Back to Journals » Journal of Inflammation Research » Volume 15
Endothelial Mechanosensors for Atheroprone and Atheroprotective Shear Stress Signals
Authors Li H, Zhou WY, Xia YY, Zhang JX
Received 21 December 2021
Accepted for publication 1 March 2022
Published 11 March 2022 Volume 2022:15 Pages 1771—1783
DOI https://doi.org/10.2147/JIR.S355158
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Hui Li,* Wen-Ying Zhou,* Yi-Yuan Xia,* Jun-Xia Zhang
Department of Cardiology, Nanjing First Hospital, Nanjing Medical University, Nanjing, Jiangsu, 210006, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jun-Xia Zhang, Department of Cardiology, Nanjing First Hospital, Nanjing Medical University, Nanjing, Jiangsu, 210006, People’s Republic of China, Tel +86 15366155682, Email [email protected]
Abstract: Vascular endothelial cells (ECs), derived from the mesoderm, form a single layer of squamous cells that covers the inner surface of blood vessels. In addition to being regulated by chemical signals from the extracellular matrix (ECM) and blood, ECs are directly confronted to complex hemodynamic environment. These physical inputs are translated into biochemical signals, dictating multiple aspects of cell behaviour and destination, including growth, differentiation, migration, adhesion, death and survival. Mechanosensors are initial responders to changes in mechanical environments, and the overwhelming majority of them are located on the plasma membrane. Physical forces affect plasma membrane fluidity and change of protein complexes on plasma membrane, accompanied by altering intercellular connections, cell-ECM adhesion, deformation of the cytoskeleton, and consequently, transcriptional responses in shaping specific phenotypes. Among the diverse forces exerted on ECs, shear stress (SS), defined as tangential friction force exerted by blood flow, has been extensively studied, from mechanosensing to mechanotransduction, as well as corresponding phenotypes. However, the precise mechanosensors and signalling pathways that determine atheroprone and atheroprotective phenotypes of arteries remain unclear. Moreover, it is worth to mention that some established mechanosensors of atheroprotective SS, endothelial glycocalyx, for example, might be dismantled by atheroprone SS. Therefore, we provide an overview of the current knowledge on mechanosensors in ECs for SS signals. We emphasize how these ECs coordinate or differentially participate in phenotype regulation induced by atheroprone and atheroprotective SS.
Keywords: shear stress, endothelial cells, mechanosensors, mechanotransduction
Introduction
At the macroscale, cells, tissues and organs are constructed as interconnected structural layers of tension-prestressed networks.1 The tensegrity systems are associated with cytoskeletal, cell–matrix and cell–cell interactions.2 They integrate physical and chemical signals before the cells respond to force locally through individual mechanosensitive molecules. Locally, blood flow produces various types of forces on the vessel wall, including hydrostatic force, tension, and shear stress (SS).3 The hydrostatic pressure exerted by fluid is perpendicular to the blood vessel wall. Traction is generated by the connection between vascular endothelial cells (ECs) during vasomotor movement. SS, the friction force exerted by blood flow on the vascular endothelium, is closely related to blood viscosity, blood flow velocity, and blood vessel geometric anatomy.
Among these forces, SS, the force exerted on the apical surface of ECs, is dominant, and the corresponding blood flow patterns are complicated.4 The fluid flow in straight arterial vessels is atheroprotective or atheroresistant, and with a well-defined direction, it produces pulsatile or laminar SS. High shear stress (HSS), ranging from 10 to 70 dynes/cm2, is believed to be atheroprotective. ECs in HSS regions maintain anti-proliferative, anti-thrombotic, and anti-inflammatory phenotypes.5 Physiologically, pulsatile or laminar flows inhibit the development of atherosclerosis (AS) and protect ECs from two other endothelial dysfunction-related diseases, hyperlipidaemia and hypertension.6 In contrast, AS develops preferentially at blood vessel bends, bifurcations, and the inner curvature of arteries, as well as downstream of the aortic valve, where the blood flow pattern is disturbed and without a defined direction. SS in these atheroprone regions is oscillating or disturbed. Generally, the force of atheroprone SS is relatively lower and less than 4 dynes/cm2. ECs in disturbed flow regions accumulate reactive oxygen species (ROS) and exhibit decreased nitric oxide (NO) synthesis and exaggerated inflammatory responses.7
How ECs responding to SS at least involves three steps: mechanosensing, mechanotransduction, and mechanoresponse. Numerous studies have shown that SS is converted into biological stimuli through specialized subcellular compartments or protein complexes, termed mechanosensors, which include junctional complexes, integrins, the cytoskeleton, caveolae, ion channels, G proteins and G protein-coupled receptors (GPCRs), the glycocalyx, primary cilia, and Plexin D1 (PLXND1).8 Mechanosensors can transmit extracellular mechanical signals into intracellular chemical signals and trigger signalling cascades upon adaptor molecule binding.
Mechanotransduction is a series of signalling cascades triggered by mechanical stimulation. Upon stimulation, the transduction of mechanical force is initiated at adjacent cell periphery and subsequently propagates throughout the whole cell.9 Almost simultaneously, multiple signalling pathways downstream of mechanosensors are activated. Then, second messengers, such as cytosolic calcium (Ca2+) and NO, are generated, and the mechanical transduction of nuclear transcription factors is activated. The transcription factors known to respond to SS are Krüppel-like Factor 2 (KLF2), nuclear factor erythroid 2-like (NRF2), and Yes-associated protein (YAP)/transcriptional coactivator with a PDZ-binding domain (TAZ). KLF2, expressed in mouse embryo vascular ECs as early as E9.5, is induced by atheroprotective flow10 and regulates gene transcription in the nucleus.11
Mechanoresponse, the process by which cells respond or adapt to their physical surroundings, is closely linked to vascular homeostasis, vascular remodelling, and angiogenesis.12 Studies in our laboratory have demonstrated that atheroprone low shear stress (LSS) induced endothelial oxidative stress, apoptosis13 and autophagic flux impairment.14
In addition, mechanoresponse includes the crosstalk of ECs with vascular smooth muscle cells (SMCs), stromal cells and immune cells in local vascular walls through microRNAs-mediated and extracellular vesicle-mediated mechanisms.15,16 ECs and SMCs are the main adjacent wall cells that form barriers. The communication between them can be achieved through physical contact, exchange of signalling cues, and extracellular matrix (ECM) deposition. Moreover, their communication is one of the major mechanisms regulating vascular tone. Dai et al demonstrated that endothelium-derived factors (PDGF-B, CXCL12, ET-1, and MIF) mediated SMC proliferation, which contributed to vascular remodelling and pulmonary arterial hypertension.17 It has been proposed that endothelial-specific alk5 overexpression in alk5−/− restores SMC proliferation in the cardiac outflow tract wall.18
However, systematic knowledge on mechanosensing and mechanotransduction of ECs in response to SS is not well established; in particular 1) the precise mechanosensors and signalling pathways that determine atheroprone and atheroprotective cellular phenotypes in arteries and 2) whether regulation of endothelial cell phenotype should be ascribed to acute or chronic effects of SS remain unclear. The purpose of this review is to provide a brief summary of the mechanosensors in ECs and the related signal transduction pathways induced by SS, with emphasis on how they coordinate or differentially participate in phenotype regulation by atheroprone and atheroprotective SS. Notably, the mechanosensors discussed herein have been reported in the literature with detailed characteristics, such as the type, magnitude, and duration of SS.
