Back to Journals » International Journal of Nanomedicine » Volume 14
Engineering of solidified glyburide nanocrystals for tablet formulation via loading of carriers: downstream processing, characterization, and bioavailability
Authors Ali HSM ![]() , Hanafy AF
, Hanafy AF ![]() , Alqurshi A
, Alqurshi A ![]()
Received 19 November 2018
Accepted for publication 29 January 2019
Published 13 March 2019 Volume 2019:14 Pages 1893—1906
DOI https://doi.org/10.2147/IJN.S194734
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Hany SM Ali,1,2 Ahmed F Hanafy,1,3 Abdulmalik Alqurshi1
1Department of Pharmaceutics and Pharmaceutical Technology, College of Pharmacy, Taibah University, Al-Madinah Al-Munawwarah, Saudi Arabia; 2Department of Pharmaceutics, Faculty of Pharmacy, Assiut University, Assiut, Egypt; 3Research and Development Department, Al Andalous Pharmaceutical Industries, Cairo, Egypt
Introduction: Presenting poorly water-soluble drugs as nanoparticles has shown to be an effective technique in enhancing drug dissolution rate, intrinsic solubility, and thus oral bioavailability. Nevertheless, working with nanoparticles introduces many challenges, one of which is their physical instability. Formulating nanoparticles into a solid dosage form may overcome such challenges and thus unlock the potential benefits of nanosizing.
Methods: The current work investigates the possibility of developing a novel solid dosage form, with enhanced dissolution rate, whereby nanocrystals (~400 nm) of the class II Biopharmaceutical Classification System drug, glyburide (GBD) were fabricated through combined precipitation and homogenization procedures. Using a novel, but scalable, spraying technique, GBD nanocrystals were loaded onto commonly used tablet fillers, water-soluble lactose monohydrate (LAC), and water insoluble microcrystalline cellulose (MCC). Conventional tableting processes were then used to convert the powders generated into a tablet dosage form.
Results: Studies of redispersibility showed considerable preservation of size characteristics of GBD nanocrystals during downstream processing with redispersibility indices of 105 and 118 for GBD–LAC and GBD–MCC, respectively. Characterization by differential scanning calorimetry, powder X-ray diffraction, and scanning electron microscopy showed that the powders generated powders contained nanosized crystals of GBD which adhered to carrier surfaces. Powder flowability was characterized using Hausner ratio (HR) and Carr’s index (CI). GBD–LAC-loaded particles exhibited poor flowability with CI and HR of 37.5% and 1.60, respectively, whilst GBD–MCC particles showed a slightly improved flowability with CI and HR of 26.47% and 1.36, respectively. The novel tablet dosage form met US Pharmacopeia specifications, including drug content, hardness, and friability.
Conclusion: Higher dissolution rates were observed from the nanocrystal-based tablets compared to the microsized and commercial drug formulations. Moreover, the novel nanocrystal tablet dosage forms showed enhanced in vivo performance with area under the plasma concentration–time curve in the first 24 hours values 1.97 and 2.24 times greater than that of marketed tablets.
Keywords: glyburide, nanocrystals, downstream, tablet, bioavailability, solidification, redispersibility
Introduction
Poor water solubility is a prominent challenge the pharmaceutical industry is currently faced with, particularly as the number of poorly water-soluble new chemical entities is growing.1 Oral bioavailability of drugs can be greatly hindered by low water solubility and dissolution rate, thus causing a delay in therapeutic responses, lack of dose proportionality, individual variations, and local irritations.2–4
In pharmaceutical research, the term nanosized is commonly applied to particle size ranging from a few nanometers to 1 μm.5 Nanonization is an attractive approach to overcoming drug solubility issues,6 whereby reduction in particle size increases the surface area by a large fold, leading to enhanced dissolution rate and intrinsic solubility. This in return can have a positive effect on oral bioavailability of class II Biopharmaceutical Classification System drugs.7,8 Furthermore, presenting a drug in its nanocrystal form can increase particle adhesiveness, causing prolonged contact to gastrointestinal mucosa and leading to enhanced drug absorption.9 Nanocrystals of drugs are generally generated and presented as nanosuspensions, in which solid nanoparticles of the drug are dispersed in liquid.10 However, nanosuspensions are notorious for their physical instability, as particle mobility promotes thermodynamic and molecular kinetic interactions11 leading to particle growth, alterations in crystal form, and may even encourage chemical degradation.12 In addition, to avoid the immediate physical instability, nanosuspensions are diluted with large volumes of solvents, making the administration of multiple doses impractical.13 Presenting nanocrystals in a conventional solid dosage form may overcome the afore mentioned problems and simplify drug administration.14
Lyophilization and spray drying are commonly used techniques to dry nanosuspensions of drugs.15 Nevertheless, powders generated, particularly using lyophilization, appear to have inadequate flowability, and therefore require further processing, such as granulation, or the addition of excipients in substantial amounts.16 In addition, the expense, time, and energy required by lyophilization are not favored in commercial-scale production.17 On the other hand, spray drying involves operations at relatively elevated temperatures. This may lead to chemical and physical degradation of drugs, and is difficult to scale up.18 So far, reported studies on the upscaling of nanocrystallization processes and their use in manufacturing conventional dosage forms are limited.8,19
Glyburide (GBD), chemical name N-[4-(β-(2-methoxy-5-chlorobenzamido)-ethyl)-benzenesulfonyl]-N-cyclohexyl-urea,20 is a class II Biopharmaceutical Classification System second-generation sulfonylurea drug that is orally administered to control blood glucose levels in type II diabetic patients.21,22 Due to its poor aqueous solubility and dissolution rate, GBD shows low and erratic oral bioavailability.23 Different strategies were investigated and published to enhance the dissolution rate and bioavailability of GBD.24–27 In spite of these efforts, overall findings were not completely satisfactory, and no GBD product which resulted from these studies have been commercialized.28 The aim of this research was to explore the feasibility of a novel spraying technique in developing powders of carriers loaded with nanocrystals of GBD suitable for the conventional tableting process, thus, resulting in a novel nanocrystal tablet dosage form, with enhanced dissolution rate and oral bioavailability.
Methodology
Materials
GBD was kindly provided from Amriya Pharmaceutical Industries, Alexandria, Egypt. Poloxamer 188 was supplied by Spectrum Chemicals (New Brunswick, NJ, USA). Dimethyl sulfoxide (DMSO) was obtained from Fisher Scientific, Loughborough, UK. Microcrystalline cellulose (MCC; MCC PH 101) was purchased from FMC, Cork, Ireland. D-lactose monohydrate was obtained from Sigma-Aldrich Chemie GmbH, Steinheim, Germany. Polyvinylpyrridone (polyplasdone XL10) was obtained from Ashland Specialty Ingredients (Wilmington, DE, USA). Colloidal silicon dioxide (Aerosil 200) was provided from Evonik Degussa AG (Essen, Germany). Other materials were of pharmaceutical grade and were used as supplied.
Nanosizing
A GBD nanosized suspension was obtained through an adapted published bottom-up/top-down procedure,29 where 5 mL of 0.4 g/mL solution of GBD in DMSO was introduced into an aqueous phase (45 mL deionized water containing poloxamer 188, 0.5%, w/v) at a rate of 0.5 mL/minute using a syringe needle (0.5 mm diameter) under sonication (Ultrasons-HD; JP Selecta, Barcelona, Spain) at 25°C. The resultant GBD dispersion was centrifuged for 30 minutes to separate solid GBD particles. Residues of DMSO were removed by washing in deionized water and recentrifugation. Half a gram of the carefully collected GBD particles was suspended into 40 mL of an aqueous solution of poloxamer 188. This was then homogenized using Ultra-Turrax T25 Digital Homogenizer (IKA–Werke, Staufen, Germany) at 10,000 rpm for 2 minutes. Following this, the obtained suspension was subjected to high-pressure homogenization (HPH) (Avestin C-5, Avestin Inc., Ottawa, Canada). First, the dispersion initially homogenized for two cycles performed at 100, 500, and 1,000 bar. Then, the GBD dispersion was homogenized at 1,500 bar until obtaining a drug nanosuspension of a constant particle size. Monitoring of particle size was performed by analyzing samples every 10 minutes.
Size characterization
The mean size distribution of the GBD nanosuspension was characterized using dynamic light scattering by Microtrac S3500 (Microtrac Inc., Montgomeryville, PA, USA). After dilution in deionized water, nanosupension samples were used for size measurement three times for 120 seconds at 25°C.
