Back to Journals » Journal of Blood Medicine » Volume 16
Emerging Therapies in Hemophilia: Improving Equitable Access to Care
Authors Lewandowska M, Nasr S, Shapiro AD
Received 8 August 2024
Accepted for publication 21 January 2025
Published 20 February 2025 Volume 2025:16 Pages 95—115
DOI https://doi.org/10.2147/JBM.S490588
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Magdalena Lewandowska,1 Sonia Nasr,2 Amy D Shapiro1
1Indiana Hemophilia & Thrombosis Center, Inc., Indianapolis, Indiana, USA; 2GLOVAL LLC, Broomfield, Colorado, USA
Correspondence: Amy D Shapiro, MD. Indiana Hemophilia and Thrombosis Center, Inc., 8326 Naab Road, Indianapolis, IN, 46260, USA, Email [email protected]
Abstract: In recent years, gene therapy and bio-engineered hemostatic molecules have revolutionized treatment for people with hemophilia. These innovative therapies aim to decrease treatment burden and improve patient quality of life. Additional novel therapies, including next-generation mimetics and agents that rebalance hemostasis, are currently being evaluated in clinical trials. Technological advances such as point-of-care musculoskeletal ultrasound and artificial intelligence may improve patient diagnostic and treatment outcomes. However, for the majority of patients with hemophilia worldwide, diagnosis and effective treatment are inaccessible. Achieving health equity for all hemophilia patients requires improved identification of barriers to optimal care, including socioeconomic status, race/ethnicity, gender, disease severity, inhibitor status, age, and use of Hemophilia Treatment Centers. Access to novel hemophilia therapies should be ensured for all patients. Approaches to improving equity include a decision-making partnership between the patient and clinician, stakeholder engagement, and pharmaceutical industry support. The development of novel hemophilia therapies should be leveraged with a patient-centered care approach to improve health equity for all patients.
Keywords: gene therapy, non-factor therapy, artificial intelligence, point-of-care musculoskeletal ultrasound, health equity
Introduction
Hemophilia A and B are X-linked bleeding disorders characterized by complete or partial deficiency of coagulation factor VIII (FVIII) or IX (FIX). Hemophilia severity is determined by residual plasma factor activity, and classified as severe (<1% of normal), moderate (1–5%), or mild (>5 - <40%).1–3 In the absence of prophylaxis, people with severe hemophilia (PwH) experience frequent spontaneous bleeding events (BEs) into joints and muscles. This results in a high risk of medical and socioeconomic morbidity, requiring significant resources for management, ultimately detracting from the patient’s quality of life (QoL).
Hemophilia treatment has progressed significantly since the 1950s, from fresh frozen plasma to the discovery of cryoprecipitate by Dr. Judith Graham Pool, which led to the development of the first FVIII concentrate.4 In the 1970s and 1980s, 60–70% of PwH became infected with human immunodeficiency virus (HIV), hepatitis B, and hepatitis C due to the widespread use of pooled contaminated plasma-derived factor concentrates.5 Ryan White, an American teenager with hemophilia who contracted HIV from an FVIII infusion, became a national advocate for increased education about and de-stigmatization of people with HIV/acquired immunodeficiency syndrome (AIDS).6 The introduction of viral testing and inactivation procedures markedly improved the safety profile of these products.7
The cloning of the F9 gene in 1982 and the F8 gene in 1984 led to the development of recombinant standard half-life factor concentrates in the 1990s.8–11 Prophylaxis for people with severe hemophilia (infusing products regularly to maintain levels in the moderate hemophilia range) was a major advancement to reduce bleeding and improve patient QoL and is now considered the standard of care.12 Still, due to the short and variable half-life of FVIII and FIX concentrates, patients experienced breakthrough bleeding events due to inadequate trough levels. Pharmacokinetic-guided personalized prophylaxis has been used to optimize these treatment regimens.13 Extended half-life (EHL) factor concentrates, which have become available over the last decade, have improved pharmacokinetic profiles and decreased treatment burden.14 More recently, three adeno-associated virus (AAV) vector-mediated gene therapies received approval from the US Food and Drug Administration (FDA) for treating adults with moderate or severe hemophilia A (HA) and B (HB), with additional gene therapies currently in clinical trials.
