Back to Journals » International Journal of Women's Health » Volume 9
Emerging treatment options for ovarian cancer: focus on rucaparib
Authors Mariappan L, Jiang XY, Jackson J, Drew Y
Received 8 September 2017
Accepted for publication 12 October 2017
Published 15 December 2017 Volume 2017:9 Pages 913—924
DOI https://doi.org/10.2147/IJWH.S151194
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Elie Al-Chaer
Lavanya Mariappan,1 Xue Yan Jiang,1 Josie Jackson,2 Yvette Drew2
1Northern Centre for Cancer Care, Freeman Hospital, 2Northern Institute for Cancer Research, Newcastle University, Newcastle-upon-Tyne, UK
Abstract: Poly(ADP-ribose) polymerase (PARP) inhibitors (PARPi) are an exciting class of anticancer drugs, which have revolutionized the management of BRCA mutant/homologous recombination-deficient recurrent high-grade serous ovarian cancer (HGSOC). With three PARPi now approved by the US Food and Drug Administration, olaparib (Lynparza™), niraparib (Zejula™), and rucaparib (Rubraca™) in 2014 (and 2017 for the tablet formulation), 2016, and 2017, respectively, these drugs have now entered routine clinical practice. The marked single-agent efficacy of PARPi either as maintenance following response to platinum-based chemotherapy or as up-front treatment in these indications is based on the well-known concept of synthetic lethality. PARPi themselves work by blocking the repair of single-strand DNA breaks by the base excision/single-strand break repair pathway and can also be directly cytotoxic by the mechanism of PARP trapping. The greatest benefit in terms of progression-free survival, in all three PARPi maintenance registration studies, was seen in women with platinum-sensitive BRCA mutation-associated HGSOC. However, it is clear that non-BRCA HGSOC can benefit from PARPi and the ongoing challenge of biomarker driven studies is how best to define these patients. PARPi are well tolerated, but more information is needed to assess the longer-term/later onset toxicities as these agents are investigated in the first-line setting. The future direction and challenges for PARPi will be to continue to expand beyond BRCA and ovarian cancer by identifying molecular or functional signatures of response; to see if the durable responses in ovarian cancer can be improved and efficacy can be achieved in other cancer sub-types by combining with novel targeted agents. This review summarizes the development of PARPi as a class in ovarian cancer with particular focus on the PARPi rucaparib.
Keywords: ovarian cancer, BRCA, PARP inhibitors, rucaparib
Introduction
Epithelial ovarian cancer (EOC) is the leading cause of death from gynecological malignancy in the Western World. It is the seventh most common cancer among women with an estimated 239,000 new cases registered in 2012.1 Women commonly present with advanced stage disease, and despite initial high responses to first-line treatment (cytoreductive surgery combined with platinum-based chemotherapy), overall 5-year survival remains poor at 28%.2
Between 2006 and 2013, there were no new drug approvals for ovarian cancer despite significant global research efforts. During the same period, major advances in the systemic management and subsequent improved survival in other cancers such as non-small-cell lung cancer and melanoma were seen. One explanation for this lack of progress in EOC treatment is the historical failure to recognize that EOC is not one but many diseases, and patient selection for clinical trials for the testing of new novel agents is key. The gyne-oncology community now accepts and understands that EOC is an umbrella term for several diseases with very distinct etiologies, molecular sub-groups, and clinical behaviors. To develop novel treatments for EOC, we must target and exploit these sub-groups. The development and subsequent US Food and Drug Administration (FDA) approvals of poly(ADP-ribose) (PAR) polymerase (PARP) inhibitors (PARPi) in breast and ovarian cancer susceptibility gene (BRCA) mutant high-grade serous ovarian cancer (HGSOC) is a successful example of this approach.
HGSOC is the most common histological sub-type of EOC (70%).3 The Cancer Genome Atlas estimated that ~50% of HGSOC has deficiencies in the DNA double-strand break repair pathway homologous recombination repair (HRR). Specifically, we know that nearly one-third of HGSOC has deleterious germline (24%) and/or somatic (9%) mutations in one or more of the 13 genes in the HRR pathway, with mutations in the BRCA1 and BRCA2 being the most prevalent (19% and 6%, respectively).4 This HRR deficiency (HRD) can be exploited by treatment with PARPi.
The FDA has now approved three PARPi olaparib capsules (Lynparza™; AstraZeneca, Cambridge, UK), niraparib (Zejula™; Tesaro Inc, Waltham, MA, USA), and rucaparib (Rubraca™, Clovis Oncology, San Francisco, CA, USA) in 2014 (and 2017 for the tablet formulation), 2016, and 2017, respectively, as treatment for BRCA mutant recurrent HGSOC.5–10 The niraparib approval includes patients without known HRD but requires patients to have a complete or partial response to prior platinum; the rucaparib and olaparib (initial) FDA approvals can be applied to patients with both platinum-resistant and -sensitive diseases. Most recently (August 2017), the FDA has approved a second indication to olaparib (using the tablet formulation) for maintenance treatment following complete or partial response to platinum-based chemotherapy. The European Medicines Agency approved olaparib in the maintenance setting in 2014. The approvals and the registration trials that led to them are shown in Table 1. The landmark approvals of all three of these agents represent decades of research based on the new well-known concept of synthetic lethality,11 as illustrated and explained by Figure 1.
