Back to Journals » International Journal of Nanomedicine » Volume 12
Engineered outer membrane vesicle is potent to elicit HPV16E7-specific cellular immunity in a mouse model of TC-1 graft tumor
Authors Wang S, Huang W, Li K, Yao Y ![]() , Yang X, Bai H, Sun W, Liu C
, Yang X, Bai H, Sun W, Liu C ![]() , Ma Y
, Ma Y ![]()
Received 5 June 2017
Accepted for publication 11 August 2017
Published 12 September 2017 Volume 2017:12 Pages 6813—6825
DOI https://doi.org/10.2147/IJN.S143264
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Dongwoo Khang
Shijie Wang,1–3,* Weiwei Huang,1–3,* Kui Li,1–3 Yufeng Yao,1–3 Xu Yang,1–3 Hongmei Bai,1–3 Wenjia Sun,1–3 Cunbao Liu,1–3 Yanbing Ma1–3
1Laboratory of Molecular Immunology, Institute of Medical Biology, Chinese Academy of Medical Sciences & Peking Union Medical College, 2Yunnan Key Laboratory of Vaccine Research & Development on Severe Infectious Diseases, 3Yunnan Engineering Research Center of Vaccine Research and Development on Severe Infectious Diseases, Kunming, People’s Republic of China
*These authors contributed equally to this work
Purpose: Currently, therapeutic tumor vaccines under development generally lack significant effects in human clinical trials. Exploring a powerful antigen delivery system is a potential approach to improve vaccine efficacy. We sought to explore engineered bacterial outer membrane vesicles (OMVs) as a new vaccine carrier for efficiently delivering tumor antigens and provoking robust antitumor immune responses.
Materials and methods: First, the tumoral antigen human papillomavirus type 16 early protein E7 (HPV16E7) was presented on Escherichia coli-derived OMVs by genetic engineering methods, acquiring the recombinant OMV vaccine. Second, the ability of recombinant OMVs delivering their components and the model antigen green fluorescent protein to antigen-presenting cells was investigated in the macrophage Raw264.7 cells and in bone marrow-derived dendritic cells in vitro. Third, it was evaluated in TC-1 graft tumor model in mice that the recombinant OMVs displaying HPV16E7 stimulated specific cellular immune response and intervened the growth of established tumor.
Results: E. coli DH5α-derived OMVs could be taken up rapidly by dendritic cells, for which vesicle structure has been proven to be important. OMVs significantly stimulated the expression of dendritic cellmaturation markers CD80, CD86, CD83 and CD40. The HPV16E7 was successfully embedded in engineered OMVs through gene recombinant techniques. Subcutaneous immunization with the engineered OMVs induced E7 antigen-specific cellular immune responses, as shown by the increased numbers of interferon-gamma-expressing splenocytes by enzyme-linked immunospot assay and interferon-gamma-expressing CD4+ and CD8+ cells by flow cytometry analyses. Furthermore, the growth of grafted TC-1 tumors in mice was significantly suppressed by therapeutic vaccination. The recombinant E7 proteins presented by OMVs were more potent than those mixed with wild-type OMVs or administered alone for inducing specific cellular immunity and suppressing tumor growth.
Conclusion: The results indicated that the nano-grade OMVs might be a useful vaccine platform for antigen delivery in cancer immunotherapy.
Keywords: outer membrane vesicles, dendritic cells, HPV immunotherapy
Introduction
Persistent infection with high-risk genotypes of human papillomavirus (HPV) is the pathogenic cause of cervical cancer, and it is also associated with some anogenital cancers as well as head and neck cancers.1,2 At the present, prophylactic HPV vaccines are commercially available and can prevent the infection of most prevalent high-risk HPVs; however, they do not exert therapeutic effects on established infections and associated lesions.3 Thus, a therapeutic vaccine is urgently needed. The HPV early protein E7 is considered one of the most potential antigen candidates for the development of a therapeutic vaccine because of its abundant and sustained expression in infected cells and tumor as well as its high conservation. E7-based vaccine candidates have been tested in HPV-associated animal models. A fusion protein combining E7, hepatitis B core antigen, and heat shock protein 65 (VR111) increased anti-HPV16 cellular immunity compared with E7 alone.4 E7 chemically coupled to a bacterial lipid moiety stimulated the immune responses toward the T-helper (Th)1 type and induced E7-specific cytotoxic lymphocyte (CTL) responses.5 The combination of the 2 fusion proteins PE(ÄIII)/E6 and PE(ÄIII)/E7 generated more potent antitumor effects than a single fusion protein.6 Nevertheless, there have been only a few protein-based vaccines that proceeded to the phase of human clinical trials, mainly attributed to weak immune responses. Recently, nanoparticles are emerging as a promising vaccine form. Based on the delivery platform, immune responses can be modulated by modifying the stability, tissue and cell targeting, and the dendritic cell (DC) activation of antigen and adjuvant, and they can improve the microenvironment to facilitate the development of anti-tumor immunity. Largely, E7 peptides, instead of whole E7 proteins, have been carried by a variety of nanoparticles such as recombinant virus-like particles7–11 or artificial particles,12 which are limited by the compatibility of the particles. A good prospect would be a long peptide or even the full-length E7 protein, which holds more epitopes, and other novel nanoparticle platforms thus need to be explored.
Outer membrane vesicles (OMVs) are naturally occurring proteoliposomes with a diameter of 20–250 nm that are released from Gram-negative bacteria by the budding of the outer membrane.13–16 The vesicles contain the bacterial outer membrane and periplasm components, including lipopolysaccharides, nucleotide acids, lipids, outer membrane proteins, periplasmic proteins, inner membrane proteins and cytoplasm proteins.13–17 OMVs were found to be highly immunogenic and elicit efficient immune responses, independent of additional adjuvants, against the bacterium from which they were isolated.16 However, their potential as a novel antigen delivery carrier has not been well studied. It has been shown that heterologous proteins can be genetically tethered to OMVs with a designated distribution. The bacterial hemolysin ClyA, which was fused to green fluorescent protein (GFP) and served as a leading polypeptide, was shown to be able to guide GFP to express on the surface of OMVs.18,19 While a twin arginine transporter anchor sequence was used, GFP was entrapped into the periplasmic space of the bacteria and thus located in the lumen of OMVs eventually.20 OMVs have been shown to contribute to the Th1-biased immune response.21 Whether OMVs are associated with an enhanced antigen-specific cellular immune response remains to be known.
In this study, the non pathogenic Escherichia coli DH5α strain was employed to prepare engineered OMVs displaying the HPV16 E7 protein. Utilizing the intrinsic immunological and structural features of OMVs, we sought to investigate whether the engineered OMVs could induce E7-specific cellular immunity and have significant therapeutic effects on established tumors in a mouse model of HPV infection, thus demonstrating the potential of using E. coli OMVs as a feasible antigen delivery platform for therapeutic tumor vaccines.