Solidification
GBD nanocrystals were solidified via adsorption on lactose monohydrate and/or MCC PH 101. This process was performed using a setting that simulates a top-driven single pot high-shear mixing machine coupled with a spray system (see Figure 1). Fifty-five milliliters of poloxamer-stabilized GBD nanosuspensions (12.5 mg/mL) were sprayed via a Glatt Midi fluid bed system nozzle (Glatt, Binzen, Germany) onto 50 g of preheated (40°C–45°C) powder bed of fillers. Spraying was optimized to keep the adsorption process continuous to avoid any significant lump formation. The experiment setting consisted of a pyrex container heated to the required temperature using a hot plate (MS-H-Pro, SCILOGEX, Berlin, CT, USA) and an external digital thermometer to monitor the bed’s temperature. Powder mixing was performed using a propeller (RW 20, IKA®–Werke GmbH & Co. KG, Staufen, Germany) fitted to the pyrex container. GBD nanosuspensions were sprayed onto the powder bed at a flow rate of 0.5 mL/minute and atomizing pressure of 0.2–0.3 MPa using a 0.1 mm (diameter) spraying nozzle fixed ~3 cm above the powder bed, designed to generate a spray pattern that covers the powder bed with minimal loss. After complete spraying, mixing was continued, at the specified temperature range, for ~10 minutes to dry the powder bed, targeting a moisture content of 0.5%–1%. This was then verified using a halogen moisture analyzer (Mettler Toledo HB43-S, Greifensee, Switzerland). Dried powders were then passed through a 500 μm sieve to break any agglomerates that may have formed in the process. Resulting powders were then stored at room temperature for further processing.
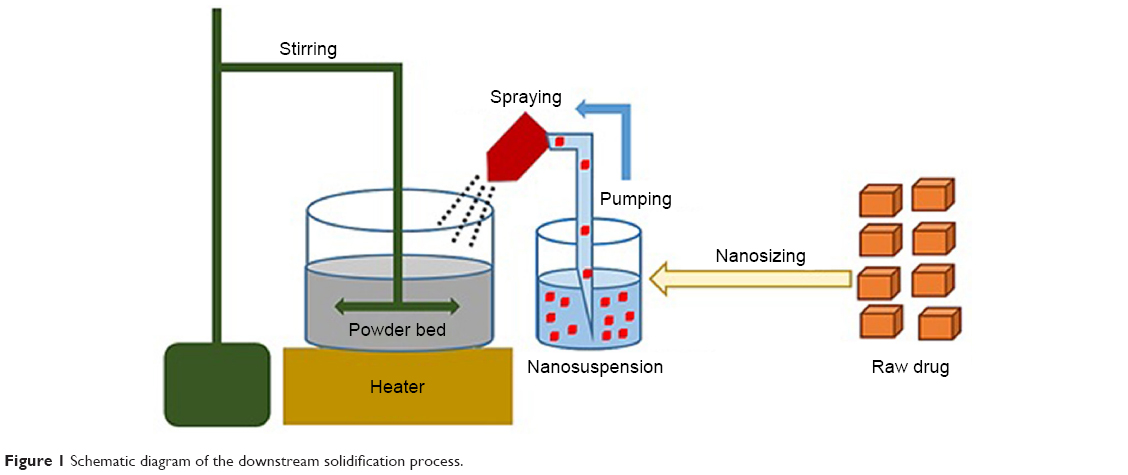 | Figure 1 Schematic diagram of the downstream solidification process. |
Redispersibility
Redispersion of the solidified GBD nanosized particles was evaluated by calculating the redispersibility index (RDI). RDI is determined as (RDI = D/D0 ×100), where D is the mean particle size of the sample powder (after redispersion), while D0 is the mean particle size of the original nanosuspension of drug alone (ie, before solidification). Thus, RDI values close to 100% indicate dried powders to have been completely redispersed, generating nanosized particles of similar sizes as produced initially.30,31
Redispersion was characterized by dispersing 50 mg of the GBD-loaded powders into 10 mL of deionized water followed by a gentle shake. Particle size measurements were then performed following the abovedescribed method (size characterization). Results were compared to particle size of the original nanosuspension of drug alone. Suspensions containing the insoluble carrier (MCC) in addition to the drug were also compared to a drug-free suspension of treated MCC particles as a control sample.
Solid state characterization
Morphology
A field emission scanning electron microscope (FESEM; LEO 1530170, Carl Zeiss SMT Inc., Oberkochen, Germany) was used for assessment of particle morphology. Samples of the generated solidified powders were taken and sputter coated with carbon to improve conductivity during FESEM processing.
Powder X-ray diffractometry (PXRD)
Shimadzu XRD 6000 diffractometer (Shimadzu Corporation, Kyoto, Japan) was applied to generate PXRD of raw unprocessed GBD, MCC, lactose monohydrate, and GBD-loaded powders. Diffraction patterns were recorded over an angular range of 10°–60° 2θ using a graphite monochromator and a copper radiation source (λ=1.5418 Å) with a scanning speed of 0.04°/minute.
Differential scanning calorimetry (DSC)
Thermal characterization was performed, using a Netsch DSC (Netsch F3 Maia®, Selb, Germany) with a cooling unit over a temperature range of 0°C–250°C. The DSC cell was purged with 50 cm3/minute of dry nitrogen gas, and the cooling unit was purged with 150 cm3/minute of dry nitrogen gas. The DSC cell was calibrated using indium and following manufacturer’s guidelines. The study was conducted on unprocessed GBD, MCC, lactose monohydrate, and GBD-loaded powders. Samples were analyzed in pin-holed aluminum hermetic pans. Experimental conditions followed an equilibration at 0°C for 5 minutes, followed by a ramp to 250°C at a rate of 10°C/minute. Sample size ranged between 3 and 8 mg.
Flow characterization
Bulk and tapped density tests were evaluated in triplicates using an automatic tapper (Erweka, Heusenstamm, Germany). Accurately weighed amounts of the MCC, lactose, and GBD-loaded carriers were placed in a 100 mL graduated cylinder. Volumes were measured before and after tapping for 500 times. Bulk (ρT) and tapped density (ρT) were calculated by dividing sample weight (g) by the volume occupied (mL). Based on the bulk and tapped densities, Carr’s compressibility index (CI) and Hausner ratio (HR) were calculated using the following equations:
|
|
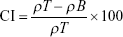
|
|
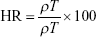
Preparation of tablet formulations
Based on preliminary trials, the drug-loaded powders (GBD–MCC and GBD–LAC) were blended with the disintegrant (polyplasdone XL10), colloidal silicon dioxide, and magnesium stearate (see Table 1).32–34 The powder mixtures were then compressed using a single-punch machine (Erweka) equipped with 8 mm biconvex punch to produce tablets of average weight 200±5 mg. Tablet hardness was adjusted to achieve a tablet friability of >1% and a disintegration time of >1 minute, thus resembling the reference commercial product (GBD–COM) Semi Daonil™. Formulations of unprocessed micronized GBD particles (GBD–MCC–MIC and GBD–LAC–MIC) were prepared similarly using raw micronized GBD suspensions instead of GBD nanosuspensions.
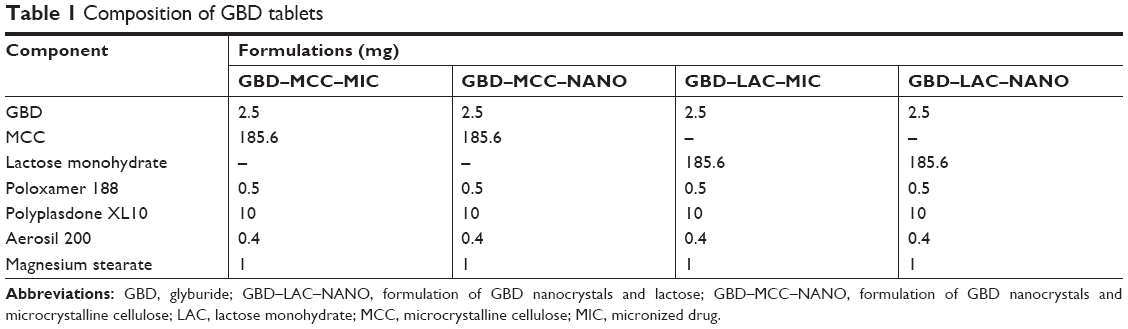 | Table 1 Composition of GBD tablets |
Tablet characterization
Samples taken from the start, middle, and end of each batch of formulated tablets were evaluated for hardness, disintegration, and friability. Hardness was evaluated using a hardness tester (Erweka GmbH, Heusentamm, Germany). Friability was determined as a percentage weight loss of tablets (n=10), using a Pharma Test friabilator (Pharma Test, Hainburg, Germany), with a total of 300 rotations. Disintegration of the GBD tablets (n=6) was analyzed in water at 37°C. Tablets were observed until complete disintegration.35
Formulated tablets were also tested for GBD content by crushing four randomly selected tablets. Following this, methanol was used to extract the drug from 200 mg of the crushed tablet powder. After filtering the resultant suspension, a HPLC assay (please see quantification procedures in section “Bioavailability”) was used to determine the content of GBD. The test was repeated on three different samples generating an average and an SD of n=3.
