Back to Journals » Journal of Inflammation Research » Volume 15
Endothelial Microparticles Derived from Primary Pulmonary Microvascular Endothelial Cells Mediate Lung Inflammation in Chronic Obstructive Pulmonary Disease by Transferring microRNA-126
Authors Ma Y, He X, Liu X, Long Y, Chen Y ![]()
Received 18 November 2021
Accepted for publication 12 February 2022
Published 28 February 2022 Volume 2022:15 Pages 1399—1411
DOI https://doi.org/10.2147/JIR.S349818
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Monika Sharma
Yiming Ma, Xue He, Xiangming Liu, Yingjiao Long, Yan Chen
Department of Respiratory and Critical Care Medicine, The Second Xiangya Hospital of Central South University, Changsha, People’s Republic of China
Correspondence: Yan Chen; Yingjiao Long, Email [email protected]; [email protected]
Background: Extracellular vesicles (EVs) are considered to new types of intercellular communication media, and microRNA is one of the most common transferring components of EVs. This study aimed to explore the potential role of endothelial microparticles (EMPs) derived from primary pulmonary microvascular endothelial cells in regulating lung inflammation of chronic obstructive pulmonary disease (COPD) through transferring microRNA-126 (miR-126).
Methods: EMPs generated from primary pulmonary microvascular endothelial cells were isolated by gradient centrifugation and characterized by transmission electron microscopy, flow cytometry and Western blotting. EMPs were treated to in vitro and in vivo COPD models induced by cigarette smoke extract (CSE). miR-126 mimics or inhibitors were transfected into EMPs by calcium chloride. Pathological changes of lung tissue, mRNA and protein levels of inflammation-related factors were measured to explore the effect of EMPs transferring miR-126 on CSE-induced inflammation.
Results: Both in vitro and in vivo studies demonstrated that mRNA and protein levels of inflammation-related factors were significantly increased in COPD group, while EMPs could dramatically reverse these increases. In vitro, overexpression of miR-126 in EMPs decreased HMGB1 expression and magnified the decreasing effect of EMPs on inflammation-related factors.
Conclusion: The present study reveals that EMPs are capable of alleviating lung inflammation and transferring miR-126 can magnify the anti-inflammatory effect of EMPs, which may provide a novel therapeutic alternative for COPD.
Keywords: chronic obstructive pulmonary disease, endothelial microparticles, primary pulmonary microvascular endothelial cell, inflammation, microRNA-126
Introduction
According to the Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2021 report,1 chronic obstructive pulmonary disease (COPD) is a preventable and treatable respiratory disease usually caused by exposure to toxic particles or gases, and it is characterized by persistent respiratory symptoms and airflow limitation due to respiratory and/or alveolar abnormalities. COPD has become a public health problem all over the world, and it caused 2.6% of global disability-adjusted life years (DALYs) during 1990 to 2015.2 The China Pulmonary Health (CPH) study demonstrated that the overall prevalence of spirometry-defined COPD in Chinese adult population was 8.6%.3
Several mechanisms are involved in the development of COPD including inflammation, imbalance between proteolytic and anti-proteolytic activity, oxidative stress and apoptosis, and there exists interaction among different mechanisms.4 Chronic inflammation of the airways plays a central role in the pathophysiology of COPD.5,6 The risk factors of COPD patients are heterogeneous in the population. Existing studies show that the risk factors of COPD mainly include genetic factors, environmental factors (smoking, air pollution, occupational exposure, infection, etc.) and bad lifestyles (eating habits and daily exercise).7 Cigarette smoke exposure is considered to be the primary risk factor for the decline of lung function and the incidence of COPD.8–10 In addition, previous studies have also proved that cigarette smoke exposure participates in the occurrence and development of COPD by inducing airway inflammatory response, oxidative stress injury, lung structural cell aging and cell death.11,12
Endothelial microparticles (EMPs) are extracellular vesicles (EVs) less than 1 μm in diameter released from the surface of endothelial cells when they are activated or apoptotic. EMPs have some antigenic properties of endothelial cells and are considered to play important biological roles in inflammation, vascular injury, endothelial dysfunction and thrombosis.13 Recently, evidence has proved that EVs can be new types of intercellular communication media, which can carry nucleic acid, protein and other components to transport between cells and participate in the pathogenesis of chronic lung diseases.14
As one of the most common transferring components of extracellular vesicle, microRNAs (miRNAs) are involved in the pathogenesis of a variety of inflammatory diseases.15–17 miRNA is a small noncoding RNA, which plays an important role in post transcriptional gene regulation, and the regulation of gene expression by miRNA may occur in three stages of transcription/translation process, including pre-translational, post-translational or co translational silencing.18,19 miRNAs participate in a variety of physiological and pathological processes in human body, including cell maturation, differentiation, and immune regulation.20,21 A recent study found that exosomal microRNA-21 (miR-21) derived from bronchial epithelial cells is involved in aberrant epithelium-fibroblast cross-talk, which suggested an alternative therapeutic target for COPD.22 However, the cellular communication between endothelial cells and bronchial epithelial cells remains unclear.
Previous studies have demonstrated that miR-126 is an endothelial “specific” microRNA, and miR-126 was found be the most abundant microRNA in endothelial cells by RNA sequencing.23,24 Van balkom et al25 found that exosomes derived from human microvascular endothelium were loaded with abundant miR-126 by gene sequencing. Through gene chip analysis, miR-126 was found to be enriched in human pulmonary microvascular endothelial cells and human pulmonary artery endothelial cell derived microparticles.26 Interestingly, previous studies reported miR-126 could suppress inflammation responses in different disease models.27–32 Nevertheless, there have been no studies investigating the relationship between miR-126 and CSE-induced inflammation. In the present study, we explored the potential role of EMPs derived from primary pulmonary microvascular endothelial cells in regulating lung inflammation of COPD through transferring miR-126.
Methods
Cell Lines and Cell Culture
Human pulmonary microvascular endothelial cells (HPMVECs), the primary microvascular endothelial cells derived from the lungs of the human foetus lungs, were purchased from Sciencell Research Laboratories (San Diego, USA). Mouse pulmonary microvascular endothelial cells (MPMVECs), the primary microvascular endothelial cells derived from mouse foetus lungs, were purchased from Procell Research Laboratories (Wuhan, China). Human bronchial epithelial cells (HBECs) were provided by Institute of Respiratory Diseases, The Second Affiliated Hospital of Zhejiang University. The use of HBECs was approved by ethics committee of Second Xiangya Hospital of Central South University. Both HPMVECs and MPMVECs were cultured with endothelial cell medium (Sciencell, San Diego, USA) under standard cell culture conditions (37°C, 5% CO2). And HBECs cultured in DMEM (Hyclone, Logan, UT, USA) in a 5% CO2 humidified incubator at 37°C.
