Back to Journals » Drug Design, Development and Therapy » Volume 9
Emerging treatment options to improve cardiovascular outcomes in patients with acute coronary syndrome: focus on losmapimod
Authors Kragholm K, Newby K, Melloni C
Received 30 April 2015
Accepted for publication 16 June 2015
Published 5 August 2015 Volume 2015:9 Pages 4279—4286
DOI https://doi.org/10.2147/DDDT.S69546
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Video presented by Kristian Kragholm
Views: 374
Kristian Kragholm, Laura Kristin Newby, Chiara Melloni
Duke Clinical Research Institute, Duke University Medical Center, Durham, NC, USA
Abstract: Each year, despite optimal use of recommended acute and secondary prevention therapies, 4%–5% of patients with acute coronary syndrome (ACS) experience relapse of ACS or other cardiovascular events including stroke, heart failure, or sudden cardiac death after the index ACS. The sudden atherosclerotic plaque rupture leading to an ACS event is often accompanied by inflammation, which is thought to be a key pathogenic pathway to these excess cardiovascular events. Losmapimod is a novel, oral p38 mitogen-activated protein kinase (MAPK) inhibitor that targets MAPKs activated in macrophages, myocardium, and endothelial cells that occur as a part of global coronary vascular inflammation following plaque rupture. This review aims to 1) discuss the pathophysiological pathways through which p38 MAPKs may play key roles in initiation and progression of inflammatory disease and how losmapimod is thought to counteract these p38 MAPKs, and 2) to describe the efficacy and safety data for losmapimod obtained from preclinical studies and randomized controlled trials that support the hypothesis that it has promise as a treatment for patients with ACS.
Keywords: p38 MAPK, p38 MAPK inhibitor, losmapimod, safety, efficacy, outcomes
Introduction
Acute coronary syndromes (ACS) include a spectrum of clinical manifestations including ST-segment elevation myocardial infarction (STEMI), non-ST-segment EMI (NSTEMI), and unstable angina. ACS is not only a primary cause of death worldwide, but is also associated with the development of heart failure, leading to significant health care utilization and economic cost.1–3 Following years of asymptomatic formation of atherosclerosis in the coronary arteries, sudden atherosclerotic plaque rupture results in a clinical manifestation of ACS.4 Atherosclerosis is a complex and incompletely understood process, but inflammation is viewed to play a pivotal role, involving low-density lipoprotein cholesterol (LDLc) deposition and oxidation, recruitment of inflammatory cells, release of cytokines, and endothelial dysfunction, the latter involving reduced nitric oxide (NO) bioavailability independently associated with increased cardiovascular risk.5–7 Atherosclerotic plaques can generally be divided into stable and unstable lesions, and while stable lesions mainly consist of extracellular matrix and smooth muscle cells with a thick fibrous cap of extracellular matrix, unstable lesions are rich in macrophages and foam cells with a thin fibrous cap that is prone to rupture. Thus, inflammation is a crucial part not only in the formation of atheroma, but also in the atherosclerotic plaque rupture leading to an ACS event.4
Markers of inflammation have been associated with prognosis among ACS patients. High-sensitivity C-reactive protein (hsCRP), a known marker of inflammation, may be elevated for weeks to months following an ACS event, even in the absence of other possible causes of systemic inflammation, and is thought to represent multiple inflamed plaques in addition to the index culprit lesion.8,9 Increased levels of hsCRP are also predictive of future cardiovascular events, suggesting that this inflammation-driven milieu represents a pathogenic pathway toward additional cardiovascular events including heart failure, stroke, and sudden cardiac death.8,9 Each year 4%–5% of ACS patients develop relapse of ACS or other cardiovascular events within weeks to months following the index ACS event, despite optimal use of acute and secondary prevention evidence-based therapies including statins, which are believed to have pleiotropic anti-inflammatory effects in addition to their lipid lowering effect.10,11 Hence, additional therapies targeting vascular inflammation following ACS are warranted.
p38 mitogen-activated protein kinases (MAPKs) are activated in macrophages, myocardium, and endothelial cells by the inflammatory milieu following plaque rupture in ACS.12 These stress-activated kinases are shown to be crucial for initiation and progression of inflammatory diseases, and inhibition of this pathway holds the potential to prevent disease relapse and progression in diseases in which inflammation plays a major pathogenic role.13–15 Losmapimod, an inhibitor of p38 MAPK α and β isoforms, is a compound developed to inhibit downstream inflammatory disease processes in ACS and other diseases. This review discusses the pathophysiological pathways in which p38 MAPKs work to initiate and amplify inflammation and how losmapimod works to counteract p38 MAPK activation. Preclinical and early phase clinical studies that have explored the safety and efficacy of losmapimod are presented.