Materials and methods
Ethics statement
The animal experimental and welfare procedures were approved by the Ethics Committee of Animal Care and Welfare, Institute of Medical Biology, CAMS (Permit Number: SYXK (dian) 2010–0007), in accordance with the animal ethics guidelines of the Chinese National Health and Medical Research Council and the Office of Laboratory Animal Management of Yunnan Province, China. All efforts were made to minimize animal suffering.
Plasmid construction
The plasmid pThioHis A was purchased from Invitrogen, Inc. DNA fragments encoding for the HPV16E7 protein were synthesized commercially by Sangong, Inc. (Shanghai, China). The E7 gene was introduced into pThioHis A between BamH I and Sal I sites using gene recombinant techniques, allowing E7 to fuse at the C-terminus of thioredoxin (trx). The recombinant plasmid was transformed into competent E. coli DH5α cells.
Expression of E7 and the preparation of OMVs and the trx-E7 fusion protein
The recombinant DH5α cells were grown in Luria-Bertani medium until the OD600 value reached 0.4–0.6, and then the expression of trx-E7 was induced by adding isopropyl β-D-1-thiogalactopyranoside (IPTG) to 0.001 mol/L. OMVs were purified in accordance with a previously established procedure.22 Briefly, 16 h after induction, the cell-free culture supernatants were collected by centrifugation at 14,000× g and 4°C for 20 min and then filtered through a 0.45-μm filter to clarify. The bacterial cells were preserved at −80°C for protein purification. The filtered supernatants were concentrated by ultrafiltration with a 500,000-nominal molecular weight cutoff (500,000 NMWC) column (QuixStand Benchtop System, GE Healthcare, Piscataway, NJ, USA). The vesicles were further collected by ultracentrifugation at 200,000× g and 4°C for 4 h (Beckman Coulter; TiSW28 rotor) and resuspended in PBS (0.02 M, 0.15 M NaCl, pH 7.2).
The gene encoding the fusion protein of trx and HPV16 E7 (trx-E7) was cloned into pThioHIsA between BamH I and Sal I. Its expression was induced with IPTG in DH5α cells, and the recombinant protein was purified by Ni2+-chelating chromatography. The amount of purified fusion protein and OMVs was identified using the total protein concentration, which was measured by the bicinchoninic acid method.
Immunoblotting analysis
OMV samples were loaded and separated on 12% sodium dodecyl sulfate (SDS)-polyacrylamide gels. The proteins were electrophoretically transferred to a polyvinylidene fluoride membrane (45 min, 20 V). The membranes were blocked with 5% skim milk in 0.02 M Tris HCl (pH 7.8) containing 0.036 M SDS and then incubated with a 1:200 dilution of the primary antibodies (goat anti-HPV16 E7 antibody; Santa Cruz Biotechnology Inc.), followed by treatment with horse reddish peroxidase-conjugated donkey anti-goat IgG antibodies (1:4,000; Santa Cruz Biotechnology Inc.). The blots were developed by the addition of an enhanced chemiluminescence substrate (Thermo Fisher Scientific).
Bone marrow-derived dendritic cells (BMDCs) isolation and culture
BMDCs were isolated from murine according to a procedure described by Inaba et al23 with minor modifications. In brief, the bone marrow was flushed from the tibias and femurs of 4- to 5-week-old male C57BL/6 mice and depleted of red blood cells (RBCs) using RBC Lysis Buffer (Sigma-Aldrich). The cells were plated in 6-well culture plates (1×106 cells per mL; 3 mL per well) in Roswell Park Memorial Institute (RPMI) 1640 supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, 100 mg/mL streptomycin and 20 ng/mL recombinant mouse granulocyte-macrophage colony-stimulating factor at 37°C in 5% CO2. On days 2 and 4 of culturing, the floating cells were gently removed, and fresh medium was added. On day 6 of culture, non-adherent cells and loosely adherent proliferating DC aggregates were harvested for analysis or stimulation.
GFP delivery into RAW 264.7 cells by OMVs
To determine if the mimetic antigen could be delivered successfully into RAW 264.7 cells and if OMVs could be taken up by RAW 264.7 cells, OMVs displaying GFP (GFP-OMVs) and Dio-labeled OMVs were used. RAW 264.7 cells are macrophages derived from Abelson murine ascites, and they were purchased from the American Type Culture Collection. Wild-type OMVs (wt OMVs, 1 mg OMV protein/mL) were labeled with 1% (v/v) fluorescence dye Dio (Molecular Probes) at 37°C for 20 min. They were then centrifuged at 5,000× g and washed with PBS for 3 times in a 100-kDa ultrafiltration centrifuge tube. A total of 1 mL of 1×106/mL cells per well was cultured in a 6-well plate. Engineered OMVs 0, 50 or 100 μg presenting GFP (GFP-OMVs) were added into the plate with 6 wells for each concentration. After 4 h, the cells were washed with PBS for 3 times (5 min each time). For each OMV concentration, 3 wells of cells were examined using a fluorescent microscope, and the other 3 wells of cells were digested with 0.25% trypsin and analyzed by flow cytometry.
Flow cytometry analysis of OMVs uptake by DCs and their maturation
1×106/mL DCs were incubated with 50 μg/mL recombinant OMVs presenting E7 (trx-E7 OMVs), wt OMVs or recombinant trx-E7 fusion protein for 24 h. The cells were resuspended in flow cytometry staining buffer and stained with the anti-CD11c-PE, anti-CD80-fluorescein isothiocyanate (FITC), anti-CD86-FITC, anti-CD83-FITC or anti-CD40-FITC antibodies (eBioscience) for 30 min at 4°C in the dark. The expression levels of the maturation markers of DCs were analyzed using a flow cytometer.
To monitor the kinetics of the OMVs taken up by DCs and assess the importance of the OMV vesicular structure, different concentrations of FITC (Isomer I; Sigma-Aldrich)-labeled OMVs or ultrasonication-treated OMVs (vesicle structure was destroyed) were added to 1×105/mL DCs and incubated at 37°C. Cell samples were taken at 1 h and stained with a PE-labeled anti-CD11c antibody. The fluorescent intensities of the cells were measured using a FACScan flow cytometer (BD Accuri™ C6; BD Biosciences, Germany).
Cell lines and mice
TC-1 tumor cells, which were derived from primary lung epithelial cells from C57BL/6 mice co-transformed with the HPV-16 oncoproteins E6 and E7 and the c-Ha-ras oncogene, were purchased from the Tumor Center of Chinese Academy of Medical Sciences. The cells were cultured in RPMI 1640 supplemented with 10% FBS.
Female C57BL/6 mice (6–8 weeks; 16–18 g; SCXK [jing] 2012-0001) were purchased from Vital River Laboratory Animal Technology, Ltd. (Beijing, China). All mice were kept under specific pathogen-free conditions in the Central Animal Care Services of the Institute of Medical Biology (CAMS and PUMC).