Dissolution
Dissolution tests were performed using the United States Pharmacopeia paddle method and United States Pharmacopeia apparatus I (Pharma Test), whereby formulated tablets were introduced into 900 mL of phosphate buffer dissolution medium (pH 6.8 and 37°C), a commonly used medium to discriminate between formulations of GBD.36,37 Paddles were set to rotate at 75 rpm. Samples of 5 mL were withdrawn at predetermined time intervals of 10, 20, 30, 45, 60, and 120 minutes and substituted with equal volumes of fresh medium. Withdrawn samples were filtered through a 0.2 μm filter (Millipore, Cork, Ireland) and quantified for GBD contents using a HPLC assay (please see quantification procedures in section “Bioavailability”). Dissolution experiments were conducted in triplicates, and an average value was calculated.
Bioavailability
Animals and samples collection
Procedures were approved by the Ethics Committee of Taibah University prior to the commencement of the study. In vivo tests were performed in the Medical Research Institute at Alexandria University in agreement with the guidelines for care and use of laboratory animals published in the Guide for the Care and Use of Laboratory Animals, 8th edition, National Academies Press, Washington, DC, USA. Male Sprague Dawley rats (250±20 g) were randomly divided into three groups (n=5 for each group). For adaptation, experimental rats were kept for 5 days in the animal house before conducting experiments. Rats were fed a standard rat diet with free access to tap water and kept under constant environmental conditions (22°C±3°C, 50%±5% relative humidity, light/dark cycle of 12 hours).38 Rats were put through an overnight fasting phase prior to the study. Tablets were dispersed in 1 mL of deionized water (10 mg of GBD per 1 kg of body weight) and were given to rats by oral gavage, using blunt intragastric tubing to ensure administration of accurate dose; this was followed by an additional 0.5 mL of deionized water for washing purposes.
Group I of Sprague Dawley rats received the commercial GBD formulation, while groups II and III received GBD–MCC–NANO (formulation of GBD nanocrystals and microcrystalline cellulose) and GBD–LAC–NANO (formulation of GBD nanocrystals and lactose), respectively. At time intervals 0.5, 1, 2, 4, 8, 12, and 24 hours, blood samples were collected in heparinized Eppendorf tubes. Samples were centrifuged instantly at 13,000 rpm for 10 minutes and stored at −20°C until required for HPLC analysis.
Processing and quantification
Precipitation of plasma protein was performed through addition of acetonitrile (0.5 mL) to the plasma samples, which was vortexed for 1 minute followed by centrifugation at 5,000 rpm for 5 minutes. Supernatant was then carefully transferred into a new Eppendorf tube of which 20 μL was used in a validated HPLC assay for GBD quantification. This was performed using a Shimadzu Prominence HPLC System (Shimadzu, Kyoto, Japan) equipped with a diode array detector (SPD-M20A).38 Calibration curve was performed using rat plasma in the concentration range 10–400 ng/mL. Gliclazide was used as an internal standard. A mobile phase, consisting of filtered and degassed mixture of 0.05 M phosphate buffer (pH 3.5): acetonitrile (40:60% v/v), was pumped at an isocratic flow rate of 1 mL/minute. The chromatographic separation was accomplished utilizing a reverse phase Hypersil™ BDS C18 Column (4.6×150 mm, 5 μm, Thermo Fisher Scientific, Waltham, MA, USA). Readings were recorded by measuring ultraviolet absorbance at 228 nm.
Pharmacokinetic analysis
The maximum plasma drug concentration (Cmax) and the time (tmax) taken to reach Cmax was observed and recorded from plasma concentration data for each rat. The area under the plasma concentration–time curve in the first 24 hours (AUC0–24 hours) was calculated using the trapezoidal rule. Percent relative bioavailability (F) was calculated by dividing AUCtest/AUCreference. One-way ANOVA was performed using SPSS 16 Statistics software (SPSS Inc., Chicago, IL, USA). P-value <0.05 was considered as statistically significant.
Results and discussion
Nanosizing
To prepare pharmaceutical nanosuspensions, the choice of type and level of stabilizer(s) was crucial. The amount of selected stabilizing agent(s) should be sufficient for complete surface coverage of the generated nanosized particles to accomplish stabilization against particle agglomeration. In the current study, the nonionic steric stabilizer, poloxamer 188, was used because of its wide range of compatibility with various compounds irrespective of their charge. Structurally, poloxamer is composed of amphiphilic block copolymers containing hydrophilic poly(ethylene oxide) (PEO) and hydrophobic poly(propylene oxide) (PPO).39 This polymer folds for attachment onto the hydrophobic surface of the drug using its PPO group, whereas the PEO tails provide a thick steric layer, which stabilizes the nanosized suspension.40 In the current study, SEM analysis showed unprocessed GBD as irregularly shaped microsized particles (Figure 2). Size characterization was confirmed by photon correlation spectroscopy analysis where the D50 of raw GBD particles was found to be 5 μm (Figure 3A). To generate nanosuspension, the raw particles were initially precipitated then subjected to HPH as described in the experimental part. The initial precipitation stage decreased the number of homogenization cycles needed for GBD nanonization markedly. The precipitation step decreased the size of raw GBD particles to 2.5 μm (Figure 3B). Moreover, the initial precipitation process helps facilitate the particle size reduction of the top-down stage by increasing friability and defects in drug crystals.29,41 The generated GBD crystals were effectively comminuted into the submicron sizes (D50 of 400 nm, Figure 3C) within five homogenization cycles at 1,500 bar. Homogenization for longer periods was not effective for further particle size reduction as the mean particle size showed very little change after the fifth cycle.