 | Table 1 Registration studies for the PARP inhibitors: FDA and EMA approval dates and indications |
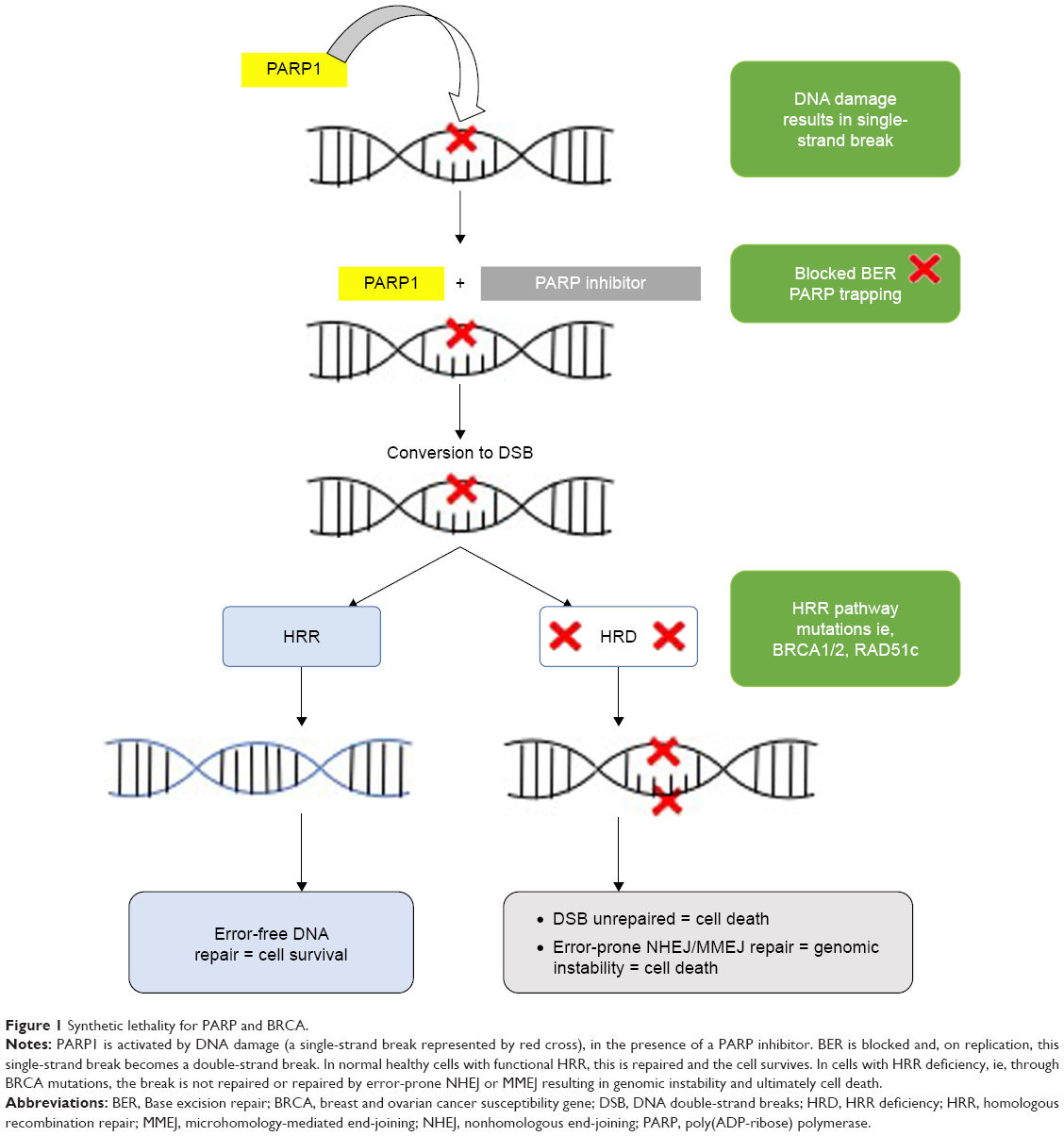 | Figure 1 Synthetic lethality for PARP and BRCA. |
The three approved PARPi are as single-agent applications in ovarian cancer only. However, other late- and early-phase studies continue to investigate the wider role of single-agent PARPi in HRD ovarian and nonovarian cancers, and registered studies are listed in Table 2 (http://www.ClinicalTrials.gov).
 | Table 2 Open and actively recruiting early/late-phase trials with single-agent PARP inhibitors in ovarian cancer (August 2017) |
The primary objective of this article is to review the current evidence for the use of PARPi in ovarian cancer focusing specifically on the clinical data and tolerability of the PARPi rucaparib to provide the reader with an overview of the background of rucaparib’s development in ovarian cancer and to discuss the future role of rucaparib and PARPi as a class in treating ovarian cancer.
DNA damage response, PARP, and the BRCA story
The accurate and efficient repair of DNA damage is essential for normal cellular function and the maintenance of genomic stability.12 In humans, acquired or inherited defects in the DNA damage response pathways (DDR) can result in an increased lifetime risk of cancer.13 The integrity of DNA faces continual threat from a variety of agents including endogenous sources, which occur as the by-products of normal cellular metabolism. For example, reactive oxygen and nitrogen species, estrogen and cholesterol metabolites, and reactive carbonyl species are generated by normal cellular activity and can damage DNA.14 Spontaneous reactions within the DNA microenvironment such as hydrolysis can also result in damage to bases, such as deaminations.15
In humans, over 130 genes have been identified that are associated with the DDR and the function of their proteins are sub-grouped into five distinct DNA repair pathways.16 They are the excision repair mechanisms: base excision repair (BER) or single-strand break repair (SSBR), nucleotide excision repair (NER), which consists of two branches, such as global genome and transcription coupled NER, and mismatch repair, and finally the double-strand DNA break repair mechanisms: nonhomologous end-joining (NHEJ) and HRR. Each pathway has evolved to deal with a specific type of DNA damage, although there is some overlap in their functions. The pathways relevant to this review are the BER/SSBR and the DNA double-strand repair pathways NHEJ and HRR.