Junctional Complexes
Within arterial vessels, endothelial cell–cell and cell–ECM junctions are crucial anchoring structures that promote architectural stability, permeability regulation, and intercellular communication. The three best-known distinct junctional domains are adherens junctions (AJs), tight junctions and gap junctions. Tight junctions and gap junctions can directly sense blood flow. The main components of these junctions are occludin/claudins and connexins, which have been proposed to be putative mechanotransducers.19
AJs, consisting of cadherin/catenin/p120 protein complexes, have been reported to colocalize with more than 170 proteins.20 The complex interactions of junctional complexes with colocalized proteins affect AJ dynamics. Tzima et al showed that platelet and endothelial cell adhesion molecule-1 (PECAM-1, also called CD31), vascular endothelial growth factor receptor 2 (VEGFR2), and vascular endothelial cell cadherin (VE-cadherin) comprise a mechanosensory complex that can directly transduce SS (12 dynes/cm2).21
VE-cadherin (also known as CDH5 and CD144), the main component of endothelial cell-to-cell AJs, plays a key role in the maintenance of vascular integrity and participates in controlling vascular permeability, inhibiting unrestricted vascular growth, and limiting cell movement.22 Dynamic SS affects the spatial distribution of VE-cadherin. VE-cadherin was primarily localized along the plasma membrane of cells exposed to HSS (10 dynes/cm2, 4 h), whereas LSS (1.8 dynes/cm2, 4 h) induced VE-cadherin endocytosis and phosphorylation (pY685 VEC).23 With fluorescence resonance energy transfer (FRET) tension biosensor measurements, Conway et al showed that VE-cadherin specifically located in cell–cell junctions caused significant myosin-dependent tension. The onset of SS (15 dynes/cm2, 24 h) triggered a rapid decrease in the tension across VE-cadherin, which paralleled a decrease in total cell–cell junctional tension.24 As reported, VE-cadherin was upstream of phosphoinositide 3-kinases (PI(3)K) and protein kinase B (Akt) during flow stimulation (12 dynes/cm2), and in VE-cadherin−/− cells, actin filaments (F-actins) failed to align along the direction of flow.21 Moreover, VE-cadherin has been found to be required for shear-induced Shc–αvβ3 and Shc-VEGFR2 complex formation.25 In addition, laminar flow induced the association of VEGFR2 with PI(3)K or PECAM-1 and β-catenin with PI(3)K in a VE-cadherin-dependent manner.21 Under laminar flow (12 dynes/cm2), VE-cadherin binds directly to VEGFR2 as well as to VEGFR3, which has been identified as another component of the junctional mechanosensory complex.26 VE-cadherin is indispensable during laminar flow-induced phosphorylation of Akt and p38 and the migration of VEGFR2 into the nucleus.27 Additionally, VE-cadherin mediated laminar shear-induced Ras-related C3 botulinum toxin substrate 1 (Rac1) activation and pTyr-occludin levels.28
PECAM-1 has been reported to maintain vascular integrity, promote cell migration and protect ECs from apoptotic and/or inflammatory stimuli.29 Xie et al proposed that LSS (2 dynes/cm2, 2 h) upregulated the expression of PECAM-1 in vitro. Furthermore, PECAM-1 knockdown inhibited LSS-induced cell apoptosis and adhesion of monocytes to human umbilical vein endothelial cells (HUVECs).30,31 Laminar SS (12 dynes/cm2, 2–120 min) induced the tyrosine phosphorylation of PECAM-1, which modulated the activation of the serine/threonine kinase Akt and eNOS in ECs.32 Laminar SS (15 dynes/cm2) mediated extracellular signal regulated kinase (ERK) phosphorylation. This process has been shown to be dependent on PECAM-1 tyrosine phosphorylation, the binding of SHP-2 to phospho-PECAM-1 and the activity of SHP-2 phosphatase.33 PECAM-1 was required for the induction of COX-2/PGI2 by laminar SS (10 dynes/cm2, 5 h).34 PECAM-1-deficient mice showed impaired vascular remodelling and reduced intima-media thickening. Regulation of ICAM-1 expression, nuclear factor kappa-B (NF-κB) pathway activation, Akt pathway activation, and leukocyte accumulation in LSS regions were involved in the protective effect on vascular remodelling.35 Instead of crossing the cell ring, actins were distributed in a circumferential ring in aortic ECs of PECAM-1−/− mice.21 Chen et al showed that PECAM-1 participated in arteriogenesis and collateral remodelling in ischemic hearts. Immediately after ligation of the superficial branch in the femoral artery, PECAM-1−/− animals showed increased perfusion due to wider collateral instead of more collateral vessels.36
VEGFR2, known as a tyrosine kinase receptor, participates in the regulation of cell migration, apoptosis, vascular permeability, vasodilatation, and angiogenesis.37–39 After the onset of flow (10 dynes/cm2), a rapid nuclear translocation of VEGFR2 occurred within 2 minutes, which was accompanied by the formation of a VEGFR2/VE-cadherin/β-catenin complex.40 Laminar SS (12 dynes/cm2, 5 min) stimulated sustained phosphorylation of VEGFR2 in a Src-independent manner without the presence of a ligand.41 VEGFR2 was increased in human umbilical vein endothelial cells (HUVECs) under laminar SS (10 dynes/cm2, 72 h).38 The expression levels of VEGFR2 were also evaluated in HUVECs under LSS (~0.5 dynes/cm2) and laminar SS (10.2–10.8 dynes/cm2) for 19 h.42 Notably, VEGFR2 activation induced by laminar flow was downstream of PECAM-1 and VE-cadherin activation.21 Activation of VEGFR2 by laminar SS (12 dynes/cm2, 30 min) sequentially mediated downstream PI(3)K-Akt-eNOS activation and NO-dependent vasodilation in vivo.27 VEGFR2 regulated laminar SS-induced p38 phosphorylation.43 In ECs subjected to laminar SS, treatment with the VEGF receptor tyrosine kinase inhibitor SU1498 or VEGFR2 neutralizing antiserum resulted in exaggerated apoptosis.38
Integrins
Integrins are a family of transmembrane protein receptors that bind adhesion molecules in the ECM and connect to the intracellular cytoskeleton via linker proteins to establish structural continuity from inside to outside a cell. Integrins facilitate cell proliferation, adhesion, migration, and differentiation.5 They are transmembrane adhesion proteins composed of two subunits, α and β. Sun et al showed that oscillating shear stress (OSS, 0.5 ± 4 dynes/cm2, 2 h) significantly elevated the level of total and activated integrin α5 in lipid rafts. EC dysfunction in the atheroprone region was integrin α5-dependent.44 Annexin A2 (ANXA2), a phospholipid-binding protein, links the F-actin cytoskeleton to the plasma membrane. ANXA2 plays a key role in many cellular biological processes, including exocytosis, endocytosis, membrane organization, and ion channel conductance.45 Zhang et al showed that ANXA2, as a potential carrier, allowed integrin α5β1 translocation to lipid rafts in response to OSS.46 Additionally, laminar SS (12 dynes/cm2, 5 min) rapidly activated integrin αvβ347 as well as integrin β3, and promoted integrin–Gα13 interaction, leading to suppression of RhoA-YAP phosphorylation in vivo and in vitro.48 OSS promoted atherosclerotic phenotypes via the CCN1-α6β1 pathway in ECs.49 A previous study has shown that integrin αvβ3 mediated Rho-dependent cytoskeletal alignment by laminar SS.47 Remarkably, integrins function as mechanotransducers through association with various proteins, including focal adhesion kinase (FAK), integrin-linked kinase (ILK), p130Cas, C-terminal Src homology 2 (SH2) domain-containing protein (Shc) and actin-binding proteins (paxillin, vinculin, and talin).5 In addition, integrin activation may be triggered downstream of the mechanosensory PECAM-1/VEGFR2/VE-cadherin complex.21
The Cytoskeleton
The cytoskeleton is described as a network system formed by the nuclear skeleton, cytoplasmic skeleton, cell membrane skeleton and ECM. Specifically, the cytoskeleton refers to the protein fibre framework, which is composed of microtubules (MTs), microfilaments (MFs) and intermediate filaments (IFs). The cytoskeleton physically connects different spatial components inside ECs to transfer forces from the apical domain to the basal or lateral domains. It is associated with changes in cell morphology and cell motility. Laminar SS induced the elongation and orientation of ECs by regulating the assembly and alignment of stress fibres.50 Avari et al used a GFP-vimentin fusion protein to observe rapid deformation of IFs in living ECs exposed to fluid laminar SS (12 dynes/cm2).51 Under static conditions, F-actin was short, thin and randomly oriented, while F-actin bands were around the cell edge. Under atheroprone conditions (1.13±0.10 dynes/cm2, 24 h), ECs presented with overall random F-actin bundle polarities, and the peripheral actin band disappeared. Under atheroprotective conditions (11.5±0.9 dynes/cm2, 24 h), F-actins aligned with the flow direction, and the cells were full of denser and thicker actin bundles.52
The LIM domain refers to an ~60 amino acid sequence that forms a double zinc finger protein-protein or protein-DNA binding interface. There are 41 LIM (Lin-11, Isl1, and MEC-3) domain-containing proteins enriched at cell adhesions and/or the actomyosin cytoskeleton.53 The LIM protein zyxin, which localized to stress fibres, F-actins, and AJs, was mobilized by laminar SS (15 dynes/cm2, 2 h) and moved from focal adhesions to F-actins.54
Caveolae
Caveolae, bulb-shaped 50–100 nm plasma membrane invaginations, are mainly composed of cholesterol, sphingolipids, and the protein caveolin-1 (Cav-1).55 They were first visualized by transmission electron microscope (TEM). Caveolae participate in endothelial transcytosis, vascular permeability, vasomotor tone control, and vascular reactivity. Cav-1 is synthesized in the endoplasmic reticulum (ER) and then transported from the Golgi complex to the plasma membrane in a cholesterol-dependent process.56 As a scaffolding protein within caveolar membranes, Cav-1 forms a stable hetero-oligomeric complex with caveolin-2 (Cav-2), which targets lipid rafts and drives caveolae formation. Cav-1 mediates the recruitment of cavin proteins (Cavin1/2/3/4) to caveolae. The cavin proteins ensure that caveolae are located only at the cell surface. As plasma membrane tension increases, cavins and caveolins dissociate, and caveolar flattening buffers changes and maintains plasma membrane integrity.56 Together with the actin cytoskeleton, dynamin-like ATPase EHD2 stabilizes caveolae on the plasma membrane.
