")
Back to Archived Journals » Core Evidence » Volume 14
Enasidenib in the treatment of relapsed/refractory acute myeloid leukemia: an evidence-based review of its place in therapy
Received 23 December 2018
Accepted for publication 31 March 2019
Published 26 April 2019 Volume 2019:14 Pages 3—17
DOI https://doi.org/10.2147/CE.S172912
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Maria Galkin,1 Brian A Jonas2
1Department of Internal Medicine, University of California Davis School of Medicine, Sacramento, CA, USA; 2Department of Internal Medicine, Division of Hematology and Oncology, University of California Davis School of Medicine, Sacramento, CA, USA
Introduction: Acute myeloid leukemia (AML) remains a disease with high mortality, especially for older patients and those with relapsed/refractory (R/R) disease. With recent advances in molecular testing, targeting particular leukemogenic mutations such as those occurring in isocitrate dehydrogenase (IDH) became possible. Enasidenib is a new small-molecule inhibitor of mutant isocitrate dehydrogenase-2 (IDH2).
Aim: The objective of this article is to review the evidence for the use of enasidenib in R/R AML, as well as to outline future directions of enasidenib therapy.
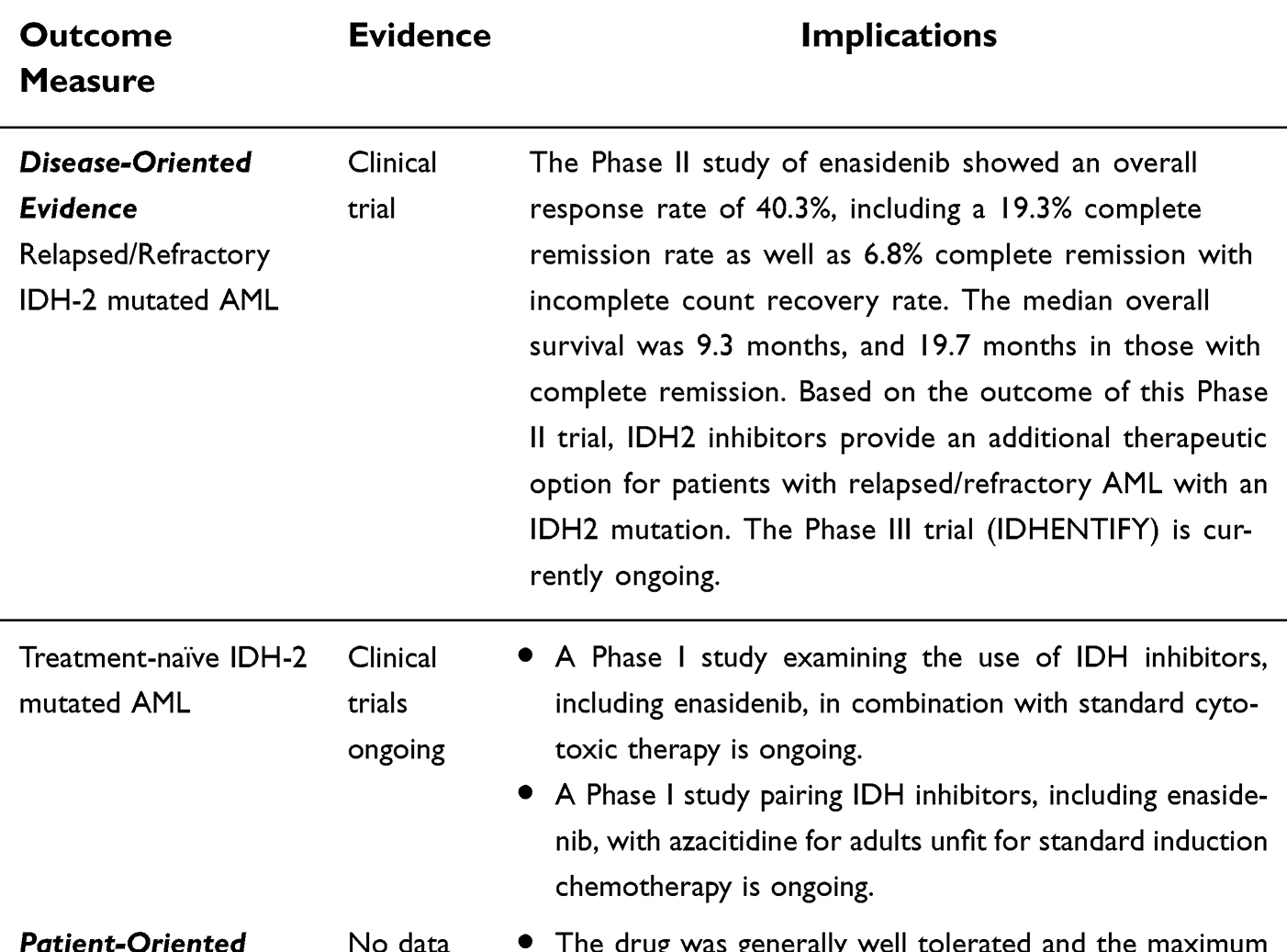
Evidence Review: Enasidenib was approved in August 2017, after a successful Phase I/II trial showing an overall response rate (ORR) of 40.3% in R/R disease, with 19.3% of patients achieving complete remission (CR). Enrollees in the trial were mostly older adults. The most prominent toxicities were hyperbilirubinemia and IDH-differentiation syndrome (IDH-DS), though the drug was generally well tolerated and the maximum tolerated dose was not reached. A Phase III trial is currently ongoing.
Conclusion: Enasidenib provides a new therapeutic option for patients with R/R AML. Further studies are ongoing to ascertain its role in combination with other agents and newly diagnosed disease.
Keywords: AML, enasidenib, IDH-2 inhibitor, Idhifa
Core Evidence Clinical Impact Summary
 |
Introduction
Acute myeloid leukemia (AML) is a heterogeneous hematologic malignancy with high mortality, characterized by excess clonal myeloid blasts that infiltrate the bone marrow and other tissues, as well as related cytopenias. Patients can initially present with constitutional symptoms, infection, bleeding or may be asymptomatic. Without therapy, this disease is fatal usually due to infection or hemorrhage.1 It therefore remains an urgent and complex disease to treat.
Despite available therapy, long-term survival rates are no higher than 45% for younger patients and 20% for older patients.2,3 In refractory disease, long-term survival rates are less than 10%.3 Thus, further research into new therapies, especially for relapsed and/or refractory (R/R) AML, are necessary.
The mainstay of initial treatment for eligible patients remains cytotoxic chemotherapy with 7+3 (cytarabine +anthracycline), which is used most commonly.4 Treatments for older patients can be more limited by performance status and organ function, but can include hypomethylating agents (HMA) such as azacitidine and decitabine.4
With growing knowledge of driver mutations in AML and new molecular testing (which is becoming standard at many centers), there is an emerging landscape of additional targets for treatment. One such target is isocitrate dehydrogenase 1/2 (IDH1/IDH2) mutations, seen in approximately 20% (some figures as high as 33%) of AML cases.5,6 Currently, there are two US Food and Drug Administration (FDA) approved IDH inhibitors—ivosidenib, an IDH1 inhibitor approved in 2018, and enasidenib (AG-221), an IDH2 inhibitor approved in 2017.7,8 Here, we aim to review enasidenib and its potential in treatment of relapsed and refractory AML.
AML background
Treatment of R/R AML
Relapsed and refractory AML poses an unfortunately common challenge to clinicians, with a typical median overall survival of approximately 6 months with active therapy.9–11 Treatment decisions depend largely on patient factors, including performance status, comorbidities and social support, as well as disease factors, such as cytogenetics, molecular analysis, relapsed/refractory status and prior treatments. Goals of therapy also differ for each patient, and may be allogeneic transplant for the fit patient, versus extending life or improving the quality of life without transplant for the unfit.