The first PARP enzyme was discovered in 1963 when researchers in Paul Mandel’s laboratory observed the synthesis of a new polyadenylic acid after adding nicotinamide mononucleotide to rat liver extracts.17 Shortly, after this discovery, independent research groups demonstrated that this new polymer, named PAR, was made up of two ribose moieties and two phosphate units.18,19 This enzyme that could generate large amounts of this PAR was later purified and designated the name ADP-ribosyl transferase (ADPRT).20 By 1980, it was known that ADPRT could be activated by both single- and double-strand DNA breaks.21 Seminal work by Sydney Shall’s group demonstrated that ADPRT was not only involved in DNA repair but inhibiting it with 3-aminobenzamide (3-AB) could enhance the cytotoxic effects of chemotherapy such as the methylating agents in mouse leukemic cells.22 They were the first to suggest that ADPRT inhibitors could act as chemosensitizers. ADPRT was later renamed PARP-1,23 and there are now 17 members of the PARP super-family.24 A true PARP is defined as an enzyme that is able to transfer ADP-ribose units from NAD+ to acceptor proteins, including itself, resulting in the formation of multiple branched and linear PAR chains, a process known as “poly(ADP-ribosyl)ation”.25 By this definition, only PARP-1, -2, -3, -4, -5, and -5a would be true PARPs. PARP-1, -2, and -3 are the only members known to be activated by DNA damage with PARP-1 playing the dominant role in DNA repair.26
PARP-1 has many functions but is best known for its role in the DDR pathways in particular BER/SSBR.24 Within BER/SSBR, PARP-1 is activated by DNA damage, which it senses through its zinc fingers, subsequently binding to the damaged DNA site undergoing poly(ADP-ribosylation) and creating a negatively charged unit to recruit other BER/SSBR proteins such as XRCC1 to promote and enable repair. PARP-1 also plays a role in the initiation of the double-strand DNA repair pathway HRR by sensing stalled replication forks and recruiting MRE11 and NBS1.27 Within HRR, it also facilitates the addition of pADPr to BRCA128 and inhibits the NHEJ pathway by preventing the binding of Ku proteins to the free damaged DNA ends.29 PARP-1 has non-DDR roles with several studies showing that it is involved in the inflammatory response to acute conditions such as cerebral ischemia, septic shock, and myocardial infarction and in chronic diseases such as diabetes mellitus.25,30 DNA double- strand breaks (DSBs) are the most lethal insult to the genome, which left unrepaired result in a genomic instability and cell death.31 DSBs can arise as a result of direct damage to both strands from anticancer treatments such as ionizing radiation or the topoisomerase inhibitors or as sequelae of unrepaired single-strand DNA breaks from normal physiology.32 DNA DSBs can be repaired by either the error-prone classical NHEJ (C-NHEJ) or HRR, which is an error-free pathway. HRR occurs only in late S/G2 phase.
The BRCA1 and BRCA2 genes were discovered in 1994 and 1995, respectively,33,34 and the BRCA1 and BRCA2 proteins are critical to the functioning of the HRR pathway. It is this deficiency in HRR that is exploited by PARPi.
Germline BRCA1 and BRCA2 mutations account for ~5%–10% of breast cancers and 10%–18% of ovarian cancers.35 Many of the other proteins involved in the DDR are now recognized to also contribute to hereditary cancer risk including ATM, CHEK2, BARD1, BRIP1, MRE11, RAD50, NBS1, RAD51C, RAD51D, and PALB2.
PARPi: mechanism of action, preclinical development, and early-phase clinical development
The first PARPi, identified over 30 years ago, was 3-aminobenzamide. It resulted in 96% PARP inhibition, but it required high intracellular concentrations to achieve this and lacked PARP specificity.36 The subsequent development of PARPi has been the focus of >20 global medicinal chemistry programs with a peak in development between the years 2001 and 2005.37 The development pipelines of PARPi have followed two routes: first combination as chemopotentiators and second as single agents in BRCA/HRR-deficient cancers. The mechanism of action of these classes of agents is that they inhibit the action of PARP and subsequent functional BER/SSBR resulting in unrepaired single-strand DNA breaks. However, more recently, it was discovered that PARPi can also promote cell death by “PARP trapping”, a process by which the PARP enzyme remains inactive but bound to the broken DNA effectively blocking DNA repair and obstructing replication forks. So the PARP–DNA complex itself becomes “cytotoxic” to the cell.38
All PARPi share a common mechanism of action of blocking BER/SSBR but differ in their bioavailability, PARP enzyme inhibition potency and specificity, and their PARP trapping capability.
The initial clinical development of PARPi focused on their utility as chemopotentiators. This was based on emerging preclinical data showing that following exposure to DNA damaging chemotherapy the addition of a PARPi would prevent functional BER/SSBR, which was essential to repair the chemotherapy-induced DNA damage, and thus, the damage was potentiated.24 Monofunctional DNA methlyating agents are the most potent activators of PARP-1 and -2, and they include dacarbazine and temozolomide. AG014361, (forerunner to rucaparib) at concentrations as low as 0.4 μM, was later shown to potentiate the antitumor effects of temozolomide causing complete tumor regressions in SW620 colorectal cancer cell line xenografts.39 In 2003, the first PARP-1 inhibitor AG014699 (forerunner to oral rucaparib: Rubraca™) entered anticancer human clinical trials.40 The study investigated the safety and efficacy of intravenous AG014699 given on days 1–5 of a 21-day cycle in combination with oral temozolomide. During this trial and subsequent PARPi – cytotoxic chemotherapy trials, a common theme emerged, which was the challenge of enhanced myelosuppression when combining these agents.
In 2005, the development of PARPi changed to focus on single-agent utility with the publication of paired Nature papers demonstrating the exquisite sensitivity of BRCA-deficient cell lines, and in vivo models to forerunners to both olaparib and rucaparib.41,42 This new knowledge about PARPi came about as the Phase I study of the oral PARPi olaparib was actively recruiting patients with advanced solid tumors to a single-agent dose escalation study.43 The study subsequently began to recruit patients with known germline (g) BRCA1/2 mutations. Of the 19 evaluable patients with gBRCA mutations (eight with advanced ovarian cancer), nine (47%) patients had an objective response by the Response Evaluation Criteria In Solid Tumors (RECIST)44 and 12 (63%) patients had clinical benefit response. These exciting first results suggested a huge potential for single-agent PARPi in BRCA mutant ovarian cancers. This preliminary efficacy signal was later confirmed in a proof of concept Phase II study in BRCA mutant ovarian cancer.45 The study investigated the efficacy of two doses of olaparib in sequential cohorts (400 mg twice daily orally; 100 mg twice daily orally every 28 days) in patients with recurrent gBRCA ovarian cancer following at least one prior line of chemotherapy. The results were a clinical benefit response of 52% in the 400 mg cohort and 21% in the 100 mg cohort, and responses were seen in patients with both BRCA1 and BRCA2 mutations and in those considered to be platinum sensitive and resistant. Olaparib was well tolerated, and nausea and fatigue were the most commonly reported adverse events (AEs). The result of this study was to take olaparib 400 mg twice daily orally forward into later-phase development, which is discussed in the next section.
