Back to Journals » Drug Design, Development and Therapy » Volume 13
Endostatin attenuates PDGF-BB- or TGF-β1-induced HSCs activation via suppressing RhoA/ROCK1 signal pathways
Authors Ren H ![]() , Li Y, Chen Y, Wang L
, Li Y, Chen Y, Wang L
Received 20 October 2018
Accepted for publication 19 December 2018
Published 11 January 2019 Volume 2019:13 Pages 285—290
DOI https://doi.org/10.2147/DDDT.S191617
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
Haitao Ren,1 Yuan Li,2 Yan Chen,3 Liang Wang4
1Department of Burns and Wound Care Center, The Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang 310009, P.R. China; 2Department of Ultrasound, Women’s Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang 310006, P.R. China; 3Emergency Department, The Second Affiliated Hospital of Dalian Medical University, Dalian, Liaoning 116023, P.R. China; 4Department of Hepatobiliary and Pancreatic Surgery, The Second Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang 310009, P.R. China
Aim: To testify the hypothesis that endostatin exerts antifibrotic effects in hepatic stellate cells (HSCs) by modulating RhoA (ras homolog gene family, member A)/ROCK 1 (Rho-associated protein kinase 1) signal pathways.
Materials and methods: HSCs-T6 of passages 3–5 were cultured in DMEM and serum starved for 48 hours. HSCs were grouped as follows: control group, TGF-β1 (transforming growth factor β1) group, endostatin+TGF-β1 group, PDGF-BB (platelet-derived growth factor-BB) group, and endostatin+PDGF-BB group. In the PDGF-BB group, HSCs were treated with PDGF-BB (200 ng/mL) for 72 hours; in the TGF-β1 group, they were treated with TGF-β1 (10 ng/mL) for 72 hours. In the Endostatin+TGF-β1 group or Endostatin+PDGF-BB group, HSCs were treated with TGF-β1 (10 ng/mL) or PDGF-BB (200 ng/mL) for 72 hours after pretreatment with endostatin (5 µg/mL) for 1 hour. In the control group, HSCs were only treated with serum-free DMEM for 72 hours. Collagen I was analyzed with ELISA. F-actin was detected with immunofluorescent staining. The mRNAs and proteins of α-smooth muscle actin, RhoA, and ROCK1 were analyzed by using real-time PCR and Western blot, respectively.
Results: TGF-β1 and PDGF-BB promote the proliferation of HSCs significantly at 48 and 72 hours. Endostatin inhibits the proliferation effect induced by TGF-β1 or PDGF-BB significantly (P<0.01). The expression of collagen I and F-actin was significantly upregulated in both TGF-β1 and PDGF-BB groups than in the control group (P<0.01). Both the collagen I and F-actin expression were downregulated significantly in the endostatin-treated groups (P<0.05). Endostatin significantly inhibited the upregulated expression of α-smooth muscle actin, RhoA, and ROCK1 induced by TGF-β1 or PDGF-BB (P<0.01).
Conclusion: These results suggested that endostatin inhibited TGF-β1- or PDGF-BB-induced fibrosis in HSCs by modulating RhoA/ROCK signal pathways.
Keywords: endostatin, liver fibrogenesis, hepatic stellate cell, signal pathways, fibrosis
Erratum for this paper has been published
Introduction
Hepatic fibrosis is a worldwide health care burden with excessive synthesis and deposition of extracellular matrix (ECM),1 which results in high mortality and morbidity. Unfortunately, no ideal therapies are effective in clinical application Therefore, the research for treating liver fibrosis is highly urgent. Hepatic stellate cells (HSCs) are recognized as liver-specific type of pericytes and can be activated by injury or inflammation. The activated HSCs differentiate into myofibroblasts and produce excessive ECM.
Endostatin is a peptide involved in multiple functions of physiological and pathological processes including angiogenesis, fibrosis, sepsis, and acute kidney injury.2–5 The antifibrotic activity has emerged as a newly attractive function. For example, endostatin was proved to have protective effects against hepatic fibrosis.6 However, the precise molecular mechanisms remain unclear.
Multiple signal pathways have relation to liver fibrosis. For example, RhoA (ras homolog gene family, member A)/ROCK (Rho-associated protein kinase) pathways play important roles in the process of liver fibrosis;7 therefore, it is hypothesized that endostatin inhibits fibrosis by modulating RhoA/ROCK1 pathways. This study was designed to investigate whether endostatin has an effect on RhoA/ROCK1 pathways in a transforming growth factor (TGF)-β1- or platelet-derived growth factor (PDGF)-induced fibrosis cell model.