The subcellular distribution and turnover of caveolae are dynamically regulated by SS. Caveolae are anchored to the cytoskeleton through filamin binding to actin and Cav-1.57 EC migration is accompanied by translocation of Cav-1 and caveolae to the rear of moving ECs exposed to laminar SS.57 Under static conditions, it has been reported that Cav-1 was predominantly located near the cell boundary. However, Cav-1 migrated towards the middle of a cell and the apical stack and did not dock on the basal stack after laminar SS exposure (15 dynes/cm2, 24 h).58 En face immunostaining showed that Cav-1 expression was higher in the inner curvature than in the outer curvature of the aortic arch. Regarding the distribution in subcellular compartments, fewer apical caveolae and more intracellular vesicles were enriched with Cav-1 in the inner curvature of the aortic arch, as indicated by TEM.59 Prolonged laminar SS (10 dynes/cm2, 6 h) resulted in increased Cav-1 expression and higher caveolae density at the plasma membranes in ECs. ECs subjected to SS displayed higher Cav-1 fluorescence intensity for along the plasma membrane. However, Cav-1 in static ECs was mainly distributed in the intracellular juxtanuclear compartment.60
Acute SS exposure induces Cav-1 phosphorylation and regulates downstream signalling pathways. Wang et al showed that OSS (4 dynes/cm2, 30 min) led to the phosphorylation of Cav-1 on tyrosine-14.111 Another group found that laminar SS (10 dynes/cm2, 5 min) stimulated Cav-1 phosphorylation (pY14), which depended on Src family kinase (SFK) activity and integrin activation.61 Laminar SS (15 dynes/cm2, seconds) evoked ATP release at caveolae and subsequently triggered Ca2+ flux at the same sites in ECs.62 Cav-1 knockdown in mice resulted in an increase in lung vascular permeability and dilation of cell–cell connections.63 Studying genetic ablation of Cav-1 in ApoE-knockout mice, Fernandez-Hernando et al discovered that the progression of atherosclerotic lesions was inhibited by a reduction in low-density lipoprotein (LDL) infiltration. Similarly, NO production and leukocyte adhesion were decreased. Re-expression of Cav-1 in the endothelium promoted lesion expansion.64 The lack of Cav-1 contributed to attenuated LDL transcytosis, intensified vascular inflammation, and fibronectin deposition independent of eNOS activation.59 Long- and short-term flow-mediated mechanotransduction has been compared in vessels of wild-type (WT) mice, Cav-1-knockout (Cav-1-KO) mice, and Cav-1-KO mice reconstituted with a transgene expressing Cav-1 specifically in ECs (Cav-1-RC mice). Ligation of the left external carotid contributed to lower blood flow in the common carotid artery, which in turn reduced the lumen diameter of carotid arteries from the WT and Cav-1-RC mice after 14 days. However, in the Cav-1-KO mice, the decrease in blood flow did not narrow the lumen but paradoxically increased wall thickness and cellular proliferation. In addition, an examination of isolated pressurized carotid arteries revealed that acute flow-mediated dilation was markedly reduced in the Cav-1-KO arteries compared with those in the WT mice.55
Ion Channels
Several ion channels are SS-responsive, and their currents can be determined by the whole-cell patch clamp method. After the application of LSS (0.5–3.5 dynes/cm2, seconds), channels that efflux chloride caused EC membrane depolarization, while flow-sensitive channels that influx potassium mediated membrane hyperpolarization.65
Adenosine triphosphate (ATP) release and subsequent activation of purinergic receptors are critical in mechanotransduction. P2X4R is a non-selective cation channel trimer dependent on ATP. P2X4 can trigger inflammation in response to high ATP release.66 P2X4 channels mediate laminar flow-induced influx of extracellular Ca2+ across the plasma membrane in the presence of extracellular ATP.67 The transient receptor potential (TRP) channel family is composed of tetrameric complexes located mainly in the plasma membrane.68 Laminar SS activated TRPV4 and TRPP1/2, which allowed Ca2+ influx.69
Recently, the transmembrane protein Piezo1 has been identified as an essential component of mechanically activated ion channel. Piezo1 mediates laminar SS-induced Ca2+ influx and also regulates sprouting angiogenesis,70 NO production, vascular tone, and blood pressure.71 Laminar SS (15 dynes/cm2) evoked Piezo1 accumulation at leading apical lamellipodia.72 Endothelial Piezo1 was required for the initiation of laminar flow-induced ATP release and vasodilation (15 dynes/cm2), thus controlling blood pressure. Knocking down Piezo1 expression strongly reduced the laminar flow-induced phosphorylation of Src kinase, Akt, and eNOS,71 which induced cell alignment. A study showed that Piezo1-deficient embryos displayed vascular remodelling defects.73 Piezo1 mediated uterine artery vasodilation during rat pregnancy.74 HSS-induced monocyte activation was dependent on Piezo1, and reduction of SS by transcatheter aortic valve implantation (TAVI) resulted in down-regulated monocyte activation.75
G Proteins and GPCRs
It has been shown that laminar SS (30 dynes/cm2, 1 min) activated and reorganized purified G protein liposomes in protein receptor-free regions, even in the absence of another potential mechanosensor, indicating that G protein is a mechanosensor.76 HSS (300 dynes/cm2, 2 min) was found to cause a ligand-independent conformational change in GPCR, as determined by FRET.77 Zeng et al showed that CXCR1 and CXCR2, well-known GPCRs, mediated EC migration induced by laminar SS.78 Gαq/11 and PECAM-1 were colocalized at the cell–cell junctions in atheroprotective regions of mouse aorta.79 Interestingly, Gαq/11 was absent from the junctions in atheroprone areas and in the whole artery of PECAM-1-knockout mice, suggesting that the Gαq/11-PECAM-1 complex is a critical mediator of vascular diseases.79
The Apical Glycocalyx
The endothelial glycocalyx (EG), composed of proteoglycans (PGs) and their associated glycosaminoglycan (GAG) side chains,80 can be recognized by ruthenium red staining. The thickness of the EG ranges from tens of nanometres to more than 1 µm.81 The EG, along with some blood-borne molecules, forms the endothelial surface layer (ESL). The ESL affects mechanotransduction, permeability, thrombogenesis, and leukocyte adhesion.82 After exposure to laminar SS (15 dynes/cm2) for 24 h, the thickness of the glycocalyx was increased.83 Yao et al showed that removal of the glycocalyx by heparinase prevented HSS-induced cell reorientation and attenuated HSS-mediated cell proliferation.83 Using a custom-built atomic force microscope (AFM) with fluorescence imaging capabilities to vertically stretch the glycocalyx, Dragovich et al demonstrated that a rapid Ca2+ influx and NO production in response to laminar SS was dependent on the EG.84 Removal of heparan sulfate by heparinase III85 and/or removal of hyaluronic acid by hyaluronidase86 led to a significant decrease in NO production. The shear-induced vessel dilation was abolished after preincubation with neuraminidase due to the elimination of a portion of the EG.87 Finally, the EG differentially mediated the effects of atheroprotective and atheroprone SS, since atheroprone disturbed flow inhibited EG expression in contrast to the effects of atheroprotective flow.88