In 2008, following the publication by the Bryant et al group of the sensitivity of BRCA2-deficient cells to AG14361 (fore-runner to rucaparib), a CRUK Phase II proof of principle trial was started to investigate the single-agent activity of the clinical candidate AG014699 based on the intravenous schedule established in the combination Phase I.40
The primary endpoint of this multicenter trial was to investigate the response rate, safety, and tolerability of AG014699 in patients with recurrent, advanced gBRCA mutation-associated breast and ovarian cancers.46 Preliminary results from the first 38 patients were disappointing with the IV PARPi dosed intermittently resulting in an objective response rate (ORR) of only 2%. In addition, the accompanying translational studies demonstrated that a more continuous dosing of the PARPi was required. At this point, the study was put on hold to recruitment and AG014699 was acquired by Clovis Oncology who foresaw the potential of the agent but recognized the need for an oral formulation. The study was subsequently re-opened in October 2011 using the oral tablet formulation now known as rucaparib (rucaparib camsylate) at higher doses, and longer schedules including continuous dosing. The study concluded that rucaparib was well tolerated up to doses of 480 mg twice daily continuous schedule, and pharmacodynamics studies demonstrated >90% inhibition of PARP sustained for 24 hours. Oral rucaparib was subsequently investigated further in a Phase I trial, which included patients with gBRCA-mutated ovarian, breast, and pancreatic cancers to establish a recommended Phase II dose of 600 mg twice a day oral continuous dosing.47 Subsequent additional parts to this study (2A) investigated the 600 mg twice daily dose in heavily pretreated platinum-sensitive gBRCA mutation-associated ovarian cancer and demonstrated that rucaparib was active with an ORR of 59.5%. The most common rucaparib-related AEs were fatigue, nausea, and anemia.48
Rucaparib and summary of the FDA approval
Rucaparib (formerly known as CO-338, AG-014699, and PF-01367338; Clovis Oncology) is a rationally designed, orally administered, small molecule inhibitor of PARP-1–3.49 Rucaparib monotherapy was granted accelerated approval by the FDA on December 19, 2016, for the treatment of patients with deleterious germline and/or somatic BRCA mutation (BRCAmut)-associated advanced ovarian cancer who have been treated with two or more chemotherapies. The recommended dose and schedule of rucaparib is 600 mg (two 300 mg tablets) taken orally twice daily with or without food. The approval was based on an integrated efficacy analysis of 106 platinum-treated patients (both sensitive and resistant) with HGSOC and a BRCAmut participating in study 1050 and ARIEL2.5 Rucaparib 600 mg twice daily was associated with a median progression-free survival (PFS) time of 10.0 months (95% CI, 7.3–12.5) and a median duration of response of 9.2 months (95% CI, 6.6–11.7). Additionally, 57/106 patients (54%) achieved a RECIST (Version 1.1) objective response (complete response, 8.5%; partial response, 45%) and 36 patients (34%) had stable disease. ORR was 66% in the platinum-sensitive patients and 25% in those with platinum-resistant disease; a respectable ORR in a group of patients with very limited treatment options.
In conjunction with this, the FDA approved the Foundation Focus companion diagnostic BRCA test (FFCDxBRCA) for use with rucaparib, which is the first next-generation sequencing (NGS)-based companion diagnostic approved by the agency. FFCDxBRCA test detects the presence of deleterious BRCA mutations in the ovarian cancer tissue.
Is there a need for a biomarker of HRD to stratify patients for PARPi treatment? What do the ARIEL2, ARIEL3, and NOVA trials teach us?
Not approved by the FDA as part of the rucaparib approval but a key part of the ARIEL2 trial (part 1) was the development of a companion diagnostic test to identify, through fresh and archival tumor biopsies, a biomarker of HRD and therefore PARPi sensitivity. As well as investigating the efficacy of rucaparib ARIEL2 attempted to address the challenging and important questions – Who else might respond to rucaparib beyond BRCA? And how can we identify these patients?
The study investigated using NGS genomic loss of heterozygosity (LOH) as a potential biomarker of HRD. A total of 192 patients were subsequently classified into three predefined HRD sub-groups, using a cut off of ≥14% = LOH high:
- BRCA mutant (germline/somatic),
- BRCA wild-type/LOH high,
- BRCA wild-type/LOH low.
The results for each group in terms of median PFS and ORR are shown in Table 3.