 | Figure 2 SEM micrograph of unprocessed GBD particles. |
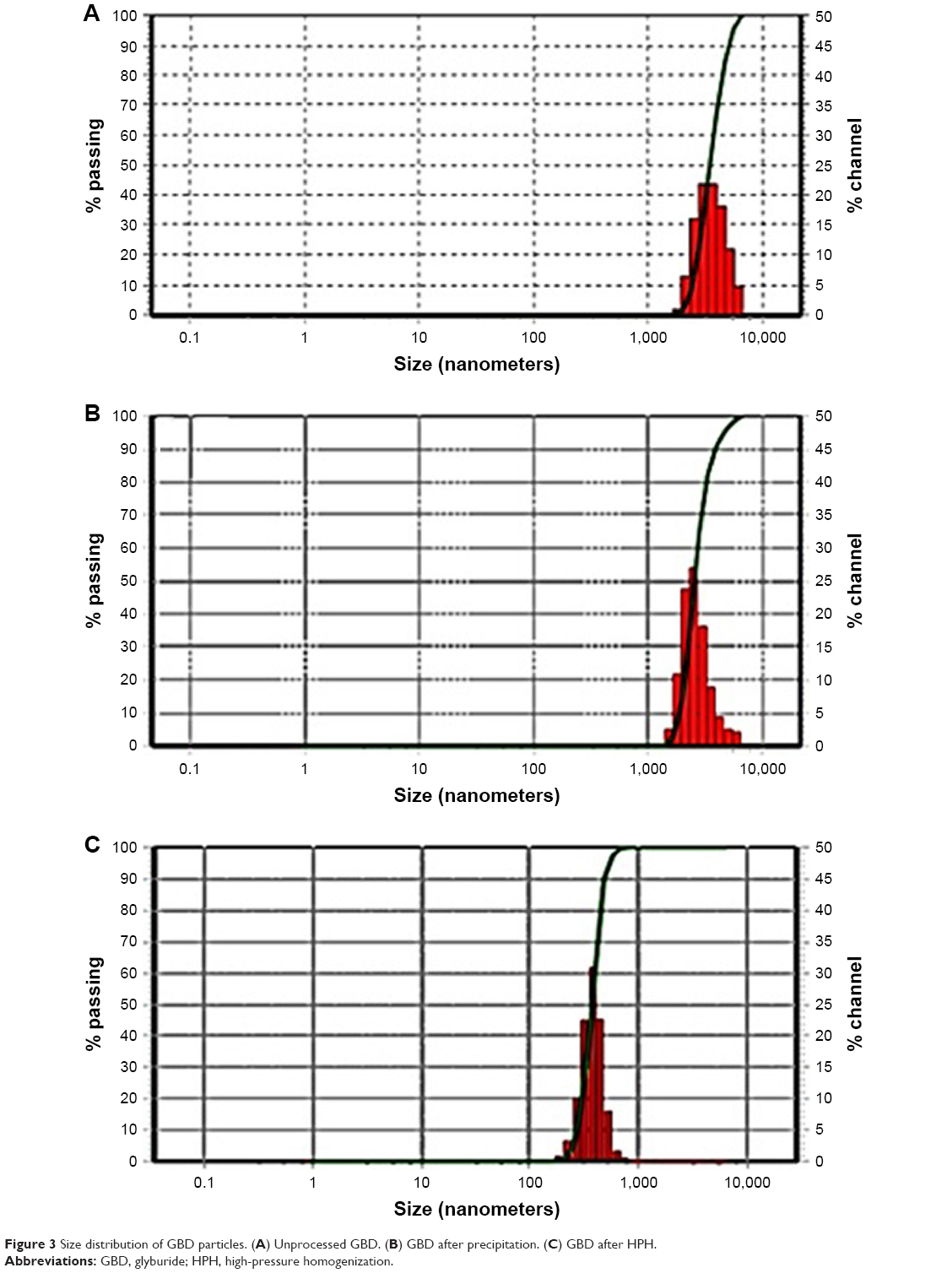 | Figure 3 Size distribution of GBD particles. (A) Unprocessed GBD. (B) GBD after precipitation. (C) GBD after HPH. |
Solidification
Instability of nanosuspension is a big challenge due to Ostwald ripening and particle aggregation. Solidification enhances stability and converts nanosuspensions to more convenient solid forms such as tablets without affecting the benefits gained from nanonization. A downstream processing procedure of spraying the generated nanosuspensions on hot solid powder beds was used to solidify drug particles in this study. Despite the advantage of being a single-step process (when compared with freeze drying), scalability remains a challenge with such a downstream process. SEM micrographs of MCC, lactose, and GBD-loaded carriers are presented in Figure 4. MCC appeared as large sized elongated particles, with an approximate length of 50 μm and width of 20 μm (Figure 4A), with an apparently smooth surface. In Figure 4B, lactose showed a mix of small and large particles of polyhedral shapes and smooth surfaces with a number of larger aggregated particles. Upon larger magnification of GBD-loaded carriers, nanosized particles were found embedded onto the surface of carrier particles (arrows in Figure 4E and F). However, such particles were not clearly noticed on the surface of the pure powders of the carriers (Figure 4C and D). These findings indicated adherence of GBD nanosized particles onto the surface of carrier particles.
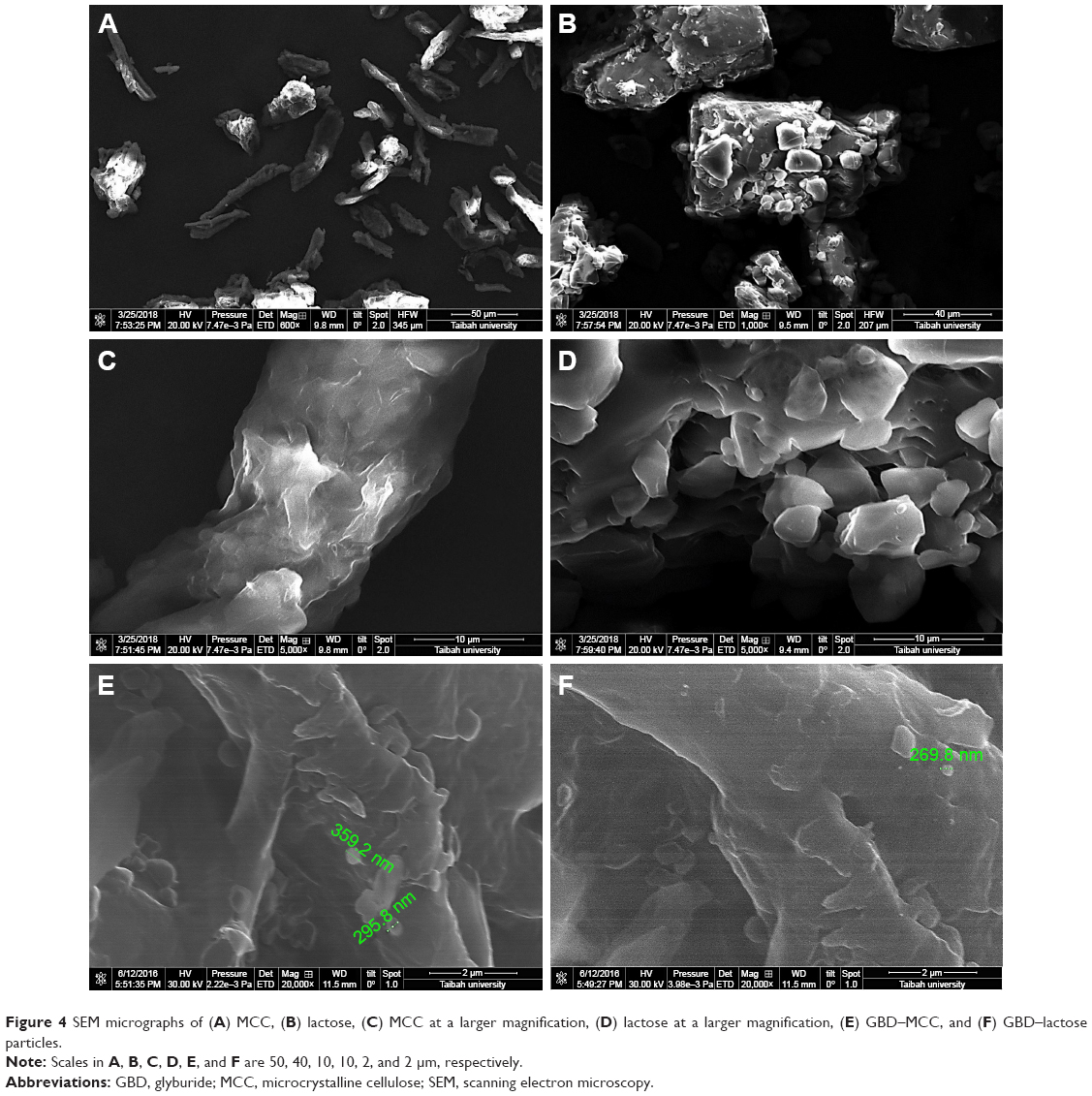 | Figure 4 SEM micrographs of (A) MCC, (B) lactose, (C) MCC at a larger magnification, (D) lactose at a larger magnification, (E) GBD–MCC, and (F) GBD–lactose particles. |
Redispersibility
Aggregation of the drug nanoparticles is reported to profoundly impact the characteristics of the products. If irreversible aggregation takes place, the benefits of large surface area of the original nanosized particles on drug dissolution and bioavailability will diminish. Figure 5 shows the particle size distribution of GBD nanosuspensions generated after redispersion from the solidified powders. Redispersion from GBD–LAC particles showed a single size distribution peak with D50 (value indicating that 50% of the particles are less than this size) of 419 nm, and no interference was detected by the water-soluble lactose (Figure 5A). However, two distinct peaks were notable in particle size distribution of GBD–MCC (Figure 5B). The first peak at 5.99 μm was most probably linked to the smallest terminal portion of MCC particles. This assumption was later confirmed by analyzing a suspension of plain MCC using the same device where a similarly located peak of D50 equal to 5.99 μm was observed (Figure 5C). The second distinct size distribution peak in the GBD–MCC graph was noticed in the submicron range with D50 of 470 nm; this was clearly attributed to the GBD nanosized particles. Generally, the small RDI values (105 and 118 for GBD–LAC and GBD–MCC, respectively) denoted greater redispersibility of the GBD-loaded powders. The minor changes observed in particle size after redispersion suggested slight aggregation of particles; this may be due to the secondary attraction of GBD particles. These results confirm that size characteristics of GBD nanosuspension are considerably preserved during downstream layering process.