Primary Cilia
The cilium is a membrane-covered and rod-like organelle with a diameter of approximately 0.2 µm. A cilium is primarily composed of a membrane layer, soluble compartment, axoneme, basal body, and ciliary tip.89 Considering the different structures of the axoneme, cilia can be divided into motile and non-motile types. Non-motile cilia, without central pairs of microtubules, are known as primary cilia, which are anchored to the basal body and thereby connected to the cytoskeletal apparatus.90 Primary cilia are more enriched and longer in low and oscillatory flow regions in vivo. However, they can barely be detected and become shorter and disassembled under laminar SS (15 dynes/cm2, 1–2 h).91 Non-ciliated cells in embryonic arteries that were subjected to laminar SS (15 dynes/cm2, 5 h) showed significantly less induction of KLF2 expression than ciliated cells in the embryonic heart.92 Primary cilia on ECs protrude into the vascular lumen and are molecular switches for Ca2+ and NO signalling.93 The communication between ECs and the ECM depends on the proper localization of polycystin-1 (PC-1) in cilia.94 The proper expression and localization of ciliary polycystin-2 (PC-2) are required for NO biosynthesis in response to laminar SS (7 dynes/cm2).40
Plexin D1
Plexin D1 (PLXND1), a subfamily of transmembrane protein plexins, is a key Ca2+-dependent cell-surface receptor in the semaphorin family with diverse structures. PLXND1 plays a crucial role in axonal guidance and vascular development. PLXND1-mutant mice demonstrated a defect in the migration and proliferation of motoneurons, indicating that de novo mutations in PLXND1 may account for Mobius syndrome.95 The expression of PLXND1 is elevated in both tumour cells and their vasculature, which makes it a marker for tumour vasculature. In contrast, it can also exert an antitumor effect by promoting apoptosis and inhibiting tumour growth.96 Homozygous PLXND1-knockout mice showed peripheral vascular malformations.97 PLXND1 might have a role in cytoskeletal rearrangements and cell motility with Rac/RhoA motifs in its intracellular domain.98 Recently, using a magnetic system, Mehta et al found that PLXND1 was a direct mechanosensor. PLXND1 formed a mechanocomplex with neuropilin-1 and VEGFR2 in response to SS (12 dynes/cm2, 2 min), and this mechanocomplex was upstream of junctional complex and integrin activation. The process of mechanotransduction requires flexion in the PLXND1 ectodomain. PLXND1-depleted bovine ECs showed no inflammatory changes [monocyte chemoattractant protein-1 (MCP1), vascular cell adhesion molecule-1 (VCAM-1) levels] in response to atheroprone flow as well as anti-inflammatory changes (KLF2 and/or KLF4 expression) in response to atheroprotective flow for 24 h. Knocking down PLXND1 expression attenuated early responses under laminar SS (12 dynes/cm2, 10 min), such as phosphorylation of the key signalling mediators Akt, ERK1/2, and eNOS. PLXND1iECKO mice showed reduced EC elongation and decreased intensity of actin stress in the descending aorta.99
Other Possible Mechanosensors
Tyrosine kinase receptors are important participants in shear stress sensing and transduction, particularly VEGFR-2 (as we mentioned above). In addition, the Tie1/Tie2 receptor system plays a pivotal role in the steady state of ECs. The total amount and phosphorylation level of Tie2 increased under laminar SS (6 dynes/cm2).100 Pulmonary Tie2 expression was suppressed in a murine model of hypertension. Idowu et al indicated that Tie2 controlled vascular barrier function in a GATA3-dependent manner.101 In addition, Tie1 was required for endothelial integrity and survival. Notably, the rapid downregulation of Tie1 expression, resulting from rapid cleavage of this molecule, is sensitive to laminar SS changes, not by force per se. The binding of the cleaved Tie1 45 kDa endodomain to Tie2 may be required for the destabilization of ECs.102 In contrast, Tie1 promoter activity was upregulated by disturbed flow.103
Mechanical stimulation directly or indirectly conveys biological information through the plasma membrane to the cytoskeleton and nucleus, which causes the nucleus to deform. The nuclear fibre layer, namely, the cytoskeleton located on the nuclear membrane, integrates proteins to form a shell that surrounds chromosomes. Therefore, mechanical stimulation acting on the nucleus may be transmitted to the chromosomes through the nuclear fibre layer. The nuclear receptors YAP and TAZ have been recently proposed to be novel nuclear relay sensors and mediators of mechanical cues.104 YAP/TAZ activity is regulated by the stiffness of the ECM and cellular geometries, which requires Rho activity and the actomyosin cytoskeleton. Since laminar SS (15 dynes/cm2, 18 h) lowered the expression of angiotensin-converting enzyme (ACE) and because this response required both extra- and the intracellular domains and ACE Ser1270, Barauna et al proposed that the ACE extracellular domain behaved as a mechanosensor, while the cytoplasmic domain transduced downstream intracellular JNK signalling.105
Perspectives
SS spans a range of spatiotemporal scales and contributes to a series of endothelial-dependent physiological and pathophysiological reactions. The spatiotemporal dynamics elicit activation and inactivation of many molecules. Endothelial mechanosensors and mechanotransduction mechanisms stimulated by atheroprone and atheroprotective SS as reported in relevant literature are summarized in Table 1 and Figure 1. In summary, there are still some challenges in drug design and translational application in this field. First, there is a lack of consensus on the definition of mechanosensors and available detection modalities. Second, information on the precise sensors and signal transduction pathways that determine atheroprone and atheroprotective phenotypes is scarce. Finally, most flow experiments are performed in vitro. Matching in vitro models with in vivo animal models will be crucial for proposing targeted therapies.