Isolation and Characterization of EMPs
As previously reported by Pan et al,33 EMPs generated from endothelial cells were isolated with minor changes. Briefly, confluent cells were placed and starved in basic medium (without endothelial growth factor or fetal bovine serum) for 24 hours to induce apoptosis. After starvation, the culture medium was collected and centrifuged at 300g for 5 minutes to remove cells. Next, the supernatant was centrifuged at 2000g for 10 minutes to remove debris. Then the collected supernatant was centrifuged at 20000g for 90minutes to pellet EMPs. Flowchart of EMPs isolation is demonstrated in Figure 1A. The pelleted EMPs were resuspended in filtered phosphate‐buffered saline (PBS). EMPs were further identified by transmission electron microscopy (TEM), flow cytometry (FCM) and Western blotting. For flow cytometry, EMPs gate was defined as microparticles with a diameter of 0.1 μm-1μm using 0.1 μm and 1μm calibration beads (Invitrogen, USA).
 |
Figure 1 Identification of endothelial microparticles (EMPs). (A) Flowchart of EMPs isolation. (B) Transmission electron microscopy (TEM) of EMPs released from human pulmonary microvascular endothelial cells (HPMVECs), scale bar: 500nm; (C) Flow cytometry (FCM) of EMPs, standard fluorescent beads of 0.1µm and 1 µm were used to define the gate; (D) Western blotting for specific biomarkers of EMPs (PECAM, E-selectin and Annexin V). |
Preparation of microRNA-126 Mimics/Inhibitors Loaded-EMPs
As stated by Zhang et al,34 a method of calcium chloride (CaCl2) transfection was used to transfect miR-126 mimics/inhibitors into EMPs. In brief, 200 pmol of microRNA mimics or inhibitors were mixed with 20 μg (protein) EMPs in filtered PBS, and then CaCl2 was added to achieve a concentration of 0.1 M. Filtered PBS was used to adjust the final volume of mixture to 300 μL. The mixture was placed on ice for 30 min. After thermal shock at 42 °C for 1 min, the mixture was placed on ice for an additional 5 min. For cell treatment, 20 μg (protein) EMPs were added to a 60-mm cell culture dish (50–70% HBECs confluency) with 5 mL EMPs-free cell culture media.
Preparation of CSE
CSE solution was prepared as previously described by Ma et al.35 Firstly, a modified syringe-driven apparatus was added with 20 mL phosphate buffered saline (PBS) or serum-free cell culture medium. Secondly, cigarettes (Furong, China Tobacco Hunan Industrial co. LTD, Hunan, China) were burned, and a vacuum pump with a pressure at –300 mmHg was used to bubble the cigarette smoke into the modified syringe-driven apparatus; of note, 10 cigarettes were burned for animal experiment and one cigarette was burned for cell experiments Thirdly, the cigarette smoke and PBS (or serum-free cell culture medium) were fully mixed to get 100% CSE solution. Fourthly, the CSE solution was filtered by the microfilter with a pore size of 0.2 μM. 100% CSE solution was used to intraperitoneally inject to mice, while 5% CSE solution was used in the in-vitro experiments.
Animal Experiment
COPD mice models were established referring to the protocol by He et al36 with minor revisions. A total of 24 BALB/C mice (six weeks old) were purchased from Hunan Slyke Jingda Laboratory Animal co. LTD. All mice were randomly divided into three groups: 1) control group, 2) COPD group, 3) COPD + EMPs group, and there were eight mice in each group. In short, the COPD mice were intraperitoneally injected with 0.3mL 100% CSE solution on days 0, 11, and 22, and they were exposed to cigarette smoke in a sealed box with ventilation holes twice a day for 28 consecutive days, except for days 0, 11, 22. For mice in control group, they were maintained in fresh air; on days 0, 11, and 22, an intraperitoneal injection of 0.3 mL PBS was given to them. Mice in COPD + EMPs group were treated with 100μg (protein) EMPs (released from primary MPMECs) in 30μL filtered PBS by intratracheal instillation on days 13 and 26, while mice in control and COPD group were intratracheally instilled with 30μL filtered PBS on days 13 and 26. The study was reviewed and approved by the Ethics Committee of Second Xiangya Hospital of Central South University. The animal experiments followed guidelines for the welfare of the laboratory animals in Second Xiangya Hospital of Central South University, and conformed to internationally accepted ethical standards. A summarized figure of the animal experiment protocol is demonstrated in the Supplementary Figure 1.
Lung Tissue Morphometry
Hematoxylin and eosin (HE) staining was applied to measure pathological changes of mouse lung tissue. Left lungs were inflated with 4% paraformaldehyde for 24 hours and then embedded in paraffin. Embedded lung tissue was sectioned (5 µm) and stained with HE for light microscopy examination. To assess emphysematous changes of lung tissue, mean linear intercept (MLI) and destructive index (DI) were calculated as stated by He et al.37 A semi-quantitative scoring system with a grading scale ranging from 0 to 3 was used to evaluate inflammation in the light of the method reported by Jang et al.38
Real-Time Quantitative Polymerase Chain Reaction Analysis (RT-qPCR)
Total RNA from HBECs was extracted using the TRIzol reagent (Invitrogen, USA). mRNA was reversely transcribed into cDNA using Revert Aid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA). RT-qPCR was performed using an All-in-One qPCR Mix kit (GeneCopoeia, Guangzhou, China) based on general SYBR Green fluorescence detection. In accordance to the comparative Ct (ΔΔCt) method, relative mRNA levels were calculated after normalization to GAPDH.
Western Blot Analysis
To extract protein from cells and lung tissues, RIPA lysis buffer (Beyotime, China) was applied. Protein concentrations were measured by a Bicinchoninic Acid (BCA) protein assay kit (Thermo Fisher Scientific, USA). Sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was used to separate total protein, and separated protein was transferred to polyvinylidene fluoride (PVDF) membranes activated by isopropanol. Sequentially, PVDF membranes were blocked in 5% non-fat milk dissolved in Tris-buffered saline with Tween (TBST) for 1 hour at room temperature. Next, PVDF membranes were incubated with primary antibodies at 4 °C for 12–16 hours. Primary antibodies included nuclear factor kappa B p65 (NF-κB p65) (Cell Signaling Technology, USA, 1:1000 dilution), phosphorylated nuclear factor kappa B p65 (p-p65) (Cell Signaling Technology, USA, 1:1000 dilution), high mobility group box 1 (HMGB1) (Abcam, USA, 1:2000 dilution), platelet endothelial cell adhesion molecule-1 (PECAM-1) (Santa Cruz, USA, 1:500 dilution), E-selectin (Santa Cruz, USA, 1:500 dilution), Annexin V (Santa Cruz, USA, 1:500 dilution), and GAPDH (Proteintech, China, 1:1000 dilution). After washing primary antibodies, PVDF members were incubated with HRP-labeled IgG secondary antibodies (Proteintech, China, 1:5000 dilution). Lastly, the ECL plus Western blotting detection system (Bio-Rad, USA) was employed to visualize labeled protein bands. And ImageJ software was used for the quantitative analysis of protein band densities.