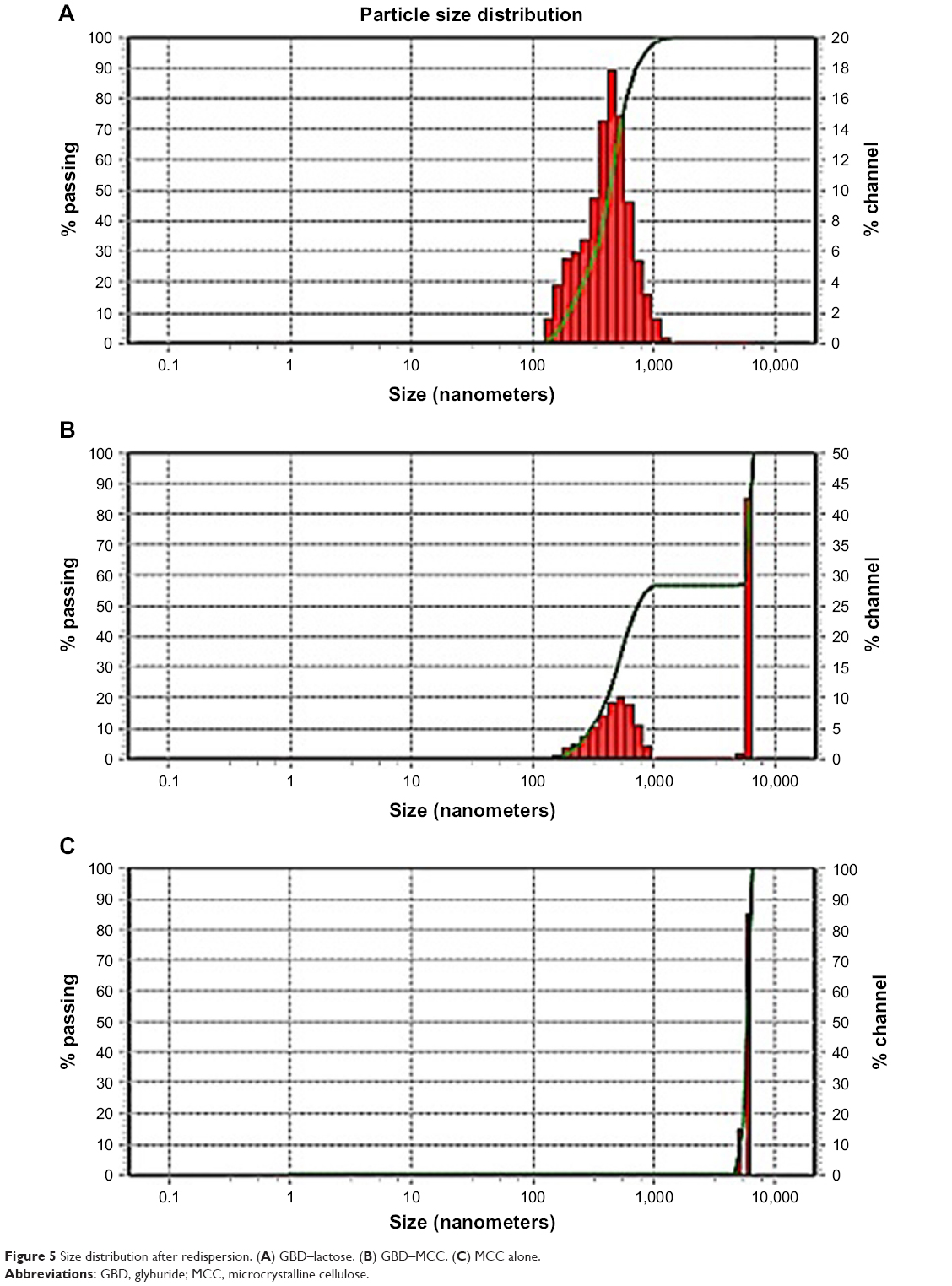 | Figure 5 Size distribution after redispersion. (A) GBD–lactose. (B) GBD–MCC. (C) MCC alone. |
Crystallinity assessment
The PXRD patterns of GBD, MCC, lactose, and GBD-loaded carriers were depicted in Figure 6. In accordance with recorded data, GBD displayed characteristic diffraction peaks at 2θ values of 10.8°, 11.5°, 19.2°, 19.6°, 21.6° and 23.7°.42 The PXRD of anhydrous lactose showed sharp diffraction peaks at 2θ of 11.30°, 18.38°, 21.62°, and 24.02°, indicating a highly crystalline compound,43 whereas MCC PH 101 showed a characteristic peak at diffraction angle of 2θ at 22.0°. The GBD–MCC or GBD–lactose PXRDs were very similar to their corresponding plain carriers and none of characteristic diffraction peaks of GBD can be clearly recorded. This might be due to the dilution effect of drug in used carriers. Furthermore, there is a possibility for a decrease in crystallinity to take place during the manufacturing process. Further assessment by DSC analysis was required to explain alterations in drug crystallinity during the processing steps.
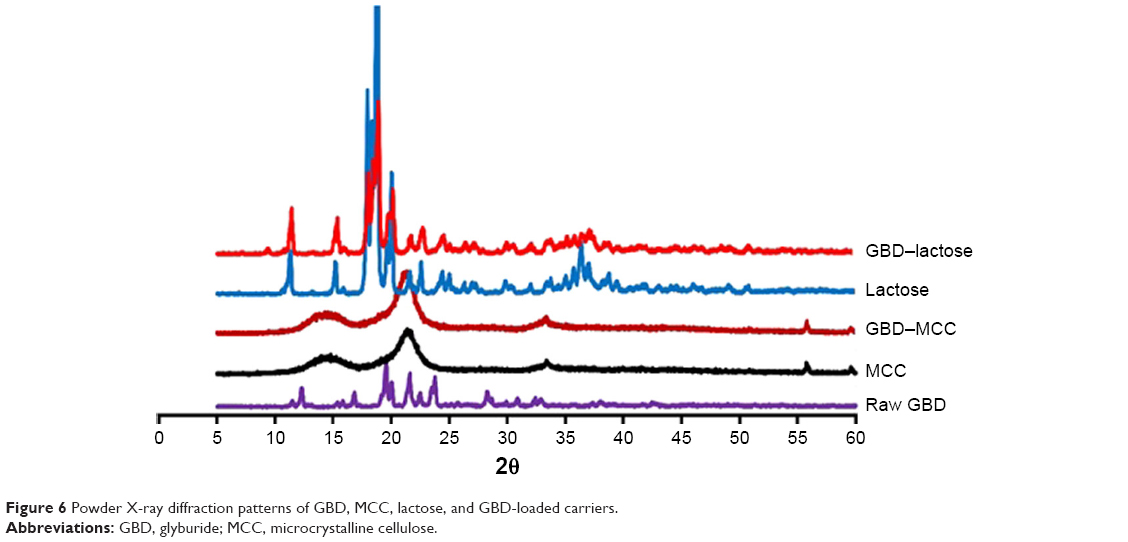 | Figure 6 Powder X-ray diffraction patterns of GBD, MCC, lactose, and GBD-loaded carriers. |
DSC thermograms of raw GBD, GBD-loaded carriers, and blank carriers are shown in Figure 7. In accordance to a previously reported analysis, the DSC thermogram of raw GBD exhibited a single sharp endothermic melting peak at 177°C indicating crystallinity of the drug.20 DSC thermogram of MCC PH101 showed a shallow broad endotherm in the scanned region between 60°C and 145°C, which corresponds to loss of adsorbed moisture.44 In GBD-loaded MCC, the GBD melting peak was detected slightly earlier to the unprocessed, 166.7°C. Such a slight shift is attributed to particle size reduction.29,45 Due to the presence of water molecules in lactose monohydrate crystals, a dehydration endothermic peak at 148°C was observed. Following this, an endothermic melting peak at 217°C was observed. These observations were in close agreement with published studies.46,47 The GBD melting peak was also observed in the GBD-loaded lactose thermogram, simultaneously with the characteristic peaks of lactose monohydrate. In conclusion, DSC results confirmed the crystalline state of GBD after nanonization and solidification processes.
