Back to Journals » International Journal of Women's Health » Volume 7
Endometriosis and ovarian cancer: links, risks, and challenges faced
Received 13 March 2015
Accepted for publication 17 April 2015
Published 1 July 2015 Volume 2015:7 Pages 663—672
DOI https://doi.org/10.2147/IJWH.S66824
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Elie Al-Chaer
Mary Ellen Pavone,1 Brianna M Lyttle2
1Division of Reproductive Endocrinology and Infertility, Department of Obstetrics and Gynecology, Northwestern University Feinberg School of Medicine, Chicago, IL, USA; 2Department of Obstetrics and Gynecology, University of Massachusetts Medical School, Worcester, MA, USA
Abstract: Endometriosis is a benign gynecological condition characterized by specific histological, molecular, and clinical findings. It affects 5%–10% of premenopausal women, is a cause of infertility, and has been implicated as a precursor for certain types of ovarian cancer. Advances in technology, primarily the ability for whole genome sequencing, have led to the discovery of new mutations and a better understanding of the function of previously identified genes and pathways associated with endometriosis associated ovarian cancers (EAOCs) that include PTEN, CTNNB1 (β-catenin), KRAS, microsatellite instability, ARID1A, and the unique role of inflammation in the development of EAOC. Clinically, EAOCs are associated with a younger age at diagnosis, lower stage and grade of tumor, and are more likely to occur in premenopausal women when compared with other ovarian cancers. A shift from screening strategies adopted to prevent EAOCs has resulted in new recommendations for clinical practice by national and international governing bodies. In this paper, we review the common histologic and molecular characteristics of endometriosis and ovarian cancer, risks associated with EAOCs, clinical challenges and give recommendations for providers.
Keywords: clinical, molecular, pathways, inflammation, premenopausal
Introduction
Endometriosis is a benign gynecological condition that affects approximately 5%–10% of reproductive aged women causing symptoms of chronic pelvic pain, dyspareunia, dysmenorrhea, and infertility.1,2 Though endometriosis is a benign disease, it has features similar to that of malignant tumors including the ability to develop distant foci through proliferation, attachment, and invasion of the endometrial glandular epithelial tissue to distant organs.1,2 The most commonly affected organs include the ovaries, fallopian tubes, bladder, rectosigmoid colon, and myometrium, and it can be challenging to both identify and treat the condition due to great variability in the age of onset, response to treatment, and clinical presentation.2,3
In 1925, Sampson was the first to describe the relationship between endometriosis and ovarian cancer and proposed a theory of malignant transformation of endometriosis.4 Czernobilsky and Morris later added to this theory by describing an “intermediate stage” in the malignant transformation called “atypical endometriosis”, which today is classified by the degree of dysplastic histologic atypia.5,6 Today it is thought that endometriosis is associated with 15%–50% of clear-cell and endometrioid ovarian tumors and there is a two- to three-fold increase in ovarian cancer in individuals with endometriosis.7–10 Modern research supports the concept of malignant transformation and the current consensus is that the histogenesis of endometriosis is multifactorial, combining genetic, hormonal, and immunological factors.
Recent advances in technology have allowed for further characterization of endometriosis associated ovarian cancers (EAOCs), leading to the identification of genetic and molecular commonalities.3,11 Studies show that the endometriotic tissue has alterations in genes such as TP53, KRAS, and PTEN, in cellular pathways such as those regulated by microsatellites, and the immune microenvironment, directly linked to the development of neoplasms.12–15 Current research is focused on how these molecular changes have allowed for proliferation outside of the uterus and the ultimate transformation to EAOCs.