p38 MAPK activation and inhibition of p38 MAPK with losmapimod in preclinical studies
MAPKs are ubiquitously expressed in cells of the human body and regulate intracellular pathways in response to extracellular stimuli including tobacco smoke and pro-inflammatory cytokines.13,14 There are four isoforms of p38 MAPK: α, β, γ, and δ. The α and β p38 isoforms are approximately 70% identical and ubiquitously expressed in all tissues. The γ and δ isoforms share around 60% sequence identity; however, the γ p38 isoform is expressed only in skeletal and cardiac tissues, and the δ p38 isoform is found in a limited number of other adult tissues, including the lung and spleen.
p38 MAPKs regulate the transcription and translation of inflammatory mediators such as tumor necrosis factor alpha (TNF-α) and interleukins 1, 6, and 8 (IL-1, IL-6, and IL-8).13,14 In atherosclerotic disease, oxidized LDLc activates the p38 MAPK cascade resulting in further activation of TNF-α, IL-1, and heat shock proteins (HSPs) (Figure 1).13–15 This p38 MAPK-activated inflammatory response not only aggravates atherosclerosis through inflammation but is also considered to be crucial in the formation of reactive oxygen species, resulting in reduced NO bioavailability leading to vasoconstriction and attenuated vasoregulation (endothelial dysfunction). Reactive oxygen species are thought to play a critical role during both myocardial ischemia and reperfusion injury.13–15
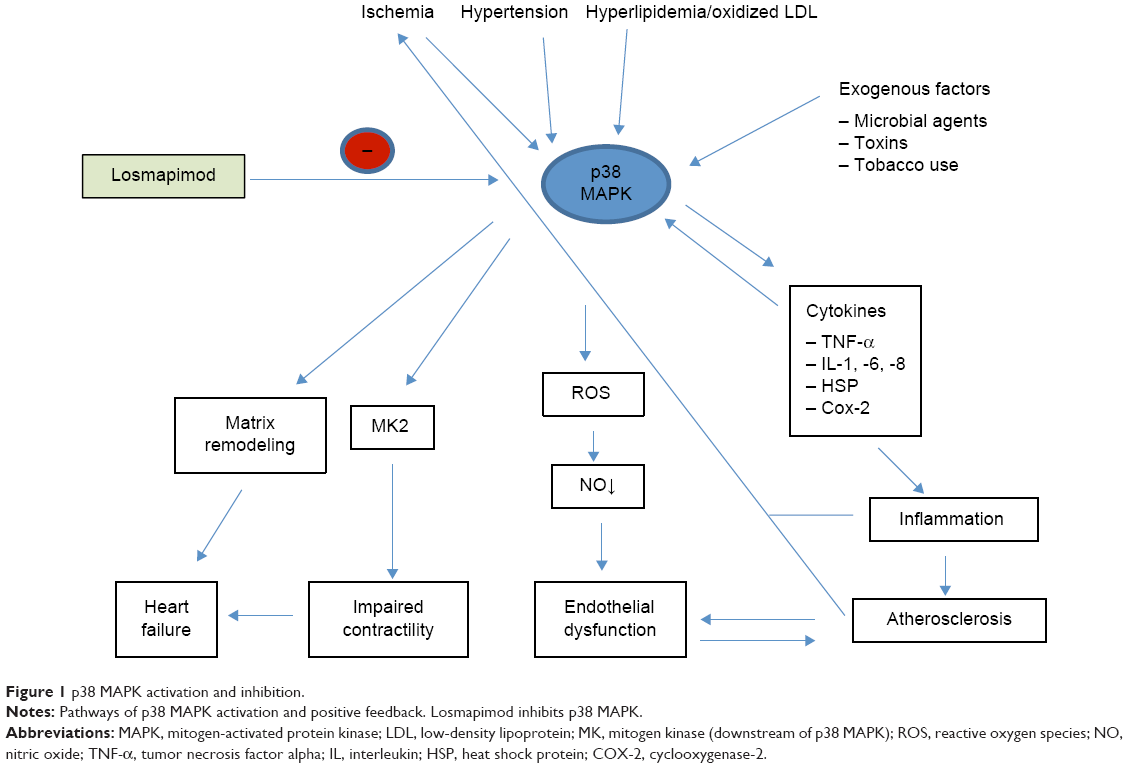 | Figure 1 p38 MAPK activation and inhibition. |
In addition to aggravating atherosclerosis, mice models have shown how activation of TNF-α by p38 MAPK can lead to cardiac remodeling with fibrosis and hypertrophy and reduced cardiac contractility.16 In a hypertensive rat model that was prone to stroke, losmapimod was shown to improve survival and endothelial function and reduce cardiac remodeling and IL-1 production.17 These outcomes were comparable to findings in studies of other compounds that inhibit the p38 MAPK pathway.13,14 Moreover, another p38 MAPK inhibitor compound similar to losmapimod has been shown to both limit infarct size in a model of acute coronary artery ligation by attenuating reperfusion injury and attenuate atherosclerosis in an animal model of chronic atherosclerosis.18 Based on these preclinical models of the role of p38 MAPK in mediating inflammation and downstream pathophysiological effects and showing the effects of compounds that inhibit p38 MAPKs, further development of p38 MAPK inhibitors, including losmapimod, was undertaken. Early clinical development of some of these compounds for use in various disease states was hindered by evidence of drug-induced liver injury.13,14 The remainder of this review will focus on losmapimod development, in particular, the rationale and supporting evidence for its development and use in ACSs.