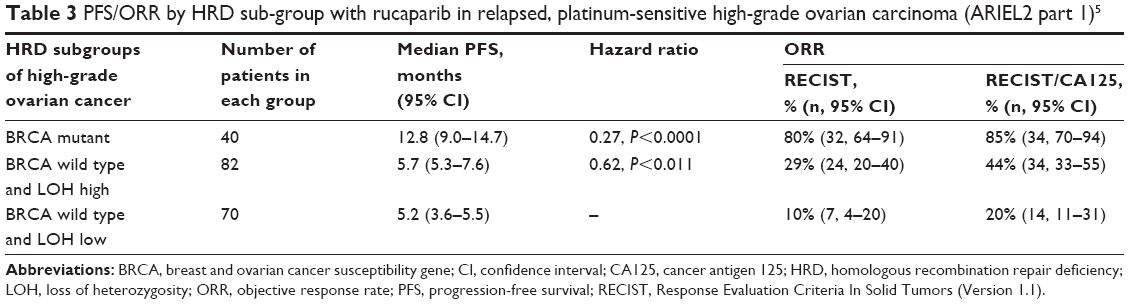 | Table 3 PFS/ORR by HRD sub-group with rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 part 1)5 |
The clinical outcomes for the BRCA wild type/LOH high group were poorer than the BRCA mutant group but still significantly better when grouped together than the BRCA wild type/LOH low group. We can learn two important things from these results. First, like study 19 and the NOVA trial, these results confirm that there is a group of patients with sporadic non-BRCA mutant ovarian cancer who will respond to PARPi. Second, the LOH signature used in ARIEL2 is not perfect in determining HRD and further work may be required to refine the LOH score to improve the predictive power of the test for response. The ARIEL3, randomized Phase III, double-blind, placebo-controlled trial of rucaparib in the maintenance setting in BRCA mutant and wild-type recurrent ovarian cancer sought to further investigate/validate this LOH score. Recently published results of ARIEL3 have shown that across all primary analysis groups (BRCA mutant, BRCA wild type/LOH high, and the intention to treat population), rucaparib significantly improved PFS following response to platinum-based chemotherapy. Similar to the NOVA trial, the greatest benefit from the PARP inhibition was seen in the BRCA mutant group with the PFS of 16.6 months in the rucaparib group vs 5.4 months in the placebo group (P<0.0001).51
The results of the NOVA and ARIEL3 trials lead us to the question: Is a predictive biomarker of PARPi response necessary given in both trials all groups, regardless of BRCA mutation or HRD status, benefited from maintenance niraparib or rucaparib, respectively, with significant improvement in PFS following response to platinum-based chemotherapy. However, it could be argued that there was a biomarker selecting patients for PARPi treatment and this was “platinum sensitivity” and we know from the original olaparib studies that response was positively associated with the surrogate marker of platinum sensitivity, the “platinum-free interval”.52 Reviewing the NOVA trial inclusion criteria published fully in the supplementary data,6 patients were required to have had a response to the most recent platinum regimen defined by the investigator as a “complete or partial response with no observable residual disease of >2 cm”. This required a degree of “extreme platinum sensitivity” that was not specified in study 197 or SOLO262 using olaparib or the ARIEL361 trial of rucaparib maintenance.
What is clear across all the registration studies is that patients whose ovarian cancer has a BRCA mutation seem to benefit the most from PARPi, and given the albeit low grade but often chronic toxicity of these agents, the authors conclude that it may not be appropriate to offer all women with HGSOC a PARPi, and each case should be managed individually discussing the benefit and toxicity. Ultimately, PARPi are likely to move into the first-line setting where the toxicity benefit ratio becomes even more of an issue in these non-BRCA groups. Using platinum sensitivity as a biomarker could work when offering maintenance treatment but what about upfront PARPi treatment? And women with platinum-resistant disease where PARPi have also been shown to be effective? It may be that it is in this setting that a biomarker of HRD is still needed to determine who would respond and so the challenge in this field is to develop other, or refine existing, signatures to be able to truly define the group of ovarian cancers that respond the best to PARPi and the best settings to use them in.
Rucaparib – safety and tolerability
PARPi as a class are generally well tolerated by ovarian cancer patients even those who have been heavily pretreated with chemotherapy. With experience, low-grade treatment-related side effects are manageable and, following the addition of supportive measures and dose reductions, are unlikely to lead to drug discontinuation. However, toxicities do occur and this section is specifically related to the tolerability of rucaparib according to the published integrated safety analysis of ARIEL2 and study 10 data, but common themes are emerging about the side effects of this class of agents as a whole.
Results from the integrated safety analysis of study 10 and the ARIEL2 trials for patients receiving the 600 mg twice daily continuous dosing regimen are summarized later. In these studies, 44% of patients had a dose reduction due to a treatment-related AE. The most common treatment-related AEs of any grade experienced by ≥20% of study patients were as follows: anemia, alanine transferase (ALT) and/or aspartate transaminase (AST) rise, serum creatinine rise, gastrointestinal (GI) related (nausea, vomiting, constipation, decreased appetite, diarrhea, abdominal pain, and dysgeusia), fatigue, and dyspnea (range 21.0%–76.9%). No patients had a rucaparib-related AE leading to death.
The most common patient-reported symptoms on rucaparib were nausea and vomiting. These were mostly low grade (1–2); <15% of patients had a treatment interruption because of nausea and vomiting, and <12% had a dose reduction. It was rare for patients to permanently stop rucaparib treatment because of nausea (1.3%) and vomiting (<1%). The authors’ experience is that the emetogenic potential of PARPi in general is at worst moderate, resulting in mild nausea, which is reported more commonly during the first cycle. In patients who present with grade >2 nausea and vomiting, other causalities should be excluded and these patients should be investigated and managed for other causes such as partial or complete bowel obstruction, which can occur in advanced ovarian cancer patients. Antiemetics can be used to improve rucaparib-related nausea and vomiting, but it is also worth considering alternative ways of managing what could be a chronic toxicity, such as changing the timing of the twice daily treatment taken. In patients who require antiemetics, the choice should be guided by the treating oncologist.
The most common investigational AE in patients taking rucaparib is anemia. Myelosuppression has been seen with all of the three approved PARPi and appears to be a class effect. Patients receiving PARPi should have regular full blood count monitoring according to the individual drug approval recommendations. Despite the high incidence of anemia (>50% of patients having grade ≥3), only 1% of patients discontinued rucaparib due to anemia. In study 10 and ARIEL2, anemia was managed by rucaparib dose interruptions/reductions and the use of supportive care according to the National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology.53
The rise in transaminases observed in the rucaparib trials is interesting and occurred with an incidence of ~41%. ALT/AST elevations occurred early and were transient, self-limiting, and not associated with other signs of liver toxicity. During the early stages of these trials, investigators were appropriately cautious about these ALT/AST elevations and patients were dose reduced appropriately. Over time, it became apparent that these elevations were rarely associated with increases in bilirubin and they normalized over time with continued rucaparib treatment. The mechanism of action or etiology of the ALT/AST elevations is not completely understood. For patients who experience rise in transaminases, no intervention is required to mitigate elevations in ALT/AST, provided all other causes have been excluded and liver function is monitored.