OMVs immunization and tumor model
Mice were injected subcutaneously (sc) on the right flank with 1×105 TC-1 tumor cells per mouse. When the tumors reached 2–3 mm in diameter, the mice were assigned to 5 groups as follows to receive different treatments: 10 μg of trx-E7 OMVs, 10 μg of wt OMVs, 10 μg of wt OMVs mixed with 0.1 μg of trx-E7 protein, 0.1 μg of trx-E7 protein combined with Freund’s adjuvant and PBS as a control. Mice were injected sc 3 times at 7-day intervals.
The tumor volume was measured every other day starting on day 13. The mice were sacrificed when the largest tumor reached an ethical point. Spleens were collected for the isolation and preparation of splenocytes according to the instructions for the use of the lymphocyte separation medium used (Dakewe Biotech Co., Ltd., Shenzhen, China). In brief, the splenocytes were dispersed using 70-μm cell strainers and suspended in 5-mL lymphocyte separation medium, covered with 500 μL RPMI 1640 medium. They were then centrifuged at 800× g for 30 min. Then, the white cell layer was removed into a new tube. The cells were washed with fresh RPMI 1640 medium and then centrifuged at 500× g for 7 min. The final splenocytes were resuspended in complete RPMI 1640 culture medium for use. For flow cytometry analysis, the cells were stimulated with an HPV16 E7 peptide (amino acids 44–62) overnight or phorbol myristate acetate (PMA) and ionomycin for 5 h. Then, the cells were stained with the anti-CD8-PE and anti-interferon-gamma (IFNγ)- antigen-presenting cell (APC) or anti-CD4-FITC and anti-IFNγ-APC or anti-IL4-PE antibodies for analyzing T-cell immune responses with flow cytometry.
Measurement of antigen-specific IgG by enzyme-linked immunosorbent assay (ELISA)
After the mice were sacrificed, the sera were collected. Ninety-six well microplates were coated with 5 μg/mL E7 protein per well. Subsequently, the serum samples were added at various dilutions, and they were incubated with biotin-conjugated anti-mouse IgG at 1:10,000. Alkaline phosphatase-conjugated avidin was added, and the reaction was developed using the alkaline phosphatase substrate. The OD405 values were detected with an ELISA reader.
Antigen-specific cellular response measured by enzyme-linked immunospot (ELISPOT)
Splenocytes were obtained as described in the OMVs immunization and tumor model section. Splenocytes at 1×105 were separated and cultured in the presence of HPV16 E7 peptide (amino acids 44–62) at 10 μg/mL for 16–20 h. The cells stimulated with PMA and ionomycin were used as positive controls, and the cells incubated with medium only were used as negative controls. The number of antigen-specific IFNγ-producing cells was measured using an ELISPOT kit according to the manufacturer’s instructions (Dakewe Biotech Co., Ltd.). The spots were counted using an automated analyzer (AID, Germany).
Statistical analysis
The significance of differences between experimental groups was analyzed using one-way analysis of variance followed by Tukey’s multiple-comparisons test, or unpaired Student’s t-test (GraphPad Prism 5.0; GraphPad Software, Inc.). Values are reported as the mean ± SD.
Results
The HPV16 E7 protein was successfully presented in engineered E. coli-derived OMVs sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis showed that the HPV16 E7 protein was efficiently expressed in E. coli DH5α cells mainly as the soluble fusion protein trx-E7 (Figure 1A). Semiquantitative analysis of the SDS/PAGE gel-band densitometry indicated that trx-E7 was ~1% of the total protein content in OMVs. Furthermore, the presence of the trx-E7 fusion protein in both E. coli whole cells and engineered OMVs samples was confirmed by immunoblotting with the detection of E7-specific antibodies (Figure 1B). The result indicated that trx-E7 was introduced into OMVs through the guidance of trx, which has the capability of moving into the cell periplasm space. Additionally, EDTA treatment can destroy the intact vesicular structure and expose the proteins in the inner lumen of OMVs; however, this did not change the content of OMVs (Figure 1B). Using proteinase K (PK) and/or EDTA treatment, followed by immunoblotting with E7-specific antibodies, E7 was shown to be partly exposed on the surface and partly encapsulated in the lumen of the engineered OMVs (Figure 1B).
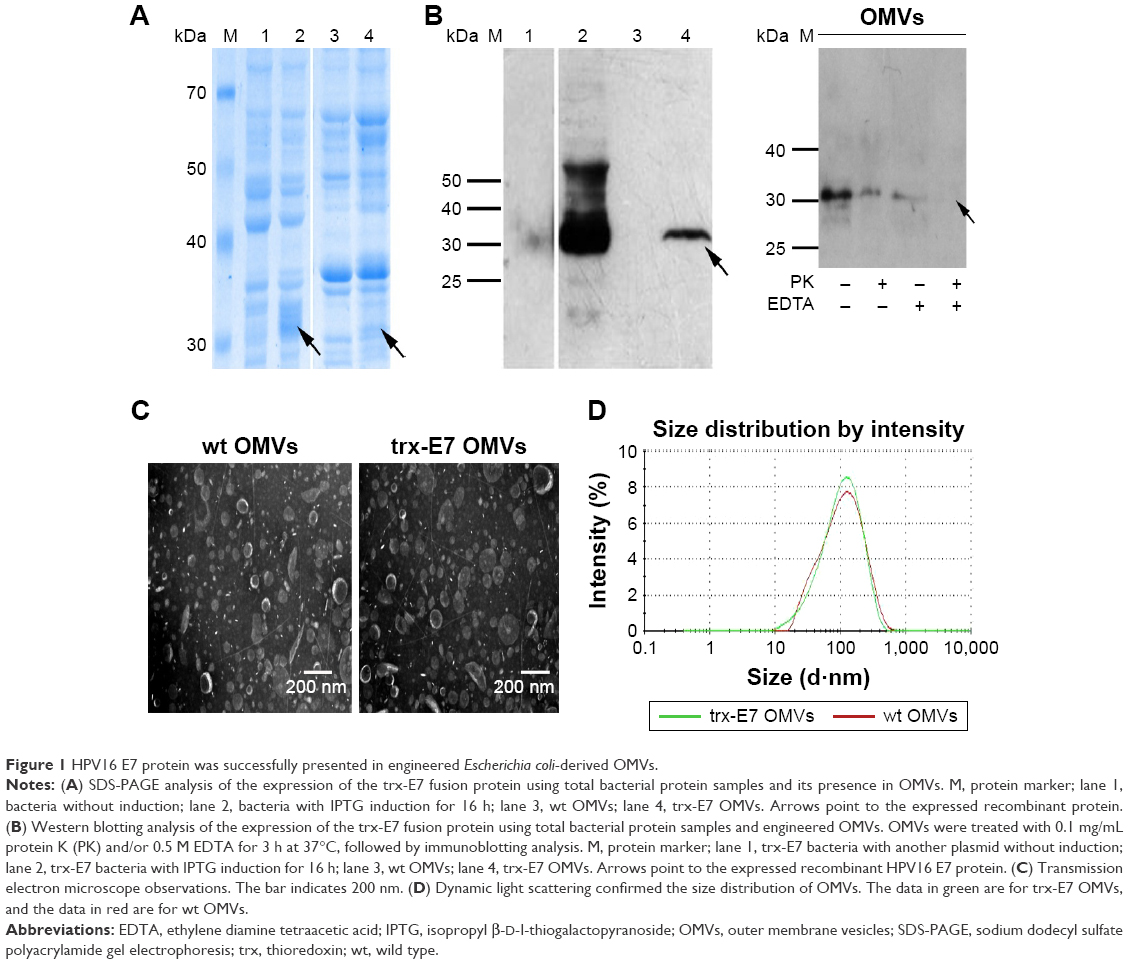 | Figure 1 HPV16 E7 protein was successfully presented in engineered Escherichia coli-derived OMVs. |
Observation under an electron microscope showed that the morphologies of the wt OMVs and trx-E7 OMVs were similar. They had a spherical vesicle structure, and the size ranged from 20 to 200 nm, with an average diameter of 100 nm (Figure 1C). Dynamic light scattering analysis confirmed the identical size distribution of wt OMVs and trx-E7 OMVs. For wt OMVs, the maximum peak values were ~131 nm, and the polydispersity index (PdI) was ~0.301. For trx-E7 OMVs, the maximum peak values were ~125 nm, and the PdI was ~0.305 (Figure 1D). This demonstrated that the morphology of the engineered OMVs was not altered by the incorporation of a heterologous protein.