Here, we review the relationship between endometriosis and ovarian cancer, including common histologic and molecular characteristics of endometriosis and ovarian cancer, the risks affiliated with EAOCs, and the clinical challenges we currently face (Tables 1 and 2).
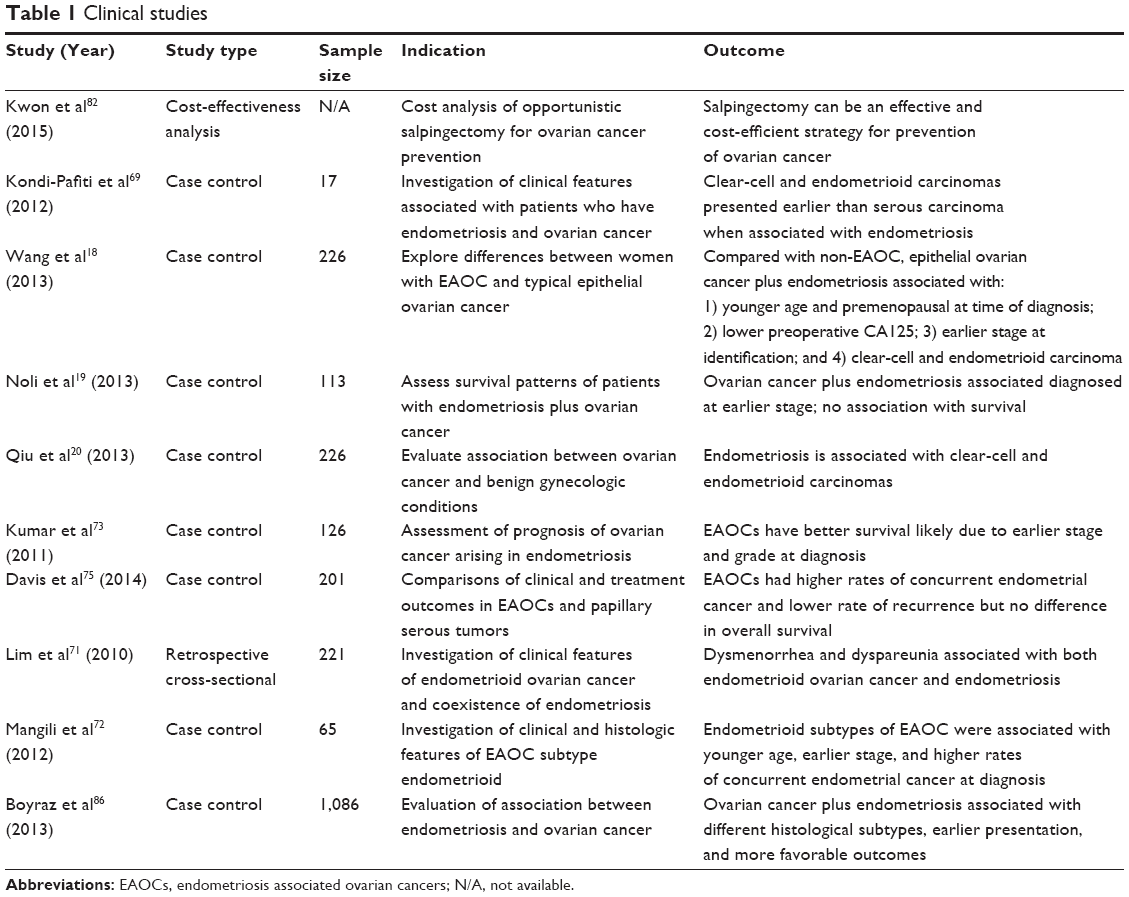 | Table 1 Clinical studies |
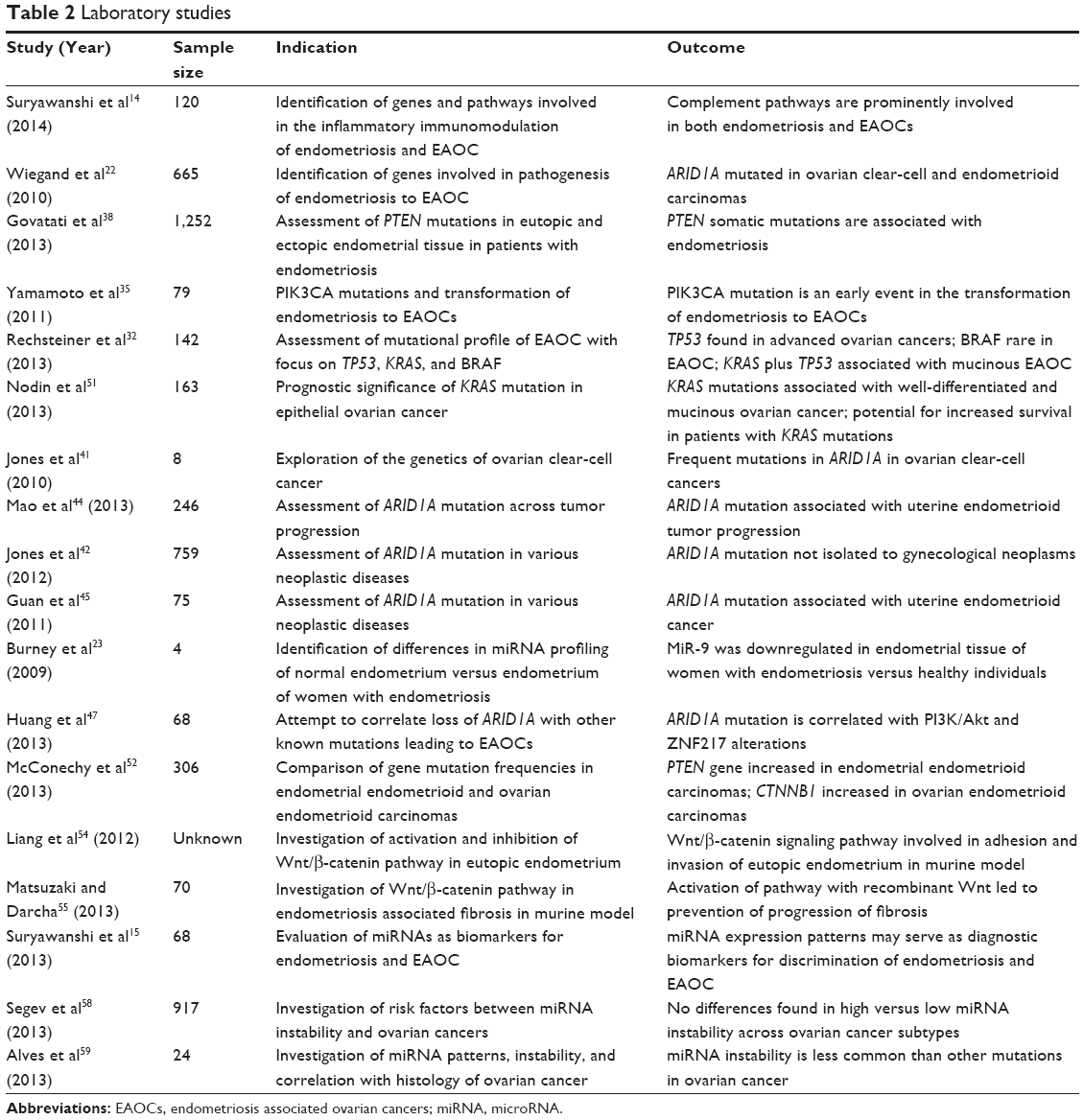 | Table 2 Laboratory studies |
Histologic and molecular characteristics of endometriosis and ovarian cancer
Epithelial ovarian cancer is divided into two subtypes: Type I and Type II. Each of these has a histological and molecular profile correlated with distinct clinical presentations, treatments, and survival outcomes.16 Type I tumors are slow growing and include low-grade serous carcinoma, mucinous carcinoma, endometrioid carcinoma, malignant Brenner tumor, and clear-cell carcinomas. Type II tumors are more aggressive and include high-grade serous carcinoma (moderately and poorly differentiated), malignant mixed mesodermal tumors (carcinosarcomas), and undifferentiated carcinoma.16
EAOCs represent a subclass of Type I ovarian neoplasms that primarily consist of endometrioid and clear-cell subtypes with endometriosis found in 30%–55% of clear-cell and 30%–40% of endometrioid ovarian cancers.7,17–20 The current understanding of the molecular aspects of endometriosis and EAOCs is that they carry distinct mutations in genes and pathways that distinguish them from non-EAOCs. These genes can be grouped into two categories: tumor suppressor genes and oncogenes. The most commonly studied in EAOCs are tumor suppressor genes including TP53, PTEN, and ARID1A while others include the oncogene KRAS.21,22 Recent investigation into pathways involved in the development of both endometriosis and ovarian cancer has provided valuable insight into the development of EAOCs, showing the similarities between the tumor-like inflammation profile of endometriosis and the gene expression of EAOCs.14,23
Tumor suppressors and EAOCs
Tumor suppressors are genes that code for proteins involved in cell cycle regulation and apoptosis. When both copies of this gene are mutated, abnormal cells are able to replicate out of control, leading to cancer.21 Tumor suppressor genes that have been identified as contributors to the development of EAOCs include TP53, PTEN, and ARID1A, which are found in 5%, 20%, and up to 50% of EAOCs, respectively.21,22
TP53 (locus 17p13.1) is one of the most well-studied tumor suppressor genes in cancer biology and encodes for nuclear protein p53.24 The role of the p53 protein is to identify abnormal or damaged cells, enhance cell cycle arrest and DNA repair, or induce apoptosis and senescence if repair is not possible.25 Mutations are related to an overexpression of the nonfunctional form that causes an accumulation of the protein in the nuclei of cells.24,26,27 Mutations in p53 are important precursors in ovarian cancer with some studies suggesting its presence in up to 50% of advanced stage ovarian carcinomas.24,28 However, controversy exists over its involvement in Type I ovarian tumors with high estimates of 10% in clear-cell and 30% in endometrioid ovarian cancers and low estimates of 5% in all Type I ovarian tumors.24 Early studies examining the role of TP53 in the transformation of endometriosis to carcinoma found mutations associated with severe/late stage endometriosis. Statistically significant changes in the rates of p53 expression in the transition from typical to atypical endometriosis have also been observed with the accumulation of the p53 protein found in endometriosis adjacent to clear-cell and endometrioid carcinomas.9,29,30 However, many of these studies had very small sample sizes and did not demonstrate statistically significant differences in these associations.