For the fit patient with relapsed/refractory disease, high-intensity options include salvage therapy with regimens such as fludarabine, cytarabine and idarubicin (FLAG-IDA), high-dose cytarabine and mitoxantrone, etoposide and cytarabine (MEC), among others, in preparation for allogeneic stem cell transplantation.4,12 For both the fit and unfit patient, enrollment in an appropriate clinical trial is recommended.2,13,14
For the unfit patient, the clinician may additionally consider treatment with low-dose cytarabine (LDAC), hypomethylating agents (HMA) or tyrosine kinase inhibitors for FMS-like tyrosine kinase internal tandem duplication (FLT3-ITD) mutated disease. Moreover, in addition to enasidenib and ivosidenib, an increasing number of targeted therapies are being approved by the FDA. Midostaurin, approved in 2017 for the treatment of FLT3-mutated AML, was the first AML drug approved since 2000.15 Gemtuzumab ozogamicin was also approved in 2017 for both newly diagnosed and relapsed/refractory CD33+ disease.16 Other agents, such as BCL-2 (B-cell lymphoma 2) directed therapies, novel FLT-3 inhibitors and other IDH inhibitors are being actively studied and incorporated into treatment algorithms.17 While the landscape of AML treatment is rapidly changing, offering new hope, patients who are not currently candidates for these therapies based on either patient or disease factors, or personal preference, may be managed with hydroxyurea, best supportive care or enter hospice programs.
IDH mutations and AML
Approximately 8–19% of AML cases carry IDH2 mutations, with another 7–14% carrying IDH1 mutations.5 IDH is an enzyme which catalyzes the conversion of isocitrate to alpha-ketoglutarate in the Tricarboxylic Acid Cycle (TCA) and peroxisomes. IDH1 is found in the cytoplasm, while IDH2 is present in mitochondria.18 The two most common mutated forms of IDH2 seen in myeloid neoplasms are IDH2-R140 and IDH2-R172, with IDH2-R140 being the more common; the mutations occur at conserved arginine residues.19,20 The mutant forms of the enzyme are able to produce an oncogenic product called R-2-hydroxyglutarate (2-HG) from alpha-ketoglutarate. 2-HG accumulates and arrests DNA and histone demethylation pathways (many of which are dependent on alpha-ketoglutarate), thereby stopping hematopoietic differentiation.21
IDH1/2 mutations are believed to occur early in leukemogenesis. In fact, they are present in 5% of myelodysplastic syndrome cases and occasionally in myeloproliferative neoplasms, further supporting that they can be present in pre-leukemic states.18,22,23 IDH1/2 mutants are frequently exclusive of each other, though myeloid malignancies with both have been described.23,24 IDH mutant cells also frequently retain a copy of the wild-type (WT) allele.22 Interestingly, IDH mutations appear to also be exclusive of mutations in TET2, another enzyme involved in epigenetic regulation.22 IDH1/2 mutations, by increasing the cell concentrations of 2-HG, inhibit TET2 enzymes and similarly cause hypermethylation.22,25 Studies have additionally shown that IDH mutations are nearly exclusive of Wilms tumor 1 mutations (WT1). WT1 is a transcription factor which physically interacts with TET2. Mutant WT1 interferes with normal TET2 function and therefore cause a similar hypermethylation phenotype.19,26,27 Thus, IDH, TET2 and WT1 mutations alter the epigenetic expression of cells in analogous ways; these are shown in Figure 1.28 Mutant WT1, specifically, is very commonly overexpressed in AML; its presence and levels may be useful markers for detection of minimal residual disease as well as prognostication.27,29,30 Given that IDH and WT1 do not frequently co-occur, this may be a less useful strategy in IDH-mutated disease.
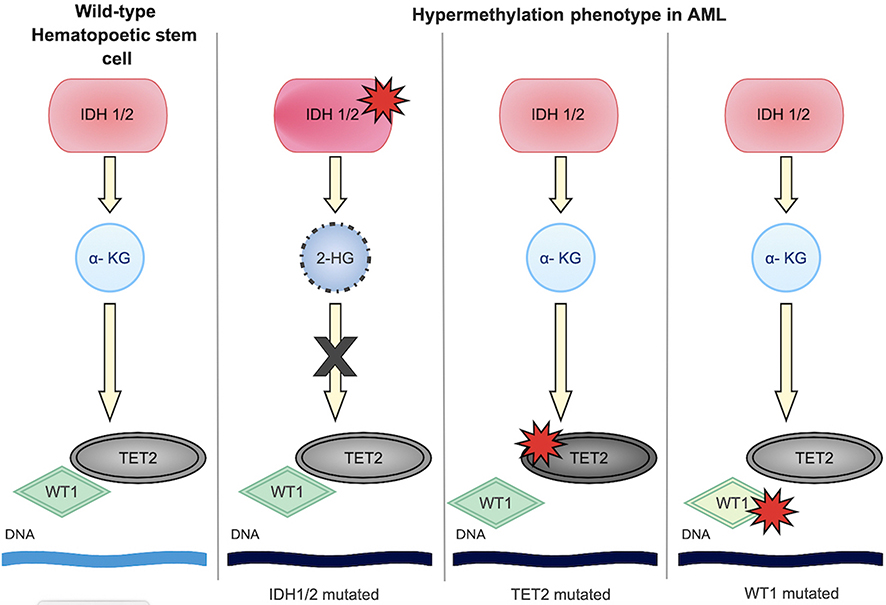 | Figure 1 Hypermethylation phenotype in acute myeloid leukemia (AML). IDH1/2, TET2 and WT1 mutations are often mutually exclusive, but similarly lead to hypermethylation and arrest of differentiation. |
2-HG has additional effects on the cell, including alterations of hypoxia-inducible factor (HIF) pathways.25 HIFs respond to low oxygen conditions and activate transcription, particularly in genes which participate in cell proliferation. Aberrant HIF expression in the absence of hypoxia has been noted in tumorigenesis. HIF is modulated by factor inhibiting HIF (FIH), which requires alpha-ketoglutarate for activity. Thus, it is proposed that 2-HG may inhibit FIH and increase HIF expression, and therefore modulate cell proliferation. However, there are various factors which may influence HIFs and this relationship continues to be studied.25
IDH mutations have been detected with increased frequency in older individuals, and appear to have increased presence in AML with normal karyotype and intermediate-risk cytogenetics groups.31 Additionally, links with co-occurring NPM1 and FLT3-ITD mutations have been reported.24 Overall, the prognostic value of IDH mutations remains unclear at this time and more data are necessary, though the prognostic impact of co-occurring NPM1 and FLT3-ITD appear to supersede IDH status.23,32 It should be noted that studies that have examined prognostic impact of IDH mutations thus far studied different patient populations and, at times, used different designations of IDH status (for example, combining IDH1/IDH2 mutations, or, combining IDH2-R140 and IDH1-R172 under a single designation). Each of these mutant IDH subtypes may need to be studied individually to truly ascertain prognostic value.24
Currently, there are two approved medications targeting IDH—Ivosidenib, targeting IDH1, and Enasidenib, targeting IDH2.7,8 We present the available data on enasidenib.