Vesicle structure was important for OMVs uptake by DCs, and OMVs significantly induced DC maturation
Using GFP as a model antigen, engineered OMVs successfully delivered the antigen of interest into RAW 264.7 cells, shown by fluorescence microscope image and flow cytometry analysis (Figure 2A). As the concentration of OMVs increased, the fluorescence intensity of the cells and the ratio of positive cells augmented. Similarly, Dio-labeled OMVs exhibited fluorescence in cells, revealing that the OMVs themselves were internalized efficiently by RAW 264.7 cells (Figure 2A). DCs are the most powerful and professional APCs, as they play critical roles in determining both the immune response direction and intensity. To assess the characteristics of the uptake of OMVs by DCs, FITC-labeled OMVs and ultrasonication-treated OMVs, which lost vesicle structure, were incubated with BMDCs. As shown in Figure 2B, original OMVs have shown to be typical vesicles, while ultrasonication-treated OMVs displayed wizened and irregular morphology. The FITC-labeled proteins have a covalent bond that is not affected when we use ultrasonication to destroy the vesicle structure of OMV, and they were used as a control. The results showed that the numbers of the cells with fluorescence were up to 23.4% and 41.1% of the total cells treated with 2 and 10 μg/mL OMVs, respectively, at 60 min, while they were 5.07% and 18.5%, respectively, in cells treated with OMVs receiving ultrasonication treatment. This indicated that the vesicle structure of OMVs significantly enhanced the uptake efficacy of DCs (Figure 2B).
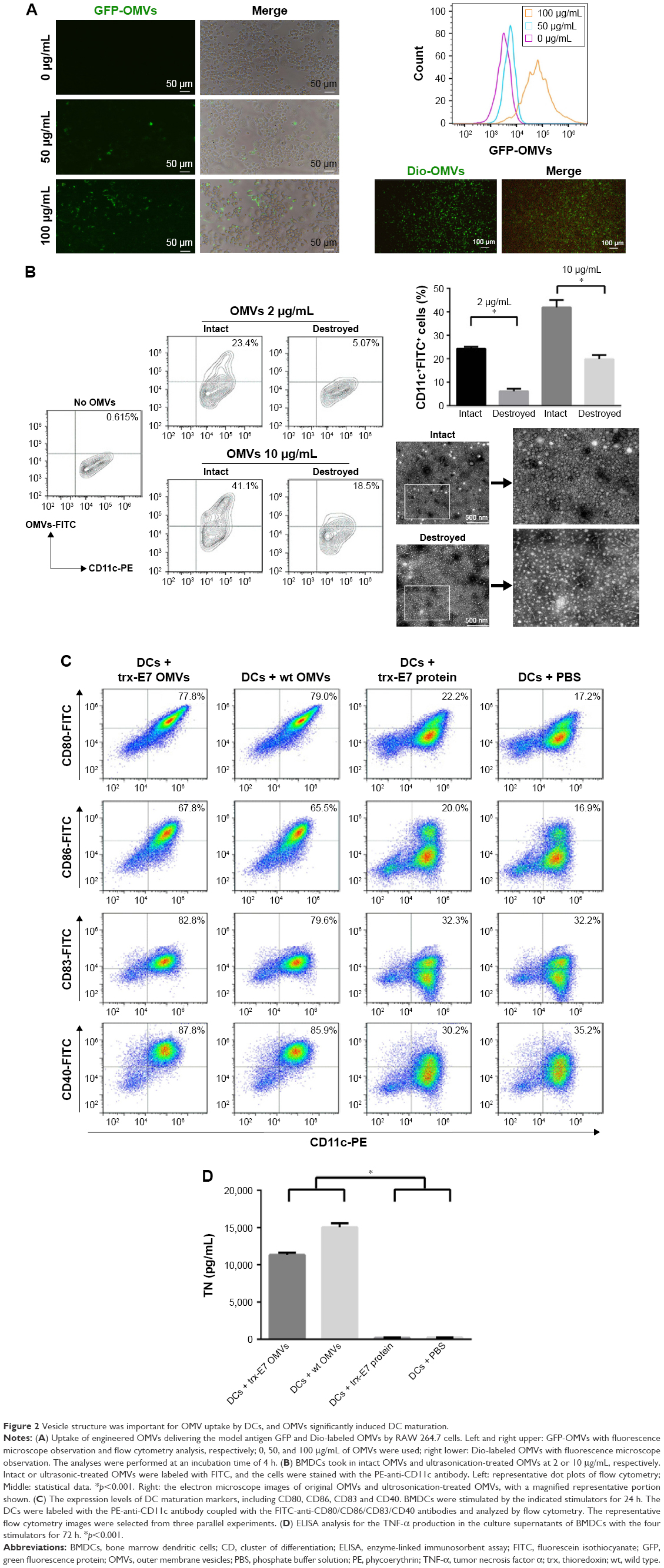 | Figure 2 Vesicle structure was important for OMV uptake by DCs, and OMVs significantly induced DC maturation. |
To assess the ability of OMVs to stimulate DC maturation, the expression levels of the surface molecules served as markers of DC maturation were measured in the cells incubated with trx-E7 OMVs, wt OMVs, trx-E7 protein and PBS. With or without trx-E7, OMVs induced the elevated expression of CD80, CD86, CD83 and CD40, which were dramatically higher than those induced by E7 protein alone and PBS (Figure 2C). Correspondingly, the production of tumor necrosis factor-alpha, a major tumor-killing cytokine, was shown to be stimulated significantly in OMVs-treated cells compared with that in trx-E7 protein and PBS-treated cells (Figure 2D).