Phase I clinical studies of losmapimod
A PubMed and clinicaltrials.gov search on March 1, 2015 for losmapimod (GW856553; GW856553X; SB856553; GSK-AHAB) revealed ten clinical studies in healthy volunteers or subjects with cardiovascular disease, of which nine have been completed (six Phase I and three Phase II studies) and one which has recently initiated recruitment (Phase III study).15,19–24 These trials are summarized in Table 1.
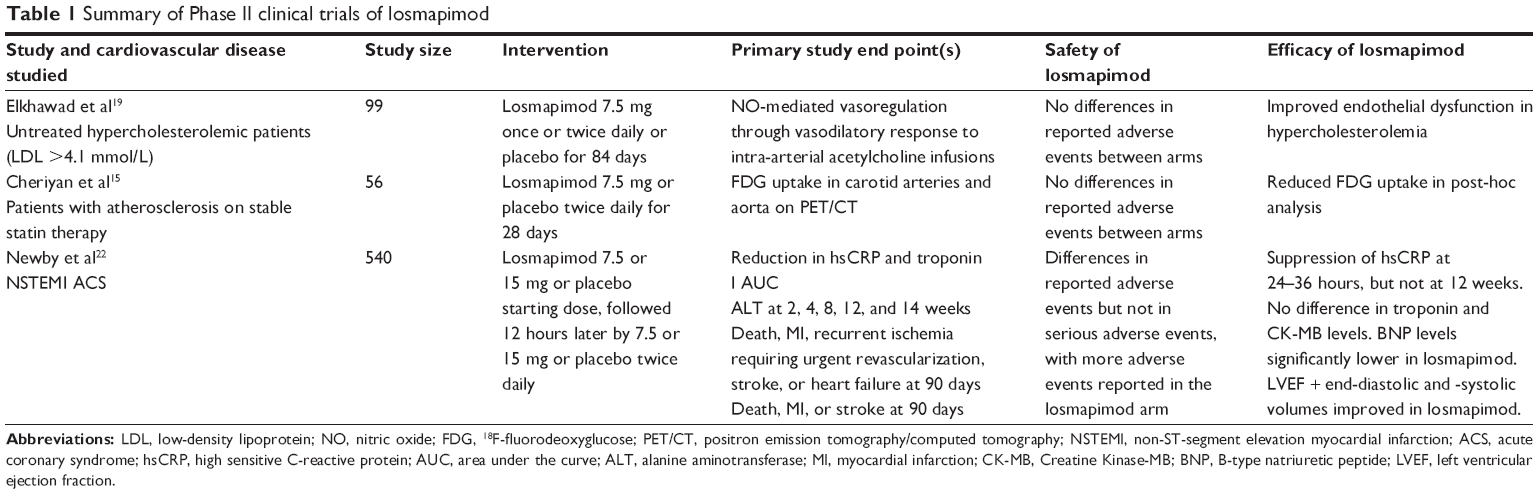 | Table 1 Summary of Phase II clinical trials of losmapimod |
Early experiences with p38 MAPK inhibitors in clinical studies were from rheumatoid arthritis and Crohn’s disease settings, in which limited efficacy and side effects of elevated liver enzymes, skin rashes, and particularly increased susceptibility to infections were noted.13,14,25,26 Other p38 MAPK inhibitor compounds, including losmapimod, were later tested in chronic obstructive pulmonary disease (COPD) patients and no serious adverse effects were noted, and none of the early Phase I studies involving losmapimod indicated any issues with elevated liver enzymes or other serious adverse events.13,14,27 A recently published population pharmacokinetic/pharmacodynamic (PK/PD) meta-analysis from six Phase I studies in healthy volunteers provided reassuring safety data showing that losmapimod plasma concentration had no significant effect on QT-interval prolongation.21 Another population PK meta-analysis that included data from three studies (30 healthy volunteers, 23 subjects with rheumatoid arthritis, and 24 subjects with COPD) showed that although some variation by age, sex, and body weight may be expected in losmapimod PK measures, there was no indication for dose adjustment according to these parameters.28
A study comparing the safety, tolerability, and PK/PD parameters of losmapimod following a single intravenous administration versus an oral dose in healthy volunteers reported no deaths, nonfatal serious adverse events, or adverse events leading to withdrawal; headache was the only adverse event reported more than once in the oral administration arm.20 The study revealed a significant temporal plasma correlation between reduction of phosphorylated HSP 27 and administration of losmapimod, both orally and intravenously.