Materials and methods
Reagents and antibodies
Primary antibodies against α-smooth muscle actin (α-SMA), RhoA, ROCK1, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were purchased from Affinity Biosciences (Cincinnati, OH, USA). The recombinant endostatin was purchased from the Simcere Pharmaceutical Company (Nanjing, P.R. China). Both PDGF-BB and TGF-β1 were purchased from the ProSpec Bio (Rehovot, Israel). The ELISA kits of human collagen I were purchased from the Abbexa Company (Cambridge, UK).
Culture of rat HSCs
The rat HSCs line, HSC-T6, was obtained from ATCC (Manassas, VA, USA). Cell passages 3–5 were cultured in DMEM with 10% fetal bovine serum (Wisent, Canada), and seeded in six-well plates at a density of 5×105 cells per well for 24 hours, then serum-starved for 48 hours before treatment.
Treatments and groups
HSCs were divided into five groups: control group, TGF-β1 group, endostatin+TGF-β1 group, PDGF-BB group, and endostatin+PDGF-BB group. In the TGF-β1 group, HSCs were treated with 10 ng/mL TGF-β1; in the PDGF-BB group, the HSCs were treated with 200 ng/mL PDGF-BB for 72 hours. In the endostatin+TGF-β1 group or endostatin+PDGF-BB group, HSCs were pretreated with endostatin (5 μg/mL) for 1 hour and then treated with TGF-β1 (10 ng/mL) or PDGF-BB (200 ng/mL) for 72 hours. Endostatin was removed before treatment with TGF-β1 or PDGF-BB. In the control group, HSCs were treated only with serum-free DMEM for 72 hours. All cells were cultured in serum-free DMEM.
Cell viability assay
Based on our previous studies of concentration determination, the aim of the cell viability assay was to choose the best working time of treatments. The cell viability was measured by CCK-8 assay. HSC-T6 cells were cultured and seeded in the 96-well microplates at a density of 1×104 per 100 mL, then treated according to the manufacturer’s guidance. The cell viability was measured at 0, 24, 48, and 72 hours. The optical density was measured by using the microplate reader at 450 nm. An additional endostatin-alone treatment group was used to analyze the potential effect of endostatin on the viability of HSCs. HSCs were incubated with endostatin (5 μg/mL) for 1 hour, and then cultured in serum-free DMEM for 72 hours. All experiments were repeated three times.
ELISA assay
HSC-T6 cells were seeded in six-well plates at a density of 5×105/mL, and treated as described above in section “Treatments and groups.” The concentrations of collagen I in the supernatants were detected using ELISA kits according to the manufacturer’s guidance. Absorbance value was measured at 450 nm with the microplate reader (Bio-Rad Laboratories Inc., Hercules, CA, USA). The concentration of type I collagen was measured consistent with the corresponding standard curves. The OD value was measured at 450 nm on a microplate reader (Bio-Rad). The concentrations of collagen I were quantified according to the standard curves. The supernatant fluid was harvested and measured three times.
Immunofluorescent staining
HSCs were treated with 4% (0.04 g/mL) paraformaldehyde solution followed by 0.2% (v/v) Triton X-100. Immunofluorescent staining with the antibody against F-actin was performed according to the manufacturer’s instruction as described previously.8 DAPI reagent (4′,6-diamidino-2-phenylindole, MP Biomedicals, 1:7,500) was used to stain the nucleus.
Real-time PCR analysis (RT-qPCR)
The total RNA was isolated with TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. The expression of target genes was assayed by using the SYBR Green Real-Time PCR system (Thermo Fisher Scientific). The target gene primer sequences are displayed in Table 1. All experiments were repeated three times.
 | Table 1 Primer probe sequences used for real-time PCR |
Western blot analysis
The procedure was performed according to our previous study.3 The proteins were transferred to a polyvinylidene fluoride membrane and then incubated overnight at 4°C with the following primary antibodies: α-SMA (1:10,000 dilution), RhoA (1:1,000 dilution), and ROCK1 (1:1,000 dilution). GAPDH (1:2,000 dilution) was used as an internal control. Secondary antibodies (goat anti-rabbit IgG, 1:2,000 dilution) were then added. The quantification of target protein bands was analyzed with ImageJ software (v.1.60) and normalized to GAPDH. All experiments were repeated three times.
Statistics
The data were analyzed with Prism 6.0 software by using the one-way analysis of variance. The differences with P<0.05 were considered significant.