Engineered OMVs presenting E7 induced a specific antitumor cellular immune response
Tumor antigen-specific cellular immune responses are predominant elements in antitumor immunotherapy. The level of IFNγ-secreting splenocytes detected by ELISPOT (Figure 3A) was significantly higher in trx-E7 OMV-vaccinated mice than in mice receiving trx-E7 protein mixed with OMVs, wt OMVs, or PBS (p<0.05). This indicated that trx-E7 OMVs were able to potently elicit an antigen-specific cellular immune response. Furthermore, E7-specific antibodies were detected by ELISA. The result showed that the specific antibodies were induced in trx-E7 OMVs-immunized mice, with a titer reaching 1×105 (Figure 3B), which is the reciprocal of the highest dilution of the sample in which the OD405 value was twice of that of the corresponding control serum. The above results indicated that engineered OMVs presenting a specific antigen were highly immunogenic and could efficiently induce both cellular and humoral immune responses.
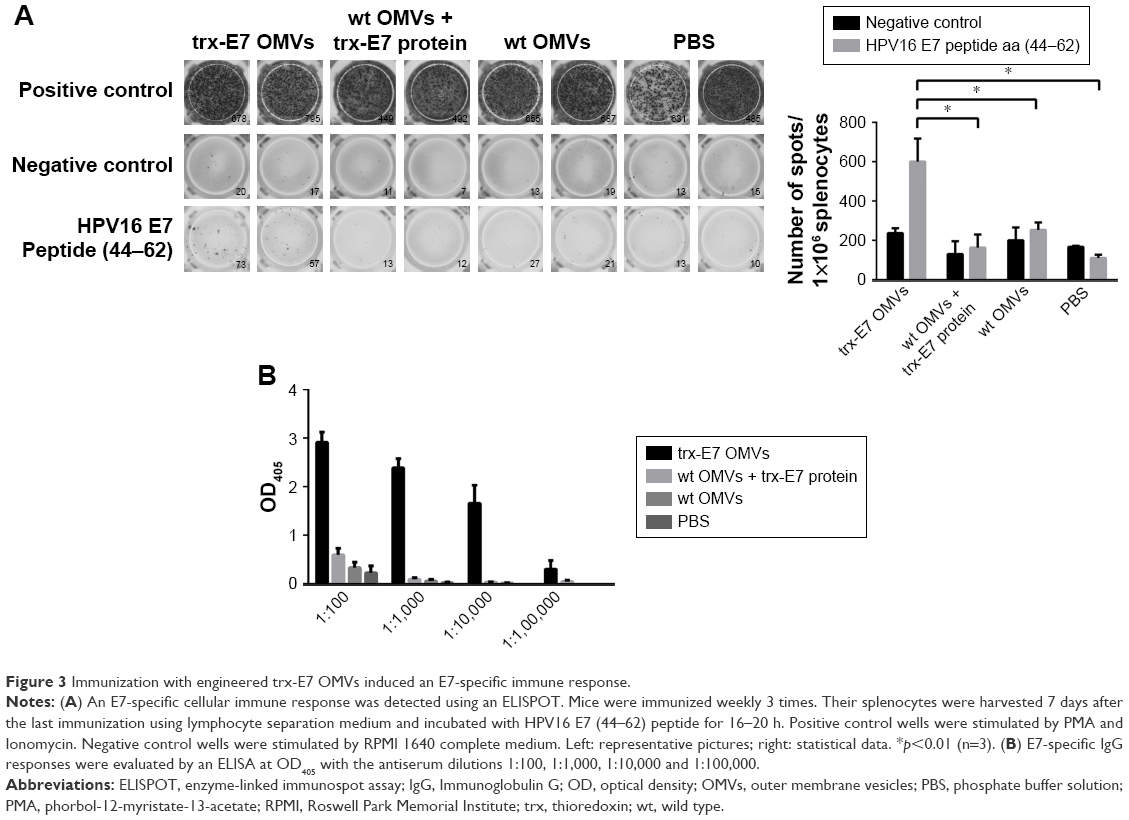 | Figure 3 Immunization with engineered trx-E7 OMVs induced an E7-specific immune response. |
Therapeutic immunization with OMVs presenting E7 inhibited TC-1 tumor growth
To investigate the therapeutic efficacy of the antitumor cellular immunity caused by the engineered OMVs, an HPV infection-associated tumor model and a treatment vaccination strategy were employed. TC-1 cells were inoculated into C57BL/6 mice first, and OMVs were immunized after the tumor was established (Figure 4A). The tumor growth trend (Figure 4B) and tumor weight at the endpoint of the experiment (Figure 4C) showed that the engineered OMVs presenting E7 suppressed tumor growth significantly comparing with the data from mice receiving wt OMVs, OMVs mixed with trx-E7 protein or trx-E7 protein along with Freund’s adjuvant. There were no apparent differences among the mice immunized with the trx-E7 protein mixed with OMVs or Freund’s adjuvant, and the mice served as model controls. The results indicated that the presence of a specific antigen E7 in OMVs, not simply adjuvanted by OMVs, or with Freund’s adjuvant were quite important for the efficient induction of antitumor immunity and suppression of tumor growth.