More recently, it has been suggested that p53 mutations exist in most of the Type II tumors and only in advanced Type I ovarian tumors. It is now most commonly associated with high-grade serous ovarian cancer and advanced ovarian cancers, with 92%–96% of these tumors expressing a mutation in this tumor suppressor gene.28,31,32 With a growing body of evidence supporting that p53 mutations are more often associated with non-EAOCs, there has been a decrease in focus on p53 and its involvement in the transition from endometriosis to EAOCs.
In contrast to the lack of supporting evidence for p53, PTEN, a tumor suppressor gene first identified by Sato et al in 2000, has continued to show involvement in the malignant transformation of endometriosis to ovarian cancer in up to 20% of EAOC. Initial studies found mutations leading to the inactivation and loss of heterozygosity of PTEN (locus 10q23.3) that were associated with endometrioid and clear-cell carcinomas in both endometrial and ovarian cancers.13,33,34 Mutations of PTEN have been found in benign ovarian endometrioid cysts and studies show that the same mutations in PTEN can be found concurrently in both the ovarian cancer and adjacent endometriosis.13,35 Further characterization of the PTEN pathway found that the PTEN mutation is involved in the PI3K signaling pathway with a loss of heterozygosity, resulting in the activation of PI3K and leading to the development of clear-cell subtypes of EAOCs.35,36 Additionally, mouse models have shown that concomitant PI3K/PTEN and Wnt signaling pathways are sufficient to induce EAOCs.37 Recent research by Govatati et al38 has further characterized the transformation of endometriosis to EAOCs. The authors demonstrated increased mutations in PI3K/PTEN and the downstream target, Akt, in the eutopic endometrial tissue of patients with endometriosis, suggesting that the initial mutations occur in eutopic endometrial cells and subsequently migrate out to become an ectopic tissue (endometriosis). There are ongoing studies on the characterization of the downstream effects of PTEN mutations with clinical studies suggesting that PTEN overexpression can enhance the antitumor effects and even reverse the resistance of some chemotherapeutic drugs.39,40
As technology advanced, the capacity to perform genome-wide analyses and the opportunity for whole exome and RNA sequencing to assess somatic mutations in ovarian cancer have developed. In 2010, two independent studies revealed that somatic mutations in ARID1A were major molecular contributors to clear-cell and endometrioid ovarian cancers.22,28,41 Subsequent research has shown that the ARID1A mutation is present in approximately 50% of clear-cell and 40% of endometrioid ovarian cancers.41,42 The majority of ARID1A mutations are either frameshift or nonsense, suggesting its role as a tumor suppressor gene. The protein it encodes, BAF250a, is part of a multiprotein SWI/SNF chromatin remodeling complex involved in the regulation of cellular processes including differentiation, proliferation, DNA repair, and tumor suppression.43,44 In a study of EAOCs, Wiegand et al found 73% of clear-cell and 50% of endometrioid ovarian cancers with an ARID1A mutation have a loss of expression of BAF250a compared to approximately 10% of non-EAOCs. They confirmed this relationship with immunohistochemical studies where they found that silencing of ARID1A resulted in a loss of BAF250a expression.22 Further investigation has found specimens where ARID1A was mutated in both the ovarian tumor and adjacent endometriosis, providing support for its involvement in the progression of endometriosis to carcinoma.
In addition to the role in ovarian cancer, loss of ARID1A has also been demonstrated in 26% of uterine endometrioid carcinomas and has been found to correlate with uterine endometrioid tumor progression from low grade to high grade.44,45 Although somatic ARID1A mutations are found in many gynecological cancers, it appears to be isolated to EAOCs and endometrioid uterine cancers.42,46
Current research is focused on correlating ARID1A mutations with other mutations in the EAOCs and ARID1A potential for use as a prognostic factor for malignant neoplasms. Huang et al recently reported that ARID1A mutations coincide with the activation of the PI3Kc-Akt pathway that has been identified in ovarian clear-cell carcinomas.47
Loss of ARID1A has not yet been identified as a prognostic factor in ovarian cancer; however, preliminary research in breast cancer has suggested that the absence of BAF250a is associated with poorer postoperative disease survival and suggests that it may be useful as a target for breast cancer treatment.48
Genetic changes in EAOCs
While the genes coding for tumor suppressors account for many of the mutations involved in the development of EAOCs, other dysregulations in both genes and cell cycle pathways have been implicated in the development of these diseases.