A rise in serum creatinine is also common in patients receiving rucaparib and was seen in 21% of patients in the registration studies. In vitro studies initiated following these observations during the clinical trials have shown that rucaparib inhibits the drug transporters MATE1 and MATE2-K, which play a role in renal secretion of creatinine; interestingly, this has also been reported with the PARPi veliparib.54 After rucaparib interruption or discontinuation, serum creatinine levels decreased and increased again with resumption of treatment. In patients taking rucaparib, an elevation in creatinine should always be investigated to exclude and treat other causes of acute kidney injury such as dehydration and obstructive uropathy, which can occur in patients with advanced ovarian cancer. Once other causes have been excluded or treated, mild-serum creatinine elevations do not require dose modification of rucaparib.
One of the most serious AEs reported in all three registration PARPi studies was the occurrence in <1% of patients of acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), and pneumonitis. No clear mechanism of action has been found to link these conditions to PARPi, and it is vital that we try to understand this more as more patients will be treated with PARPi following new approvals and patients receive PARPi earlier on in their treatment, such as in the adjuvant, curative setting. What we do know is that, in the placebo-controlled maintenance studies, MDS/AML was seen in the placebo arms too; one theory is that this is not related to the PARPi but as a result of prior lines of DNA damaging chemotherapies received by these patients. All patients taking PARPi should be counseled about this risk and given all the up-to-date information available, prior to commencing treatment.
Future directions and challenges for rucaparib and the class of PARPi
There is no doubt that the development and subsequent approval of PARPi have changed the management of BRCA mutant HGSOC, and with the NOVA and ARIEL3 data, this is being extended to ovarian cancers beyond BRCA. Ongoing work continues to best define these non-BRCA patients, but what next for these agents?
The first avenue is to broaden the single-agent use of these agents to other malignancies. Olaparib has already shown promise in metastatic castrate-resistant prostate cancer.
The TOPARP-A trial was a Phase II single-arm study of olaparib 400 mg twice daily in unselected metastatic castrate-resistant prostate cancer patients. Patients with tumors underwent NGS to identify mutations in DNA repair genes including BRCA1/2, ATM, Fanconi’s anemia genes, and CHEK2. These were found in 16 of the 49 evaluable patients (33%), and of these 16 patients, 14 (88%) patients had a response to olaparib.55 Rucaparib is also currently under investigation in prostate cancer in the TRITON studies and in BRCA/PALB mutant pancreatic cancer. Olaparib has also been shown to be effective in germline BRCA mutant HER2-negative breast cancer with the publication of the Phase III Olympiad trial in 2017, showing significantly improved PFS over standard of care.56
Another emerging strategy for rucaparib and PARPi as a class is to use them in combination. Prior clinical trial experience combining PARPi (and other inhibitors of the DNA damage) response with chemotherapy has been difficult with increased myelosuppression being the main challenge; the optimal dose and scheduling of these combinations require further research, and we await the results of the ongoing trials.57 PARPi as a radiosensitizer to targeted radiotherapy is a very attractive combination in cancer treatment given PARPs role in the DDR, and this concept is well supported by the preclinical data.58 Clinical studies are now underway investigating novel PARPi – radiotherapy combinations in multiple tumor types including glioblastoma and head and neck, pancreatic, breast, and rectal cancers.
The third exploration is the combination of PARPi with other novel/targeted agents. Table 4 lists the registered ongoing rucaparib/other PARPi cytotoxic and noncytotoxic combinations in clinical trials. Reviewing the current clinical trials these combination studies can be grouped as follows: PARPi + anti-angiogenics, PARPi + immune checkpoint inhibitors (IO), and PARPi + other inhibitors of the DDR. The rationale for these studies is based on several hypotheses explained below.
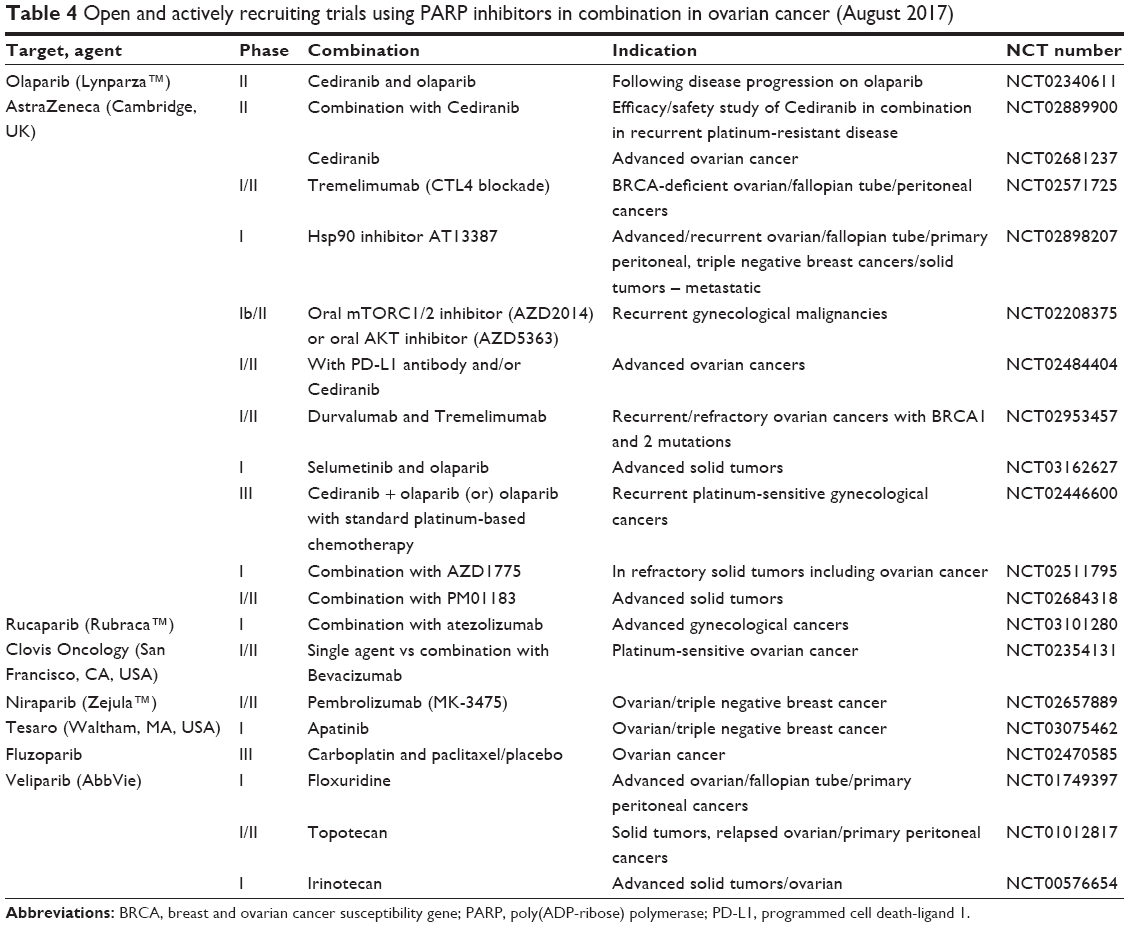 | Table 4 Open and actively recruiting trials using PARP inhibitors in combination in ovarian cancer (August 2017) |
Anti-angiogenesis + PARPi
Inefficient tumor vasculature and the high metabolic demand for oxygen mean that most cancers operate under a degree of hypoxia. Hypoxic conditions can result in chemo/radio-resistance, increased invasion, and metastasis. However, increasing hypoxia through for example antiangiogenics adversely affects DDR pathways. Cells experiencing moderate/severe hypoxia and/or re-oxygenation are sensitive to loss or inhibition of components of the DDR including Chk1, ATM, ATR, and PARP, therefore providing opportunities to exploit this with PARPi and anti-VEGF(R) combinations.