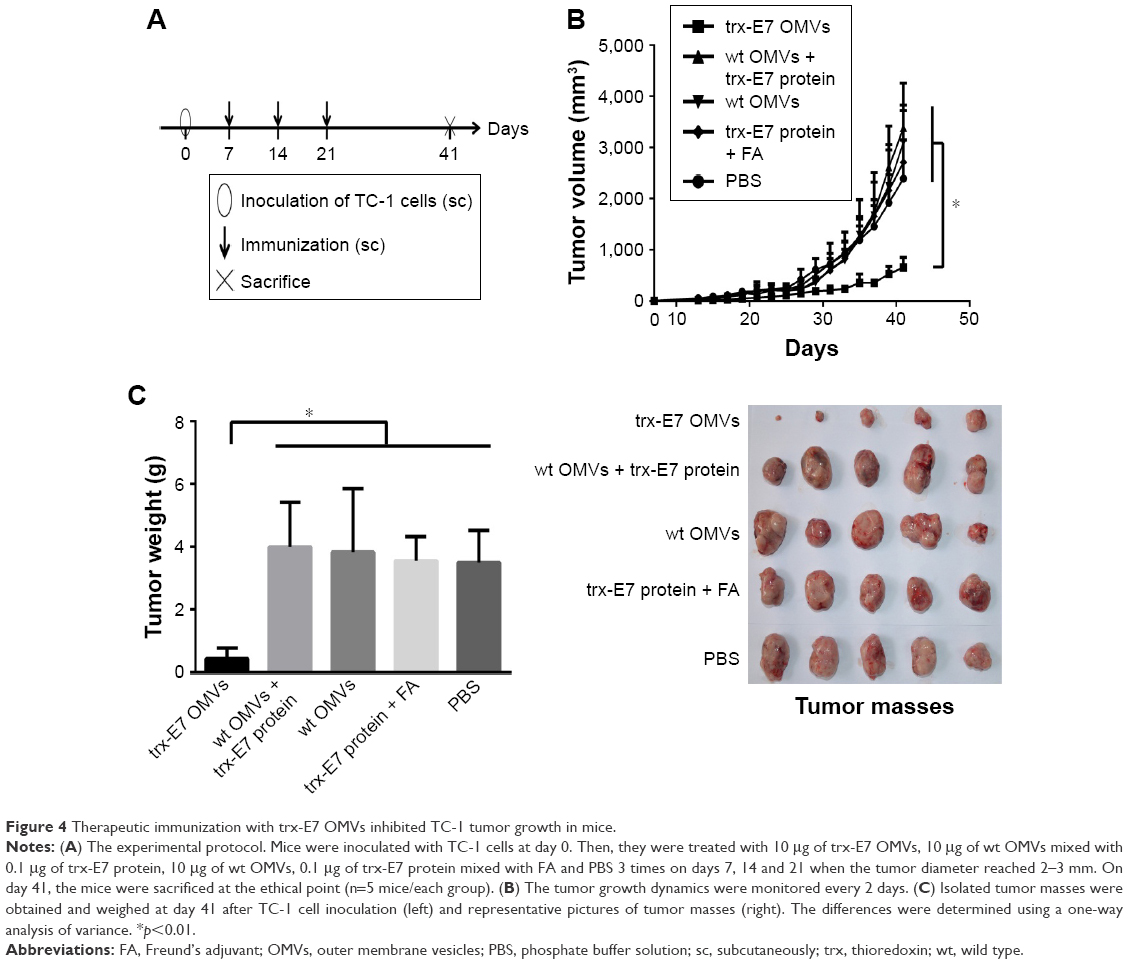 | Figure 4 Therapeutic immunization with trx-E7 OMVs inhibited TC-1 tumor growth in mice. |
Correspondingly, when analyzed by flow cytometry, the level of CD8+ IFNγ+ T cells in trx-E7 OMV-immunized mice was significantly elevated, while the level in mice receiving trx-E7 protein mixed with OMVs or Freund’s adjuvant remained at a similar level to the control mice (Figure 5A). Furthermore, it is known that CD4+ T cells can be polarized into Th1 cells functionally to assist in the production and maintenance of the key antitumor effector cells, CTLs, while Th2 cells may contribute to immunosuppressive microenvironments.24 To verify the spleen-derived Th-cell bias, in vitro-stimulated splenic lymphocytes were analyzed using flow cytometry, and the results showed that compared with the PBS-treated and trx-E7 protein-treated groups, the levels of IFNγ-expressing CD4+ T cells in trx-E7 OMV-immunized mice were significantly higher than those of the mice receiving wt OMVs, trx-E7 protein mixed with wt OMVs, trx-E7 protein mixed with Freund’s adjuvant or PBS (Figure 5B). The response of IL4-expressing CD4+ T cells showed no significant differences in all the groups (Figure 5C). Taken together, OMVs promoted T-cell responses to polarize the Th1/CTL subtypes.
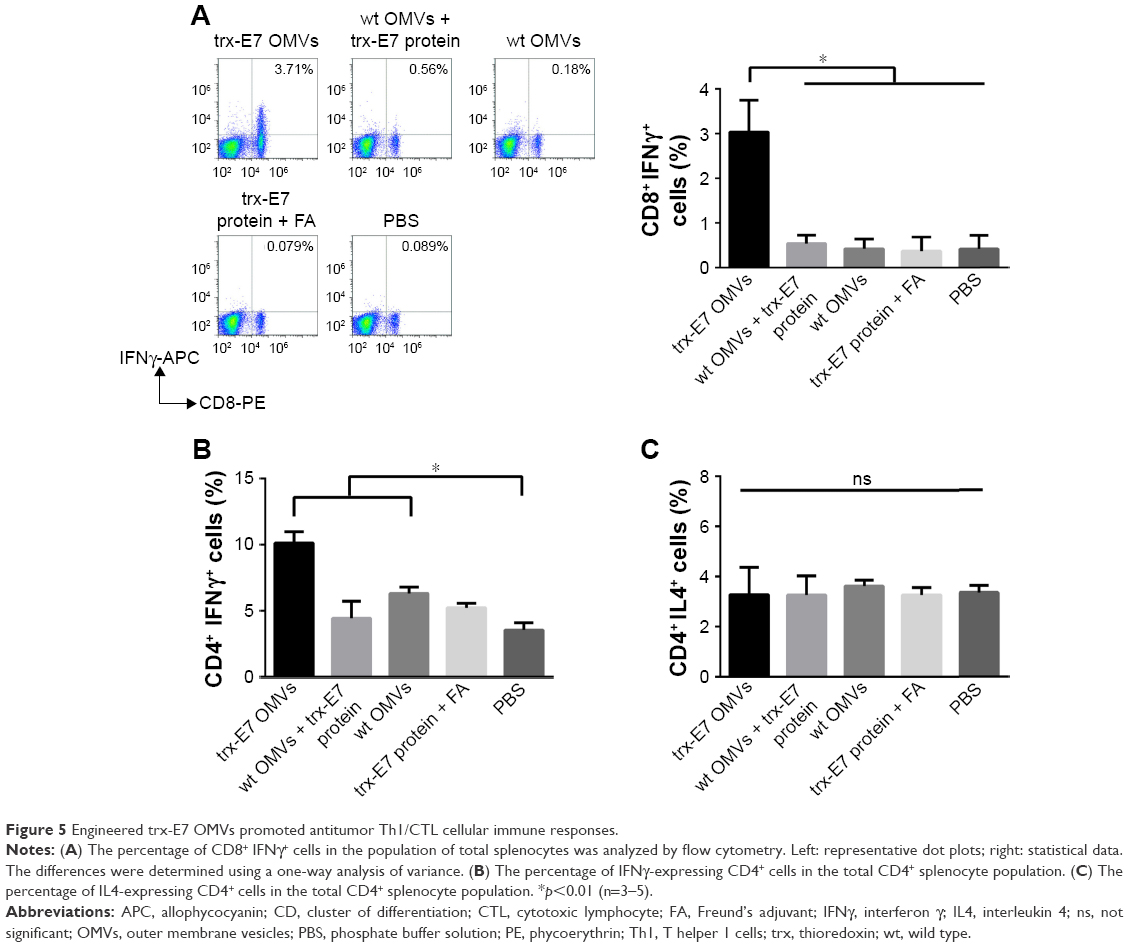 | Figure 5 Engineered trx-E7 OMVs promoted antitumor Th1/CTL cellular immune responses. |
Discussion
In this study, a novel nano-vesicle antigen delivery platform of E. coli-derived OMV was investigated to assess its potential in eliciting cellular immunity and developing a therapeutic cancer vaccine. Recently, OMVs have received increased attention as an emerging and feasible vaccine carrier25 because of their distinct immunological and structural features, including the nanometer-scale vesicle structure, self-adjuvant effectiveness, ability to be genetically modified, tolerance of presenting exogenous large proteins and ability to carry immune stimulators.25 However, the related studies are just in their infancy, especially research concerning their application potentials in stimulating specific cellular immunity. Using OMVs as a carrier, even large antigen proteins could be delivered, and more importantly, antigen and immune stimulators that enhance the intensity of immune responses and modulate the direction of the response could be delivered to the same APCs simultaneously and facilitate the production of a robust and proper type of antigen-specific immune response.