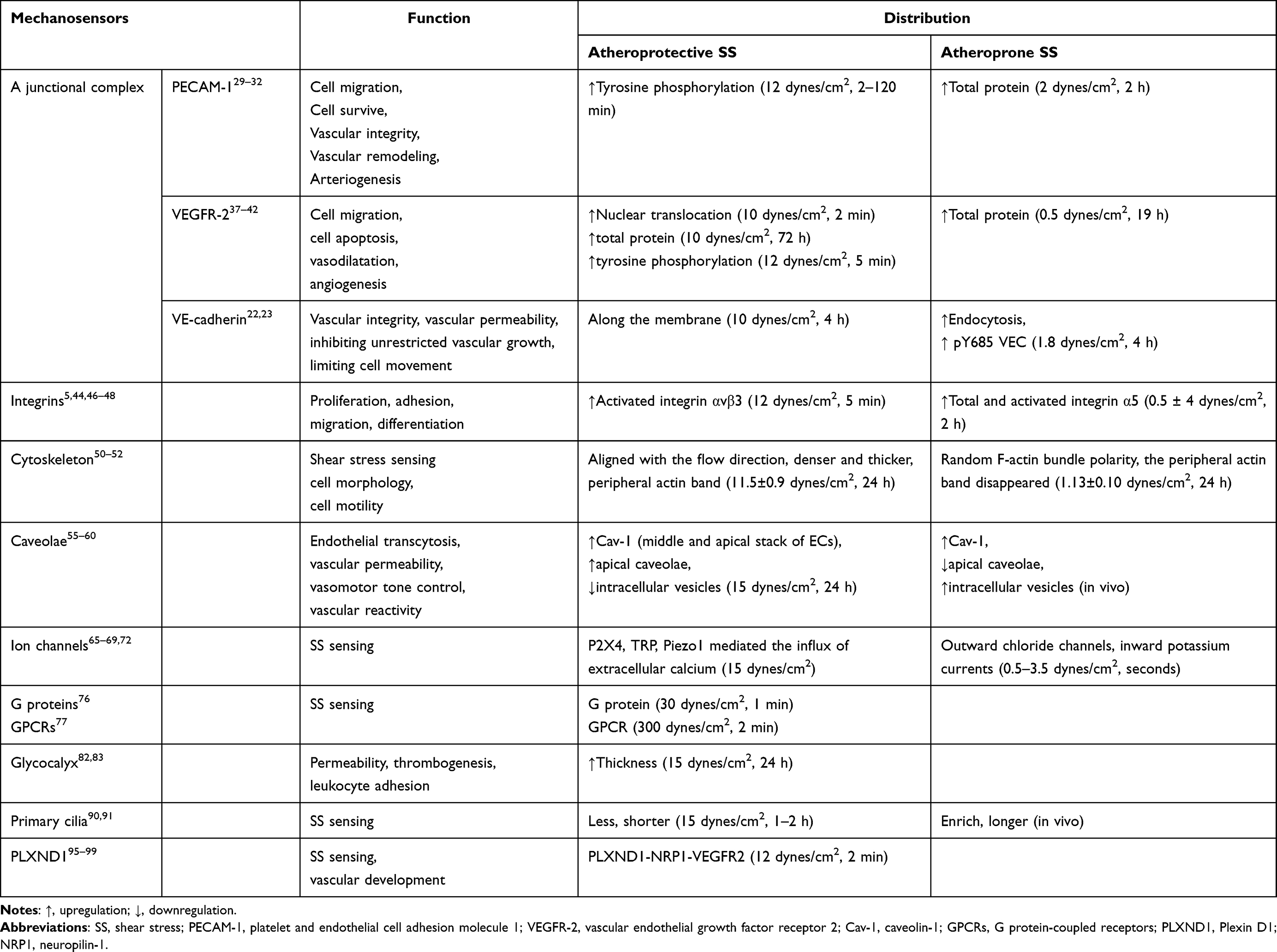 |
Table 1 The Function and Distribution of Endothelial Mechanosensors |
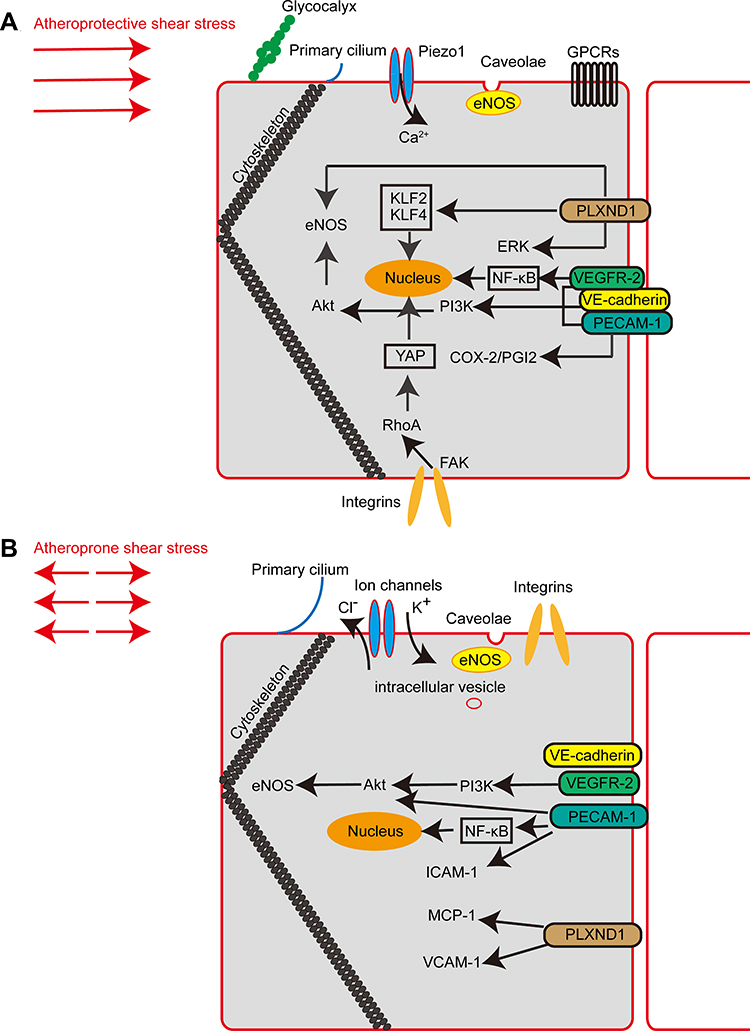 |
Figure 1 Major mechanosensors and signalling pathways involved in endothelial mechanotransduction. (A) Schematic diagram showing endothelial mechanosensors and signalling pathways in atheroprotective shear stress. (B) Schematic diagram showing endothelial mechanosensors and signalling pathways in atheroprone shear stress. |
However, advances in biophysics, molecular cell biology, and genetics have led to new insights into vascular diseases as they relate to hemodynamics and have suggested candidate targets for therapy. As multi-level and multi-dimensional strategies are applied to explore flow-responsive networks, potential mechano-sensitive genes are being discovered, such as SRGN, which was found through an analysis of two high-throughput datasets.106 Performing bioinformatics analyses, Sainz-Jaspeado et al indicated that palmdelphin (PALMD) regulated nuclear actin cap formation in response to mechanical stress. PALMD deficiency impaired nuclear resilience in ECs exposed to flow forces.107 Epigenetic mechanisms, such as DNA de/methylation, histone modifications, and non-coding RNAs, deserve more attention because they are associated with hemodynamic-directed EC dysfunction and the early stages of vascular diseases.108 From the perspective of clinical applications, mechanosensors are involved in the plaque complications in AS. HSS can independently predict sites of acute coronary plaque rupture and erosion.109 The association of plaque progression with impaired EG has been thought to be due to enhanced EG degradation. Nemoto et al showed a correlation between plaque vulnerability and the serum concentration of EG components.110 To determine whether the protection against EG degradation or the restoration of damaged EG is a promising therapeutic strategy, further research is needed. Other mechanosensors may also be involved in different stages of atherosclerosis with other factors. In the future, we can further explore this field.