Thus, these early phase studies provided the PK/PD foundation for losmapimod use in humans and reassuring data on the safety and tolerability of losmapimod administered both intravenously and orally, setting the potential for its use in the context of ACS.
Phase II clinical studies of losmapimod
The clinical Phase II randomized controlled trials of losmapimod in cardiovascular disease are summarized in Table 1. A Phase II randomized controlled trial of 56 hypercholesterolemic patients with LDLc >4.1 mmol/L treated with 7.5 mg of losmapimod twice daily for 28 days or matching placebo revealed no major adverse effects of losmapimod.15 Specifically, no study participants had alanine aminotransferase (ALT) or aspartate aminotransferase values above the upper limits of normal, and there were no other liver function test abnormalities reported. Headache was the most frequent adverse effect, but it was equally distributed between losmapimod- and placebo-treated participants.15 In this study, losmapimod administration compared with placebo improved NO-mediated vasodilation in response to intra-arterial acetylcholine infusion. Treatment with losmapimod for 28 days not only improved the endothelium-dependent vasodilatory responses to acetylcholine but also the endothelium-independent vasodilatory responses to nitroprusside compared with placebo. The improved endothelium-dependent and -independent findings were evident without alteration in LDLc levels.15 In addition, losmapimod-treated participants had greater lowering of phosphorylated HSP27, a previously described well-established downstream marker of p38 MAPK activity, than placebo-treated participants (Figure 1). A prespecified secondary analysis found hsCRP to be significantly lower in the losmapimod arm compared with placebo.
Another randomized Phase II clinical trial of 99 patients with documented atherosclerosis on stable doses of a statin medication assessed vascular inflammation using 18F-fluorodeoxyglucose positron emission tomography/computed tomography (PET/CT) imaging of carotid arteries and aorta.19 It also showed no difference in the frequency of adverse events between the losmapimod and placebo arms. This study was powered to detect a significant difference between the index vessel with the highest tissue-to-background ratio at baseline and at 84 days but failed to meet this end point, although a trend toward attenuation of inflammation area was observed for the losmapimod groups (7.5 mg once or twice daily) compared with placebo. However, in a prespecified secondary analysis there was significantly less uptake of 18F-fluorodeoxyglucose in visceral fat in the losmapimod groups compared with the placebo arm, suggesting a systemic anti-inflammatory effect of losmapimod.19
Based on the promising findings of efficacy without major safety concerns regarding the administration of losmapimod in these initial Phase II studies, the Study of LoSmapimod Treatment on Inflammation and infarCt sizE (SOLSTICE) trial was undertaken to confirm the safety profile of the drug and explore potential clinical benefit in NSTEMI patients.22,29 A total of 535 NSTEMI patients were randomized to an initial dose of 7.5 or 15.0 mg of oral losmapimod followed 12 (±4) hours later by 7.5 mg twice daily or matching placebo for 12 weeks.