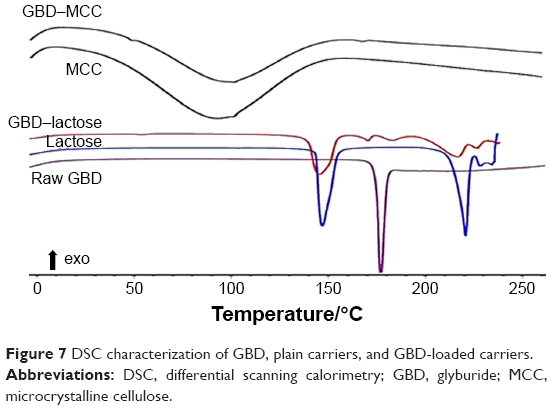 | Figure 7 DSC characterization of GBD, plain carriers, and GBD-loaded carriers. |
Flowability
The extra cohesion displayed by very fine particles has hindered powder flowability and thus challenged their industrial applicability. However, loading such particles onto carriers could enhance, to a certain degree, the flow of solidified nanosized particles and simplify further manufacturing. In this study, flowability of the loaded carrier powders was characterized using HR and CI. As shown in Table 2, lactose particles exhibited poor flow property with CI and HR of 37.5% and 1.60, respectively. Whereas MCC particles showed a slightly improved flowability with CI and HR of 26.47% and 1.36, respectively. The difference in particle size between lactose and MCC is expected to be the main cause of variation of their flowability patterns. In the case of GBD-loaded carriers, the lower values of CI and HR have shown powder flow improvement (see Table 2). The enhanced flowability of GBD-loaded carriers was a result of the granulating effect, which has occurred during the downstream processing. Powder flowability could be enhanced further by proper selection of an additional glidant in the optimized formulations.
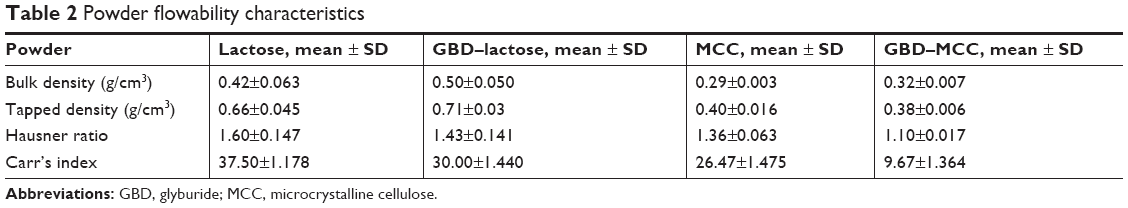 | Table 2 Powder flowability characteristics |
Tablet characteristics
Another advantage offered by solid carriers is the improved flowability of drug-loaded powders. Nanoparticles alone are well known for their enhanced adhesiveness. However, mixing nanoparticles with carriers is expected to enhance their flowability. Finally formed GBD-loaded powders showed residual moisture content <2.5%, which demonstrates that water in the drug suspensions was mostly removed during the solidification processes. The resultant GBD-loaded powders were blended with selected excipients and further processed into tablet solid dosage forms (Table 1). As mentioned, the ionic steric stabilizer poloxamer 188 was used, because of its compatibility with other ingredients, irrespective of their charge. Produced tablets were evaluated for hardness, friability, drug content, and disintegration time (see Table 3). Results show all parameters to be within pharmacopoeial limits. All tested tablets showed friability values <1% and hardness ranging between 4 and 6 kg/cm2.
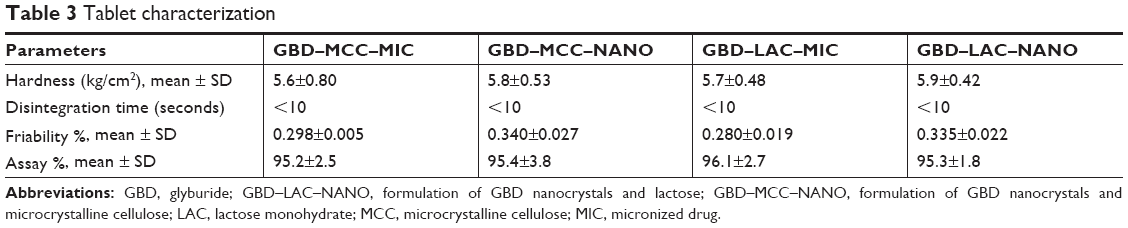 | Table 3 Tablet characterization |
Dissolution
One of the main challenges during processing into solid dosage forms is to preserve the favorable characteristics of the generated nanosized particles. Drug nanocrystals should retain their initial properties with minimal aggregation upon release from the solidified formulations.48 Accordingly, maintaining a high degree of dispersibility in solution by suitable dispersing agents (eg, surfactants, sugars, and/or disintegrants) is a crucial factor for efficient utilization of nanoparticles in tablets.49 Figure 8 demonstrates the in vitro dissolution profiles of different GBD formulations in comparison to unprocessed GBD powder. In general, GBD showed higher dissolution rates with the water-soluble lactose carrier compared to those formulated with MCC-based powders. Higher dissolution rates were also observed by GBD nanocrystals formulations (GBD–MCC–NANO and GBD–LAC–NANO) relative to micro-based formulations (GBD–MCC–MIC and GBD–LAC–MIC). During the first 10 minutes of dissolution, 56% was dissolved from GBD–LAC–NANO compared to 33% from GBD–LAC–MIC. On the other hand, the percentages dissolved within 10 minutes were 34% and 19% for GBD–MCC–NANO and GBD–MCC–MIC, respectively. The crystallinity assessment studies verified preservation of the GBD crystalline state after processing and the enhancement of dissolution rate was mostly due to the reduction of GBD particles rather than generating the drug in its amorphous state. Rapid dissolution is an evident benefit in drug therapy, while maintenance of the initial crystalline state is also an advantage for long-term stability.