Acknowledgment
This study is supported by the National Natural Science Foundation of China (NSFC) Grant Nos. 81970309 and 81700398.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Ingber DE. Cellular mechanotransduction: putting all the pieces together again. FASEB J. 2006;20(7):811–827. doi:10.1096/fj.05-5424rev
2. Ingber DE, Wang N, Stamenovic D. Tensegrity, cellular biophysics, and the mechanics of living systems. Rep Prog Phys. 2014;77(4):046603. doi:10.1088/0034-4885/77/4/046603
3. Ando J, Yamamoto K. Vascular mechanobiology: endothelial cell responses to fluid shear stress. Circ j. 2009;73(11):1983–1992. doi:10.1253/circj.CJ-09-0583
4. Hahn C, Schwartz MA. Mechanotransduction in vascular physiology and atherogenesis. Nat Rev Mol Cell Biol. 2009;10(1):53–62. doi:10.1038/nrm2596
5. Zaragoza C, Marquez S, Saura M. Endothelial mechanosensors of shear stress as regulators of atherogenesis. Curr Opin Lipidol. 2012;23(5):446–452. doi:10.1097/MOL.0b013e328357e837
6. Davies PF. Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat Clin Pract Cardiovasc Med. 2009;6(1):16–26. doi:10.1038/ncpcardio1397
7. Zhou J, Li YS, Chien S. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler Thromb Vasc Biol. 2014;34(10):2191–2198. doi:10.1161/ATVBAHA.114.303422
8. Chatterjee S, Fisher AB. Mechanotransduction in the endothelium: role of membrane proteins and reactive oxygen species in sensing, transduction, and transmission of the signal with altered blood flow. Antioxid Redox Signal. 2014;20(6):899–913. doi:10.1089/ars.2013.5624
9. Vogel V, Sheetz M. Local force and geometry sensing regulate cell functions. Nat Rev Mol Cell Biol. 2006;7(4):265–275. doi:10.1038/nrm1890
10. Dekker RJ, van Soest S, Fontijn RD, et al. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Kruppel-like factor (KLF2). Blood. 2002;100(5):1689–1698. doi:10.1182/blood-2002-01-0046
11. Dekker RJ, van Thienen JV, Rohlena J, et al. Endothelial KLF2 links local arterial shear stress levels to the expression of vascular tone-regulating genes. Am J Pathol. 2005;167(2):609–618. doi:10.1016/S0002-9440(10)63002-7
12. Yamashiro Y, Yanagisawa H. The molecular mechanism of mechanotransduction in vascular homeostasis and disease. Clin Sci (Lond). 2020;134(17):2399–2418. doi:10.1042/CS20190488
13. Zhang J, Wang Z, Zhang J, et al. Rapamycin attenuates endothelial apoptosis induced by low shear stress via mTOR and sestrin1 related redox regulation. Mediators Inflamm. 2014;2014:769608. doi:10.1155/2014/769608
14. Zhang JX, Qu XL, Chu P, et al. Low shear stress induces vascular eNOS uncoupling via autophagy-mediated eNOS phosphorylation. Biochim Biophys Acta Mol Cell Res. 2018;1865(5):709–720. doi:10.1016/j.bbamcr.2018.02.005
15. Hergenreider E, Heydt S, Treguer K, et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol. 2012;14(3):249–256. doi:10.1038/ncb2441
16. Li M, Qian M, Kyler K, et al. Endothelial-vascular smooth muscle cells interactions in atherosclerosis. Front Cardiovasc Med. 2018;5:151. doi:10.3389/fcvm.2018.00151
17. Dai Z, Zhu MM, Peng Y, et al. Endothelial and smooth muscle cell interaction via FoxM1 signaling mediates vascular remodeling and pulmonary hypertension. Am J Respir Crit Care Med. 2018;198(6):788–802. doi:10.1164/rccm.201709-1835OC
18. Boezio GL, Bensimon-Brito A, Piesker J, et al. Endothelial TGF-beta signaling instructs smooth muscle cell development in the cardiac outflow tract. Elife. 2020;9. doi:10.7554/eLife.57603
19. Deng Q, Huo Y, Luo J. Endothelial mechanosensors: the gatekeepers of vascular homeostasis and adaptation under mechanical stress. Sci China Life Sci. 2014;57(8):755–762. doi:10.1007/s11427-014-4705-3
20. Zaidel-Bar R. Cadherin adhesome at a glance. J Cell Sci. 2013;126(Pt 2):373–378. doi:10.1242/jcs.111559
21. Tzima E, Irani-Tehrani M, Kiosses WB, et al. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437(7057):426–431. doi:10.1038/nature03952
22. Giannotta M, Trani M, Dejana E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell. 2013;26(5):441–454. doi:10.1016/j.devcel.2013.08.020
23. Caolo V, Peacock HM, Kasaai B, et al. Shear stress and VE-Cadherin. Arterioscler Thromb Vasc Biol. 2018;38(9):2174–2183. doi:10.1161/ATVBAHA.118.310823
24. Conway DE, Breckenridge MT, Hinde E, et al. Fluid shear stress on endothelial cells modulates mechanical tension across VE-cadherin and PECAM-1. Curr Biol. 2013;23(11):1024–1030. doi:10.1016/j.cub.2013.04.049
25. Liu Y, Sweet DT, Irani-Tehrani M, et al. Shc coordinates signals from intercellular junctions and integrins to regulate flow-induced inflammation. J Cell Biol. 2008;182(1):185–196. doi:10.1083/jcb.200709176
26. Coon BG, Baeyens N, Han J, et al. Intramembrane binding of VE-cadherin to VEGFR2 and VEGFR3 assembles the endothelial mechanosensory complex. J Cell Biol. 2015;208(7):975–986. doi:10.1083/jcb.201408103
27. Shay-Salit A, Shushy M, Wolfovitz E, et al. VEGF receptor 2 and the adherens junction as a mechanical transducer in vascular endothelial cells. Proc Natl Acad Sci U S A. 2002;99(14):9462–9467. doi:10.1073/pnas.142224299
28. Walsh TG, Murphy RP, Fitzpatrick P, et al. Stabilization of brain microvascular endothelial barrier function by shear stress involves VE-cadherin signaling leading to modulation of pTyr-occludin levels. J Cell Physiol. 2011;226(11):3053–3063. doi:10.1002/jcp.22655
29. Lertkiatmongkol P, Liao D, Mei H, et al. Endothelial functions of platelet/endothelial cell adhesion molecule-1 (CD31). Curr Opin Hematol. 2016;23(3):253–259. doi:10.1097/MOH.0000000000000239
30. Xie X, Wang F, Zhu L, et al. Low shear stress induces endothelial cell apoptosis and monocyte adhesion by upregulating PECAM-1 expression. Mol Med Rep. 2020;21(6):2580–2588. doi:10.3892/mmr.2020.11060
31. Qin WD, Mi SH, Li C, et al. Low shear stress induced HMGB1 translocation and release via PECAM-1/PARP-1 pathway to induce inflammation response. PLoS One. 2015;10(3):e0120586. doi:10.1371/journal.pone.0120586
32. Fleming I, Fisslthaler B, Dixit M, et al. Role of PECAM-1 in the shear-stress-induced activation of Akt and the endothelial nitric oxide synthase (eNOS) in endothelial cells. J Cell Sci. 2005;118(Pt 18):4103–4111. doi:10.1242/jcs.02541
33. Osawa M, Masuda M, Kusano K, et al. Evidence for a role of platelet endothelial cell adhesion molecule-1 in endothelial cell mechanosignal transduction: is it a mechanoresponsive molecule? J Cell Biol. 2002;158(4):773–785. doi:10.1083/jcb.200205049
34. Russell-Puleri S, Dela Paz NG, Adams D, et al. Fluid shear stress induces upregulation of COX-2 and PGI2 release in endothelial cells via a pathway involving PECAM-1, PI3K, FAK, and p38. Am J Physiol Heart Circ Physiol. 2017;312(3):H485–H500. doi:10.1152/ajpheart.00035.2016
35. Chen Z, Tzima E. PECAM-1 is necessary for flow-induced vascular remodeling. Arterioscler Thromb Vasc Biol. 2009;29(7):1067–1073. doi:10.1161/ATVBAHA.109.186692
36. Chen Z, Rubin J, Tzima E. Role of PECAM-1 in arteriogenesis and specification of preexisting collaterals. Circ Res. 2010;107(11):1355–1363. doi:10.1161/CIRCRESAHA.110.229955
37. Edirisinghe I, Rahman I. Cigarette smoke-mediated oxidative stress, shear stress, and endothelial dysfunction: role of VEGFR2. Ann N Y Acad Sci. 2010;1203:66–72. doi:10.1111/j.1749-6632.2010.05601.x
