC. The magnetophoretic mobility and superparamagnetism of core-shell iron oxide nanoparticles with dual targeting and imaging functionality. Biomaterials. 2010;31:5842–5848. | |
Gueguen Y, Chemardin P, Labrot P, Arnaud A, Galzy P. Purification and characterization of an intracellular β-glucosidase from a new strain of Leuconostoc mesenteroides isolated from cassava. J Appl Microbiol. 1997;82:469–476. | |
Chertok B, David AE, Yang VC. Polyethyleneimine-modified iron oxide nanoparticles for brain tumor drug delivery using magnetic targeting and intra-carotid administration. Biomaterials. 2010;31:6317–6324. | |
Liu S, Bae Y, Leggas M, et al. Pharmacologic properties of polyethylene glycol-modified Bacillus thiaminolyticus thiaminase I enzyme. J Pharmacol Exp Ther. 2012;341:775–783. | |
Sekiguchi S, Yasukawa K, Inouye K. Effects of polyethylene glycol on bovine intestine alkaline phosphatase activity and stability. Biosci Biotechnol Biochem. 2011;75:2132–2137. | |
Welsch N, Wittemann A, Ballauff M. Enhanced activity of enzymes immobilized in thermoresponsive core-shell microgels. J Phys Chem B. 2009;113:16039–16045. | |
Yu Y, Yuan J, Wang Q, Fan X, Wang P. Covalent immobilization of cellulases onto a water-soluble-insoluble reversible polymer. Appl Biochem Biotechnol. 2012;166:1433–1441. | |
Ni F, Jiang L, Yang R, Chen Z, Qi X, Wang J. Effects of PEG length and iron oxide nanoparticles size on reduced protein adsorption and non-specific uptake by macrophage cells. J Nanosci Nanotechnol. 2012;12:2094–2100. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.