Primary safety end points included evidence of liver injury (ALT concentrations) and two composite ischemic end points: 1) death, MI, recurrent ischemia requiring urgent revascularization, stroke, or heart failure, and 2) death, MI, or stroke.22,29 Primary efficacy end points were inflammation (hsCRP concentration at week 12) and infarct size (area under the curve [AUC] for troponin I within 72 hours or by discharge, whichever was first). Secondary efficacy measures were hsCRP level at week 14 and IL-6 level at 24 hours and week 12, in addition to Creatine Kinase-MB AUC and peak troponin I through 72 hours or discharge, whichever was first. B-type natriuretic peptide (BNP) at 72 hours or discharge and at 12 weeks was also assessed in addition to a substudy of cardiac magnetic resonance imaging (MRI) among patients with CK-MB twice the upper limit of normal.22,29
Safety outcomes in the SOLSTICE trial
Primary results were prespecified to be presented for the combined losmapimod groups versus placebo. Protocol-specified discontinuation of the study drug occurred equally between the combined losmapimod groups (7%) and the placebo arm (6%).22 However, in contrast to earlier Phase II trials, in which almost none of the patients discontinued their treatment regimen, 37.5% of patients discontinued their randomly assigned treatment in the SOLSTICE trial, 39.2% in the 7.5 mg losmapimod loading dose group, 39.6% in the 15 mg losmapimod loading dose group, and 31.9% in the placebo group. Patient’s decision to withdraw was the most frequently reported reason for discontinuation. Adverse events was the second most reported reason for the discontinuation of the study drug and was more frequently observed among losmapimod-treated patients than placebo (9% vs 4%, P=0.0412). Report of at least one adverse event was equally distributed across treatment arms (69% in the combined losmapimod arms vs 71% in placebo), as were serious adverse events (24% vs 24%). For the safety analyses at 90 days, for which fewer than 1% of patients were lost to follow-up, no differences were seen between treatment groups in death, MI, recurrent ischemia requiring urgent revascularization, stroke, or heart failure or death, MI, or stroke (Table 2). Elevation of ALT to or above three times the upper limit of normal was infrequent, but was nonsignificantly higher among the losmapimod-treated patients. Serum creatinine levels at 12 weeks were significantly higher in the losmapimod arm than those in the placebo group (P=0.0008), although the absolute difference was small.22
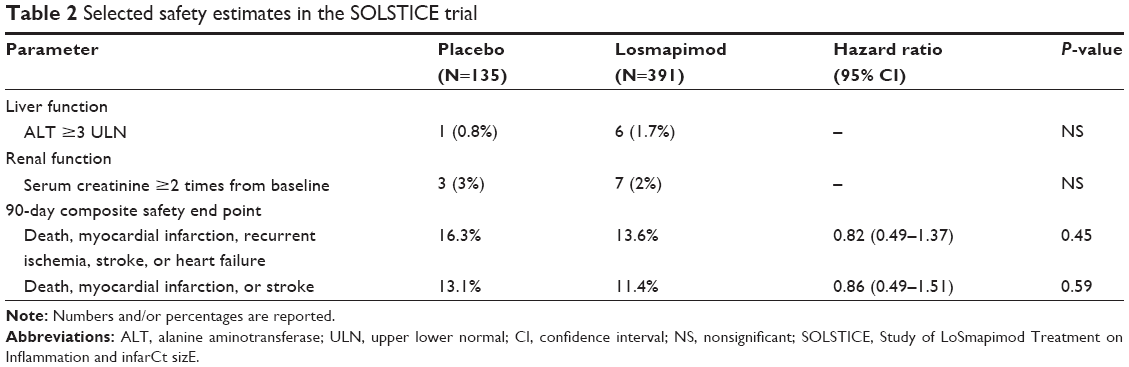 | Table 2 Selected safety estimates in the SOLSTICE trial |
Efficacy outcomes in the SOLSTICE trial
Relative to placebo, losmapimod significantly suppressed hsCRP at the 24–36 hour assessments, but levels of hsCRP were similar at the 12-week assessment (the primary efficacy assessment point). A rebound effect was observed at 2 weeks after treatment ended, showing significantly higher hsCRP levels in the losmapimod groups compared with placebo (Table 3). A similar observation was also made in an earlier Phase II study of losmapimod, although in that study, the levels of hsCRP in high- and low-dose