Results
Endostatin suppressed PDGF-BB- or TGF-β1-induced HSC proliferation
There is no significant difference among these groups at 0 and 24 hours. TGF-β1, 10 ng/mL, or PDGF-BB, 200 ng/mL, significantly promoted HSC-T6 proliferation at 48 and 72 hours (P<0.01, Figure 1). Endostatin significantly suppressed the proliferation effect at 48 and 72 hours in both endostatin-pretreated and endostatin-alone groups (P<0.01). Therefore, we selected 72 hours as the working time in the subsequent experiments.
 | Figure 1 Endostatin inhibits HSC-T6 cells proliferation. |
Effect of endostatin on the expression of collagen I
As shown in Figure 2, PDGF-BB and TGF-β1 induced a significant increase in collagen I protein level. The upregulated expression of collagen I induced by PDGF-BB and TGF-β1 was significantly inhibited by endostatin (P<0.01, Figure 2).
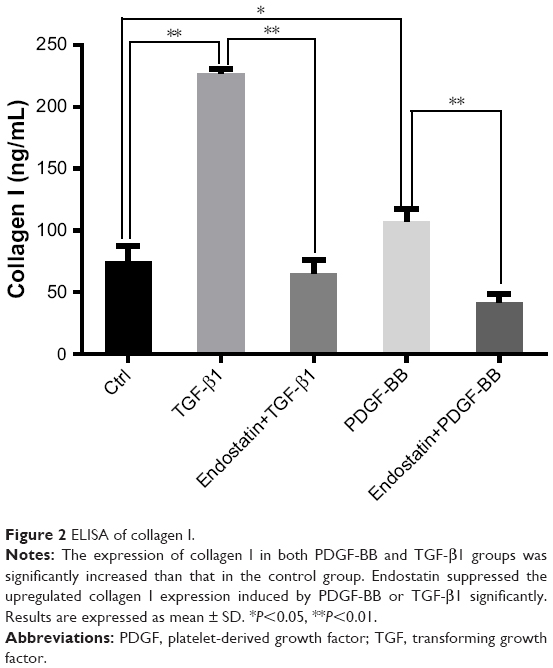 | Figure 2 ELISA of collagen I. |
Endostatin inhibited the expression of F-actin
The expression of F-actin in TGF-β1- or PDGF-BB-induced HSC-T6 cells was significantly upregulated than that in the control group. Endostatin reduced the TGF-β1- or PDGF-BB-induced expression of F-actin significantly (Figure 3, P<0.01).
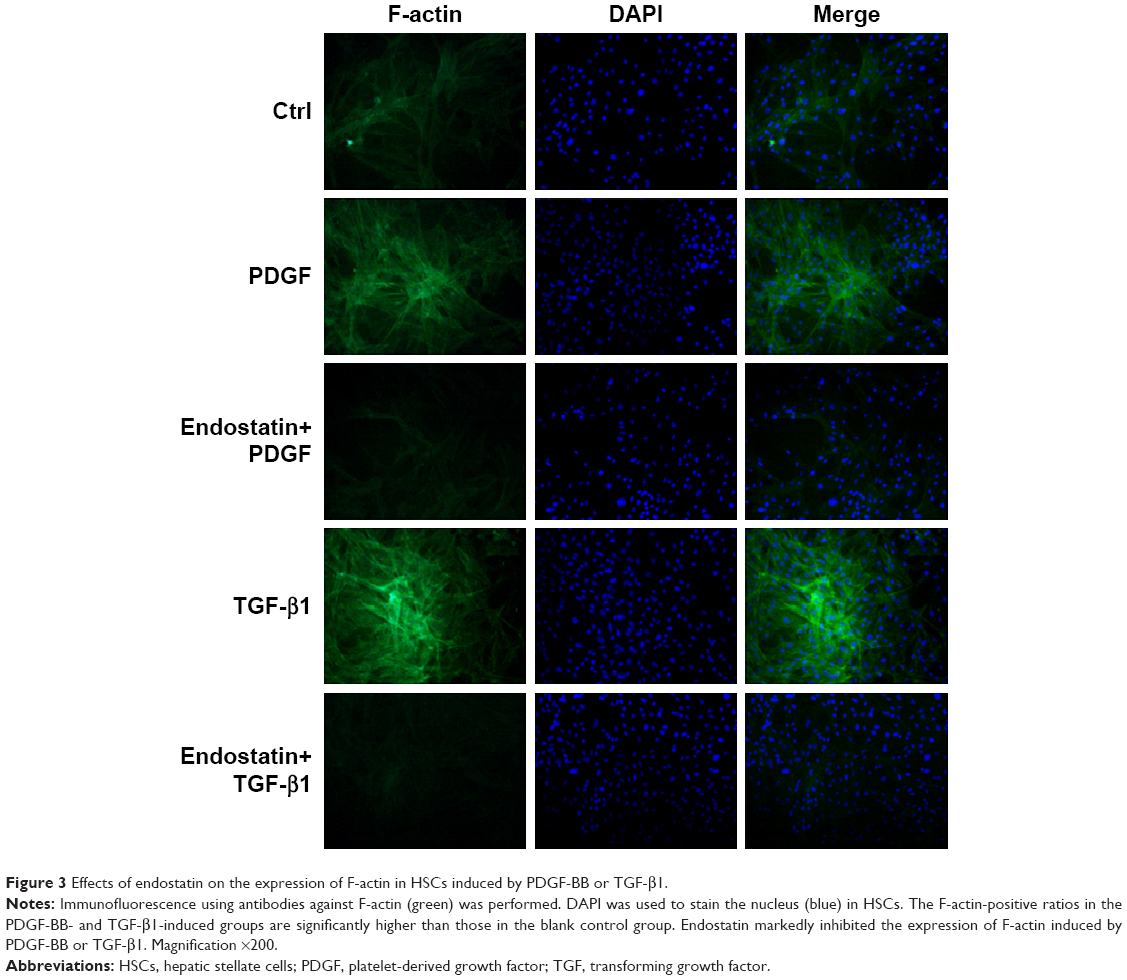 | Figure 3 Effects of endostatin on the expression of F-actin in HSCs induced by PDGF-BB or TGF-β1. |
Endostatin inhibited the expressions of α-SMA, RhoA, and ROCK1 at the mRNA level
The expression of α-SMA, RhoA, and ROCK1 at mRNA level in the PDGF-BB- or TGF-β1-induced groups was significantly higher than that in the control group. Endostatin significantly inhibited the expression of α-SMA, RhoA, and ROCK1 at the mRNA level (P<0.01, Figure 4A–C).
 | Figure 4 Endostatin inhibits the expression of α-SMA, RhoA, and ROCK1 at mRNA level. |
Endostatin inhibited the expressions of α-SMA, RhoA, and ROCK1 at the protein level
The expressions of α-SMA, RhoA, and ROCK1 at protein level were significantly upregulated in the PDGF-BB- and TGF-β1-induced groups. Endostatin significantly inhibited the expressions of α-SMA, RhoA, and ROCK1 at the protein level (P<0.01, Figure 5A–D).