Enasidenib
Preclinical data
In addition to further elucidating the pathogenic mechanisms of mutant IDH1/2, preclinical data on enasidenib offered new potential in AML therapy. Using 2-HG levels as a surrogate of drug activity, Yen et al were first able to use enasidenib to lower 2-HG levels and induce differentiation in an IDH2 mutant TF-1 erythroleukemia cell line, suggesting that the presence of an IDH2 mutation blocked erythropoietin-dependent cellular maturation. This was then expanded to show that enasidenib reduced 2-HG levels in AML blasts ex vivo. Further, in mouse xenograft models, enasidenib was able to lower 2-HG by more than 90% and successfully reestablish myeloblast differentiation. In primary human AML xenografts, enasidenib-treated mice showed dramatic results, including 2-HG levels which approached normal, with 2 to 35-fold reduction in blasts on bone marrow analysis on day 38 of treatment, while untreated mice displayed human AML cells disseminated to both hematopoietic and nonhematopoietic organs. It was also noted that enasidenib is not cytotoxic to IDH2 mutant cells; rather, it induces a differentiation effect, and mature neutrophils which develop in cells treated with enasidenib continue to carry the mutant allele.33
Based on the successes of preclinical studies, a multi-center Phase I/II trial was initiated by Agios and Celgene, ultimately leading to approval of enasidenib.7 Below, we review the prominent findings, including results and toxicities. The Phase III trial is currently ongoing.34
Formulation
Enasidenib is an oral, small molecule agent, currently approved to be taken at a dose of 100 mg daily.6,7,35
Pharmacokinetics/pharmacodynamics
Enasidenib has a half-life of 137 hrs after multiple doses and reached steady state by cycle 2 within the Phase I/II study (28-day cycles); this study additionally established 100 mg as efficacious in maintaining steady-state drug concentrations. Patients treated with 100 mg daily achieved a median 93% plasma reduction of 2-HG in those with IDH2-R140 mutations, and a median of 28% (though with a maximum of 94% plasma reduction of 2-HG) in IDH2-R172 mutated AML.6
Enasidenib is metabolized via CYP and UGT enzymes. It can notably induce enzymes CYP3A4 and CYP2B6. However, the overall effects on other medications are not clear at this time, as it also inhibits some hepatic enzymes. As such, caution should be exercised in regard to medication interactions. For example, enasidenib can both increase and decrease oral contraception concentrations. In terms of cardiac pharmacodynamics, enasidenib is not QT prolonging.36
Trial description
The Phase I/II trial enrolled 239 patients. 113 patients were in the dose-escalation phase, during which doses of either 30, 50, 75, 100 or 150 mg were administered twice per day, or doses of 50, 75, 100, 150, 200, 300, 450 and 650 mg once per day; all were administered in continuous 28-day cycles. 126 patients were enrolled in the 4-arm expansion phase using the 100 mg once daily dose, which was chosen due to good steady-state drug concentrations as well as ample clinical activity. The expansion phase compared patients who were 60 years or older with relapsed/refractory AML (including those who have relapsed after allogeneic hematopoietic stem cell transplant), patients younger than 60 who were not transplanted but who had relapsed/refractory AML, age 60 or older and not candidates for induction chemotherapy, or patients who were not eligible for the other expansion arms. Notably, the median age among all patients on the trial was 70.6
In the overall study enrollment, 27% of patients were older than 75, with 24% among those who received 100 mg enasidenib daily. Notably, 26% of patients received greater than 3 prior AML treatments (22% in the enasidenib 100 mg subgroup). 18% of patients had an ECOG score of 2 (15% in the enasidenib 100 mg subgroup); most patients had an ECOG of 0 or 1.37 Though the authors did not report additional medical comorbidities for these patients, the represented group included many elderly and relapsed/refractory patients (including 16% of those who relapsed post-transplant), thus studying a vulnerable population of AML patients.37
Efficacy
The results of the Phase I/II trial of Enasidenib are summarized in Table 1. The overall response rate (ORR, defined as complete remission, complete remission with incomplete count recovery, partial remission and morphological leukemia free state) was 40.3% in relapsed/refractory disease; for the group which received 100 mg daily, the ORR was 38.5%. The overall complete remission rate in patients with relapsed/refractory disease was 19.3%. Clinical activity was seen at all enasidenib dose levels tested.
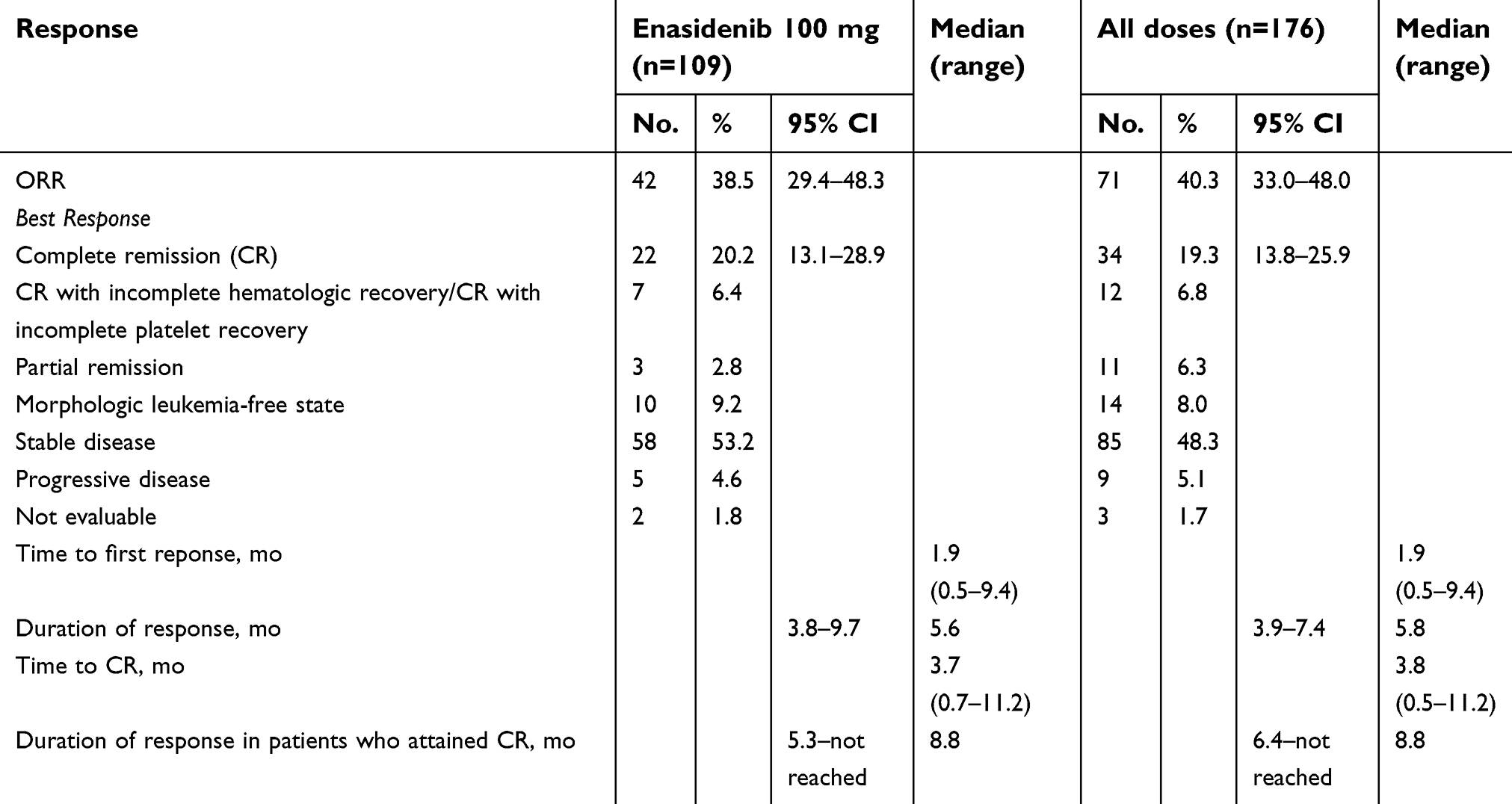 | Table 1 Hematologic response, time to response and response duration of patients with relapsed/refractory acute myeloid leukemia (AML) in the Phase II trial of Enasidenib |
Across the entire study population, those with IDH2-R140 mutations had an ORR of 35.4%, and those with IDH2-R172 mutations had an ORR of 53.3%. The CR rate for IDH2-R140 disease was 17.7% and IDH2-R172 disease, 24.4%. Approximately three-fourths of the study population had IDH2-R140 mutations, and one-fourth had IDH2-R172 mutations.6
87.3% of patients who ultimately responded to therapy saw a response by cycle 5, while 82.4% of patients who achieved complete remission did so by cycle 7. The median time to first response was 1.9 months, notably with a wide range of 0.5–9.4 months. This suggested the importance of continuing enasidenib therapy for at least 5–6 cycles, as more than half of the patients in this study achieved their best response after cycle 5.37
Importantly, enasidenib showed an improvement in survival. Among patients with R/R leukemia, the overall median survival was 9.3 months, while for those who achieved CR or PR, survival was 19.7 and 14.7 months, respectively.6 Among patients who received >3 prior AML therapies, estimated overall median survival was 7 months.37 This is compared with a median survival of 4–8 months in all relapsed/refractory disease, depending on therapy.11,38 Moreover, approximately 9% of R/R AML patients proceeded to transplant at the time of data cut-off; median overall survival for this group was 23.6 months.6,37
Predictive biomarkers
2-HG, as the oncometabolite of mutant IDH, was investigated as a potential biomarker. In preclinical studies, Yen et al monitored 2-HG level reduction through various models, noting in the ex vivo model of blasts that reduction of 2-HG levels was less robust in IDH2-R172 mutants versus R140. Similarly, the Phase I/II study for enasidenib found that patients with IDH2-R140 mutations had a median 93% reduction in 2-HG levels (maximum 99%), while patients with IDH2-R172 had a median 28% reduction (maximum 94%). However, as stated previously, both patients with IDH2-R140 disease and IDH2-R172 showed a CR of 17.7% and 24.4%, respectively, initially suggesting that 2-HG levels did not correlate with response. Further analysis of patients with IDH2-R140 mutated disease showed that all patients achieved robust 2-HG reductions, even non-responders, making 2-HG levels an unreliable biomarker in these patients. Conversely, greater reductions of 2-HG levels correlated with improved response in patients with IDH2-R172 mutations.37 2-HG levels, therefore, may be a better predictive biomarker for IDH2-R172 disease than for IDH2-R140, although enasidenib may also have other mechanisms that are of clinical importance independent of 2-HG reduction.6,37 This represents an active area of investigation.