losmapimod arms were consistently and significantly lower than in the placebo arm throughout the 84-day study period.19 As with hsCRP, IL-6 levels were significantly lower in the losmapimod arms than in the placebo arm 24 hours post-randomization, but no significant difference was seen at 12 weeks (Table 3). No significant difference between study groups were seen in the infarct size at any time as evaluated by troponin I AUC and through 72 hours or discharge or by CK-MB AUC or peak troponin I level. BNP levels at 12 weeks were significantly lower in the combined losmapimod arms than placebo, but there were no significant differences at baseline or at 72-hour/discharge time point.22
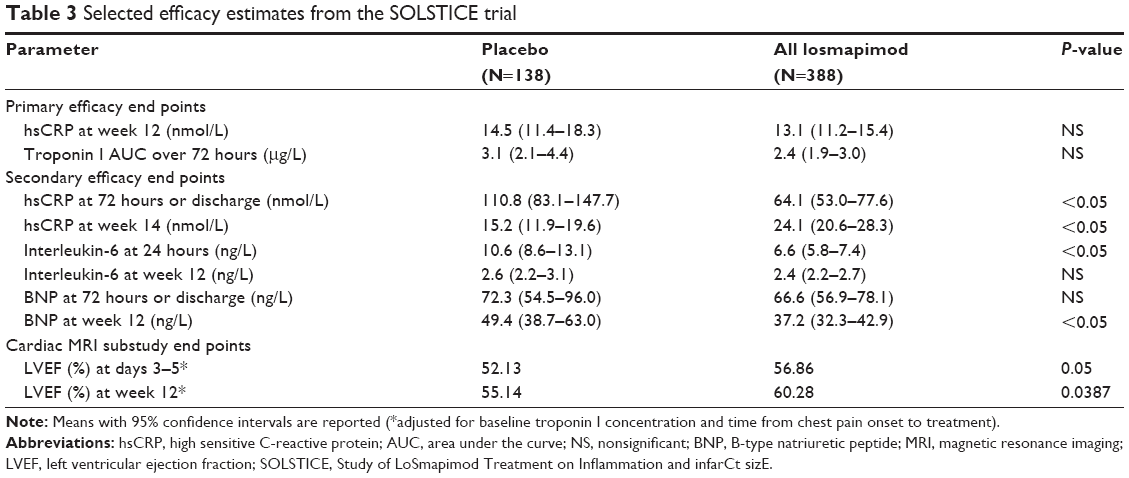 | Table 3 Selected efficacy estimates from the SOLSTICE trial |
In the cardiac MRI substudy, infarct size (the percentage of left ventricle) was significantly lower and left ventricular ejection fraction was significantly higher at 3–5 days and 12 weeks post-randomization in the losmapimod arms compared with placebo.22 Consistent with the higher left ventricular ejection fraction, left ventricular end-diastolic and end-systolic volumes were significantly lower among losmapimod-treated patients at both time points. Changes between 3–5 days and 12 weeks post-randomization were not significant for any of these MRI parameters.22
Interpretation and potential future implications
Findings from these clinical studies of losmapimod show potential for its incorporation in treatment and preventive strategies in atherosclerotic disease, hypercholesterolemia, and ACS and heart failure populations, but this potential remains to be confirmed in adequately powered Phase III randomized clinical trials. The findings of losmapimod-improved vasoregulation and reduced 18F-fluorodeoxyglucose uptake on PET/CT scans in pilot Phase II clinical studies of untreated hypercholesterolemic patients and atherosclerotic patients on stable statin therapy indicated efficacy of losmapimod in attenuating inflammation, potentially counteracting atherosclerotic pathogenic pathways without accruing any notable safety concerns.15,19
SOLSTICE was limited by a total of 37.5% of patients who had not completed 12 weeks of the randomly assigned treatment.22 However, the safety and efficacy findings from this trial look promising as well. In addition, the relatively large drop out in the SOLSTICE trial was not primarily due to adverse events, and although a higher proportion of patients in the combined losmapimod arms had elevated ALT and higher creatinine levels relative to placebo, differences were nonsignificant.
While inflammation measured by hsCRP was significantly lower in the losmapimod arms relative to placebo early after treatment initiation, there was a normalization of hsCRP in both groups by week 12. This normalization of hsCRP by 12 weeks in both groups is not surprising based on what is known of the time course of hsCRP after ACS. The translation of the early suppression of inflammation as measured by hsCRP and IL-6 to clinical outcomes (efficacy and safety) remains to be demonstrated in Phase III trials that are adequately powered for safety and efficacy clinical events.