Immunohistochemistry (IHC)
Lung tissue fixed in 4% formaldehyde for more than 24 hours were embedded in paraffin, and cut into 3.5-μm-thick sections. 0.3% hydrogen peroxide was used to fixed lung sections for 10 min after antigen retrieval in citrate buffer (pH 6.0) for 10 min in microwave. Then lung sections were incubated with anti-HMGB1 (Abcam, USA, 1:200 dilution) at 4 °C overnight. Next, lung sections were incubated with goat anti-rabbit IgG antibody conjugated with peroxidase for 30 min at room temperature. Finally, diaminobenzidine (DAB) was added and hematoxylin was applied for counterstaining.
Statistical Analysis
Data were presented as mean ± standard error (SEM) and analyzed by GraphPad Prism (GraphPad Prism 7.04, San Diego, CA, USA). One-way analysis of variance (ANOVA) combined with post hoc test was used to perform statistical comparisons. P values less than 0.05 were considered statistically significant.
Results
Identification of EMPs
Figure 1A demonstrated the flowchart for isolating EMPs from HPMVECs. As shown in Figure 1B, TEM analysis showed that EMPs released from HPMVECs ranged from 100 to 500 nm in diameter. As the size of released EMPs varied, FCM analyses were conducted to further measure the size of isolated vesicles. (Figure 1C) FCM revealed that the size of isolated vesicles majorly ranged from 0.1μm to 1μm. Furthermore, Western blotting results confirmed the expression of specific endothelial markers (PECAM-1, E-selectin, and Annexin V) in EMPs. (Figure 1D) Above results suggest a proper isolation of EMPs.
Effect of EMPs on Inflammation in Human HBECs Induced by CSE
The flowchart of using EMPs to treat HBECs was illustrated in Figure 2A. In order to demonstrate EMPs involving in HBECs, we used DAPI to dye the nucleus of HBECs and CFSE to dye EMPs. Figure 2B illustrated that EMPs distributed in or around the nucleus of HBECs. RT-qPCR results indicated that pro-inflammatory gene (TNF-α, IL-6, and IL-1β) mRNA levels in CSE group significantly increased in contrast with control group, and EMPs treatment markedly reversed increased pro-inflammatory gene (TNF-α, IL-6, and IL-1β) mRNA levels induced by CSE (Figure 2C). Furthermore, the relative p-p65 protein level to p65 in CSE group significantly increased compared with control group, and treatment of EMPs could significantly diminish the protein level of p-p65 to p65 (Figure 2D and E). Figure 2F and G showed protein levels of TNF-α and IL-1β in cell supernatant of HBECs measured by ELISA. CSE dramatically increased TNF-α and IL-1β protein levels in cell supernatant, and elevated TNF-α and IL-1β protein levels were significantly reversed by EMPs. These results reveal that EMPs released from primary human microvascular endothelial cells are able to inhibit CSE-induced inflammation in vitro.
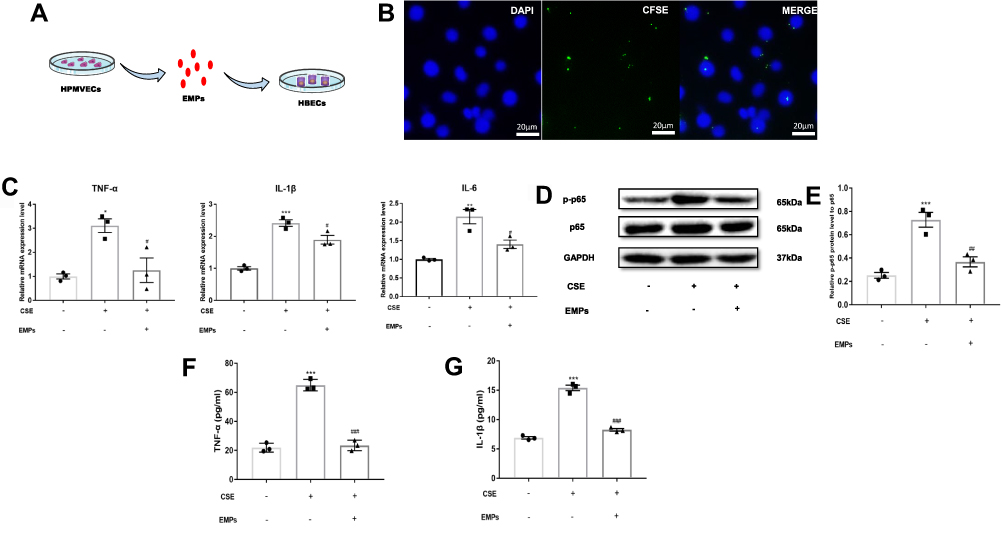 |
Figure 2 Effect of endothelial microparticles (EMPs) on cigarette smoke extract (CSE) induced inflammation in human bronchial epithelium cells (HBECs). (A) Treating diagram of EMPs from human pulmonary microvascular endothelial cells (HPMVECs) on HBECs; (B) Representative images of CFSE‐labeled EMPs merging with HBECs (green: CFSE; blue: DAPI), scale bar: 20µm; (C) Levels of TNF-α, IL-1β, and IL-6 mRNA in HBECs with RT-qPCR; (D) Western blotting analyses of NF-κB p65 and phosphorylated-NF-κB p65 (p-p65) in HBECs; (E) Statistical analysis for relative p-p65 protein level in HBECs; (F) Levels of TNF-α in the cell supernatant of HBECs with ELISA. (G) Levels of IL-1β in the cell supernatant of HBECs with ELISA. Data are presented as the mean ± SEM. *p<0.05 in comparison with control group, **p<0.01 in comparison with control group, ***p<0.001 in comparison with control group, #p<0.05 in comparison with CSE group, ##p<0.01 in comparison with CSE group, ###p<0.001 in comparison with CSE group. |
Effect of EMPs on Inflammation in COPD Mice
To measure the effect of EMPs on inflammation in vivo, we intratracheally instilled EMPs to COPD mice. (Figure 3A) Histological changes in mouse lung sections stained with HE were shown in Figure 3B. Pathological changes of emphysema including enlarged alveolar space and destroyed lung parenchyma were observed in COPD mouse lung tissue. Values of MLI and DI in COPD group were significantly increased compared to control group, while treatment of EMPs dramatically decreased MLI and DI values (Figure 3C and D). The scores of lung inflammation in COPD mice were significantly increased compared with mice in control group, and the enhanced lung inflammation in COPD mice was markedly attenuated by intratracheal instillation of EMPs (Figure 3E and F). Besides, we found that pre-treatment of EMPs remarkably decreased the number of total cells (Figure 3G), neutrophils (Figure 3H), and macrophages (Figure 3I) in mouse bronchoalveolar lavage fluid (BALF). In lung tissue of COPD mice, pro-inflammatory gene (TNF-α, IL-6, and IL-1β) mRNA levels significantly increased when comparing with control group; and EMPs could significantly diminish increased pro-inflammatory gene mRNA levels (Figure 4A). Western blotting results showed that the protein level of HMGB1 and the relative p-p65 protein level to p65 in COPD group was markedly increased compared with control group, and the increase was significantly reversed by EMPs instillation (Figure 4B–E). Also, IHC demonstrated that pre-treatment EMPs could remarkably reduce the increased protein level of HMGB1 in COPD group. (Figure 4F and G) Protein levels of TNF-α and IL-1β in mouse BALF measured by ELISA were illustrated in Figure 4H and I. TNF-α and IL-1β protein levels in COPD mouse BALF were significantly increased in contrast with control group, while EMPs dramatically decreased the elevated levels of TNF-α and IL-1β. Collectively, these results indicate that EMPs released from primary mouse microvascular endothelial cells can reduce mouse lung inflammation induced by CSE.