38. Dela Paz NG, Walshe TE, Leach LL, et al. Role of shear-stress-induced VEGF expression in endothelial cell survival. J Cell Sci. 2012;125(Pt 4):831–843. doi:10.1242/jcs.084301
39. Holmes K, Roberts OL, Thomas AM, et al. Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signal. 2007;19(10):2003–2012. doi:10.1016/j.cellsig.2007.05.013
40. AbouAlaiwi WA, Takahashi M, Mell BR, et al. Ciliary polycystin-2 is a mechanosensitive calcium channel involved in nitric oxide signaling cascades. Circ Res. 2009;104(7):860–869. doi:10.1161/CIRCRESAHA.108.192765
41. Jin ZG, Ueba H, Tanimoto T, et al. Ligand-independent activation of vascular endothelial growth factor receptor 2 by fluid shear stress regulates activation of endothelial nitric oxide synthase. Circ Res. 2003;93(4):354–363. doi:10.1161/01.RES.0000089257.94002.96
42. Schacher NM, Raaz-Schrauder D, Pasutto F, et al. Impact of single nucleotide polymorphisms in the VEGFR2 gene on endothelial cell activation under non‑uniform shear stress. Int J Mol Med. 2019;44(4):1366–1376. doi:10.3892/ijmm.2019.4301
43. Gee E, Milkiewicz M, Haas TL. p38 MAPK activity is stimulated by vascular endothelial growth factor receptor 2 activation and is essential for shear stress-induced angiogenesis. J Cell Physiol. 2010;222(1):120–126. doi:10.1002/jcp.21924
44. Sun X, Fu Y, Gu M, et al. Activation of integrin alpha5 mediated by flow requires its translocation to membrane lipid rafts in vascular endothelial cells. Proc Natl Acad Sci U S A. 2016;113(3):769–774. doi:10.1073/pnas.1524523113
45. Bharadwaj A, Bydoun M, Holloway R, et al. Annexin A2 heterotetramer: structure and function. Int J Mol Sci. 2013;14(3):6259–6305. doi:10.3390/ijms14036259
46. Zhang C, Zhou T, Chen Z, et al. Coupling of integrin alpha5 to annexin A2 by flow drives endothelial activation. Circ Res. 2020;127(8):1074–1090. doi:10.1161/CIRCRESAHA.120.316857
47. Tzima E, Del Pozo MA, Shattil SJ, et al. Activation of integrins in endothelial cells by fluid shear stress mediates Rho-dependent cytoskeletal alignment. EMBO j. 2001;20(17):4639–4647. doi:10.1093/emboj/20.17.4639
48. Wang L, Luo JY, Li B, et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature. 2016;540(7634):579–582. doi:10.1038/nature20602
49. Hsu PL, Chen JS, Wang CY, et al. Shear-induced CCN1 promotes atheroprone endothelial phenotypes and atherosclerosis. Circulation. 2019;139(25):2877–2891. doi:10.1161/CIRCULATIONAHA.118.033895
50. McCue S, Noria S, Langille BL. Shear-induced reorganization of endothelial cell cytoskeleton and adhesion complexes. Trends Cardiovasc Med. 2004;14(4):143–151. doi:10.1016/j.tcm.2004.02.003
51. Helmke BP, Goldman RD, Davies PF. Rapid displacement of vimentin intermediate filaments in living endothelial cells exposed to flow. Circ Res. 2000;86(7):745–752. doi:10.1161/01.RES.86.7.745
52. Avari H, Rogers KA, Savory E. Quantification of morphological modulation, F-actin remodeling and PECAM-1 (CD-31) re-distribution in endothelial cells in response to fluid-induced shear stress under various flow conditions. J Biomech Eng. 2019;141(4). doi:10.1115/1.4042601
53. Anderson CA, Kovar DR, Gardel ML, et al. LIM domain proteins in cell mechanobiology. Cytoskeleton (Hoboken). 2021;78(6):303–311. doi:10.1002/cm.21677
54. Yoshigi M, Hoffman LM, Jensen CC, et al. Mechanical force mobilizes zyxin from focal adhesions to actin filaments and regulates cytoskeletal reinforcement. J Cell Biol. 2005;171(2):209–215. doi:10.1083/jcb.200505018
55. Yu J, Bergaya S, Murata T, et al. Direct evidence for the role of caveolin-1 and caveolae in mechanotransduction and remodeling of blood vessels. J Clin Invest. 2006;116(5):1284–1291. doi:10.1172/JCI27100
56. Parton RG, Tillu VA, Collins BM. Caveolae. Curr Biol. 2018;28(8):R402–R405. doi:10.1016/j.cub.2017.11.075
57. Navarro A, Anand-Apte B, Parat MO. A role for caveolae in cell migration. FASEB J. 2004;18(15):1801–1811. doi:10.1096/fj.04-2516rev
58. Zeng Y, Tarbell JM. The adaptive remodeling of endothelial glycocalyx in response to fluid shear stress. PLoS One. 2014;9(1):e86249. doi:10.1371/journal.pone.0086249
59. Ramirez CM, Zhang X, Bandyopadhyay C, et al. Caveolin-1 regulates atherogenesis by attenuating low-density lipoprotein transcytosis and vascular inflammation independently of endothelial nitric oxide synthase activation. Circulation. 2019;140(3):225–239. doi:10.1161/CIRCULATIONAHA.118.038571
60. Rizzo V, Morton C, DePaola N, et al. Recruitment of endothelial caveolae into mechanotransduction pathways by flow conditioning in vitro. Am J Physiol Heart Circ Physiol. 2003;285(4):H1720–1729. doi:10.1152/ajpheart.00344.2002
61. Radel C, Rizzo V. Integrin mechanotransduction stimulates caveolin-1 phosphorylation and recruitment of Csk to mediate actin reorganization. Am J Physiol Heart Circ Physiol. 2005;288(2):H936–945. doi:10.1152/ajpheart.00519.2004
62. Yamamoto K, Furuya K, Nakamura M, et al. Visualization of flow-induced ATP release and triggering of Ca2+ waves at caveolae in vascular endothelial cells. J Cell Sci. 2011;124(Pt 20):3477–3483. doi:10.1242/jcs.087221
63. Miyawaki-Shimizu K, Predescu D, Shimizu J, et al. siRNA-induced caveolin-1 knockdown in mice increases lung vascular permeability via the junctional pathway. Am J Physiol Lung Cell Mol Physiol. 2006;290(2):L405–413. doi:10.1152/ajplung.00292.2005
64. Fernandez-Hernando C, Yu J, Suarez Y, et al. Genetic evidence supporting a critical role of endothelial caveolin-1 during the progression of atherosclerosis. Cell Metab. 2009;10(1):48–54. doi:10.1016/j.cmet.2009.06.003
65. Gautam M, Shen Y, Thirkill TL, et al. Flow-activated chloride channels in vascular endothelium. Shear stress sensitivity, desensitization dynamics, and physiological implications. J Biol Chem. 2006;281(48):36492–36500. doi:10.1074/jbc.M605866200
66. Suurvali J, Boudinot P, Kanellopoulos J, et al. P2X4: a fast and sensitive purinergic receptor. Biomed J. 2017;40(5):245–256. doi:10.1016/j.bj.2017.06.010
67. Yamamoto K, Korenaga R, Kamiya A, et al. Fluid shear stress activates Ca(2+) influx into human endothelial cells via P2X4 purinoceptors. Circ Res. 2000;87(5):385–391. doi:10.1161/01.RES.87.5.385
68. Liu C, Montell C. Forcing open TRP channels: mechanical gating as a unifying activation mechanism. Biochem Biophys Res Commun. 2015;460(1):22–25. doi:10.1016/j.bbrc.2015.02.067
69. Yao X, Garland CJ. Recent developments in vascular endothelial cell transient receptor potential channels. Circ Res. 2005;97(9):853–863. doi:10.1161/01.RES.0000187473.85419.3e
70. Kang H, Hong Z, Zhong M, et al. Piezo1 mediates angiogenesis through activation of MT1-MMP signaling. Am J Physiol Cell Physiol. 2019;316(1):C92–C103. doi:10.1152/ajpcell.00346.2018
71. Wang S, Chennupati R, Kaur H, et al. Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release. J Clin Invest. 2016;126(12):4527–4536. doi:10.1172/JCI87343
72. Li J, Hou B, Tumova S, et al. Piezo1 integration of vascular architecture with physiological force. Nature. 2014;515(7526):279–282. doi:10.1038/nature13701
73. Ranade SS, Qiu Z, Woo SH, et al. Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proc Natl Acad Sci U S A. 2014;111(28):10347–10352. doi:10.1073/pnas.1409233111