The limited effect of losmapimod in the SOLSTICE trial on circulating biomarkers of infarct size (troponin I and CK-MB) may reflect an issue of lateness in the timing of losmapimod administration, since it was administered on average15 hours after the onset of the NSTEMI event, potentially beyond a point that p38 MAPK inhibition could realistically influence infarct size. Nevertheless, the MRI substudy did show a trend toward reduction of the infarct size on MRI at 3–5 days and 12 weeks post-randomization among losmapimod-treated patients compared with placebo. Further, left ventricular ejection fraction was significantly higher and left ventricular end-diastolic and end-systolic volumes and BNP were significantly lower among losmapimod-treated patients versus placebo.22
The observation of BNP lowering and improved MRI parameters of remodeling post-infarct among losmapimod-treated patients in SOLSTICE is interesting. This potential protective effect of p38 MAPK inhibition on heart failure is supported by preclinical work in mice which showed that p38 MAPK activation led to TNF-α-activated cardiac remodeling with fibrosis and hypertrophy and that reduced cardiac contractility could be mitigated by treatment with losmapimod.16
Phase III clinical trials of losmapimod
Although the Phase II SOLSTICE study suggested promise for losmapimod treatment of patients with acute MI, safety and efficacy data from adequately powered Phase III clinical trials are needed. The LATITUDE (LosmApimod To Inhibit p38 MAP kinase as a TherapeUtic target and moDify outcomes after an acute coronary syndromE)-TIMI 60 trial recently began recruitment and will include 25,500 patients hospitalized with NSTEMI or STEMI who will be randomized to either oral losmapimod (7.5 mg twice daily) or placebo.24 The primary end point is a composite of cardiovascular death, MI, or severe recurrent ischemia requiring urgent revascularization. The main secondary end point is the composite of cardiovascular death or MI. The trial design uses a stepwise approach to address safety and efficacy. Part A consists of a leading cohort of 3,500 patients to assess safety and explore efficacy before enrolling 22,000 patients in an event-driven Part B that is designed to address efficacy. An independent safety review will be conducted after Part A is completed to decide whether patients in Part B need to have a more intense schedule of clinic visits and laboratory assessments. Furthermore, an independent effect on attenuating platelet aggregation has been previously seen following p38 MAPK inhibition; this represents another concept that needs to be searched and examined in a randomized controlled trial to prove its effectiveness in the management of ACS patients.30–32
Conclusion
The effect of losmapimod on vasoregulation, inflammation, and cardiac function without notable safety concerns in early phase studies suggests promise for p38 MAPK inhibition as a novel strategy to improve outcomes of patients with ACS. This promise may include treatment in the ACS/post-ACS phase as well as for primary and secondary prevention to slow progression of atherosclerosis and prevent future cardiovascular events. The safety and efficacy of losmapimod will be confirmed in few years by the LATITUDE-TIMI 60 study, a larger-scale and longer-duration randomized trial adequately powered to assess clinical outcomes, and will inform the potential role of losmapimod in the treatment of cardiovascular disease.
Disclosure
Kragholm, Melloni and Newby have no conflicts of interest relevant to this paper.
References
Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3(11):e442. | ||
Lim SS, Vos T, Flaxman AD, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2224–2260. | ||
Weir RA, McMurray JJV, Velazquez EJ. Epidemiology of heart failure and left ventricular systolic dysfunction after acute myocardial infarction: prevalence, clinical characteristics, and prognostic importance. Am J Cardiol. 2006;97(10A):13F–25F. | ||
Finn AV, Nakano M, Narula J, Kolodgie FD, Virmani R. Concept of vulnerable/unstable plaque. Arterioscler Thromb Vasc Biol. 2010;30(7):1282–1292. | ||
Hingorani AD, Cross J, Kharbanda RK, et al. Acute systemic inflammation impairs endothelium-dependent dilatation in humans. Circulation. 2000;102(9):994–999. | ||
Kinlay S, Libby P, Ganz P. Endothelial function and coronary artery disease. Curr Opin Lipidol. 2001;12(4):383–389. | ||
Vanhoutte PM. Endothelial dysfunction: the first step toward coronary arteriosclerosis. Circ J. 2009;73(4):595–601. | ||
Mueller C. Inflammation and long-term mortality after non-ST elevation acute coronary syndrome treated with a very early invasive strategy in 1,042 consecutive patients. Circulation. 2002;105(12):1412–1415. | ||
Suleiman M, Khatib R, Agmon Y, et al. Early inflammation and risk of long-term development of heart failure and mortality in survivors of acute myocardial infarction predictive role of C-reactive protein. J Am Coll Cardiol. 2006;47(5):962–968. | ||