Molecular remission
Recently, IDH2 variant allele frequency (VAF) was analyzed for patients in the Phase I/II trial.37 Data were available for 101 R/R AML patients. IDH2 VAF mean values were similar for patients with IDH2-R140 and IDH2-R172 mutations. Of the 101, 12 patients (11.9%), all with IDH2-R140 mutations, achieved molecular remission during enasidenib treatment. Of these 12, 83.3% achieved CR, 8.3% achieved CRp and 8.3% achieved PR. Among all patients on the trial who attained CR, survival was not different for those who attained molecular remission versus those who did not. Among the 23 patients with IDH2-R172 (22.7% of total cohort available for analysis), 2 patients (8.7%) achieved IDH2-R172 VAF levels <2%, both of whom had CR. These data suggest that while molecular remission is not necessary to achieve CR, its presence may be predictive, and also underscores the importance of molecular monitoring over time.37 Moreover, it will be important to elucidate how enasidenib, as a non-cytotoxic agent, is able to induce molecular remissions in certain patients. Knowing this mechanism may help identify patients who may gain greater benefit from enasidenib, as well as suggest additional synergistic therapies.
Safety
During the dose-escalation portion of the trial, the maximum tolerated dose was not reached. At higher doses, the drug was less well tolerated, and 5 of 7 patients in the 650 mg group were dose modified or reduced; these events did not qualify as a dose limiting toxicity (DLT). 238 of 239 patients experienced a treatment-emergent adverse event (TEAE), with 192 (82%) experiencing a treatment-related TEAE.
The most common treatment-related TEAEs were indirect hyperbilirubinemia (38%) and nausea (23%). Other common TEAEs included fatigue, diarrhea, decreased appetite and vomiting. We review selected TEAEs in Table 2. 41% of patients had grade 3 to 4 TEAEs, again, most commonly indirect hyperbilirubinemia, but additionally, grade 3 to 4 IDH-inhibitor-associated differentiation syndrome (IDH-DS) was seen in 6%. IDH-DS of any grade occurred in 10% of patients. Despite these, enasidenib was discontinued in only 5% of patients.
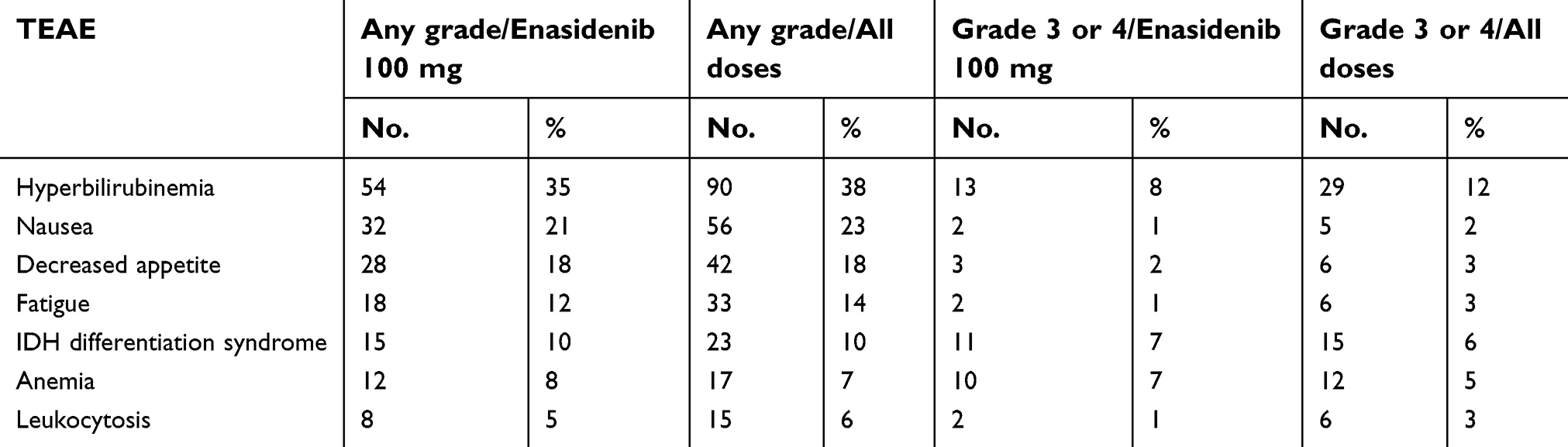 | Table 2 Select treatment-related TEAEs from the Phase II trial of Enasidenib |
Of note, enasidenib-induced hyperbilirubinemia does not reflect intrinsic liver injury. The proposed mechanism is that enasidenib inhibits UGT1A1 enzyme, producing an effect similar to patients with Gilbert syndrome (congenital deficiency of the enzyme).6 Notably, there is a new trial that is not yet recruiting whose purpose is to determine pharmacokinetics of enasidenib in patients with pre-existing liver dysfunction compared with healthy controls (NCT03290443).39
IDH differentiation syndrome and leukocytosis
IDH differentiation syndrome (IDH-DS) is a clinical diagnosis and requires a high index of suspicion from the treating physician. The diagnosis is made by excluding other explanations for signs and symptoms, which include: dyspnea and hypoxemia, radiological evidence of bilateral pulmonary infiltrates not responsive to antimicrobials, worsening peripheral edema, renal failure, fever, rash and lymphadenopathy.40 It is similar to the syndrome described in patients receiving all-trans retinoic acid or arsenic trioxide for acute promyelocytic leukemia (APL). IDH-DS appears to have a later onset, with a median of 30 days, as compared to 10–12 days for differentiation syndrome in patients with APL.40,41
10% of patients enrolled in the trial experienced IDH-DS, with 2 deaths on the trial initially associated with, but not directly linked to, the syndrome. For the purposes of the study, IDH-DS was termed as “retinoic acid syndrome” due to the fact that IDH-DS was not yet an established preferred term. The syndrome has also been described with the IDH1 inhibitor ivosidenib.42
Further retrospective analysis of study participants and suspected cases was performed by a differentiation syndrome review committee, who concluded that it affected approximately 12% of patients in the study. The patient characteristics linked with IDH-DS were as follows: male sex, fewer previous cancer-directed therapies and less likely to have less than 20% bone marrow blasts. Of those affected, 45% of patients interrupted treatment due to the syndrome, but none stopped permanently. Of note, 30% of patients with IDH-DS required admission to the intensive care unit, but no death was directly attributed to IDH-DS. Recommended management of recognized IDH-DS is dexamethasone 10 mg twice daily, until clinical improvement. Notably, IDH-DS may be associated with a positive clinical response.40,42
Leukocytosis was also independently noted in patients receiving enasidenib, and did not always signify IDH-DS. Of the patients retrospectively identified with IDH-DS, 39% had concurrent leukocytosis.40 Overall, treatment-related leukocytosis occurred in 6% of patients.6 Leukocytosis alone can be successfully treated with hydroxyurea or drug interruption.36
Recently, the FDA conducted a systematic analysis of differentiation syndrome associated with both ivosidenib and enasidenib using the patient data from the Phase I/II trial, specifically looking at patients who received currently approved doses of the agents.43 By utilizing an algorithm as well as individualized review of algorithm-identified cases, the analysis established a higher incidence of IDH-DS than previously reported. For enasidenib, the rate was approximately 19%, higher than the reported rates in the Phase I/II trial rate (10%) and the retrospective review (12%). Of the cases identified, 66% were considered at least grade 3, and 5% were fatal. This suggests that the main reason for the discrepancy between the initial reported rate of IDH-DS and the rate in the FDA review was enhanced detection of previously unidentified cases, including fatal incidences. Leukocytosis was associated with 61% of cases. Interestingly, this review also suggested that patients with IDH-DS may have lower response rates.43 Overall, this indicates that continued vigilance regarding detection of IDH-DS is necessary. Furthermore, its impact on predicting clinical response is unclear at this time, and should remain a focus of active surveillance and research.