74. John L, Ko NL, Gokin A, et al. The Piezo1 cation channel mediates uterine artery shear stress mechanotransduction and vasodilation during rat pregnancy. Am J Physiol Heart Circ Physiol. 2018;315(4):H1019–h1026. doi:10.1152/ajpheart.00103.2018
75. Baratchi S, Zaldivia MTK, Wallert M, et al. Transcatheter aortic valve implantation represents an anti-inflammatory therapy via reduction of shear stress-induced, piezo-1-mediated monocyte activation. Circulation. 2020;142(11):1092–1105. doi:10.1161/CIRCULATIONAHA.120.045536
76. Gudi S, Nolan JP, Frangos JA. Modulation of GTPase activity of G proteins by fluid shear stress and phospholipid composition. Proc Natl Acad Sci U S A. 1998;95(5):2515–2519. doi:10.1073/pnas.95.5.2515
77. Chachisvilis M, Zhang YL, Frangos JA. G protein-coupled receptors sense fluid shear stress in endothelial cells. Proc Natl Acad Sci U S A. 2006;103(42):15463–15468. doi:10.1073/pnas.0607224103
78. Zeng Y, Sun HR, Yu C, et al. CXCR1 and CXCR2 are novel mechano-sensors mediating laminar shear stress-induced endothelial cell migration. Cytokine. 2011;53(1):42–51. doi:10.1016/j.cyto.2010.09.007
79. Otte LA, Bell KS, Loufrani L, et al. Rapid changes in shear stress induce dissociation of a G alpha(q/11)-platelet endothelial cell adhesion molecule-1 complex. J Physiol. 2009;587(Pt 10):2365–2373. doi:10.1113/jphysiol.2009.172643
80. Song JW, Goligorsky MS. Perioperative implication of the endothelial glycocalyx. Korean J Anesthesiol. 2018;71(2):92–102. doi:10.4097/kjae.2018.71.2.92
81. Pries AR, Secomb TW, Gaehtgens P. The endothelial surface layer. Pflugers Arch. 2000;440(5):653–666. doi:10.1007/s004240000307
82. Sieve I, Munster-Kuhnel AK, Hilfiker-Kleiner D. Regulation and function of endothelial glycocalyx layer in vascular diseases. Vascul Pharmacol. 2018;100:26–33. doi:10.1016/j.vph.2017.09.002
83. Yao Y, Rabodzey A, Dewey CF
84. Dragovich MA, Chester D, Fu BM, et al. Mechanotransduction of the endothelial glycocalyx mediates nitric oxide production through activation of TRP channels. Am J Physiol Cell Physiol. 2016;311(6):C846–C853. doi:10.1152/ajpcell.00288.2015
85. Florian JA, Kosky JR, Ainslie K, et al. Heparan sulfate proteoglycan is a mechanosensor on endothelial cells. Circ Res. 2003;93(10):e136–142. doi:10.1161/01.RES.0000101744.47866.D5
86. Mochizuki S, Vink H, Hiramatsu O, et al. Role of hyaluronic acid glycosaminoglycans in shear-induced endothelium-derived nitric oxide release. Am J Physiol Heart Circ Physiol. 2003;285(2):H722–726. doi:10.1152/ajpheart.00691.2002
87. Pohl U, Herlan K, Huang A, et al. EDRF-mediated shear-induced dilation opposes myogenic vasoconstriction in small rabbit arteries. Am J Physiol. 1991;261(6 Pt 2):H2016–2023. doi:10.1152/ajpheart.1991.261.6.H2016
88. Harding IC, Mitra R, Mensah SA, et al. Pro-atherosclerotic disturbed flow disrupts caveolin-1 expression, localization, and function via glycocalyx degradation. J Transl Med. 2018;16(1):364. doi:10.1186/s12967-018-1721-2
89. Pala R, Alomari N, Nauli SM. Primary cilium-dependent signaling mechanisms. Int J Mol Sci. 2017;18(11):2272. doi:10.3390/ijms18112272
90. Poelmann RE, Van der Heiden K, Gittenberger-de Groot A, et al. Deciphering the endothelial shear stress sensor. Circulation. 2008;117(9):1124–1126. doi:10.1161/CIRCULATIONAHA.107.753889
91. Iomini C, Tejada K, Mo W, et al. Primary cilia of human endothelial cells disassemble under laminar shear stress. J Cell Biol. 2004;164(6):811–817. doi:10.1083/jcb.200312133
92. Hierck BP, Van der Heiden K, Alkemade FE, et al. Primary cilia sensitize endothelial cells for fluid shear stress. Dev Dyn. 2008;237(3):725–735. doi:10.1002/dvdy.21472
93. Pala R, Jamal M, Alshammari Q, et al. The roles of primary cilia in cardiovascular diseases. Cells. 2018;7(12):233. doi:10.3390/cells7120233
94. Nauli SM, Kawanabe Y, Kaminski JJ, et al. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation. 2008;117(9):1161–1171. doi:10.1161/CIRCULATIONAHA.107.710111
95. Tomas-Roca L, Tsaalbi-Shtylik A, Jansen JG, et al. De novo mutations in PLXND1 and REV3L cause Mobius syndrome. Nat Commun. 2015;6:7199. doi:10.1038/ncomms8199
96. Luchino J, Hocine M, Amoureux MC, et al. Semaphorin 3E suppresses tumor cell death triggered by the plexin D1 dependence receptor in metastatic breast cancers. Cancer Cell. 2013;24(5):673–685. doi:10.1016/j.ccr.2013.09.010
97. Zhang Y, Singh MK, Degenhardt KR, et al. Tie2Cre-mediated inactivation of plexinD1 results in congenital heart, vascular and skeletal defects. Dev Biol. 2009;325(1):82–93. doi:10.1016/j.ydbio.2008.09.031
98. Roodink I, Verrijp K, Raats J, et al. Plexin D1 is ubiquitously expressed on tumor vessels and tumor cells in solid malignancies. BMC Cancer. 2009;9:297. doi:10.1186/1471-2407-9-297
99. Mehta V, Pang KL, Rozbesky D, et al. The guidance receptor plexin D1 is a mechanosensor in endothelial cells. Nature. 2020;578(7794):290–295. doi:10.1038/s41586-020-1979-4
100. Chlench S, Mecha Disassa N, Hohberg M, et al. Regulation of Foxo-1 and the angiopoietin-2/Tie2 system by shear stress. FEBS Lett. 2007;581(4):673–680. doi:10.1016/j.febslet.2007.01.028
101. Idowu TO, Etzrodt V, Pape T, et al. Flow-dependent regulation of endothelial Tie2 by GATA3 in vivo. Intensive Care Med Exp. 2021;9(1):38. doi:10.1186/s40635-021-00402-x
102. Woo KV, Baldwin HS. Role of Tie1 in shear stress and atherosclerosis. Trends Cardiovasc Med. 2011;21(4):118–123. doi:10.1016/j.tcm.2012.03.009
103. Porat RM, Grunewald M, Globerman A, et al. Specific induction of tie1 promoter by disturbed flow in atherosclerosis-prone vascular niches and flow-obstructing pathologies. Circ Res. 2004;94(3):394–401. doi:10.1161/01.RES.0000111803.92923.D6
104. Dupont S, Morsut L, Aragona M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474(7350):179–183. doi:10.1038/nature10137
105. Barauna VG, Campos LC, Miyakawa AA, et al. ACE as a mechanosensor to shear stress influences the control of its own regulation via phosphorylation of cytoplasmic Ser(1270). PLoS One. 2011;6(8):e22803. doi:10.1371/journal.pone.0022803
106. Ma Q, Gu W, Li T, et al. SRGN, a new identified shear-stress-responsive gene in endothelial cells. Mol Cell Biochem. 2020;474(1–2):15–26. doi:10.1007/s11010-020-03830-7
107. Sainz-Jaspeado M, Smith RO, Plunde O, et al. Palmdelphin regulates nuclear resilience to mechanical stress in the endothelium. Circulation. 2021;144(20):1629–1645. doi:10.1161/CIRCULATIONAHA.121.054182
108. Karthika CL, Ahalya S, Radhakrishnan N, et al. Hemodynamics mediated epigenetic regulators in the pathogenesis of vascular diseases. Mol Cell Biochem. 2021;476(1):125–143. doi:10.1007/s11010-020-03890-9
109. Thondapu V, Mamon C, Poon EKW, et al. High spatial endothelial shear stress gradient independently predicts site of acute coronary plaque rupture and erosion. Cardiovasc Res. 2021;117(8):1974–1985. doi:10.1093/cvr/cvaa251
110. Nemoto T, Minami Y, Yamaoka-Tojo M, et al. Endothelial glycocalyx and severity and vulnerability of coronary plaque in patients with coronary artery disease. Atherosclerosis. 2020;302:1–7. doi:10.1016/j.atherosclerosis.2020.04.014
111. Wang Z, Wang F, Kong X, et al. Oscillatory Shear Stress Induces Oxidative Stress via TLR4 Activation in Endothelial Cells. Mediators Inflamm. 2019;2019:7162976. doi:10.1155/2019/7162976
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.