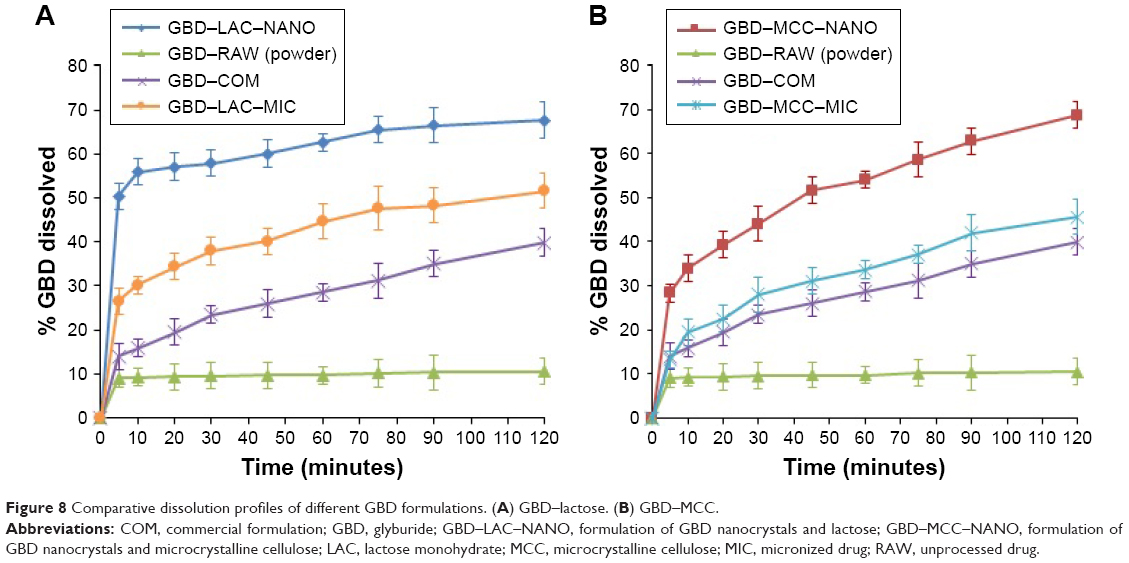 | Figure 8 Comparative dissolution profiles of different GBD formulations. (A) GBD–lactose. (B) GBD–MCC. |
Bioavailability
The plasma drug concentration–time curves after oral administration of GBD formulations to experimental rats are presented in Figure 9. Pharmacokinetic parameters including maximum peak concentration of the drug in plasma (Cmax), the time to reach maximum concentration (tmax), and AUC for tested formulations were shown in Table 4. Cmax values for GBD–MCC–NANO and GBD–LAC–NANO were 2.03±0.06 and 2.21±0.18 μg/mL, respectively. Both of these values were significantly higher (P<0.05) than that of GBD–COM, 0.39±0.10. The absorption rate (Tmax) was 2 hours for GBD–COM; however, it was reached at 1 hour in the case of GBD–MCC–NANO and GBD–LAC–NANO. Results of Cmax and Tmax confirmed fast absorption of GBD into blood, following rapid dissolution in the gastrointestinal tract. AUC0–24 hours values of GBD were 4.58±0.62, 9.05±0.41, and 10.27±0.50 for GBD–COM, GBD–MCC–NANO, and GBD–LAC–NANO, respectively. Correspondingly, the relative bioavailability values were 100.00%, 197.60%, and 224.23% confirming enhancement of GBD bioavailability. Here, nanosized drug particles are supposed to dissolve faster in gastrointestinal tract (GIT) than large GBD counterparts. In addition, small-sized particle of 300 nm displayed superior transport by enterocytes and M cells and systemic biodistribution than larger-sized particle sizes, 600 and 1,000 nm.50 Interestingly, detectable amounts of GBD were recorded after 24 hours for GBD nanoformulations. Previous studies reported that small nanoparticles could penetrate deeper into the gastrointestinal mucous gel layer in rats and sustain the drug in the body for a longer time.36,51 Overall findings of bioavailability studies confirmed the success of the nanosizing-solidification technique to improve bioavailability of GBD. Particle size reduction together with distribution of nanosized particles on the carriers was effective to increase the surface area for dissolution enhancement and thereby improving the bioavailability of poor water-soluble drugs. However, aggregation of nanoparticles in GIT may occur and thus result in a decreased effective surface area. Stabilizers are also beneficial for in vivo pharmacokinetic behaviors of nanosized drugs. Ionic stabilizers may not be sufficient to stabilize drug nanocrystals alone, as they may induce aggregation and agglomeration of drug crystals when passing through the gastrointestinal tract, experiencing the different pH levels.52 In the current study, presence of poloxamer, a nonionic stabilizer, minimizes particle aggregation and enhances wettability and facilitates the GBD dissolution process. Furthermore, nanoparticles were proven to possess higher adhesiveness to absorption tissues in the GIT. An increase in adhesion surface area between nanoparticles and intestinal epithelium of villi provides a direct contact with the absorbing membranes of the gut and immediate release of drug, which makes it readily available at absorption sites.53
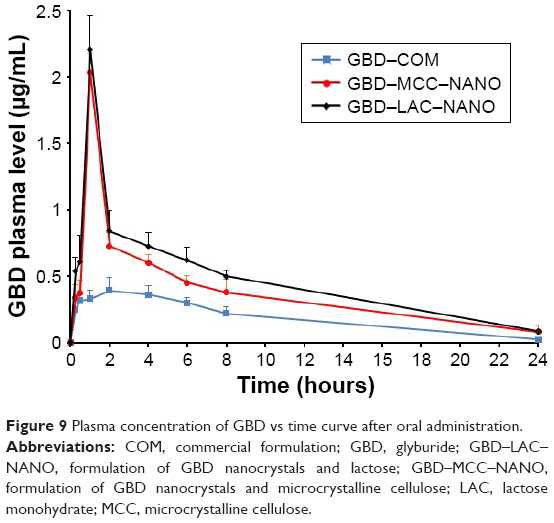 | Figure 9 Plasma concentration of GBD vs time curve after oral administration. |
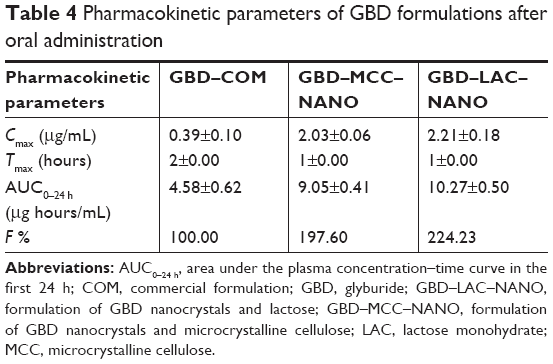 | Table 4 Pharmacokinetic parameters of GBD formulations after oral administration |
Conclusion
In this study, spraying of nanosuspension onto hot powder beds was examined to generate solid systems of nanosized particles. This technique was successfully employed to incorporate nanoparticles of glyburide into solid tablet formulations. Although it is a fast, single-step process when compared to the commonly used lyopholization procedures, scalability as a downstream process needs further examinations. Tablets obtained with nanoglyburide-loaded tablets showed faster dissolution and improved bioavailability, reflected by greater AUC values, when compared to a commercial formulation.
Acknowledgment
The authors would like to acknowledge Taibah University for providing support and facilities to carry out this study.
Disclosure
Ahmed F Hanafy is employed by Al Andalous Pharmaceutical Industries. The authors report no other conflicts of interest in this work.
References
Fu Q, Li B, Zhang D, et al. Comparative studies of the in vitro dissolution and in vivo pharmacokinetics for different formulation strategies (solid dispersion, micronization, and nanocrystals) for poorly water-soluble drugs: a case study for lacidipine. Colloids Surf B: Biointerf. 2015;132(Supplement C):171–176. | ||
Jinno J, Kamada N, Miyake M, et al. Effect of particle size reduction on dissolution and oral absorption of a poorly water-soluble drug, cilostazol, in beagle dogs. J Control Release. 2006;111(1–2):56–64. | ||
Salazar J, Ghanem A, Müller RH, Möschwitzer JP. Nanocrystals: comparison of the size reduction effectiveness of a novel combinative method with conventional top-down approaches. Eur J Pharm Biopharm. 2012;81(1):82–90. | ||
Junghanns JU, Müller RH. Nanocrystal technology, drug delivery and clinical applications. Int J Nanomed. 2008;3(3):295–310. | ||
Assadpour E, Jafari SM. A systematic review on nanoencapsulation of food bioactive ingredients and nutraceuticals by various nanocarriers. Crit Rev Food Sci Nutr. 2018;6(3):1–23. | ||
Gadadare R, Mandpe L, Pokharkar V. Ultra rapidly dissolving repaglinide nanosized crystals prepared via bottom-up and top-down approach: influence of food on pharmacokinetics behavior. AAPS PharmSciTech. 2015;16(4):787–799. | ||
Romero GB, Brysch W, Keck CM, Müller RH. Nanocapsule formation by nanocrystals. In: Jafari SM, editor. Nanoencapsulation Technologies for the Food and Nutraceutical Industries. Cambridge, MA: Academic Press; 2017:165–186. | ||
Raghava Srivalli KM, Mishra B. Drug nanocrystals: a way toward scale-up. Saudi Pharm J. 2016;24(4):386–404. | ||
Müller RH, Gohla S, Keck CM. State of the art of nanocrystals – special features, production, nanotoxicology aspects and intracellular delivery. Eur J Pharm Biopharm. 2011;78(1):1–9. | ||
Müller RH, Shegokar R, Keck CM. 20 years of lipid nanoparticles (SLN and NLC): present state of development and industrial applications. Curr Drug Discov Technol. 2011;8(3):207–227. | ||
Pawar VK, Singh Y, Meher JG, Gupta S, Chourasia MK. Engineered nanocrystal technology: in-vivo fate, targeting and applications in drug delivery. J Control Release. 2014;183(Supplement C):51–66. | ||
Wu L, Zhang J, Watanabe W. Physical and chemical stability of drug nanoparticles. Adv Drug Deliv Rev. 2011;63(6):456–469. | ||
D’Addio SM, Prud’homme RK. Controlling drug nanoparticle formation by rapid precipitation. Adv Drug Deliv Rev. 2011;63(6):417–426. | ||
Rabinow BE. Nanosuspensions in drug delivery. Nat Rev Drug Discov. 2004;3(9):785–796. | ||
Arpagaus C, Collenberg A, Rütti D, Assadpour E, Jafari SM. Nano spray drying for encapsulation of pharmaceuticals. Int J Pharm. 2018;546(1–2):194–214. | ||
Cal K, Sollohub K. Spray drying technique. I: hardware and process parameters. J Pharm Sci. 2010;99(2):575–586. | ||
Walters RH, Bhatnagar B, Tchessalov S, Izutsu KI, Tsumoto K, Ohtake S. Next generation drying technologies for pharmaceutical applications. J Pharm Sci. 2014;103(9):2673–2695. | ||