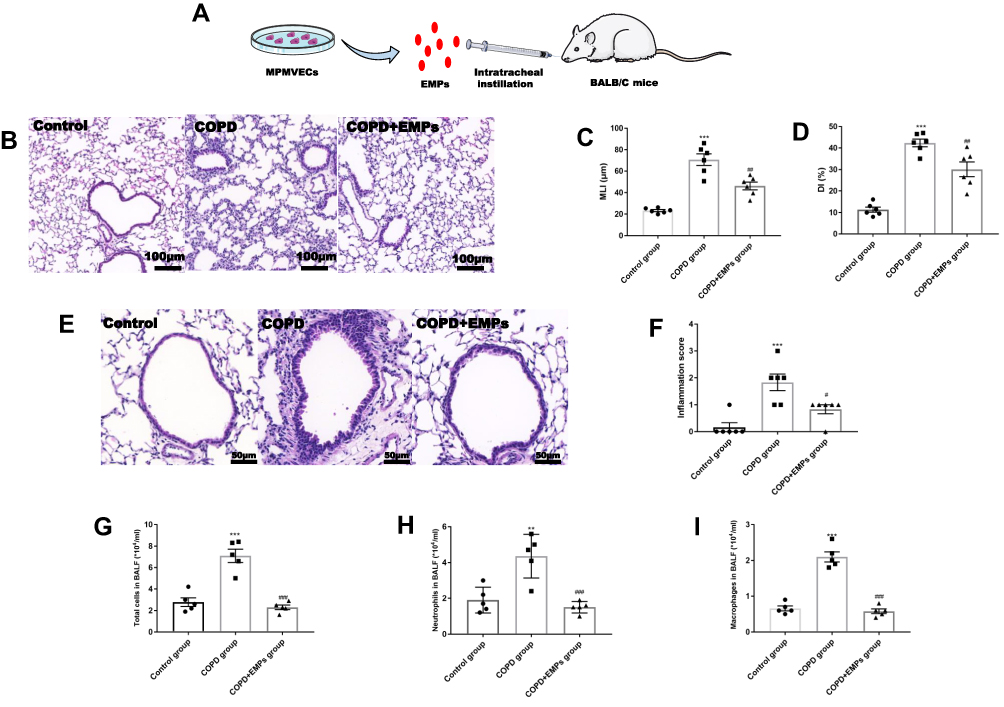 |
Figure 3 Effect of endothelial microparticles (EMPs) on pathological changes in COPD mice. (A) Treating diagram of EMPs from mouse pulmonary microvascular endothelial cells (MPMVECs) on COPD mice; (B) Pathological changes in mouse lung sections with hematoxylin and eosin (HE) staining; (C) Morphometric measurements of MLI (μm); (D) Morphometric measurements of DI (%); (E) Pathological changes in mouse airway with HE staining; (F) Lung inflammation scores in mice; (G) The number of total cells in mouse bronchoalveolar lavage fluid (BALF); (H) The number of neutrophils in mouse BALF; (I) The number of macrophages in mouse BALF. Data are presented as the mean ± SEM. **p<0.01 in comparison with control group, ***p<0.001 in comparison with control group, #p<0.05 in comparison with COPD group, ##p<0.01 in comparison with COPD group, ###p<0.001 in comparison with COPD group. |
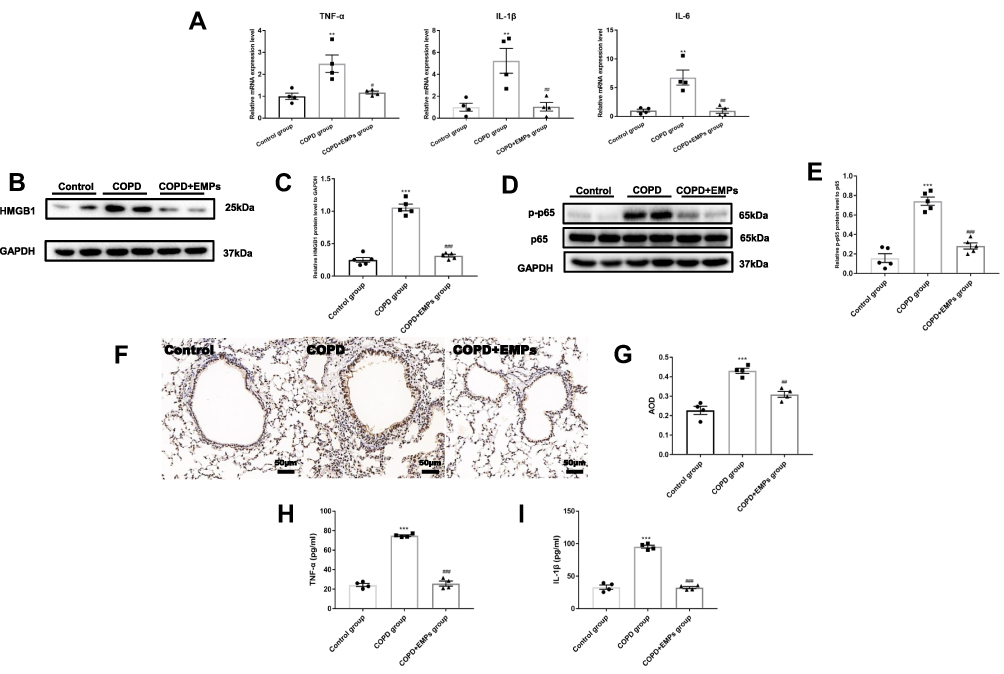 |
Figure 4 Effect of endothelial microparticles (EMPs) on lung inflammation in COPD mice. (A) Levels of TNF-α, IL-1β, and IL-6 mRNA in lung tissue with RT-qPCR; (B) Western blotting analyses of HMGB1 in lung tissue; (C) Statistical analysis for relative HMGB1 protein level in lung tissue; (D) Western blotting analyses of NF-κB p65 and phosphorylated-NF-κB p65 (p-p65) in lung tissue; (E) Statistical analysis for relative p-p65 protein level in lung tissue; (F) Immunohistochemistry (IHC) for HMGB1 in lung tissue; (G) Statistical analysis for average optical density (AOD) of HMGB1 in lung tissue; (H) Levels of TNF-α in mouse BALF with ELISA; (I) Levels of IL-1β in mouse BALF with ELISA. Data are presented as the mean ± SEM. **p<0.01 in comparison with control group, ***p<0.001 in comparison with control group, #p<0.05 in comparison with COPD group, ##p<0.01 in comparison with COPD group, ###p<0.001 in comparison with COPD group. |
EMPs-Derived miR-126 Mediates CSE-Induced Inflammation in HBECs
To verify the mechanism of protecting effect of EMPs on CSE-induced inflammation, we transfected miR-126 mimics or inhibitors into EMPs and then examined the effect of EMPs-derived miR-126 on CSE-induced inflammation in HBECs. (Figure 5A) We found that EMPsmiR−126mimic dramatically increased miR-126 level in HBECs, while EMPsmiR−126mimic significantly reduced miR-126 level in HBECs. (Figure 5B and C) Pre-treatment of EMPsmiR−126mimic could significantly decrease the protein level of HMGB1 and relative protein level of p-p65 to p65 (Figure 5D–G). Moreover, the protein level of HMGB1 and relative protein level of p-p65 to p65 significantly increased after the treatment of EMPsmiR−126inhibitor (Figure 5H–K). Above findings suggest that the protecting effect of EMPs on CSE-induced inflammation may result from the transferred microRNA in EMPs.