Oncogenes and cell pathways
Oncogenes are mutant gene forms that when activated contribute to the development of cancer by causing uncontrolled cellular growth and division. KRAS is an oncogene that encodes a GTPase transducer protein involved in regulating cell division by relaying external signals to the cell nucleus. KRAS mutations disrupt the ability of the GTPase transducer protein to switch between active and inactive forms leading to dysregulated cell proliferation.49 Mutations have been found to occur in 3.7%–36.4% of endometrioid cancers, with most studies suggesting a mutation rate of approximately 10%.34 A mouse model comparing the activation of KRAS in endometrial cells deposited in the peritoneum to the activation of KRAS in peritoneal cells showed that the KRAS and PTEN mutations concurrently progressed to endometrioid ovarian cancer.32,34 However, recent evidence suggests that although mutations in KRAS occur in both endometrioid and clear-cell ovarian tumors, they appear to be most prevalent in mucinous, low grade, and well-differentiated ovarian cancers.50,51 Further studies are warranted to determine whether prognosis is impacted by the presence of KRAS, as some research has suggested that survival may be increased in patients with this mutation.51
Another gene involved in the regulation of the cell cycle, CTNNB1, is the gene that encodes the β-catenin protein involved in the Wnt/β-catenin pathway and regulation of cellular processes including proliferation, motility, and survival.37 It is found in up to 40% of endometrioid ovarian carcinomas and both accumulation and depletion of β-catenin may be associated with dysregulation, leading to malignancy.52,53 CTNNB1 has been associated with squamous differentiation, low tumor grade, and a good prognosis in endometrioid ovarian cancers.31,52 Recent in vivo murine models have shown that alterations in the Wnt/β-catenin signaling pathway promote eutopic endometrial cell invasion and adhesion, providing evidence for the involvement of this pathway in the transformation of endometriosis to ovarian cancer.54 Another study investigated the effects of the signaling pathway in the mechanism of endometriosis associated fibrosis and reported that the activation of the Wnt/β-catenin pathway by treatment with recombinant Wnt led to the prevention of fibrosis associated with endometriosis.55
miRNA
miRNAs are endogenous 22-nucleotide-long, highly conserved, noncoding RNAs thought to be primarily involved in the negative regulation of target genes in cell cycle progression, proliferation, and differentiation.23 Studies on miRNA expression have shown downregulation of specific miRNAs in clear-cell and endometrioid ovarian cancers, decreased expression in recurrent versus primary ovarian cancers, and up to 25-fold reductions in moderate-to-severe endometriosis when compared to normal endometrium.23,56,57 miRNA profiling in the plasma of healthy individuals compared to that of individuals with endometriosis and ovarian cancer found that novel miRNA markers could distinguish between these groups with good sensitivity and specificity.15,23 However, subsequent studies comparing expression of high, low, or stable microsatellite instability have failed to find any differences that can distinguish between specific histologic subtypes of ovarian cancers or provide prognostic value.58,59
Microsatellite instability
Microsatellite instability is defined as a size change in short tandem repeat sequences in a tumor compared to normal tissue of the same patient, reflecting the inactivation of the mismatch repair genes. This alteration has been implicated in 7%–22% of sporadic ovarian cancers and is commonly associated with the BRCA1 or BRCA2 germ-line mutations found in inherited ovarian cancers.60,61
Inflammation and the immune system
The role of inflammation in the development of cancers is well known, but in the case of EAOCs it may be of particular relevance. Endometriosis is the site of local inflammatory reactions that are associated with an increase in cytokines and growth factors, leading to angiogenesis, dysregulation of cellular proliferation and apoptosis, invasion, metastasis, and reactive oxygen species known for their damage to DNA.27 This inflammation is thought to promote the growth and invasion of endometriotic tissue, possibly contributing to the histological change from benign endometriosis to what is known as an intermediate stage or “atypical” endometriosis.62 Atypical endometriosis is characterized by large, pleiomorphic nuclei, increased nuclear-to-cytoplasmic ratio, cellular crowding, stratification, and tufting of cells.14 With 60%–80% of EAOCs found to be associated with atypical endometriosis, efforts have been made to better understand the role of inflammation and its contribution to the immune microenvironment. Studies have shown that the peritoneal fluid of women with endometriosis has increased levels of pro-inflammatory cytokines and growth factors such as TNF-α, IL-1, and IL-6 and have serum profiles of inflammatory markers comparable to those found in women with ovarian cancer.63,64 Furthermore, when both eutopic and ectopic endometrial tissue were exposed to upregulators of the inflammatory response, the ectopic tissue was 100 times more sensitive, suggesting a much different molecular profile than its eutopic counterpart.65 Most recently, the evaluation of the tissue immune microenvironment has revealed a specific role for complement proteins in the malignant transformation of endometrial tissue.14 Suryawanshi et al demonstrated that when the KRAS and PTEN pathways were activated, upregulation of complement proteins in epithelial cells were observed. They also found that different immune “profiles” exist for each of eutopic endometrium, endometriotic tissue, and EAOCs. Because of the sensitivity and specificity these immune profiles have for detecting early molecular changes in cells undergoing malignant transformation, it is possible that immune system profiling could lead to early detection of women at risk for EAOCs.14 Continued research into inflammation and the role of the immune system in the development of endometriosis and EAOCs is underway and represents exciting possibilities for new management and treatment strategies of these diseases.
Risks associated with endometriosis and EAOCs
Despite the evidence to support the malignant transformation of endometriosis, the risk of developing an ovarian carcinoma from endometriosis is relatively low with frequencies cited between 0.3% and 0.8%.66,67 However, with endometriosis affecting approximately 10%–15% of all premenopausal women and from 30% to 50% of women with a diagnosis of infertility, it is important to understand the clinical characteristics of EAOCs in an attempt to provide earlier diagnosis and more targeted treatments, and develop prevention strategies for these women.3,68
EAOCs represent a subclass of Type I ovarian neoplasms primarily including endometrioid and clear-cell histology. They have unique clinical characteristics that include age at time of diagnosis, International Federation of Gynecology and Obstetrics (FIGO) stage, CA125 levels, and survival outcomes.69
Age at diagnosis
Studies indicate that at the time of diagnosis, patients who are diagnosed with EAOC are younger, present at an earlier stage, and have a lower grade tumor than those with other ovarian carcinomas.17,18,70,71 One recent study found that patients diagnosed with EAOCs were 6 years younger and 35% more likely to be premenopausal than women with other ovarian cancers. This notion was further supported by Mangili et al who found that the average age at diagnosis for women with EAOCs was 55 years while the average age of women diagnosed with non-EAOCs was 62 years (P=0.03).72
Stage at diagnosis
FIGO stage and grade of ovarian cancer are significantly correlated with 5-year survival rates in ovarian cancer and there is strong evidence to suggest EAOCs are more likely to be diagnosed at both an earlier stage and lower grade53,54 Wang et al18 stated that 88.2% of EAOCs were diagnosed at Stage I versus 15.8% of non-EAOCs. Kumar et al73 found 49% to be FIGO I or II; and Erzen et al74 reported that 67% of EAOCs were diagnosed at Stage I versus 27.6% for non-EAOCs. Similarly, grade I/II tumors were found more commonly in patients who had EAOCs than in those who had other ovarian tumors.18,75 Earlier grade and stage in ovarian cancer predict increased survival at 5 years; thus the diagnosis of an EAOC could mean better clinical outcomes for patients. Davis et al recently compared clinical outcomes of the most common type of ovarian tumor, papillary serous, and outcomes of EAOCs.75 The results showed that compared to papillary serous subtypes, EAOCs were associated with an earlier stage (I/II), lower grade and younger age at time of diagnosis, as well as a lower rate of recurrence.