He W, Lu Y, Qi J, Chen L, Yin L, Wu W. Formulating food protein-stabilized indomethacin nanosuspensions into pellets by fluid-bed coating technology: physical characterization, redispersibility, and dissolution. Int J Nanomed. 2013;8:3119–3128. | ||
Tuomela A, Laaksonen T, Laru J, et al. Solid formulations by a nanocrystal approach: critical process parameters regarding scale-ability of nanocrystals for tableting applications. Int J Pharm. 2015;485(1–2):77–86. | ||
Yu L, Li C, Le Y, Chen J-F, Zou H. Stabilized amorphous glibenclamide nanoparticles by high-gravity technique. Mater Chem Phys. 2011;130(1–2):361–366. | ||
Guan J, Han J, Zhang D, et al. Increased dissolution rate and oral bioavailability of hydrophobic drug glyburide tablets produced using supercritical CO2 silica dispersion technology. Eur J Pharm Biopharm. 2014;86(3):376–382. | ||
Wei H, Löbenberg R. Biorelevant dissolution media as a predictive tool for glyburide a class II drug. Eur J Pharm Sci. 2006;29(1):45–52. | ||
Liu H, Shang K, Liu W, et al. Improved oral bioavailability of glyburide by a self-nanoemulsifying drug delivery system. J Microencapsul. 2014;31(3):277–283. | ||
Betageri G, Makarla KR. Enhancement of dissolution of glyburide by solid dispersion and lyophilization techniques. Int J Pharm. 1995;126(1–2):155–160. | ||
Savolainen J, Järvinen K, Taipale H, Jarho P, Loftsson T, Järvinen T. Co-administration of a water-soluble polymer increases the usefulness of cyclodextrins in solid oral dosage forms. Pharm Res. 1998;15(11):1696–1701. | ||
Nnamani PO, Attama AA, Ibezim EC, Adikwu MU. SRMS142-based solid lipid microparticles: application in oral delivery of glibenclamide to diabetic rats. Eur J Pharm Biopharm. 2010;76(1):68–74. | ||
Shah SR, Parikh RH, Chavda JR, Sheth NR. Application of Plackett–Burman screening design for preparing glibenclamide nanoparticles for dissolution enhancement. Powder Technol. 2013;235(Supplement C):405–411. | ||
Gonçalves LM, Maestrelli F, di Cesare Mannelli L, Ghelardini C, Almeida AJ, Mura P. Development of solid lipid nanoparticles as carriers for improving oral bioavailability of glibenclamide. Eur J Pharm Biopharm. 2016;102(Supplement C):41–50. | ||
Ali HSM, Hanafy AF. Glibenclamide nanocrystals in a biodegradable chitosan patch for transdermal delivery: engineering, formulation, and evaluation. J Pharm Sci. 2017;106(1):402–410. | ||
Zhang X, Guan J, Ni R, Li LC, Mao S. Preparation and solidification of redispersible nanosuspensions. J Pharm Sci. 2014;103(7):2166–2176. | ||
Yue PF, Wan J, Wang Y, et al. D-alpha-tocopherol acid polyethylene glycol 1000 succinate, an effective stabilizer during solidification transformation of baicalin nanosuspensions. Int J Pharm. 2013;443(1–2):279–287. | ||
Osamura T, Takeuchi Y, Onodera R, et al. Formulation design of granules prepared by wet granulation method using a multi-functional single-punch tablet press to avoid tableting failures. Asian J Pharm Sci. 2018;13(2):113–119. | ||
Jaipakdee N, Limpongsa E, Sripanidkulchai BO, Piyachaturawat P. Preparation of Curcuma comosa tablets using liquisolid techniques: in vitro and in vivo evaluation. Int J Pharm. 2018;553(1–2):157–168. | ||
Salazar J, Müller RH, Möschwitzer JP. Application of the combinative particle size reduction technology H 42 to produce fast dissolving glibenclamide tablets. Eur J Pharm Sci. 2013;49(4):565–577. | ||
Sallam NM, Sanad RA, Kharshoum RM, Zineldin MA. Development of salbutamol sulphate fast disintegrating sublingual tablets with enhanced bioavailability and improved clinical efficacy for potential treatment of asthma. J Drug Deliv Sci Technol. 2017;41:78–89. | ||
Yang W, Wang Y, Fu Q, et al. The role of particle size of glyburide crystals in improving its oral absorption. Drug Deliv Transl Res. 2017;7(3):428–438. | ||
Lucio D, Irache JM, Font M, Martínez-Ohárriz MC. Supramolecular structure of glibenclamide and β-cyclodextrins complexes. Int J Pharm. 2017;530(1–2):377–386. | ||
Elbahwy IA, Ibrahim HM, Ismael HR, Kasem AA. Enhancing bioavailability and controlling the release of glibenclamide from optimized solid lipid nanoparticles. J Drug Deliv Sci Technol. 2017;38:78–89. | ||
Moghimi SM, Hunter AC. Poloxamers and poloxamines in nanoparticle engineering and experimental medicine. Trends Biotechnol. 2000;18(10):412–420. | ||
Liu P, Viitala T, Kartal-Hodzic A, et al. Interaction studies between indomethacin nanocrystals and PEO/PPO copolymer stabilizers. Pharm Res. 2015;32(2):628–639. | ||
Morakul B, Suksiriworapong J, Leanpolchareanchai J, Junyaprasert VB. Precipitation-lyophilization-homogenization (PLH) for preparation of clarithromycin nanocrystals: influencing factors on physicochemical properties and stability. Int J Pharm. 2013;457(1):187–196. | ||
Lucio D, Martínez-Ohárriz MC, Jaras G, et al. Optimization and evaluation of zein nanoparticles to improve the oral delivery of glibenclamide in vivo study using C. elegans. Eur J Pharm Biopharm. 2017;121:104–112. | ||
Lee HJ, Lee HG, Kwon YB, et al. The role of lactose carrier on the powder behavior and aerodynamic performance of bosentan microparticles for dry powder inhalation. Eur J Pharm Sci. 2018;117:279–289. | ||
Emami J, Kazemali MR. Design and in vitro evaluation of a novel controlled onset extended-release delivery system of metoprolol tartrate. Res Pharm Sci. 2016;11(1):81–92. | ||
Lai SL, Guo JY, Petrova V, Ramanath G, Allen LH. Size-dependent melting properties of small tin particles: nanocalorimetric measurements. Phys Rev Lett. 1996;77(1):99–102. | ||
Moolchandani V, Augsburger LL, Gupta A, Khan M, Langridge J, Hoag SW. Characterization and selection of suitable grades of lactose as functional fillers for capsule filling: Part 1. Drug Dev Ind Pharm. 2015;41(9):1452–1463. | ||
Fu Q, Li B, Zhang D, et al. Comparative studies of the in vitro dissolution and in vivo pharmacokinetics for different formulation strategies (solid dispersion, micronization, and nanocrystals) for poorly water-soluble drugs: a case study for lacidipine. Colloids Surf B Biointerfaces. 2015;132:171–176. | ||
Möschwitzer JP, Müller RH. Factors influencing the release kinetics of drug nanocrystal-loaded pellet formulations. Drug Dev Ind Pharm. 2013;39(5):762–769. | ||
Heng D, Ogawa K, Cutler DJ, et al. Pure drug nanoparticles in tablets: what are the dissolution limitations? J Nanopart Res. 2010;12(5):1743–1754. | ||
He C, Yin L, Tang C, Yin C. Size-dependent absorption mechanism of polymeric nanoparticles for oral delivery of protein drugs. Biomaterials. 2012;33(33):8569–8578. | ||
Baek IH, Kim JS, Ha ES, et al. Dissolution and oral absorption of pranlukast nanosuspensions stabilized by hydroxypropylmethyl cellulose. Int J Biol Macromol. 2014;67:53–57. | ||
Mu S, Li M, Guo M, et al. Spironolactone nanocrystals for oral administration: different pharmacokinetic performances induced by stabilizers. Colloids Surf B Biointerfaces. 2016;147:73–80. | ||
Jiang T, Han N, Zhao B, Xie Y, Wang S. Enhanced dissolution rate and oral bioavailability of simvastatin nanocrystal prepared by sonoprecipitation. Drug Dev Ind Pharm. 2012;38(10):1230–1239. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.