Quality of life
No specific analyses on the effect of enasidenib on quality of life exist. However, as it is generally well tolerated even in unfit patients, and is an oral agent, it can be postulated that patients treated with enasidenib may have a better quality of life than those undergoing standard cytotoxic chemotherapy. Additionally, in the Phase I/II trial, 43.1% of RBC transfusion-dependent patients and 40.2% of platelet-dependent patients attained transfusion independence, which is another important factor that may improve quality of life in the R/R AML population.37
Cost and cost-effectiveness
To date, there has not been a full cost-effectiveness analysis published. The whole-sale cost of enasidenib 100 mg tablets starts from about $25,000 for 30 tablets.44,45 Insurance coverage for the drug is available based on insurance-specific guidelines. Patients who qualify for the Celgene patient assistance program can obtain the drug for a $25 co-pay.46
Resistance
Acquired resistance to enasidenib therapy has been reported. Intelkofer et al published a report of two patients who developed resistance to therapy after initial response. They identified a new somatic missense mutation in each patient, Q316E in the first, and I319M in the second, and then evaluated the effect of these mutations in WT and IDH2-R140 mutants. In WT IDH, mutations in Q316E and I319M alone did not induce production of 2-HG. However, IDH2-R140 mutants with the presence of either the Q316E or 1319M mutations continued to produce 2-HG when exposed to enasidenib. Both Q316E and I319M are second site mutations which occur in the binding site of enasidenib and mediate resistance to enasidenib.47
Harding et al recently described isoform switching, where another form of mutant IDH1/2 emerges after treatment with a targeted inhibitor. In one example, a patient with myelodysplastic syndrome, subtype refractory anemia with excess blasts 2, initially completely responded to enasidenib for seventeen cycles. She then once again developed excess blasts and elevated 2-HG levels, with molecular testing identifying a new IDH1-R132C mutation. Further analysis showed that this mutation arose early during enasidenib treatment.48 Isoform switching was similarly noted by Quek et al previously, as well as additional mechanisms of resistance including gain of function mutations in proliferation genes and transcription factor mutations.49 Notably, the described patient was then started on a dual IDH1/IDH2 inhibitor and initially responded, but subsequently progressed despite adequate 2-HG suppression, indicating that there are likely more resistance mechanisms that are yet to be identified.48 This also argues for strong consideration of combination therapy, as resistance to monotherapy can clearly develop.
Future directions
Current trials
Enasidenib continues to be studied in several active and planned clinical trials. Many of these are outlined in Table 3. In the following section, we review select enasidenib studies.
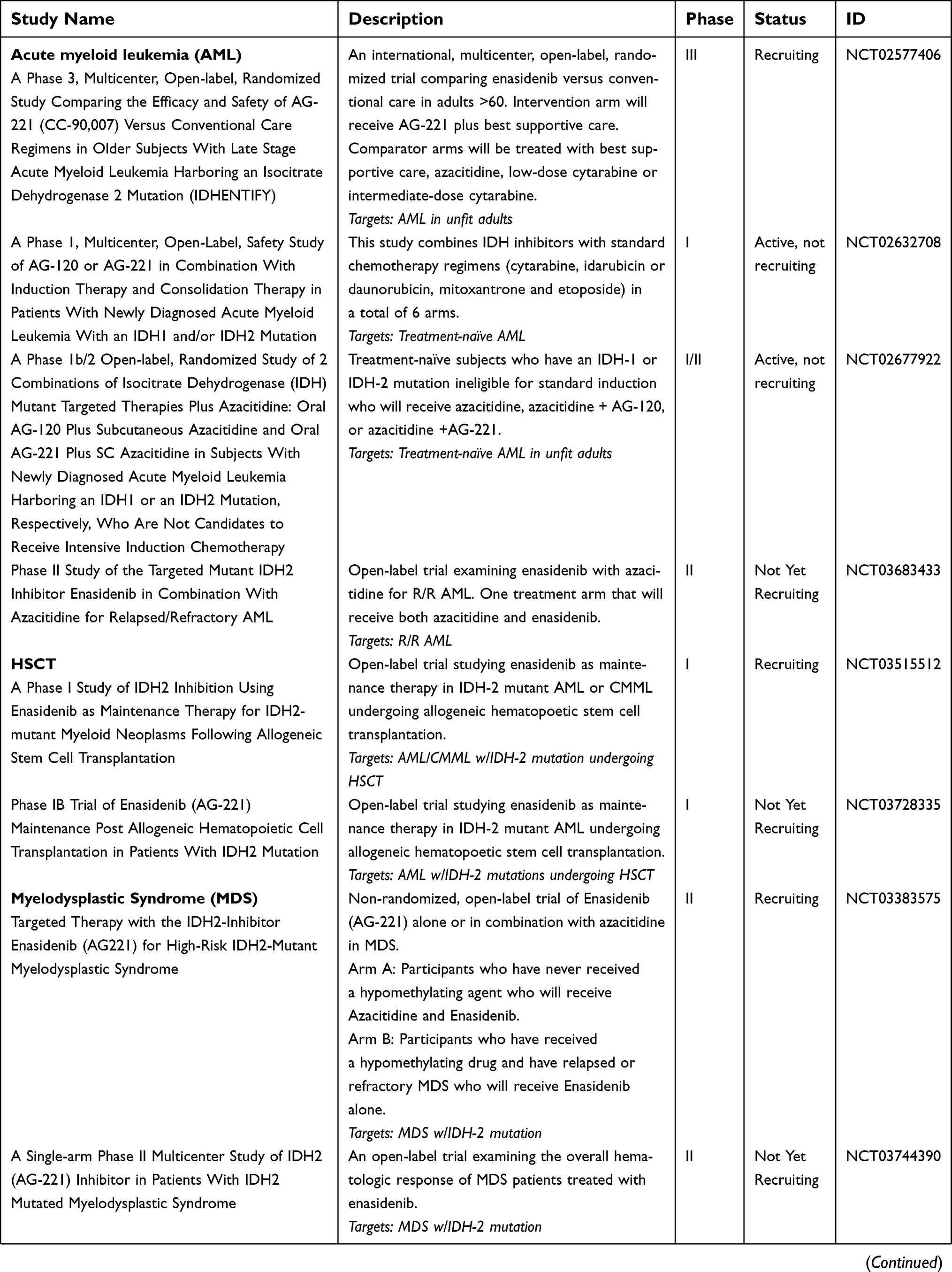 | Table 3 Active and recently completed studies of Enasidenib |
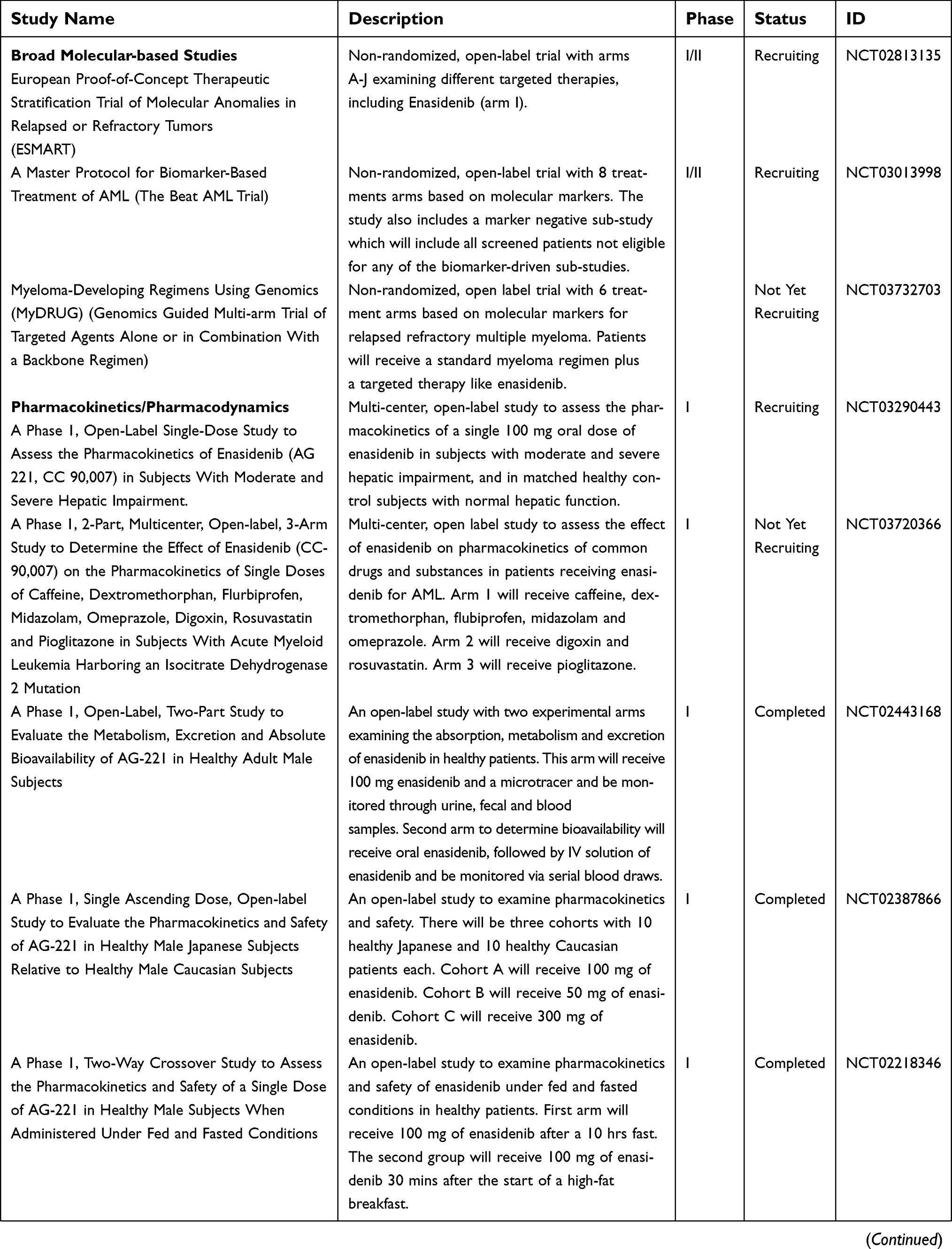 | Table 3 (Continued). |