As reviewed above, most studies confirm that EAOCs are associated with younger ages, earlier stages, and lower grades at diagnosis. However, despite these seemingly beneficial prognostic factors, research has not demonstrated a difference in 5-year survival rates between patients with EAOCs and non-EAOCs when stage was controlled.18,19,72,75
Concurrent cancers at diagnosis
One identified risk for women diagnosed with EAOC is the increased incidence of concurrent malignancy at the time of diagnosis. In up to 10% of women diagnosed with EAOCs, a secondary cancer diagnosis has been made.76 Davis et al found that 23.8% of EAOCs had a concurrent primary cancer diagnosis and that 94.1% were endometrial cancer.75 This finding was replicated by Mangili et al who reported that 40% of patients with EAOCs also had a diagnosis of endometrial cancer.72 Furthermore, of these patients, 33% of them had an endometrioid EAOC subtype and 94% had the same histology in both the ovarian and uterine malignancy. This relationship between EAOCs and endometrial cancer has led to much debate over the molecular mechanisms that dictate the transformation of endometriosis to ovarian cancer.
Screening for endometriosis and EAOCs
Because an earlier stage at diagnosis of ovarian cancer is associated with a better prognosis, women diagnosed with endometriosis represent a unique population that could benefit from a reliable screening tool that helps in earlier diagnosis and treatment. CA125 is a marker used in the surveillance of ovarian tumors and has a high sensitivity, but poor specificity for diagnosis of ovarian cancers. Its relationship in EAOCs has been studied and results are controversial. A recent study by Wang et al18 found that patients with EAOCs had significantly lower levels of presurgical CA125 (122.9 vs 1,377.5). In contrast, most other studies have found no significant difference between patients with EAOCs and non-EAOCs.17,71,72,75
Though molecular advancements have allowed for the identification of unique immune profiles, these have yet to be validated in the clinical setting. Ongoing efforts continue with a goal of identifying markers that can reliably predict if a woman with endometriosis is at a high risk for the development of an EAOC.
Conclusion
Over the last several decades, technology has advanced and allowed for further characterization of the link between endometriosis and EAOCs. Genetic profiling has defined two main categories of epithelial ovarian cancers: Type I and Type II. Type I tumors are the focus of this review and include the less aggressive endometrioid and clear-cell histologies, associated with low-grade, earlier stage at diagnosis and, as some studies suggest, better overall prognosis. Evidence exists to support a unique etiology of these tumors compared to other types of ovarian cancers. Specifically, these tumors are thought to arise as a result of mutations in genes and pathways involved in cell signaling including molecules such as PTEN, ARID1A, and KRAS as well as the CTNNB1 signaling pathway of benign endometriotic tissue. These mutations cause histologic changes that lead to what is thought to be the intermediary stage known as atypical endometriosis and finally to the malignant form of EAOC.31 Though this theory is supported by strong research, we do not yet have conclusive evidence to say that endometriosis is definitively the precursor to EAOCs.
While efforts continue to focus on solidifying this theory, other areas of research include the development of prevention strategies. Though we have not yet found a reliable method to screen for ovarian cancer, research into prevention strategies are promising and provide further insight into the pathogenesis of ovarian cancer.
Two predominant theories surrounding the location of initial malignant transformation of endometrial cells leading to EAOCs have been established. One theory suggests that invasive endometriosis leading to ovarian carcinoma results from phenotypically normal endometrial cells that are displaced into the pelvis by retrograde menstrual flow and develop the adhesive and proliferative properties of endometriosis. Over time, these cells acquire the mutations discussed above and become invasive subsequently leading to ovarian cancer. Another theory proposes that the inciting event is the mutation in eutopic endometrium that allows for endometrial cell migration through the fallopian tubes and proliferation outside of the uterus resulting in an invasive endometriosis phenotype and subsequent carcinoma. This theory is supported by molecular differences originally thought to be isolated to endometriotic tissue, but were later found to also be present in eutopic endometrial tissue of women with endometriosis and absent in endometrial tissue of disease-free women.2,77
Both the above theories propose an endometrial origin of the cells of EAOC that has been most convincingly supported through evidence collected from research into prevention strategies. It is well established that the use of combined oral contraceptives for at least 5 years can reduce the risk of developing ovarian cancer by up to 50%.78 One of the main reasons for this protective effect was thought to be the reduction in the number of ovulatory cycles over a woman’s lifetime.
However, other theories have proposed that it is a thickening of the cervical mucosa that prevents an influx of inflammatory agents leading to the damage of cells that protects against ovarian cancer.79 This latter idea is supported by evidence from studies such as the large meta-analysis by Sieh et al demonstrating a 50% risk reduction in both clear-cell and endometrioid ovarian cancers with tubal ligation by all methods.80
Recently, an emerging body of evidence exists to suggest that the removal of the distal one-third of the fallopian tube significantly reduces a woman’s chance of developing ovarian cancer.81 Though the idea that most ovarian cancers actually originate from the fallopian tube was first postulated in 1896 by Alban Doran, new research is supporting a return to the idea that many of the cancers thought to originate from the ovary are actually of fallopian tube origin. With this in mind, it has been proposed that by removing the tube, the connection between the uterus and the peritoneal cavity, the pathway for endometriotic tissue and carcinogenic inflammants leading to the development of ovarian cancer is eliminated.
A collaborative effort from the British Columbia Ovarian Cancer Research Group operating out of Canada has been one of the biggest research and educational proponents of what is known as the “opportunistic salpingectomy” for prevention of ovarian cancer.82 This procedure, described as the removal of fallopian tubes in a woman undergoing pelvic surgery for another indication, is supported by initial studies that project the reduction in ovarian cancer by this strategy alone to be up to 40%.82,83 If this continues to be supported, then careful contraceptive counseling and efforts to remove fallopian tubes during appropriate procedures by providers can help to reduce the incidence of ovarian cancers, including EAOCs, by preventative methods. Governing bodies, including the American College of Obstetrics and Gynecology and the Society of Gynecology Oncology, currently endorse that risk-reducing salpingectomy should be discussed at the time of abdominal or pelvic surgery, hysterectomy, or lieu of tubal ligation.84,85 Further studies on the long-term effects of these recommendations are underway and it will be interesting to see how this new strategy contributes to the knowledge of both pathogenesis of ovarian cancer and methods in the prevention and protection against this disease.