 |
Figure 5 Effect of endothelial microparticles (EMPs)-derived microRNA-126 (miR-126) on cigarette smoke extract (CSE) induced inflammation in human bronchial epithelium cells (HBECs). (A) Treating diagram of EMPsmiR−126 on HBECs; (B) Effect of EMPsmiR−126mimic on miR-126 levels in HBECs with RT-qPCR; (C) Effect of EMPsmiR−126inhibitor on miR-126 levels in HBECs with RT-qPCR; (D) Effect of EMPsmiR−126mimic on relative HMGB1 protein level in HBECs with Western blotting; (E) Statistical analysis for the effect of EMPsmiR−126mimic on relative HMGB1 protein level in HBECs; (F) Effect of EMPsmiR−126mimic on relative phosphorylated-NF-κB p65 (p-p65) protein level in HBECs with Western blotting; (G) Statistical analysis for the effect of EMPsmiR−126mimic on relative p-p65 protein level in HBECs; (H) Effect of EMPsmiR−126inhibitor on relative HMGB1 protein level in HBECs with Western blotting; (I) Statistical analysis for the effect of EMPsmiR−126inhibitor on relative HMGB1 protein level in HBECs; (J) Effect of EMPsmiR−126inhibitor on relative p-p65 protein level in HBECs with Western blotting; (K) Statistical analysis for the effect of EMPsmiR−126inhibitor on relative p-p65 protein level in HBECs. EMPsmiR−126mimic represents EMPs transfected with miR-126 mimics; EMPsmiR−126inhibitor represents EMPs transfected with miR-126 inhibitors; EMPsmiR-NC represents EMPs transfected with miR-NC. Data are presented as the mean ± SEM. *p<0.05 in comparison with CSE group, **p<0.01 in comparison with CSE group, ***p<0.001 in comparison with CSE group, #p<0.05 in comparison with CSE+EMPs group, ##p<0.01 in comparison with CSE+EMPs group, ###p<0.001 in comparison with CSE+EMPs group, &p<0.05 in comparison with CSE+EMPsmiR-NC group, &&p<0.01 in comparison with CSE+EMPsmiR-NC group, &&&p<0.001 in comparison with CSE+EMPsmiR-NC group. |
Discussion
This study demonstrates that EMPs released from primary pulmonary microvascular endothelial cells could reduce lung inflammation induced by CSE in vitro and in vivo. Moreover, we proved that the protecting effect of EMPs on CSE-induced inflammation might result from the transferred miR-126 in EMPs, as miR-126 from EMPs could target HMGB1 and inhibit its expression in HBECs. Furthermore, the inhibition of HMGB1 mitigated the activation of downstream typical inflammatory pathway (NF-κB p65). The mechanism diagram of summarized results is illustrated in Figure 6.
 |
Figure 6 The mechanism diagram of summarized findings. HPMVECs-human pulmonary microvascular endothelial cells, EMPs-endothelial microparticles, HMGB1-high mobility group box 1, HBECs-human bronchial epithelium cells. |
Chronic airway inflammation is an important pathophysiological mechanism in the pathogenesis of COPD, which is characterized by excessive mucus secretion, small airway stenosis and emphysema; and inflammatory cells and inflammatory mediators are involved in the pathogenesis of COPD.6 As a major risk factor for COPD, cigarette smoke augments a progressive biphasic infiltration of inflammatory cells in BALF, airways and lung parenchyma, including dendritic cells (DCs), macrophages, and lymphocytes etc.39 Besides, cigarette smoke could stimulate the release of numerous pro-inflammatory cytokines and inhibit the production of anti-inflammatory cytokines.40 Of note, higher level of inflammatory cytokine is related to poorer prognosis of COPD patients.41 Thus, searching strategies targeting inflammatory responses seems crucial for the treatment of COPD.
EVs as therapeutic tools in treating lung diseases have drawn broad attention, as biologically derived EVs have a better safety profile compared with synthetic carriers.42 Particularly, EVs from mesenchymal stem cells (MSCs) become the research focus due to anti-inflammatory and regenerative capability of MSCs.43 EVs derived from MSCs could downregulate the expression of proinflammatory cytokines IL-1β and IL-6 in epithelial cells of cystic fibrosis.44 Xu et al45 reported that bone marrow mesenchymal stem cells (BMSCs)-derived exosomes can effectively rescue smoke inhalation lung injury by inhibiting the HMGB1/NF-κB pathway, suggesting the potential therapeutic role of EVs in treating smoke inhalation lung injury. Moreover, a recent study by Ridzuan et al46 revealed that human umbilical cord mesenchymal stem cells (HUCMSCs) derived-EVs reduced the COPD-induced inflammation in part by the expression of NF-κB subunits p65 and p50. Recently, there have been novel findings to prove that EVs derived from non-mesenchymal stem cells also had therapeutic effects. In 2021, Kadota et al47 confirmed that extracellular vesicles derived from normal human bronchial epithelium can inhibit TGF-β/Wnt pathway improves pulmonary fibrosis both in vitro and in vivo. Interestingly, a study previously published by Kadota and his team showed that extracellular vesicles derived from human bronchial epithelial cells stimulated by cigarette smoke can significantly promote the differentiation of pulmonary myofibroblasts through autophagy, and finally participate in the pathogenesis of COPD.11 Furthermore, their team proposed that extracellular vesicles derived from normal human bronchial epithelium can “treat” diseases, while the extracellular vesicles derived from human bronchial epithelium stimulated by pathogenic factors (such as cigarette smoke) may participate in the “pathogenesis” of diseases. Same as their conclusion, our study proved that EVs from another kind of non-mesenchymal stem cell (endothelial cell) also have therapeutic effects. Researches also have demonstrated that EVs from endothelial progenitor cells (EPCs) are capable of reducing inflammation in lipopolysaccharide-induced acute lung injury48 and sepsis.49 Our study proved that primary pulmonary microvascular endothelial cells derived-EMPs were able to reduce CSE-induced inflammation responses in vitro and in vivo for the first time, which might provide a novel therapeutic choice for COPD.