IO + PARPi
The rationale for this combination is that inhibition of PARP will result in an increase in mutational load and modification of tumor immunogenicity,59,60 resulting in an increased sensitivity to immune checkpoint inhibitors of CTLA-4, programmed cell death-1, and programmed cell death-ligand 1 (PD-L1). This is supported by preclinical data, but we are yet to see the final results of the current studies. One of the main issues with some of these studies such as the MEDIOLA study (Phase Ib trial of olaparib + Durvalumab; NCT 02734004) is that the ovarian dose expansion cohort was limited to patients with germline BRCA mutations who one would expect to respond well to the olaparib alone, so the duration of response results will be interesting when the study finally reports. Rucaparib is being investigated in combination with the inhibitor PD-L1 blocking antibody atezolizumab (MPDL3280A) in a Phase I trial (NCT03101280) in patients with HGSOCs not limited to BRCA mutation carriers.
DDR inhibitors + PARPi
The third group of agents being investigated in combination with PARPi is other inhibitors of the DDR such as ATR and Wee1 inhibitors. The rationale here is that either the dual inhibition can lead to a BRCA-like phenotype, which is then exploited by the PARPi, or as in the case of inhibitors of Wee1 kinase (the gate keeper of the G2–M cell-cycle checkpoint) an increase in replication stress will be particularly detrimental in HGSOC and other cancers that have lost their G1–S checkpoint due to P53 mutations. Early-phase studies are underway with only preliminary or no results reported (Table 4).
The key to establish in the design of all these combination studies is whether PARPi combinations can benefit patients, either by broadening their efficacy in patients who would not respond to PARPi alone, or by prolonging response and preventing resistance in the responders. The other important issue is the tolerability of any PARPi combination.
We eagerly await the results of the ongoing studies.
Disclosure
YD is an investigator on clinical trials of olaparib and rucaparib, has participated in advisory boards for Clovis Oncology and AstraZeneca, has received research grant support from Clovis Oncology, and was involved in the preclinical and clinical development of rucaparib. Newcastle University receives royalties for rucaparib. The authors report no other conflicts of interest in this work.
References
IARC. World Cancer Report 2014. Geneva, Switzerland: World Health Organisation; 2014. | ||
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62(1):10–29. | ||
Seidman JD, Horkayne-Szakaly I, Haiba M, Boice CR, Kurman RJ, Ronnett BM. The histologic type and stage distribution of ovarian carcinomas of surface epithelial origin. Int J Gynecol Pathol. 2004;23(1):41–44. | ||
Pennington KP, Walsh T, Harrell MI, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014;20(3):764–775. | ||
Swisher EM, Lin KK, Oza AM, et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18(1):75–87. | ||
Mirza MR, Monk BJ, Herrstedt J, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154–2164. | ||
Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366(15):1382–1392. | ||
fda.gov [webpage on the Internet]. FDA Approves Olaparib Tablets for Maintenance Treatment in Ovarian Cancer. Available from: https://www.fda.gov/drugs/informationondrugs/approveddrugs/ucm572143.htm. Accessed September 3, 2017. | ||
fda.gov [webpage on the Internet]. FDA Grants Accelerated Approval to New Treatment for Advanced Ovarian Cancer. Available from: https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm533873.htm. Accessed September 3, 2017. | ||
fda.gov [webpage on the Internet]. FDA Approves Maintenance Treatment for Recurrent Epithelial Ovarian, Fallopian Tube or Primary Peritoneal Cancers. Available from: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm548948.htm. Accessed September 3, 2017. | ||
Kaelin WG Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5(9):689–698. | ||
Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366–374. | ||
Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361(15):1475–1485. | ||
De Bont R, van Larebeke N. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis. 2004;19(3):169–185. | ||
Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362(6422):709–715. | ||
Jeggo PA, Pearl LH, Carr AM. DNA repair, genome stability and cancer: a historical perspective. Nat Rev Cancer. 2016;16(1):35–42. | ||
Chambon P, Weill JD, Mandel P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem Biophys Res Commun. 1963;11:39–43. | ||
Sugimura T, Fujimura S, Hasegawa S, Kawamura Y. Polymerization of the adenosine 5′-diphosphate ribose moiety of NAD by rat liver nuclear enzyme. Biochim Biophys Acta. 1967;138(2):438–441. | ||
Nishizuka Y, Ueda K, Nakazawa K, Hayaishi O. Studies on the polymer of adenosine diphosphate ribose. I. Enzymic formation from nicotinamide adenine dinuclotide in mammalian nuclei. J Biol Chem. 1967;242(13):3164–3171. | ||
Okayama H, Edson CM, Fukushima M, Ueda K, Hayaishi O. Purification and properties of poly(adenosine diphosphate ribose) synthetase. J Biol Chem. 1977;252(20):7000–7005. | ||
Benjamin RC, Gill DM. ADP-ribosylation in mammalian cell ghosts. Dependence of poly(ADP-ribose) synthesis on strand breakage in DNA. J Biol Chem. 1980;255(21):10493–10501. | ||