In conclusion, we have made great strides in our understanding of the association between endometriosis and EAOCs. Through characterization of what was at one time thought to be two independent diseases, we have now proven that endometriosis may be the precursor for Type I epithelial ovarian cancers. Women with endometriosis should be considered at an increased risk for the development of ovarian cancer and providers should pay close attention to changes or worsening of symptoms. Furthermore, major governing bodies support new recommendations for prevention strategies of ovarian cancer through opportunistic salpingectomy and attempts should be made to adopt these into clinical practice. Future research should attempt to answer why only some patients with endometriosis over time will develop an EAOC and how we can identify this subgroup of patients. Our challenge is to continue to build on current research leading to future discoveries that will help us to understand enough about ovarian cancer so that we can significantly reduce the morbidity and mortality associated with this disease.
Acknowledgment
Dr Pavone is partially funded by National Institute of Health grant P50HD076188 and UH3TR001207.
Disclosure
The authors report no conflicts of interest in this work.
References
Somigliana E, Vigano P, Parazzini F, Stoppelli S, Giambattista E, Vercellini P. Association between endometriosis and cancer: a comprehensive review and critical analysis of clinical and epidemiological evidence. Gynecol Oncol. 2006;101(2):331–341. | ||
Bulun SE. Endometriosis. N Engl J Med. 2009;360(3):268–279. | ||
Giudice LC. Clinical practice. Endometriosis. N Engl J Med. 2010;362(25):2389–2398. | ||
Sampson JA. Endometrial carcinoma of the ovary, arising in endometrial tissue in that organ. Arch Surg. 1925;10(1):111–114. | ||
Czernobilsky B, Morris W. A histologic study of ovarian endometriosis and emphasis on hyperplastic and atypical changes. Obstet Gynecol. 1979;53:1080–1084. | ||
Wei J-J, William J, Bulun SE. Endometriosis and ovarian cancer: a review of clinical, pathologic and molecular aspects. Int J Gynecol Pathol. 2011;30(6):553–568. | ||
Brinton LA, Sakoda LC, Sherman ME, et al. Relationship of benign gynecologic diseases to subsequent risk of ovarian and uterine tumors. Cancer Epidemiol Biomarkers Prev. 2005;14:2929–2935. | ||
Rossing MA, Cushing-Haugen KL, Wicklund KG, Doherty JA, Weiss NS. Risk of epithelial ovarian cancer in relation to benign ovarian conditions and ovarian surgery. Cancer Causes Control. 2008;19(10):1357–1364. | ||
Sainz de la Cuesta R, Eichhorn JH, Rice LW, Fuller AFJ, Nikrui N, Goff BA. Histologic transformation of benign endometriosis to early epithelial ovarian cancer. Gynecol Oncol. 1996;60(2):238–244. | ||
Forte A, Cipollaro M, Galderisi U. Genetic, epigenetic and stem cell alterations in endometriosis: new insights and potential therapeutic perspectives. Clin Sci. 2014;126:123–138. | ||
Gazvani R, Templeton A. New considerations for the pathogenesis of endometriosis. Int J Gynaecol Obstet. 2002;76(2):117–126. | ||
Jiang X, Hitchcock A, Bryan EJ, et al. Microsatellite analysis of endometriosis reveals loss of heterozygosity at candidate ovarian tumor suppressor gene loci. Cancer Res. 1996;56:3534–3539. | ||
Sato N, Tsunoda H, Nishida M, et al. Loss of heterozygosity on 10q23.3 and mutation of the tumor suppressor gene PTEN in benign endometrial cyst of the ovary: possible sequence progression from benign endometrial cyst to enodmetrioid carcinoma and clear cell carcinoma of the ovary. Cancer Res. 2000;60(24):7052–7056. | ||
Suryawanshi S, Huang X, Elishaev E, et al. Complement pathway is frequently altered in endometriosis and endometriosis-associated ovarian cancer. Clinical Cancer Research. 2014;20(23):6163–6174. | ||
Suryawanshi S, Vlad AM, Lin HM, et al. Plasma MicroRNAs as novel biomarkers for endometriosis and endometriosis-associated ovarian cancer. Clin Cancer Res. 2013;19(5):1213–1224. | ||
Shih I-M, Kurman RJ. Ovarian tumorigenesis: a proposed model based on morphological and molecular genetic analysis. Am J Pathol. 2004;164(5):1511–1518. | ||
Komiyama S, Aoki D, Tominaga E, Nobuyuki S, Udagawa Y, Nozawa S. Prognosis of Japanese patients with ovarian clear cell carcinoma associated with pelvic endometriosis: clinicopathologic evaluation. Gynecol Oncol. 1999;72(3):342–346. | ||
Wang S, Qiu L, Lang JH, et al. Clinical analysis of ovarian epithelial carcinoma with coexisting pelvic endometriosis. Am J Obstet Gynecol. 2013;208:e.411–e.415. | ||
Noli S, Cipriani S, Scarfone G, et al. Long term survival of ovarian endometriosis associated clear cell and endometrioid ovarian cancers. Int J Gynecol Cancer. 2013;23(2):244–248. | ||
Qiu L, Wang S, Lang JH, et al. The occurrence of endometriosis with ovarian carcinoma is not purely coincidental. Eur J Obstet Gynecol Reprod Biol. 2013;170(1):225–228. | ||
Singer G, Kurman RJ, Chang H-W, Cho SK, Shih I-M. Diverse tumorigenic pathways in ovarian serous carcinoma. Am J Pathol. 2002;160(4):1223–1228. | ||
Wiegand KC, Shah SP, Al-Agha OM, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363:1532–1543. | ||
Burney R, Hamilton A, Aghajanova L, et al. MicroRNA expression profiling of eutopic secretory endometrium in women with versus without endometriosis. Mol Hum Reprod. 2009;15(10):625–631. | ||
Schuijer M, Berns EM. TP53 and ovarian cancer. Hum Mutat. 2003;21(3):285–291. | ||