Trx-like proteins have a critical role in the redox biology of cells. The system is of particular significance in the periplasm of pathogenic bacteria, where disulfide bond formation is essential for both the motility and folding of a variety of toxins and secreted enzymes.26,27 Therefore, trx is widely used as a molecular chaperone for the efficient expression and proper folding of heterogenous proteins in the form of fusion proteins. In addition, trx has the ability to transport into the periplasm space. Thus, we deduced that trx ought to be used as a rational leader protein to mediate soluble antigen expression and translocation to the periplasm, allowing the further encapsulation into the lumen during OMV production. Our results confirmed that trx was an effective leading protein when preparing engineered OMVs, and SDS-PAGE and western blot analyses showed E7 was successfully enclosed in OMVs. We used a method described by Kim et al to determine the location of heterogeneous proteins on OMVs.19 When roughly analyzed with PK and/or EDTA treatment, combined with immuno-blotting analyses, we found that E7 was presented both on the surface and in the lumen of OMVs.
Some of the OMV components are pathogen-associated molecular patterns, which are recognized by the innate arm or the first defensive line of the immune system and sensed by pattern recognition receptors, such as Toll-like receptors, thus driving the inflammatory response in conjunction with complement system activation.28–30 It has been reported that the immunological characteristics of OMVs endow them with distinct capabilities to stimulate both innate and adaptive immunity in vitro and in vivo.31–35 In addition, OMVs have a bilayer biomembrane structure containing proteins, polysaccharides, and lipids, allowing multiple possible mechanisms to mediate the entry of OMVs into APCs, including receptor-mediated entrance, and it has been reported that OMVs can transport materials into cells through membrane fusion. GFP is a protein with structure-dependent fluorescent activity, and uptaken GFP is subjected to proteinase digestion in the endosome/lysosome. OMVs labeled with Dio, which is a small molecule dye, highly retained their fluorescence in a long-lasting pattern, as shown in Figure 2A. We isolated mouse BMDCs and compared the capacity of OMVs and non-vesicle OMV components (the vesicle structure was destroyed by ultrasonic treatment) to deliver antigens into DCs and stimulate DC maturation. Our results showed that OMVs were taken up by DCs more quickly than non-vesicle OMV, which strongly indicated the importance of the vesicle structure of engineered OMVs.
In this study, we used a vaccination treatment strategy to assess the efficacy of engineered OMVs on eliciting anti-tumor immunity. We found that the OMVs presenting E7, even without the use of a conventional adjuvant, significantly suppressed TC-1 growth, whereas recombinant E7 protein mixed with OMVs or Freund’ adjuvant did not show tumor suppression when compared with the model control and wt OMVs control. We also assessed E7-specific IgG, and again, the results confirmed that the presentation of E7 by OMVs was critical to induce specific antibody responses. We further analyzed IFNγ expressing lymphocytes in the spleen by ELISPOT and CD8+ cells and CD8+ IFNγ+ cells in the spleen by flow cytometry, and we found that they were significantly enhanced in engineered trx-E7 OMV-immunized mice. In addition, we found the level of CD4+ IFNγ+ cells were elevated, whereas that of CD4+ IL4+ cells in the spleen was not affected significantly, indicating trx-E7 OMV immunization induced Th1/CTL-biased responses. The enhanced Th1/CTLs responses in trx-E7 OMV-immunized mice were probably due to the increased uptake of E7, the elevated cross-presentation through vesicle delivery and the simultaneous delivery of immune stimulators and antigens in APCs. The results strongly indicated the importance of E7 to be carried by the vesicles rather than mixed with OMVs or a strong adjuvant, such Freund’s adjuvant, indicating that presence in the OMVs endows the high immunogenicity of the tumor antigen.
In this study, the engineered trx-E7 OMVs contained only ~1% of E7 out of the total OMV proteins, which is similar to the level reported by other groups (ranging from 0.32% to 0.8%).20,36 However, E7-specific immune responses were induced successfully, which strongly indicates the potent immunological effects of OMVs to serve as a vaccine carrier. There were some concerns raised about using OMVs as a conventional vaccine platform. First, immune responses directed to the carrier itself may affect the repeated use of a vaccine based on the same antigen delivery platform. It was reported that preexisting anti-carrier antibodies may mediate antibody-dependent phagocytosis and promote antigen clearance, thus reducing antigen uptake by APCs and weakening the subsequent immune responses. However, anti-carrier antibodies may not produce significant adverse effects, and there are also reports showing that preexisting antibodies enhanced immune responses by promoting the antigen uptake of APCs. Second, undesired responses to OMV components may excessively consume the responses of the immune system and thus attenuate the specific responses to the target antigen. Although we did not investigate how long the antibodies against the carrier and anti-carrier immune memory will persist and whether a preexisting antibody or how strongly the antibody responses will affect the repeated use of the vaccine, we clearly demonstrated that an OMV-based vaccine efficiently induced both specific humoral and cellular immune responses in the current study. It is also worth mentioning that even if preexisting anti-carrier antibodies may have adverse influences on the repeated use of OMV-based vaccines with different antigens, considering the unique features of OMVs as a vaccine platform, it will still be of value to employ an OMV-based vaccine in a prime-boost strategy for vaccination as well as other situations where other vaccine forms do not work well.
Conclusion
In summary, OMVs could present the complete E7 protein. Immunization with trx-E7 OMVs produced a strong E7-specific cellular immunity response in mice, even without the use of conventional adjuvants, and trx-E7 OMV immunization suppressed the growth of grafted tumors in mice. These results indicated that OMVs could be utilized as a new and feasible antigen delivery platform for vaccines to induce therapeutic cellular immunity.
Acknowledgments
This research was financially supported by the CAMS Initiative for Innovative Medicine (2016-I2M-1-019), the Science and Technology Project of Yunnan Province (2016FA049), the Fundamental Research Funds for the Central Universities of China (2012N08, 3332015197, 3332016117 and 2016ZX350073), the National Natural Science Foundation of China (81773270, 81573206), Fundamental Research Funds for Institute of Pathogen Biology of PUMC (2014IPB107) and the Peking Union Medical College (PUMC) Postgraduates Innovative Funds Project (10023-0710-1003). The authors thank Mr Jingxian Zhou for excellent assistance in electron microscopy and Ms Suping Dai for technology supports.
Disclosure
The authors report no conflicts of interest in this work.