Durkacz BW, Omidiji O, Gray DA, Shall S. (ADP-ribose)n participates in DNA excision repair. Nature. 1980;283(5747):593–596. | ||
de Murcia G, Menissier de Murcia J. Poly(ADP-ribose) polymerase: a molecular nick-sensor. Trends Biochem Sci. 1994;19(4):172–176. | ||
Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10(4):293–301. | ||
Schreiber V, Dantzer F, Ame JC, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7(7):517–528. | ||
Ame JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26(8):882–893. | ||
Bryant HE, Petermann E, Schultz N, et al. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009;28(17):2601–2615. | ||
Hu Y, Petit SA, Ficarro SB, et al. PARP1-driven poly-ADP-ribosylation regulates BRCA1 function in homologous recombination-mediated DNA repair. Cancer Discov. 2014;4(12):1430–1447. | ||
Wang M, Wu W, Wu W, et al. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34(21):6170–6182. | ||
Oliver FJ, Menissier-de Murcia J, de Murcia G. Poly(ADP-ribose) polymerase in the cellular response to DNA damage, apoptosis, and disease. Am J Hum Genet. 1999;64(5):1282–1288. | ||
Huertas P. DNA resection in eukaryotes: deciding how to fix the break. Nat Struct Mol Biol. 2010;17(1):11–16. | ||
Neale MJ, Keeney S. Clarifying the mechanics of DNA strand exchange in meiotic recombination. Nature. 2006;442(7099):153–158. | ||
Wooster R, Bignell G, Lancaster J, et al. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378(6559):789–792. | ||
Miki Y, Swensen J, Shattuck-Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266(5182):66–71. | ||
Alsop K, Fereday S, Meldrum C, et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: a report from the Australian Ovarian Cancer Study Group. J Clin Oncol. 2012;30(21):2654–2663. | ||
Milam KM, Cleaver JE. Inhibitors of poly(adenosine diphosphate-ribose) synthesis: effect on other metabolic processes. Science. 1984;223(4636):589–591. | ||
Ferraris DV. Evolution of poly(ADP-ribose) polymerase-1 (PARP-1) inhibitors. From concept to clinic. J Med Chem. 2010;53(12):4561–4584. | ||
Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol. 2011;5(4):387–393. | ||
Calabrese CR, Almassy R, Barton S, et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst. 2004;96(1):56–67. | ||
Plummer R, Jones C, Middleton M, et al. Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res. 2008;14(23):7917–7923. | ||
Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. | ||
Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. | ||
Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. | ||
Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228–247. | ||
Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376(9737):245–251. | ||
Drew Y, Ledermann J, Hall G, et al. Phase 2 multicentre trial investigating intermittent and continuous dosing schedules of the poly(ADP-ribose) polymerase inhibitor rucaparib in germline BRCA mutation carriers with advanced ovarian and breast cancer. Br J Cancer. 2016;114(12):e21. | ||
Rebecca Sophie Kristeleit M. PhD phase 1/2 study of oral rucaparib: FINAL phase 1 results. Abstract presented at: ASCO; June 1; 2014; Alexandria, VA. | ||
Kristeleit R, Shapiro GI, Burris HA, et al. A phase I-II study of the oral PARP inhibitor rucaparib in patients with germline BRCA1/2-mutated ovarian carcinoma or other solid tumors. Clin Cancer Res. 2017;23(15):4095–4106. | ||
Wahlberg E, Karlberg T, Kouznetsova E, et al. Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nat Biotechnol. 2012;30(3):283–288. | ||
The study of oral rucaparib in patients with a solid tumor (Phase I) or with gBRCA mutation ovarian cancer (Phase II). Available from: https://clinicaltrials.gov/ct2/show/NCT01482715. NLM identifier: NCT01482715. Accessed September 3, 2017. | ||
Coleman RL, Oza AM, Lorusso D, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. Epub 2017 Sep 12. | ||
Fong PC, Yap TA, Boss DS, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28(15):2512–2519. | ||
National Comprehensive Cancer Network I, [US] [webpage on the Internet]. NCCN Guidelines for Supportive Care. Available from: https://www.nccn.org/professionals/physician_gls/f_guidelines.asp#supportive. Accessed September 3, 2017. | ||
Kikuchi R, Lao Y, Bow DA, et al. Prediction of clinical drug-drug interactions of veliparib (ABT-888) with human renal transporters (OAT1, OAT3, OCT2, MATE1, and MATE2K). J Pharm Sci. 2013;102(12):4426–4432. | ||
Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373(18):1697–1708. | ||
Robson M, Im SA, Senkus E, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377(6): 523–533. | ||
Brown JS, O’Carrigan B, Jackson SP, Yap TA. Targeting DNA repair in cancer: beyond PARP inhibitors. Cancer Discov. 2017;7(1):20–37. | ||
Lesueur P, Chevalier F, Austry JB, et al. Poly-(ADP-ribose)-polymerase inhibitors as radiosensitizers: a systemic review of pre-clinical and clinical human studies. Oncotarget. 2017;8(40):69105–69124. | ||
Tang ML, Khan MK, Croxford JL, Tan KW, Angeli V, Gasser S. The DNA damage response induces antigen presenting cell-like functions in fibroblasts. Eur J Immunol. 2014;44(4):1108–1118. | ||
Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72. | ||
Coleman RL, Oza AM, Lorusso DA, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10106):1949–1961. | ||
Pujade-Lauraine E, Ledermann JA, Selle F, et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18(9):1274–1284. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.