Brachova P, Mueting SR, Carlson MJ, et al. TP53 oncomorphic mutations predict resistance to platinum- and taxane-based standard chemotheraphy in patients diagnosed with advanced serous ovarian carcinoma. Int J Oncol. 2015;46(2):607–618. | ||
Høgdall EV, Christensen L, Høgdall CK, et al. Distribution of p53 expression in tissue from 774 Danish ovarian tumour patients and its prognostic significance in ovarian carcinomas. APMIS. 2008;116(5):400–409. | ||
Munksgaard PS, Blaakaer J. The association between endometriosis and ovarian cancer: a review of the histological, genetic and molecular alterations. Gynecol Oncol. 2011;124:164–169. | ||
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. | ||
Bischoff FZ, Heard M, Simpson JL. Somatic DNA alterations in endometriosis: high frequency of chromosome 17 and p53 in late-stage endometriosis. J Reprod Immunol. 2002;55(1–2):49–64. | ||
Nezhat F, Cohen C, Rahaman J, Gretz H, Cole P, Kalir T. Comparative immunohistochemical studies of bcl-2 and p53 proteins in benign and malignant ovarian endometriotic cysts. Cancer. 2002;94(11):2935–2940. | ||
Kurman RJ, Shih I-M. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer – shifting the paradigm. Hum Pathol. 2011;42(7):918–931. | ||
Rechsteiner M, Zimmermann AK, Wild PJ, et al. TP53 are common in all subtypes of epithelial ovarian cancer and occur concomitantly with KRAS mutations in the mucinous type. Diagn Pathol. 2013;95(2):235–241. | ||
Martini M, Ciccarone M, Garganese G, et al. Possible involvement of hMLH1, p16(INK4a) and PTEN in the malignant transformation of endometriosis. Int J Cancer. 2002;102(4):398–406. | ||
Dinulescu DM, Ince TA, Quade BJ, Shafer SA, Crowley D, Jacks T. Role of K-ras and Pten in the development of mouse models of endometriosis and endometrioid ovarian cancer. Nat Med. 2005;11:63–70. | ||
Yamamoto S, Tsuda H, Takano M, Iwaya K, Seiichi T, Matsubara O. PIK3CA mutation is an early even in the development of endometriosis-associated ovarian clear cell adenocarcinoma. J Pathol. 2011;225(2):189–194. | ||
Ramaswamy S, Nakamura N, Vazquez F, et al. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci U S A. 1999;96(5):2110–2115. | ||
Wu R, Hendrix-Lucas N, Kuick R, et al. Mouse model of human ovarian endometrioid adenocarcinoma based on somatic defects in the Wnt/beta-catenin and PI3K/Pten signaling pathways. Cancer Cell. 2007;11:321–333. | ||
Govatati S, Kodati VL, Deenadayal M, Chakravarty B, Shivaji S, Bhanoori M. Mutations in the PTEN tumor gene and risk of endometriosis: a case-control study. Hum Reprod. 2014;29(2)324–336. | ||
Li D, Zhang Y, Xie Y, Xiang J, Zhu Y, Yang J. Enhanced tumor suppression by adenoviral PTEN gene therapy combined with cisplatin chemotherapy in small-cell lung cancer. Cancer Gene Ther. 2013;20(4):251–259. | ||
Wu H, Wang K, Liu W, Hao Q. PTEN overexpression improves cisplatin-resistance of human ovarian cancer cells through upregulation of KRT10 expression. Biochem Biophys Res Commun. 2014;444(2):141–146. | ||
Jones S, Wang T-L, Shih I-M, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–231. | ||
Jones S, Li M, Parsons DW, et al. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Hum Mutat. 2012;33(1):100–103. | ||
Reisman D, Glaros S, Thompson E. The SWI/SNF complex and cancer. Oncogene. 2009;28:1653–1668. | ||
Mao TL, Ardighieri L, Ayhan A, et al. Loss of ARID1A expression correlates with stages of tumor progression in uterine endometrioid carcinoma. Am J Surg Pathol. 2013;37(9):1342–1348. | ||
Guan B, Mao TL, Panuganti PK, et al. Mutation and loss of expression of ARID1A in uterine low-grade endometrioid carcinoma. Am J Surg Pathol. 2011;35:625–632. | ||
Kuhn E, Wu RC, Guan B, et al. Identification of molecular pathway aberrations in uterine serous carcinoma by genome-wide analyses. J Natl Cancer Inst. 2012;104(19):1503–1513. | ||
Huang H-N, Lin M-C, Huang W-C, Chiang Y-C, Kuo K-T. Loss of ARID1A expression and its relationship with PI3K-Akt pathway alterations and ZNF217 amplification in ovarian clear cell carcinoma. Modern Pathol. 2013;213:1–8. | ||
Zhao J, Liu C, Zhao Z. ARID1A: a potential prognostic factor for breast cancer. Tumor Biol. 2014;35(5):4813–4819. | ||
Chetty R, Govender D. Gene of the month: KRAS. J Clin Pathol. 2013;66:548–550. | ||
Auner V, Kriegshäuser G, Tong D, et al. KRAS mutation analysis in ovarian samples using a high sensitivity biochip assay. BMC Cancer. 2009;9:111. | ||
Nodin B, Zendehrokh N, Sundstrom M, Jirstrom K. Clinicopathological correlates and prognostic significance of KRAS mutation status in a pooled prospective cohort of epithelial ovarian cancer. Diagn Pathol. 2013;8:106. | ||
McConechy MK, Ding J, Senz J, et al. Ovarian and endometrial endometrioid carcinomas have distinct CTNNB1 and PTEN mutation profiles. Modern Pathol. 2013;27:128–134. | ||
Arend RC, Londono-Joshi AI, Straughn JMJ, Buchsbaum DJ. The Wnt/β-catenin pathway in ovarian cancer: a review. Gynecol Oncol. 2013;131(3):772–779. | ||
Liang J, LI C, ZHang W. Effects of activating and inhibiting Wnt/β-catenin signaling pathway on murine model of eutopic endometrium and endometriosis. Zhonghua Yi Xue Za Zhi. 2012;92(19):1352–1355. | ||
Matsuzaki S, Darcha C. Involvement of the Wnt/β-catenin signaling pathway in the cellular and molecular mechanisms of fibrosis in endometriosis. PLoS One. 2013;8(10):e76808. | ||
Iorio MV, Visone R, Di Leva G, et al. MicroRNA signatures in human ovarian cancer. Cancer Res. 2007;67(18):8699–8707. | ||
Laios A, O’Toole S, Flavin R, et al. Potential role of miR-9 and miR-223 in recurrent ovarian cancer. Mol Cancer. 2008;7:35–48. | ||