Recently, microRNAs have been found to transfer from a donor cell into a recipient cell via exosomes and microparticles; emerging evidence have suggested that microRNAs are delivered with intact functionality in a paracrine fashion.50 In addition, miRNAs regulate the translation of the target genes and the function of the target cells after being delivered by EVs.51 In particular, enrichment of certain microRNAs in EVs is able to establish a targeted cross-talk in many inflammation-related diseases. Zhang et al52 reported that sensory neurons transfered EVs-encapsulated miR-23a to promote inflammatory M1 macrophages by binding to A20 and enhance neuropathic pain following the peripheral nerve injury. Interestingly, a recent study manifested that inhibition of miR-10a expression in endothelial cells weakened their anti-inflammatory effects on monocytes.53 Zheng et al54 found that human umbilical vascular endothelial cells (HUVECs)-derived EVs carrying miR-129 could mitigate myocardial I/R injury by downregulating TLR4 and disrupting the NF-κB signaling and NLRP3 inflammasome. In this study, we found that overexpression of miR-126 in EMPs was capable of decreasing HMGB1 expression in HBECs and further magnifying anti-inflammatory effect of EMPs in treating airway inflammation of COPD. Our results are consistent with previous studies. Two studies by Zhou et al48,49 revealed that EPCs derived exosomes were beneficial in lipopolysaccharide-induced acute lung injury and sepsis potentially through the delivery of miR-126. Moreover, exosomes derived from MSCs were able to transfer miR-126 and ameliorate hyperglycemia-induced retinal inflammation.30
miR-126, as an endothelial cell-restricted microRNA, were proved to be associated with the inflammatory responses. Hu et al55 found that miR-126 promoted angiogenesis and inhibited inflammation after contusion spinal cord injury in rats. In sepsis, regulation of miR-126 could mediate inflammatory response, differentiation of T lymphocyte subsets, and apoptosis of lymphocytes.28 HMGB1 is a non-histone nuclear protein, which serves as an alarmin to drive the pathogenesis of inflammatory and autoimmune diseases.56 HMGB1 is a late inflammatory mediator that can promote NF-κB nuclear translocation that leads to the release of inflammatory cytokines, and inflammatory cytokines further enhance the release of HMGBl, leading to a positive feedback loop that amplifies the inflammatory cascade.57,58 An earlier research by Ferhani et al59 proved that elevated expression of HMGB1 existed in proximal and distal airways of smokers with COPD, and HMGB1 also showed correlations with impaired lung function and increased level of IL-1β. A systematic review further revealed the association between HMGB1 and COPD: cigarette smoke could induce neutrophils necrosis and the necrosis of neutrophil cells resulted in HMGB1 release, suggesting that HMGB1 was an intervention target of inflammation in COPD.60 In this study, HMGB1 in HBECs was also found to be a potential target of miR-126, which was consistent with previous studies.27,30,48,49,61,62
Conclusion
In summary, our results demonstrate that EMPs released from primary pulmonary microvascular endothelial cells can reduce airway inflammation of COPD in vitro and in vivo. Moreover, overexpression of miR-126 in EMPs inhibits HMGB1 expression and further magnifies anti-inflammatory effect of EMPs. The present study may provide a novel therapeutic alternative for COPD.
Funding
This work was supported by National Natural Science Foundation of China [No. 81873410, No. 81800043, and No. 82070049], and Natural Science Foundation of Hunan Province [No. 2020JJ5818].
Disclosure
The authors report no conflicts of interest for this work and declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease 2021 report. Available from: https://goldcopd.org/wp-content/uploads/2020/11/GOLD-REPORT-2021-v1.1-25Nov20_WMV.pdf.
2. Soriano JB, Abajobir AA, Abate KH, et al. Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Respir Med. 2017;5(9):691–706. doi:10.1111/imr.12055
3. Wang C, Xu J, Yang L, et al. Prevalence and risk factors of chronic obstructive pulmonary disease in China (the China Pulmonary Health [CPH] study): a national cross-sectional study. Lancet. 2018;391(10131):1706–1717. doi:10.1016/S0140-6736(18)30841-9
4. Plataki M, Tzortzaki E, Rytila P, Demosthenes M, Koutsopoulos A, Siafakas NM. Apoptotic mechanisms in the pathogenesis of COPD. Int J Chron Obstruct Pulmon Dis. 2006;1(2):161–171. doi:10.2147/copd.2006.1.2.161
5. Brightling C, Greening N. Airway inflammation in COPD: progress to precision medicine. Eur Respir J. 2019;54(2):1900651. doi:10.1183/13993003.00651-2019
6. Angelis N, Porpodis K, Zarogoulidis P, et al. Airway inflammation in chronic obstructive pulmonary disease. J Thorac Dis. 2014;6:S167–S72. doi:10.3978/j.issn.2072-1439.2014.03.07
7. Postma DS, Bush A, van den Berge M. Risk factors and early origins of chronic obstructive pulmonary disease. Lancet. 2015;385(9971):899–909. doi:10.1016/S0140-6736(14)60446-3
8. Hancock DB. Compelling interaction of cigarette smoking and polygenetic risk emerges for lung function and COPD. JAMA netw open. 2021;4(12):e2140347. doi:10.1001/jamanetworkopen.2021.40347
9. Wheaton AG, Liu Y, Croft JB, et al. Chronic obstructive pulmonary disease and smoking status - United States, 2017. MMWR Morb Mortal Wkly Rep. 2019;68(24):533–538. doi:10.15585/mmwr.mm6824a1
10. Wang B, Xiao D, Wang C. Smoking and chronic obstructive pulmonary disease in Chinese population: a meta-analysis. Clin Respir J. 2015;9(2):165–175. doi:10.1111/crj.12118