Braunwald E, Antman EM, Beasley JW, et al. ACC/AHA guidelines for the management of patients with unstable angina and non-ST-segment elevation myocardial infarction: executive summary and recommendations : a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2000;102(10):1193–1209. | ||
Grech ED, Ramsdale DR. Acute coronary syndrome: unstable angina and non-ST segment elevation myocardial infarction. BMJ. 2003;326(7401):1259–1261. | ||
Muslin AJ. MAPK signalling in cardiovascular health and disease: molecular mechanisms and therapeutic targets. Clin Sci. 2008;115(7):203–218. | ||
Martin ED, Bassi R, Marber MS. p38 MAPK in cardioprotection – are we there yet? Br J Pharmacol. 2015;172(8):2101–2013. | ||
Fisk M, Gajendragadkar PR, Mäki-Petäjä KM, Wilkinson IB, Cheriyan J. Therapeutic potential of p38 MAP kinase inhibition in the management of cardiovascular disease. Am J Cardiovasc Drugs. 2014;14(3):155–165. | ||
Cheriyan J, Webb AJ, Sarov-Blat L, et al. Inhibition of p38 mitogen-activated protein kinase improves nitric oxide-mediated vasodilatation and reduces inflammation in hypercholesterolemia. Circulation. 2011;123(5):515–523. | ||
Bellahcene M, Jacquet S, Cao XB, et al. Activation of p38 mitogen-activated protein kinase contributes to the early cardiodepressant action of tumor necrosis factor. J Am Coll Cardiol. 2006;48(3):545–555. | ||
Willette RN, Eybye ME, Olzinski AR, et al. Differential effects of p38 mitogen-activated protein kinase and cyclooxygenase 2 inhibitors in a model of cardiovascular disease. J Pharmacol Exp Ther. 2009;330(3):964–970. | ||
Ma XL, Kumar S, Gao F, et al. Inhibition of p38 mitogen-activated protein kinase decreases cardiomyocyte apoptosis and improves cardiac function after myocardial ischemia and reperfusion. Circulation. 1999;99(13):1685–1691. | ||
Elkhawad M, Rudd JHF, Sarov-Blat L, et al. Effects of p38 mitogen-activated protein kinase inhibition on vascular and systemic inflammation in patients with atherosclerosis. JACC Cardiovasc Imaging. 2012;5(9):911–922. | ||
Barbour AM, Sarov-Blat L, Cai G, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of losmapimod following a single intravenous or oral dose in healthy volunteers. Br J Clin Pharmacol. 2013;76(1):99–106. | ||
Yang S, Beerahee M. Losmapimod concentration-QT relationship in healthy volunteers: meta-analysis of data from six clinical trials. Eur J Clin Pharmacol. 2013;69(6):1261–1267. | ||
Newby LK, Marber MS, Melloni C, et al. Losmapimod, a novel p38 mitogen-activated protein kinase inhibitor, in non-ST-segment elevation myocardial infarction: a randomised phase 2 trial. Lancet. 2014;384(9949):1187–1195. | ||
Barbour AM, Magee M, Shaddinger B, et al. Utility of concentration-effect modeling and simulation in a thorough QT study of losmapimod. J Clin Pharmacol. 2015;55(6):661–670. | ||
O’Donoghue ML, Glaser R, Aylward PE, et al. Rationale and Design of the LATITUDE-TIMI 60 (LosmApimod To Inhibit p38 MAP kinase as a TherapeUtic target and moDify outcomes after an acute coronary syndromE) Trial. Am Heart J. 2015;169(5):622–630. | ||
Cohen SB, Cheng T-T, Chindalore V, et al. Evaluation of the efficacy and safety of pamapimod, a p38 MAP kinase inhibitor, in a double-blind, methotrexate-controlled study of patients with active rheumatoid arthritis. Arthritis Rheum. 2009;60(2):335–344. | ||
Genovese MC, Cohen SB, Wofsy D, et al. A 24-week, randomized, double-blind, placebo-controlled, parallel group study of the efficacy of oral SCIO-469, a p38 mitogen-activated protein kinase inhibitor, in patients with active rheumatoid arthritis. J Rheumatol. 2011;38(5):846–854. | ||
Watz H, Barnacle H, Hartley BF, Chan R. Efficacy and safety of the p38 MAPK inhibitor losmapimod for patients with chronic obstructive pulmonary disease: a randomised, double-blind, placebo-controlled trial. Lancet Respir Med. 2014;2(1):63–72. | ||
Yang S, Lukey P, Beerahee M, Hoke F. Population pharmacokinetics of losmapimod in healthy subjects and patients with rheumatoid arthritis and chronic obstructive pulmonary diseases. Clin Pharmacokinet. 2013;52(3):187–198. | ||
Melloni C, Sprecher DL, Sarov-Blat L, et al. The study of LoSmapimod treatment on inflammation and InfarCtSizE (SOLSTICE): design and rationale. American Am Heart J. 2012;164(5):646–653.e3. | ||
Rawlinson L. Role for p38 Mitogen-activated protein kinase in platelet aggregation caused by collagen or a thromboxane analogue. J Biol Chem. 1996;271(12):6586–6589. | ||
Flevaris P, Li Z, Zhang G, Zheng Y, Liu J, Du X. Two distinct roles of mitogen-activated protein kinases in platelets and a novel Rac1-MAPK-dependent integrin outside-in retractile signaling pathway. Blood. 2009;113(4):893–901. | ||
Kuliopulos A, Mohanlal R, Covic L. Effect of selective inhibition of the p38 MAP kinase pathway on platelet aggregation. Thromb Haemost. 2004;64701:1387–1393. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.