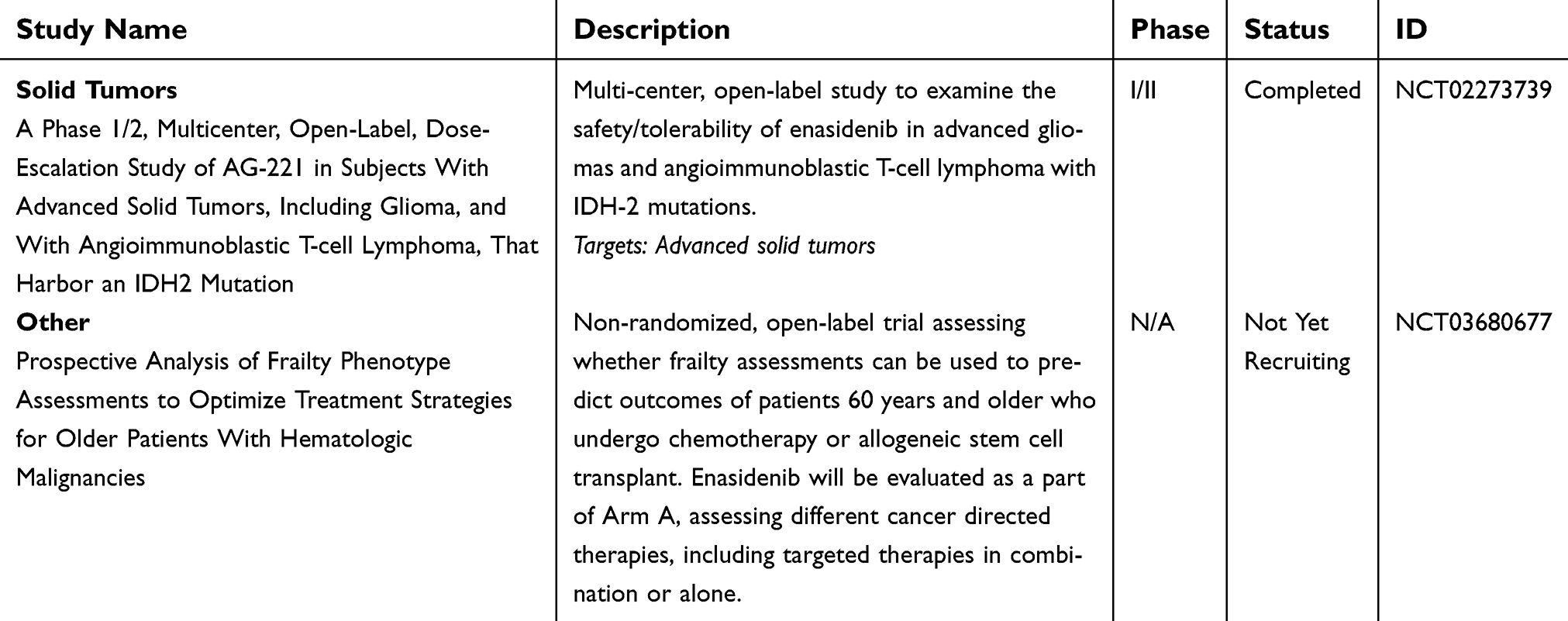 | Table 3 (Continued). |
Monotherapy, R/R AML
The open-label, randomized Phase III trial for enasidenib (IDHENTIFY, NCT02577406) is currently ongoing, comparing enasidenib with standard therapies in adults older than 60; estimated recruitment will be approximately 300 participants. The experimental arm is enasidenib alone, and the conventional care regimen arm includes best supportive care alone, azacitidine alone, as well as low and intermediate dose cytarabine. The primary endpoint is overall survival.34
Combination with induction chemotherapy, treatment-naïve AML
Approximately 134 participants, eligible for standard chemotherapy, are currently enrolled in the Phase 1 trial examining treatment-naïve AML with an IDH1/2 mutation (NCT02632708). The trial will examine the following combinations: AG-120 (ivosidenib) + cytarabine + daunorubicin or idarubicin; enasidenib + cytarabine + daunorubicin or idarubicin; enasidenib starting day 8+ cytarabine + daunorubicin or idarubicin. This study aims to determine the safety and primary efficacy of combining IDH inhibitors with induction agents.50 Preliminary outcomes from this trial are showing robust results. Among patients with de-novo AML treated with enasidenib, 73% have achieved CR, CR with incomplete count recovery (CRi) or CR with incomplete platelet recovery (CRp). Among those with secondary AML, 63% have achieved CR, CRi or CRp. Among all patients treated with enasidenib, 42% have proceeded to HSCT.51 In regard to adverse events, in the induction period, 91% of patients in the enasidenib arm had 1 or greater grade 3 or higher TEAE. The most common was febrile neutropenia, occurring in 61% of patients. The next most common were hypophosphatemia and lung infection, each occurring in approximately 10% of patients. Approximately 8% of patients experienced hyperbilirubinemia, which was the most common TEAE in the Phase I/II enasidenib trial. Additionally, approximately 30% of enasidenib-treated patients had mutation clearance after day 28 of induction. Overall, the combination of enasidenib with induction chemotherapy is showing promising results with acceptable toxicity.51
Combination with HMA, treatment-naïve AML
A Phase 1/2 randomized for subjects with newly diagnosed AML harboring either an IDH1 or IDH2 mutation who are ineligible for standard cytotoxic chemotherapy is expected to enroll approximately 125 participants (NCT02677922). These patients will receive azacitidine alone or in combination with an appropriate IDH inhibitor. Primary outcomes will be dose-limiting toxicities and adverse events, with secondary outcomes of overall response rate.52 Preliminary enasidenib data for 6 patients (3 receiving the approved dose of 100 mg daily, and 3 receiving 200 mg) showed CR in 2 patients of 4 responders; 2 patients discontinued due to progression of disease and 1 other for unavailable reasons. Most common adverse events were nausea and hyperbilirubinemia.53 Given that mutant IDH-2 induces a hypermethylation phenotype, the concurrent use of a hypomethylating agent such as azacitidine with enasidenib may potentially be synergistic, with further data pending at this time.