11. Fujita Y, Araya J, Ito S, et al. Suppression of autophagy by extracellular vesicles promotes myofibroblast differentiation in COPD pathogenesis. J Extracell Vesicles. 2015;4(1):28388. doi:10.3402/jev.v4.28388
12. Zhang J, Xu Q, Sun W, Zhou X, Fu D, Mao L. New insights into the role of NLRP3 inflammasome in pathogenesis and treatment of chronic obstructive pulmonary disease. J Inflamm Res. 2021;14:4155–4168. doi:10.2147/JIR.S324323
13. Combes V, Simon AC, Grau GE, et al. In vitro generation of endothelial microparticles and possible prothrombotic activity in patients with lupus anticoagulant. J Clin Invest. 1999;104(1):93–102. doi:10.1172/JCI4985
14. Trappe A, Donnelly SC, McNally P, Coppinger JA. Role of extracellular vesicles in chronic lung disease. Thorax. 2021;76(10):1047–1056. doi:10.1136/thoraxjnl-2020-216370
15. Oglesby IK, McElvaney NG, Greene CM. MicroRNAs in inflammatory lung disease - master regulators or target practice? Respir Res. 2010;11. doi:10.1186/1465-9921-11-11
16. Yang QB, Li LQ, Zhang QB, He YL, Mi QS, Zhou JG. microRNA-223 deficiency exacerbates acute inflammatory response to monosodium urate crystals by targeting NLRP3. J Inflamm Res. 2021;14:1845–1858. doi:10.2147/JIR.S307796
17. Feng X, Hu J, Zhan F, Luo D, Hua F, Xu G. MicroRNA-138-5p regulates hippocampal neuroinflammation and cognitive impairment by NLRP3/Caspase-1 signaling pathway in rats. J Inflamm Res. 2021;14:1125–1143. doi:10.2147/JIR.S304461
18. Bushati N, Cohen SM. microRNA functions. Annu Rev Cell Dev Biol. 2007;23(1):175–205. doi:10.1146/annurev.cellbio.23.090506.123406
19. Alipoor SD, Adcock IM, Garssen J, et al. The roles of miRNAs as potential biomarkers in lung diseases. Eur J Pharmacol. 2016;791:395–404. doi:10.1016/j.ejphar.2016.09.015
20. Baumjohann D, Ansel KM. MicroRNA regulation of the germinal center response. Curr Opin Immunol. 2014;28:6–11. doi:10.1016/j.coi.2014.01.003
21. Pagani M, Rossetti G, Panzeri I, et al. Role of microRNAs and long-non-coding RNAs in CD4(+) T-cell differentiation. Immunol Rev. 2013;253(1):82–96.
22. Xu H, Ling M, Xue J, et al. Exosomal microRNA-21 derived from bronchial epithelial cells is involved in aberrant epithelium-fibroblast cross-talk in COPD induced by cigarette smoking. Theranostics. 2018;8(19):5419–5433. doi:10.7150/thno.27876
23. Kuosmanen SM, Kansanen E, Sihvola V, Levonen A-L. MicroRNA profiling reveals distinct profiles for tissue-derived and cultured endothelial cells. Sci Rep. 2017;7(1). doi:10.1038/s41598-017-11487-4
24. Wang S, Aurora AB, Johnson BA, et al. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell. 2008;15(2):261–271. doi:10.1016/j.devcel.2008.07.002
25. van Balkom BW, Eisele AS, Pegtel DM, Bervoets S, Verhaar MC. Quantitative and qualitative analysis of small RNAs in human endothelial cells and exosomes provides insights into localized RNA processing, degradation and sorting. J Extracell Vesicles. 2015;4(1):26760. doi:10.3402/jev.v4.26760
26. Serban KA, Rezania S, Petrusca DN, et al. Structural and functional characterization of endothelial microparticles released by cigarette smoke. Sci Rep. 2016;6(1):13. doi:10.1038/srep31596
27. Tang S-T, Wang F, Shao M, Wang Y, Zhu H-Q. MicroRNA-126 suppresses inflammation in endothelial cells under hyperglycemic condition by targeting HMGB1. Vascul Pharmacol. 2017;88:48–55. doi:10.1016/j.vph.2016.12.002
28. Zou Q, Yang M, Yu M, Liu C. Influences of regulation of miR-126 on inflammation, Th17/Treg subpopulation differentiation, and lymphocyte apoptosis through caspase signaling pathway in sepsis. Inflammation. 2020;43(6):2287–2300. doi:10.1007/s10753-020-01298-7
29. Cerutti C, Edwards LJ, de Vries HE, Sharrack B, Male DK, Romero IA. MiR-126 and miR-126* regulate shear-resistant firm leukocyte adhesion to human brain endothelium. Sci Rep. 2017;7(1):45284. doi:10.1038/srep45284
30. Zhang W, Wang Y, Kong YC. Exosomes derived from mesenchymal stem cells modulate miR-126 to ameliorate hyperglycemia-induced retinal inflammation via targeting HMGB1. Invest Ophthalmol Vis Sci. 2019;60(1):294–303. doi:10.1167/iovs.18-25617
31. Coulson DJ, Bakhashab S, Latief JS, Weaver JU. MiR-126, IL-7, CXCR1/2 receptors, inflammation and circulating endothelial progenitor cells: the study on targets for treatment pathways in a model of subclinical cardiovascular disease (type 1 diabetes mellitus). J Transl Med. 2021;19(1):140. doi:10.1186/s12967-021-02785-7
32. Qiu B, Qi X, Wang J. CircTLK1 downregulation attenuates high glucose-induced human mesangial cell injury by blocking the AKT/NF-κB pathway through sponging miR-126-5p/miR-204-5p. Biochem Genet. 2021. doi:10.1007/s10528-021-10146-8
33. Pan Q, Ma C, Wang Y, et al. Microvesicles-mediated communication between endothelial cells modulates, endothelial survival, and angiogenic function via transferring of miR-125a-5p. J Cell Biochem. 2019;120(3):3160–3172. doi:10.1002/jcb.27581
34. Zhang D, Lee H, Zhu ZW, Minhas JK, Jin Y. Enrichment of selective miRNAs in exosomes and delivery of exosomal miRNAs in vitro and in vivo. Am J Physiol Lung Cell Mol Physiol. 2017;312(1):L110–L21. doi:10.1152/ajplung.00423.2016
35. Ma Y, Luo L, Liu X, et al. Pirfenidone mediates cigarette smoke extract induced inflammation and oxidative stress in vitro and in vivo. Int Immunopharmacol. 2021;96:107593. doi:10.1016/j.intimp.2021.107593
36. He S, Li L, Sun S, Zeng Z, Lu J, Xie L. A novel murine chronic obstructive pulmonary disease model and the pathogenic role of microRNA-21. Front Physiol. 2018;9:503. doi:10.3389/fphys.2018.00503