Segev Y, Tuya P, Barry R, et al. Risk factors for ovarian cancer with and without microsatellite instability. Int J Gynecol Cancer. 2013;23(6):1010–1015. | ||
Alves LN, Wolfgramm EV, de Castro Neto AK, Louro ID. Analysis of microsatellite instability and loss of heterozygosity in ovarian cancer: a study in the population of Espirito Santo, Brazil. Genet Mol Res. 2013;12(2):1996–2001. | ||
Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–5257. | ||
Pal T, Wey-Permuth J, Kumar A, Sellers TA. Systematic review and meta-analysis of ovarian cancers: estimation of microsatellite-high frequency and characterization of mismatch repair deficient tumor histology. Clin Cancer Res. 2008;14(21):6847–6854. | ||
Ness RB. Endometriosis and ovarian cancer: thoughts on shared pathophysiology. Am J Obstet Gynecol. 2003;189:280–294. | ||
Hou Z, Sun L, Gao L, Liao L, Mao Y, Liu J. Cytokine array analysis of peritoneal fluid between women with endometriosis of different stages and those without endometriosis. Biomarkers. 2009;14:604–618. | ||
Darai E, Detchev R, Hugol D, Quang NT. Serum and cyst fluid levels of interleukin (IL)-6, IL-8 and tumour necrosis factor-alpha in women with endometriomas and benign and malignant cystic ovarian tumours. Hum Reprod. 2003;18(8):1681–1685. | ||
Wu M, Wang C, Lin C, Chen L, Chang W, Tsai J. Distinct regulation of cyclo-oxygenase-2 by interleukin-1beta in normal and endometriotic stromal cells. J Clin Endocrinol Metab. 2005;90:286–295. | ||
Moll U, Chumas J, Chalas E, Mann W. Ovarian carcinoma arising in atypical endometriosis. Obstet Gynecol. 1990;75(3 pt 2):537–539. | ||
Heaps J, Nieberg R, Berek J. Malignant neoplasms arising in endometriosis. Obstet Gynecol. 1990;75:1023–1028. | ||
Eskenazi B, Warner M. Epidemiology of endometriosis. Obstetrics Gynecology Clin North Am. 1997;24:235–258. | ||
Kondi-Pafiti A, Papakonstantinou E, Lavazzo C, Grigoriadis SN, Gregoriou O. Clinicopathological characteristics of ovarian carcinomas associated with endometriosis. Arch Gynecol Obstet. 2012;285(2):479–483. | ||
McMeekin DS, Burger RA, Manetta A, DiSaia P, Berman ML. Endometrioid adenocarcinoma of the ovary and its relationship to endometriosis. Gynecol Oncol. 1995;59(1):81–86. | ||
Lim MC, Chun KC, Shin SJ, et al. Clinical presentation of endometrioid epithelial ovarian cancer with concurrent endometriosis: a multicenter retrospective study. Cancer Epidemiol Biomarkers Prev. 2010;19:398–404. | ||
Mangili G, Bergamini A, Taccagni G, et al. Unraveling the two entities of endometrioid ovarian cancer: a single center clinical experience. Gynecol Oncol. 2012;126(3):403–407. | ||
Kumar S, Munkarah A, Aravi, H, et al. Prognostic analysis of ovarian cancer associated with endometriosis. Am J Obstet Gynecol. 2011; 204(1)63. | ||
Erzen M, Rakar S, Klancnik B, Syrjanen K. Endometriosis-associated ovarian carcinoma (EAOC): an entity distinct from other ovarian carcinomas as suggested by a nested case-control study. Gynecol Oncol. 2001;83(1)100–108. | ||
Davis M, Rauh-Hain JA, Andrade C, et al. Comparison of clinical outcomes of patients with clear cell and endometrioid ovarian cancer associated with endometriosis to papillary serous carcinoma of the ovary. Gynecol Oncol. 2014;132(3):760–766. | ||
Zaino R, Whitney C, Brady MF, DeGeest K, Burger RA, Buller RE. Simultaneously detected endometrial and ovarian carcinomas – a prospective clinicopathologic study of 74 cases: a Gynecologic Oncology Group Study. Gynecol Oncol. 2001;83(2):355–362. | ||
Tseng JF, Ryan IP, Milam TD, et al. Interleukin-6 secretion in vitro is up-regulated in ectopic and eutopic endometrial stromal cells from women with endometriosis. J Clin Endocrinol Metab. 1996;81(3):1118–1122. | ||
Risch H, Weiss NS, Lyon J, Daling J, Liff J. Events of reproductive life and the incidence of epithelial ovarian cancer. Am J Epidemiol. 1983;117:128–139. | ||
Ness RB, Cottreau C. Possible role of ovarian epithelial inflammation in ovarian cancer. J Natl Cancer Inst. 1999;91(17):1459–1467. | ||
Sieh W, Salvador S, McGuire V, et al; Ovarian Cancer Association Consortium. Tubal ligation and risk of ovarian cancer subtypes: a pooled analysis of case-control studies. Int J Epidemiol. 2013;42(2):579–589. | ||
Lessard-Anderson CR, Handlogten KS, Molitor RJ, et al. Effect of tubal sterilization technique on risk of serous epithelial ovarian and primary peritoneal carcinoma. Gynecol Oncol. 2014;135(3):423–427. | ||
Kwon JS, McAlpine JN, Hanley GE, et al. Costs and benefits of opportunistic salpingectomy as an ovarian cancer prevention strategy. Obstet Gynecol. 2015;125(2):338–345. | ||
McAlpine JN, Hanley G, Woo M. Opportunistic salpingectomy: uptake, risks, and complications of a regional initiative for ovarian cancer prevention. Am J Obstet Gynecol. 2014;210(471):e1–e11. | ||
Salpingectomy for ovarian cancer prevention. Committee Opinion No 620. American College of Obstetricians and Gynecologists. Obste Gynecol. 2015;125:279–281. | ||
SGO Clinical Practice Statement: Salpingectomy for Ovarian Cancer Prevention [webpage on the Internet]. Society of Gynarcologic Oncology [cited November 2013]. Available from: https://www.sgo.org/clinical-practice/guidelines/sgo-clinical-practice-statement-salpingectomy-for-ovarian-cancer-prevention/. Accessed June 2, 2015. | ||
Boyraz G, Selcuk I, Yaziciouglu A, Tuncer ZS. Ovarian carcinoma associated with endometriosis. Eur J Obstet Gynecol Reprod Biol. 2013;170(1):211–213. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.