Combination with HMA, R/R AML
For R/R AML, a Phase II clinical trial is exploring enasidenib and azacitidine combination, recruiting about 50 participants (NCT03683433). Patients will receive the medications concurrently. Primary objectives will be overall response rate, with secondary objectives of determining event-free survival and overall survival.54
Maintenance post-allo HSCT
A small Phase I trial aims to examine the role of enasidenib in maintenance therapy or patients receiving transplant for IDH-2 mutant AML or chronic myelomonocytic leukemia (CMML) (NCT03728335). Primary outcomes will be maximum tolerated dose and dose-limiting toxicities. Secondary outcomes will be enasidenib-related adverse events, as well as cumulative incidence of both acute and chronic graft versus host disease.55
Future preclinical and clinical investigations
In addition to combinations with induction chemotherapy and HMA, trials examining the role of enasidenib in combination with other targeted agents or as a maintenance agent are of interest.
Preclinical data showed that mutations in IDH cause dependence on the anti-apoptotic gene BCL-2 in leukemic cells, and the BCL-2 inhibitor, venetoclax, demonstrated significant preclinical activity in mIDH disease.56 In a recent clinical trial of venetoclax + HMA in treatment-naïve older AML patients, patients with IDH1/2 mutations in the venetoclax 400 mg + azacitidine cohort attained a 90% CR/CRi rate and 100% CR/CRi in the venetoclax 400 mg + decitabine cohort.57 The overall survival was 24.4 months for those with IDH1/2 mutations, but did not reach statistical significance to predict outcome for this group.58 Given such robust responses, it may be of interest to study combinations of enasidenib with venetoclax + HMA or venetoclax alone. Interestingly, some data suggest that pairing an mIDH inhibitor with venetoclax may lead to lower response rates by reversing the pathways that render mIDH AML sensitive.59 However, given the complex molecular landscape of AML, there may be additional pathways by which a combination of IDH inhibition and venetoclax may induce durable responses, potentially in combination with HMA. IDH inhibitors combined with targeted therapies against FLT3 may be another viable option. With early success of combining HMA with enasidenib, addition of FLT3 targeted therapy may prove to be an even more potent combination. While enasidenib is currently being examined as maintenance post-transplant (NCT03728335), it could also have a role as maintenance agent after intensive chemotherapy in patients not going to transplant, in patients with minimal/measurable residual disease (MRD) or in patients with molecular relapses. Additional preclinical and clinical investigations are likely to address such additional questions to further define the potential role of enasidenib in the treatment of AML bearing IDH2 mutations.
Advancing the role of IDH2 molecular testing
Given the availabilities of new targeted therapies, molecular testing should be standard in all newly diagnosed AML and include testing for IDH2.12 Not only the availability, but also the timeliness of such testing is becoming increasingly important. The BEAT AML trial, designed in part to assess whether molecular testing could be used to assign patients to treatment arms within 7 days, has recently found this to be feasible.60 Of the 210 patients enrolled, 52% were successfully entered into an appropriate sub-study based on molecular testing and received treatment. Further, 81 additional patients pursued alternative therapies after treatment assignment, suggesting that molecular testing could be obtained and used for clinical decision making within 7 days for a total of 91% of patients enrolled.60 In the enasidenib arm of this study, 96% of consented patients received treatment.61 Overall, this demonstrates that rapid identification of targetable mutations in AML is already both possible and practical, and further advancements in testing have the potential to make targeted therapies even more accessible.
Rapid, easily available testing could also accelerate the use of IDH2 mutations as a marker of MRD. Detection of certain genetic abnormalities (including IDH2) after treatment has been associated with worse outcomes and an increased risk of relapse.62 However, the prognostic significance of persistent IDH2 mutations in the setting of specific treatments and/or subsets of AML remains to be fully elucidated as does the optimal method and cutoff for detecting IDH2 mutations and the impact of prospectively treating such patients with enasidenib or other treatment modalities, such as allotransplant.
Conclusion
The availability of molecular testing and targeted drugs is transforming the approach to treating newly diagnosed AML as well as R/R disease, making precision and personalized medicine a possibility. The emergence of molecular-based therapy in AML offers new hope, especially to patients who cannot tolerate standard cytotoxic regimens. Molecular testing should be done for all newly diagnosed AML cases, and continue during both treatment and relapse to identify response, MRD and treatment-emergent targetable mutations.
Enasidenib is FDA approved for R/R AML, a promising therapy with a generally favorable side effect profile that should be considered for patients with IDH2-mutated AML. Enasidenib’s role in upfront AML therapy with induction, combination with HMA, combination with other targeted therapies, as well as post-consolidation or transplant maintenance remains to be determined, but additional clinical investigations with enasidenib are likely to continue to evolve the landscape of AML therapy.
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
Maria Galkin has no conflicts of interest to disclose in this work. Brian A Jonas has served as a consultant or advisory board member for AbbVie, Amgen, Celgene, Jazz and Tolero; has received research funding to his institution from AbbVie, Accelerated Medical Diagnostics, AROG, Celgene, Daiichi Sankyo, Esanex, Forma, Genentech/Roche, Glycomimetics, Incyte, Kalobios, LP Therapeutics and Pharmacyclics.
References
1. Hoffman R. Hematology : Basic Principles and Practice.
2. Longo DL, Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;12(373):1136–1152. doi:10.1056/NEJMra1406184
3. Bose P, Vachhani P, Cortes JE. Treatment of relapsed/refractory acute myeloid leukemia. Curr Treat Options Oncol. 2017;18:3. doi:10.1007/s11864-017-0456-2
4. Thol F, Schlenk RF, Heuser M, Ganser A. How I treat refractory and early relapsed acute myeloid leukemia. Blood. 2015;126(3):319–327. doi:10.1182/blood-2014-10-551911
5. Buege MJ, DiPippo AJ, DiNardo CD. Evolving treatment strategies for elderly leukemia patients with IDH mutations. Cancers (Basel). 2018;10(6):1–20. doi:10.3390/cancers10060187
6. Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722–732. doi:10.1182/blood-2017-04-779405
7. Scott LJ, Kim ES. Enasidenib: first global approval. Drugs. 2017;77:1705–1711. doi:10.1007/s40265-018-0861-2
8. Dhillon S. Ivosidenib: first global approval. Drugs. 2018;78(14):1509–1516. doi:10.1007/s40265-018-0978-3
9. Bergua JM, Montesinos P, Martinez-Cuadrón D, et al. A prognostic model for survival after salvage treatment with FLAG-Ida +/− gemtuzumab-ozogamicine in adult patients with refractory/relapsed acute myeloid leukaemia. Br J Haematol. 2016;174(5):700–710. doi:10.1111/bjh.14107
10. Fridle C, Medinger M, Wilk MC, et al. Cladribine, cytarabine and idarubicin (CLA-Ida) salvage chemotherapy in relapsed acute myeloid leukemia (AML). Leuk Lymphoma. 2017;58(5):1068–1075. doi:10.1080/10428194.2016.1235274
11. Itzykson R, Thépot S, Berthon C, et al. Azacitidine for the treatment of relapsed and refractory AML in older patients. Leuk Res. 2015;39(2):124–130. doi:10.1016/J.LEUKRES.2014.11.009
12. Abboud CN, Altman JK, Lurie RH, et al. NCCN guidelines version 2.2018 acute myeloid leukemia; 2018. Available from:
13. Estey E, Grimwade D, Amadori S, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447. doi:10.1182/blood-2016-08
14. Ossenkoppele G, Owenberg BL. How I treat the older patient with acute myeloid leukemia. Blood. 2015;125(5):767–773. doi:10.1182/blood-2014
15. Levis M. Midostaurin approved for FLT3-mutated AML. Blood. 2017;129(26):3403–3406. doi:10.1182/blood-2017-05-782292
16. Appelbaum FR, Bernstein ID. Gemtuzumab ozogamicin for acute myeloid leukemia. Blood. 2017;130(22):2373–2376. doi:10.1182/blood-2017-09-797712
17. DeStefano C, Hourigan CS. Personalizing initial therapy in acute myeloid leukemia: incorporating novel agents into clinical practice. Ther Adv Hematol. 2018;9(5):109–121. doi:10.1177/https
18. Nassereddine S, Lap CJ, Haroun F, Tabbara I. The role of mutant IDH1 and IDH2 inhibitors in the treatment of acute myeloid leukemia. Ann Hematol. 2017;96(12):1983–1991. doi:10.1007/s00277-017-3161-0
19. Marcucci G, Maharry K, Wu Y-Z, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a cancer and leukemia group B study. J Clin Oncol. 2010;28(14):2348–2355. doi:10.1200/JCO.2009.27.3730
20. Molenaar RJ, Thota S, Nagata Y, et al. Clinical and biological implications of ancestral and non-ancestral IDH1 and IDH2 mutations in myeloid neoplasms. Leukemia. 2015;29(11):2134–2142. doi:10.1038/leu.2015.91
21. Rakheja D, Konoplev S, Jeffrey Medeiros L, Chen W. IDH mutations in acute myeloid leukemia. Hum Pathol. 2012;43(10):1541–1551. doi:10.1016/j.humpath.2012.05.003
22. Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi:10.1016/j.ccr.2010.11.015
23. Abbas S, Lugthart S, Kavelaars FG, et al. Acquired mutations in the genes encoding IDH1 and IDH2 both are recurrent aberrations in acute myeloid leukemia: prevalence and prognostic value. Blood. 2010;116(12):2122–2126. doi:10.1182/blood-2009-11-250878
24. Dinardo CD, Ravandi F, Agresta S, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol. 2015;90(8):732–736. doi:10.1002/ajh.24072
25. Gagné LM, Boulay K, Topisirovic I, Huot M-É, Mallette FA. Oncogenic activities of IDH1/2 mutations: from epigenetics to cellular signaling. Trends Cell Biol. 2017;27(10):738–752. doi:10.1016/j.tcb.2017.06.002
26. Rampal R, Alkalin A, Madzo J, et al. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 2014;9(5):1841–1855. doi:10.1016/J.CELREP.2014.11.004
27. Rossi G, Minervini MM, Carella AM, Melillo L, Cascavilla N. Wilms’ tumor gene (WT1) expression and minimal residual disease in acute myeloid leukemia. In: van den Heuvel-Elbrink MM, editor. Wilms Tumor. Brisbane (AU): Codon Publications; 2016. Chapter 16. doi10.15586/CODON.WT.2016.CH16
28. Galkin M. Hypermethylation phenotype diagram. Available from:
29. Martínez-Laperche C, Kwon M, Franco-Villegas AC, et al. Wilms tumor 1 gene expression levels improve risk stratification in AML patients. Results of a multicentre study within the Spanish group for molecular biology in haematology. Br J Haematol. 2018;181(4):542–546. doi:10.1111/bjh.14635
30. Frairia C, Aydin S, Audisio E, et al. Post-remissional and pre-transplant role of minimal residual disease detected by WT1 in acute myeloid leukemia: a retrospective cohort study. Leuk Res. 2017;61:10–17. doi:10.1016/J.LEUKRES.2017.08.008
31. Patel KP, Ravandi F, Ma D, et al. Acute myeloid leukemia with IDH1 or IDH2 mutation frequency and clinicopathologic features. Am J Clin Pathol. 2011;135:35–45. doi:10.1309/AJCPD7NR2RMNQDVF
32. Medeiros BC, Fathi AT, DiNardo CD, Pollyea DA, Chan SM, Swords R. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia. 2017;31:272–281. doi:10.1038/leu.2016.275
33. Yen K, Travins J, Wang F, et al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov. 2017;7(5):478–493. doi:10.1158/2159-8290.CD-16-1034
34.
35. Dalle IA, Dinardo CD. The role of enasidenib in the treatment of mutant IDH2 acute myeloid leukemia. Ther Adv Hematol. 2018;9(7):163–173. doi:10.1177/https
36.
37. Stein EM, DiNardo CD, Fathi AT, et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood. 2019;133(7):676–687. doi:10.1182/blood-2018-08-869008
38. Oran B, Weisdorf DJ. Survival for older patients with acute myeloid leukemia: a population-based study. Haematologica. 2012;97(12):1916–1924. doi:10.3324/haematol.2012.066100
39.
40. Fathi AT, DiNardo CD, Kline I, et al. Differentiation syndrome associated with enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2. JAMA Oncol. 2018:1–5. doi:10.1001/jamaoncol.2017.4695.
41. Rogers JE, Yang D. Differentiation syndrome in patients with acute promyelocytic leukemia. J Oncol Pharm Pract. 2011;18(1):109–114. doi:10.1177/1078155211399163
42. Kc B, Dinardo CD. Evidence for clinical differentiation and differentiation syndrome in patients with acute myeloid leukemia and IDH1 mutations treated with the targeted mutant IDH1 inhibitor, AG-120. Clin Lymphoma Myeloma Leuk. 2016;16(8):460–465. doi:10.1016/j.clml.2016.04.006
43. Norsworthy KJ, Mulkey F, Ward AF, et al. Incidence of differentiation syndrome with Ivosidenib (IVO) and Enasidenib (ENA) for treatment of patients with Relapsed or Refractory (R/R) isocitrate dehydrogenase (idh)1- or idh2-mutated acute myeloid leukemia (aml): a systematic analysis by the U.S. food and drug administration (FDA). Blood. 2018;132(Suppl1):288. doi:10.1182/BLOOD-2018-99-117426
44.
45. Prices I, Coupons & patient assistance programs. Drugs.com. Available from:
46.
47. Intlekofer AM, Shih AH, Wang B, et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature. 2018;559(7712):125–129. doi:10.1038/s41586-018-0251-7
48. Harding JJ, Lowery MA, Shih AH, et al. Isoform switching as a mechanism of acquired resistance to mutant isocitrate dehydrogenase inhibition. Cancer Discov. 2018;8(12):1540–1547. doi:10.1158/2159-8290.CD-18-0877
49. Quek L, David M, Kennedy A, et al. Clonal heterogeneity in differentiation response and resistance to the IDH2 inhibitor Enasidenib in acute myeloid leukemia. Blood. 2017;130(Suppl1). Available from:
50.
51. Stein E, DiNardo CD, Mims AS, et al. Ivosidenib or Enasidenib combined with induction and consolidation chemotherapy in patients with newly diagnosed AML with an IDH1 or IDH2 mutation is safe, effective, and leads to MRD-negative complete remissions. Blood. 2018;132(Suppl 1):560.
52.
53. Dinardo CD, Stein AS, Stein EM, et al. Mutant IDH (mIDH) inhibitors, ivosidenib or enasidenib, with azacitidine (AZA) in patients with acute myeloid leukemia (AML). J Clin Oncol. 2018;36(15_suppl):7042. doi:10.1200/JCO.2018.36.15_suppl.7042
54.
55.
56. Chan SM, Thomas D, Corces-Zimmerman MR, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med. 2015;21(2):178–184. doi:10.1038/nm.3788
57. Pollyea DA, Pratz KW, Jonas BA, et al. Venetoclax in combination with hypomethylating agents induces rapid, deep, and durable responses in patients with AML ineligible for intensive therapy. Blood. 2018;132(Suppl 1):285. doi:10.1182/BLOOD-2018-99-117179
58. DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133(1):7–17. doi:10.1182/blood-2018-08-868752
59. Molenaar RJ, Maciejewski JP, Wilmink JW, van Noorden CJF. Wild-type and mutated IDH1/2 enzymes and therapy responses. Oncogene. 2018;37(15):1949–1960. doi:10.1038/s41388-017-0077-z
60. Burd A, Levine RL, Shoben A, et al. Initial report of the beat AML umbrella study for previously untreated AML: evidence of feasibility and early success in molecularly driven phase 1 and 2 studies. Blood. 2018;132(Suppl1):559. doi:10.1182/BLOOD-2018-99-118494
61. Stein EM, Shoben A, Borate U, et al. Enasidenib is highly active in previously untreated idh2 mutant AML: early results from the beat AML master trial. Blood. 2018;132(Suppl1):287. doi:10.1182/BLOOD-2018-99-118287
62. Jongen-Lavrencic M, Grob T, Hanekamp D, et al. Molecular minimal residual disease in acute myeloid leukemia. N Engl J Med. 2018;378(13):1189–1199. doi:10.1056/NEJMoa1716863
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.