 | Figure 5 Endostatin inhibits the expressions of α-SMA, RhoA, and ROCK1 at protein level. |
Discussion
Hepatic fibrosis is actually the result of a wound-healing process. HSCs are located in the Disse space between hepatocytes and sinusoidal endothelial cells. HSCs have been recognized as liver-specific pericytes.9 HSCs could be activated by stimulations and differentiated into myofibroblasts.10–12 In our study, an in vitro TGF-β1- or PDGF-BB-induced HSCs fibrosis model was used. TGF-β1 and PDGF-BB promote the expressions of collagen I, α-SMA, and F-actin significantly.
Endostatin is the C-terminal of collagen type XVIII present in the liver sinusoidal and basement membrane, and discovered to ameliorate liver fibrosis.13 Our previous studies found that endostatin ameliorates scar formation in the rabbit ear model14 and inhibits fibrosis in human fibroblasts.8,15 In the present study, endostatin significantly inhibited the upregulated expressions of collagen I, α-SMA, and F-actin induced by PDGF-BB and TGF-β1 in HSCs.
Hepatic fibrosis is a complex pathological process that involves a variety of signaling pathways. RhoA/ROCK1 pathways participated in liver fibrosis.16 The transformation from fibroblasts into myofibroblasts has a close relationship with the RhoA/ROCK1 signal pathways.17 RhoA regulates the cell cytoskeleton and activates HSCs.18 The actin filament production is directed by the RhoA/ROCK1 signal pathways.19 RhoA/ROCK1 signaling pathways control cell morphology, proliferation, and adhesion. In addition, ROCK1 plays important roles in PDGF-BB-induced cell proliferation in human aortic vascular smooth muscle cells.20
We have uncovered here a novel mechanism of endostatin to inhibit HSC activation by selectively modulating the RhoA/ROCK pathways. For the limitations, we investigated only the effect of endostatin on RhoA and ROCK1 expressions in mRNA and protein levels. Further research is needed to clarify the molecular mechanisms behind these effects.
Conclusion
Endostatin attenuates PDGF-BB- or TGF-β1-induced HSC activation via suppressing RhoA/ROCK1 signal pathways.