37. He ZH, Chen Y, Chen P, et al. 5-Aza-2’-deoxycytidine protects against emphysema in mice via suppressing p16(Ink4a) expression in lung tissue. Int J Chron Obstruct Pulmon Dis. 2017;12:3149–3158. doi:10.2147/COPD.S131090
38. Jang H-Y, Kwon O-K, Oh S-R, Lee H-K, Ahn K-S, Chin Y-W. Mangosteen xanthones mitigate ovalbumin-induced airway inflammation in a mouse model of asthma. Food Chem Toxicol. 2012;50(11):4042–4050. doi:10.1016/j.fct.2012.08.037
39. D’Hulst AI, Vermaelen KY, Brusselle GG, Joos GF, Pauwels RA. Time course of cigarette smoke-induced pulmonary inflammation in mice. Eur Respir J. 2005;26(2):204–213. doi:10.1183/09031936.05.00095204
40. Arnson Y, Shoenfeld Y, Amital H. Effects of tobacco smoke on immunity, inflammation and autoimmunity. J Autoimmun. 2010;34(3):J258–J65. doi:10.1016/j.jaut.2009.12.003
41. Li H, Ma Y, Xue J, et al. C-reactive protein to serum albumin ratio as a novel biomarker to predict prognosis in patients with chronic obstructive pulmonary disease. Clin Lab. 2021;67(3):755–763. doi:10.7754/Clin.Lab.2020.200630
42. Elsharkasy OM, Nordin JZ, Hagey DW, et al. Extracellular vesicles as drug delivery systems: Why and how? Adv Drug Deliv Rev. 2020;159:332–343. doi:10.1016/j.addr.2020.04.004
43. Khatri M, Richardson LA, Meulia T. Mesenchymal stem cell-derived extracellular vesicles attenuate influenza virus-induced acute lung injury in a pig model. Stem Cell Res Ther. 2018;9(1). doi:10.1186/s13287-018-0774-8
44. Zulueta A, Colombo M, Peli V, et al. Lung mesenchymal stem cells-derived extracellular vesicles attenuate the inflammatory profile of Cystic Fibrosis epithelial cells. Cell Signal. 2018;51:110–118. doi:10.1016/j.cellsig.2018.07.015
45. Xu B, Gan C-X, Chen -S-S, Li J-Q, Liu M-Z, Guo G-H. BMSC-derived exosomes alleviate smoke inhalation lung injury through blockade of the HMGB1/NF-kappa B pathway. Life Sci. 2020;257:118042. doi:10.1016/j.lfs.2020.118042
46. Ridzuan N, Zakaria N, Widera D, et al. Human umbilical cord mesenchymal stem cell-derived extracellular vesicles ameliorate airway inflammation in a rat model of chronic obstructive pulmonary disease (COPD). Stem Cell Res Ther. 2021;12(1). doi:10.1186/s13287-020-02088-6.
47. Kadota T, Fujita Y, Araya J, et al. Human bronchial epithelial cell-derived extracellular vesicle therapy for pulmonary fibrosis via inhibition of TGF-β-WNT crosstalk. J Extracell Vesicles. 2021;10(10):e12124. doi:10.1002/jev2.12124
48. Zhou Y, Li P, Goodwin AJ, et al. Exosomes from endothelial progenitor cells improve outcomes of the lipopolysaccharide-induced acute lung injury. Crit Care. 2019;23(1). doi:10.1186/s13054-019-2339-3.
49. Zhou Y, Li P, Goodwin AJ, et al. Exosomes from endothelial progenitor cells improve the outcome of a murine model of sepsis. Mol Ther. 2018;26(5):1375–1384. doi:10.1016/j.ymthe.2018.02.020
50. Das S, Halushka MK. Extracellular vesicle microRNA transfer in cardiovascular disease. Cardiovasc Pathol. 2015;24(4):199–206. doi:10.1016/j.carpath.2015.04.007
51. Tkach M, Thery C. Communication by extracellular vesicles: where we are and where we need to go. Cell. 2016;164(6):1226–1232. doi:10.1016/j.cell.2016.01.043
52. Zhang YM, Liu JY, Wang X, Zhang JF, Xie CC. Extracellular vesicle-encapsulated microRNA-23a from dorsal root ganglia neurons binds to A20 and promotes inflammatory macrophage polarization following peripheral nerve injury. Aging-US. 2021;13(5):6752–6764. doi:10.18632/aging.202532
53. Liang JQ, Gu SY, Mao XL, et al. Endothelial Cell morphology regulates inflammatory cells through microRNA transferred by extracellular vesicles. Front Bioeng Biotechnol. 2020;8:10. doi:10.3389/fbioe.2020.00369
54. Zheng SY, Wang LS, Ma HY, Sun F, Wen FX. microRNA-129 overexpression in endothelial cell-derived extracellular vesicle influences inflammatory response caused by myocardial ischemia/reperfusion injury. Cell Biol Int. 2021;45:1743–1756.
55. Hu JZ, Zeng L, Huang JH, Wang G, Lu HB. miR-126 promotes angiogenesis and attenuates inflammation after contusion spinal cord injury in rats. Brain Res. 2015;1608:191–202. doi:10.1016/j.brainres.2015.02.036
56. Harris HE, Andersson U, Pisetsky DS. HMGB1: a multifunctional alarmin driving autoimmune and inflammatory disease. Nat Rev Rheumatol. 2012;8(4):195–202. doi:10.1038/nrrheum.2011.222
57. Qu L, Chen C, Chen Y, et al. High-Mobility Group Box 1 (HMGB1) and Autophagy in Acute Lung Injury (ALI): a review. Med Sci Monitor. 2019;25:1828–1837. doi:10.12659/MSM.912867
58. Le Y, Wang Y, Zhou L, et al. Cigarette smoke-induced HMGB1 translocation and release contribute to migration and NF-κB activation through inducing autophagy in lung macrophages. J Cell Mol Med. 2020;24(2):1319–1331. doi:10.1111/jcmm.14789
59. Ferhani N, Letuve S, Kozhich A, et al. Expression of High-Mobility Group Box 1 and of receptor for advanced glycation end products in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;181(9):917–927. doi:10.1164/rccm.200903-0340OC
60. Gangemi S, Casciaro M, Trapani G, et al. Association between HMGB1 and COPD: a systematic review. Mediators Inflamm. 2015;2015:164913. doi:10.1155/2015/164913
61. Chen Z, Pan X, Sheng Z, Yan G, Chen L, Ma G. Baicalin suppresses the proliferation and migration of Ox-LDL-VSMCs in atherosclerosis through upregulating miR-126-5p. Biol Pharm Bull. 2019;42(9):1517–1523. doi:10.1248/bpb.b19-00196
62. Ma XG, Liu Y, Xue MX. [Dexmedetomidine alleviates hepatic ischemia-reperfusion injury by regulating MALAT1/miR-126-5p/HMGB1 axis]. Sheng Li Xue Bao. 2021;73(2):253–262. Chinese.
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.