References
Munoz N, Bosch FX, de Sanjose S, et al. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003;348(6):518–527. | ||
Schiller JT, Lowy DR. Understanding and learning from the success of prophylactic human papillomavirus vaccines. Nat Rev Microbiol. 2012;10(10):681–692. | ||
Yang MC, Yang A, Qiu J, et al. Buccal injection of synthetic HPV long peptide vaccine induces local and systemic antigen-specific CD8+ T-cell immune responses and antitumor effects without adjuvant. Cell Biosci. 2016;6:17. | ||
Zhou CM, Zhang GX, Ma XX. Characterization and evaluation of the immune responses elicited by a novel human papillomavirus (HPV) therapeutic vaccine: HPV 16E7-HBcAg-Hsp65 fusion protein. J Virol Methods. 2014;197:1–6. | ||
Appel S, Huang CY, Chen JJW, et al. Recombinant lipidated HPV E7 induces a Th-1-biased immune response and protective immunity against cervical cancer in a mouse model. PLoS One. 2012;7(7):e40970. | ||
Cheng WF, Chang MC, Sun WZ, et al. Fusion protein vaccines targeting two tumor antigens generate synergistic anti-tumor effects. PLoS One. 2013;8(9):e71216. | ||
Cerovska N, Hoffmeisterova H, Pecenkova T, et al. Transient expression of HPV16 E7 peptide (aa 44–60) and HPV16 L2 peptide (aa 108–120) on chimeric potyvirus-like particles using Potato virus X-based vector. Protein Expr Purif. 2008;58(1):154–161. | ||
Liu WJ, Liu XS, Zhao KN, Leggatt GR, Frazer IH. Papillomavirus virus-like particles for the delivery of multiple cytotoxic T cell epitopes. Virology. 2000;273(2):374–382. | ||
Sharma C, Dey B, Wahiduzzaman M, Singh N. Human papillomavirus 16 L1-E7 chimeric virus like particles show prophylactic and therapeutic efficacy in murine model of cervical cancer. Vaccine. 2012;30(36):5417–5424. | ||
Chu X, Li Y, Long Q, et al. Chimeric HBcAg virus-like particles presenting a HPV 16 E7 epitope significantly suppressed tumor progression through preventive or therapeutic immunization in a TC-1-grafted mouse model. Int J Nanomedicine. 2016;11:2417–2429. | ||
Peng S, Frazer IH, Fernando GJ, Zhou J. Papillomavirus virus-like particles can deliver defined CTL epitopes to the MHC class I pathway. Virology. 1998;240(1):147–157. | ||
Sharma C, Khan MA, Mohan T, Shrinet J, Latha N, Singh N. A synthetic chimeric peptide harboring human papillomavirus 16 cytotoxic T lymphocyte epitopes shows therapeutic potential in a murine model of cervical cancer. Immunol Res. 2014;58(1):132–138. | ||
Kuehn MJ, Kesty NC. Bacterial outer membrane vesicles and the host-pathogen interaction. Genes Dev. 2005;19(22):2645–2655. | ||
Lee EY, Choi DS, Kim KP, Gho YS. Proteomics in gram-negative bacterial outer membrane vesicles. Mass Spectrom Rev. 2008;27(6):535–555. | ||
Mashburn-Warren L, McLean RJ, Whiteley M. Gram-negative outer membrane vesicles: beyond the cell surface. Geobiology. 2008;6(3):214–219. | ||
Rosenthal JA, Chen L, Baker JL, Putnam D, DeLisa MP. Pathogen-like particles: biomimetic vaccine carriers engineered at the nanoscale. Curr Opin Biotechnol. 2014;28:51–58. | ||
Huang W, Wang S, Yao Y, et al. Employing Escherichia coli-derived outer membrane vesicles as an antigen delivery platform elicits protective immunity against Acinetobacter baumannii infection. Sci Rep. 2016;6:37242. | ||
Chen DJ, Osterrieder N, Metzger SM, et al. Delivery of foreign antigens by engineered outer membrane vesicle vaccines. Proc Natl Acad Sci U S A. 2010;107(7):3099–3104. | ||
Kim JY, Doody AM, Chen DJ, et al. Engineered bacterial outer membrane vesicles with enhanced functionality. J Mol Biol. 2008;380(1):51–66. | ||
Kesty NC, Kuehn MJ. Incorporation of heterologous outer membrane and periplasmic proteins into Escherichia coli outer membrane vesicles. J Biol Chem. 2003;279(3):2069–2076. | ||
Rosenthal JA, Huang CJ, Doody AM, et al. Mechanistic insight into the TH1-biased immune response to recombinant subunit vaccines delivered by probiotic bacteria-derived outer membrane vesicles. PLoS One. 2014;9(11):e112802. | ||
Huang W, Yao Y, Long Q, et al. Immunization against multidrug-resistant acinetobacter baumannii effectively protects mice in both pneumonia and sepsis models. PLoS One. 2014;9(6):e100727. | ||
Inaba K, Inaba M, Romani N, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176(6):1693–1702. | ||
Kennedy R, Celis E. Multiple roles for CD4+ T cells in anti-tumor immune responses. Immunol Rev. 2008;222:129–144. | ||
Collins BS. Gram-negative outer membrane vesicles in vaccine development. Discov Med. 2011;12(62):7–15. | ||
Heras B, Shouldice SR, Totsika M, Scanlon MJ, Schembri MA, Martin JL. DSB proteins and bacterial pathogenicity. Nat Rev Microbiol. 2009;7(3):215–225. | ||
Sinha S, Langford PR, Kroll JS. Functional diversity of three different DsbA proteins from Neisseria meningitidis. Microbiology. 2004;150(Pt 9):2993–3000. | ||
Amano A, Takeuchi H, Furuta N. Outer membrane vesicles function as offensive weapons in host-parasite interactions. Microbes Infect. 2010;12(11):791–798. | ||
Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. 2006;440(7085):808–812. | ||
Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R. Toll-like receptors control activation of adaptive immune responses. Nat Immunol. 2001;2(10):947–950. | ||
Dalseg R, Wedege E, Holst J, Haugen IL, Hoiby EA, Haneberg B. Outer membrane vesicles from group B meningococci are strongly immunogenic when given intranasally to mice. Vaccine. 1999;17(19):2336–2345. | ||
Keenan J, Day T, Neal S, et al. A role for the bacterial outer membrane in the pathogenesis of Helicobacter pylori infection. FEMS Microbiol Lett. 2000;182(2):259–264. | ||
Kesavalu L, Ebersole JL, Machen RL, Holt SC. Porphyromonas gingivalis virulence in mice: induction of immunity to bacterial components. Infect Immun. 1992;60(4):1455–1464. | ||
Saunders NB, Shoemaker DR, Brandt BL, Moran EE, Larsen T, Zollinger WD. Immunogenicity of intranasally administered meningococcal native outer membrane vesicles in mice. Infect Immun. 1999;67(1):113–119. | ||
Schild S, Nelson EJ, Bishop AL, Camilli A. Characterization of Vibrio cholerae outer membrane vesicles as a candidate vaccine for cholera. Infect Immun. 2009;77(1):472–484. | ||
Muralinath M, Kuehn MJ, Roland KL, Curtiss R 3rd. Immunization with salmonella enterica serovar typhimurium-derived outer membrane vesicles delivering the pneumococcal protein PspA confers protection against challenge with streptococcus pneumoniae. Infect Immun. 2011;79(2):887–894. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.