Acknowledgment
This research was supported by Zhejiang Provincial Natural Science Foundation of China under Grant Nos LY15H150004 and LY17H160035, Foundation of Teaching Department of Zhejiang Province (No Y201330073), and Foundation of Health Department of Zhejiang Province (No 2013KYB132).
Disclosure
The authors report no conflicts of interest in this work.
References
Lim YS, Kim WR. The global impact of hepatic fibrosis and end-stage liver disease. Clin Liver Dis. 2008;12(4):733–746. | ||
Mårtensson J, Jonsson N, Glassford NJ, et al. Plasma endostatin may improve acute kidney injury risk prediction in critically ill patients. Ann Intensive Care. 2016;6(1):1–9. | ||
Mårtensson J, Vaara ST, Pettilä V, et al. Assessment of plasma endostatin to predict acute kidney injury in critically ill patients. Acta Anaesthesiol Scand. 2017;61(10):1286–1295. | ||
Peng Y, Gao M, Jiang Y, et al. Angiogenesis inhibitor endostatin protects mice with sepsis from multiple organ dysfunction syndrome. Shock. 2015;44(4):357–364. | ||
Wan YY, Tian GY, Guo HS, et al. Endostatin, an angiogenesis inhibitor, ameliorates bleomycin-induced pulmonary fibrosis in rats. Respir Res. 2013;14(1):56–56. | ||
You Q, Kong LJ, Li FD, et al. Human recombinant endostatin Endostar attenuates hepatic sinusoidal endothelial cell capillarization in CCl4-induced fibrosis in mice. Mol Med Rep. 2015;12(4):5594–5600. | ||
Zhang CG, Zhang B, Deng WS, Duan M, Chen W, Wu ZY. Role of estrogen receptor β selective agonist in ameliorating portal hypertension in rats with CCl4-induced liver cirrhosis. World J Gastroenterol. 2016;22(18):4484–4500. | ||
Li Y, Ren HT. Endostatin inhibits fibrosis by modulating the PDGFR/ERK signal pathway: an in vitro study. J Zhejiang Univ Sci B. 2017;18(11):994–1001. | ||
Greenhalgh SN, Iredale JP, Henderson NC. Origins of fibrosis: pericytes take centre stage. F1000Prime Rep. 2013;5:37. | ||
Aguilera KY, Brekken RA. Recruitment and retention: factors that affect pericyte migration. Cell Mol Life Sci. 2014;71(2):299–309. | ||
Geevarghese A, Herman IM. Pericyte-endothelial crosstalk: implications and opportunities for advanced cellular therapies. Transl Res. 2014;163(4):296–306. | ||
Tacke F, Weiskirchen R. Update on hepatic stellate cells: pathogenic role in liver fibrosis and novel isolation techniques. Expert Rev Gastroenterol Hepatol. 2012;6(1):67–80. | ||
Chen J, Liu DG, Yang G, et al. Endostar, a novel human recombinant endostatin, attenuates liver fibrosis in CCl4-induced mice. Exp Biol Med. 2014;239(8):998–1006. | ||
Ren HT, Hu H, Li Y, Jiang HF, Hu XL, Han CM. Endostatin inhibits hypertrophic scarring in a rabbit ear model. J Zhejiang Univ Sci B. 2013;14(3):224–230. | ||
Ren HT, Li Y, Wang SD, Han CM. Effects of endostatin pretreatment on fibrosis of human skin fibroblasts and the mechanisms. Zhonghua Shao Shang ZaZhi. 2017;33(11):694–698. | ||
You K, Li SY, Gong J, et al. MicroRNA-125b promotes hepatic stellate cell activation and liver fibrosis by activating RhoA signaling. Mol Ther Nucleic Acids. 2018;12:57–66. | ||
Deng H, Xu H, Zhang X, et al. Protective effect of Ac-SDKP on alveolar epithelial cells through inhibition of EMT via TGF-β1/ROCK1 pathway in silicosis in rat. Toxicol Appl Pharmacol. 2016;294:1–10. | ||
Gan DK, Zhu X. Role of RhoA in occurrence and development of liver fibrosis. World Chin J Digestol. 2016;24(11):1682. | ||
Haudek SB, Gupta D, Dewald O, et al. Rho kinase-1 mediates cardiac fibrosis by regulating fibroblast precursor cell differentiation. Cardiovasc Res. 2009;83(3):511–518. | ||
Tang L, Dai F, Liu Y, et al. RhoA/ROCK signaling regulates smooth muscle phenotypic modulation and vascular remodeling via the JNK pathway and vimentin cytoskeleton. Pharmacol Res. 2018;133:201–212. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.