Approximately 30% of people with severe HA and up to 10% of persons with severe HB develop antibodies directed against the replacement factor, termed inhibitors.15,16 Depending on the titer of the neutralizing antibody, FVIII and FIX concentrates may be rendered ineffective. Management of BEs in PwH with inhibitors requires repeated doses of recombinant activated factor VII (rFVIIa) or activated prothrombin complex concentrate (aPCC).17 Since becoming available in 2017, FVIII mimetic therapy with emicizumab has revolutionized prophylaxis for persons with hemophilia A (PwHA) with or without inhibitors.18,19 Ongoing clinical trials are evaluating new rebalancing agents for hemophilia regardless of inhibitor status. The mechanism of action of these agents involves decreasing the activity of the naturally occurring clotting factors that down-regulate coagulation. The agents currently under evaluation are all administered subcutaneously.
The development of novel therapeutics for treating hemophilia with and without inhibitors has been met with enthusiasm from both patients and providers. Beyond the one currently available novel agent, emicizumab (Hemlibra®, Hoffman-La Roche), for persons with hemophilia A with or without inhibitors, marstacimab (Pfizer), a low-affinity human IgG1 antibody that binds the K2 domain of TFPI, was approved in October 2024 by the FDA for prophylaxis in adult and adolescent patients (≥12 years of age) with hemophilia A or B without inhibitors.20 Further novel therapeutics in the pipeline are in various stages of development. Once a full complement of these therapies becomes available, health metrics including life expectancy, annualized bleeding rate (ABR), joint health, and health-related QoL may be positively impacted.19,21–24 However, there are still many barriers that will limit access to these new therapies including socioeconomic status, race/ethnicity, gender, disease severity, inhibitor status, age, and access to hemophilia treatment centers (HTCs). In 2022, the World Federation of Hemophilia (WFH) estimated there are 830,895 cases of hemophilia globally (with 282,266 severe cases); a total of 271,359 cases of hemophilia had been identified, suggesting that almost 70% of cases, most from low- and middle-income countries, are undiagnosed and untreated.21,25,26 To deliver on the promise of emerging hemophilia therapies, optimized and equitable access to care must be ensured worldwide, where all hemophilia patients have a fair opportunity to attain the highest level of possible health.
This review highlights the safety and efficacy of recently licensed hemophilia treatments and novel therapies currently in clinical trials or late-stage preclinical development. We also describe how currently available treatments have improved health equity in this population and the emerging challenges to ensure equity and optimize patient care with new treatments and technologies.
Replacement Therapies
Replacement therapy has been the mainstay of treatment for PwH for the control and prevention of bleeding events. Currently available FVIII and FIX replacement therapies used for prophylaxis and management of acute BEs provide many choices for patients and clinicians (Figure 1). Obstacles to effective prophylactic treatment may include frequent need for intravenous infusions, depending upon the product utilized, and socioeconomic factors related to the high cost of treatment, which can result in imperfect adherence.27 Recent advances in the extension of coagulation factor half-life have significantly reduced the treatment burden for many patients.
 |
Figure 1 Procoagulant therapies: Available and investigational. Procoagulant hemophilia therapies including factor concentrates, gene therapy, cellular therapy, and gene editing can replace the missing coagulation factors FVIII and FIX in patients without inhibitors. For patients with inhibitors, recombinant FVIIa and activated prothrombin complex concentrates can bypass the missing coagulation factors for the treatment of acute bleeding events. Rebalancing agents and FVIIIa memetics can provide prophylaxis for hemophilia A patients with and without inhibitors. SuperVa, a variant of Factor Va resistant to APC, has been shown to enhance thrombin generation in preclinical bleeding models. This figure was adapted and updated with permission from: Lewandowska M, Nasr S, Shapiro AD. Therapeutic and technological advancements in haemophilia care: Quantum leaps forward. Haemophilia. 2022;28 Suppl 4:77–92. © 2022 John Wiley & Sons Ltd.102 Abbreviations: HA, hemophilia A; I, inhibitor; AAV, adeno-associated virus; HB, hemophilia B; FVa, activated factor V; FVIIa, activated factor VII; FVIII, factor VIII; FVIIIa, activated factor VIII; FIX, factor IX; FIXa, activated factor IX; FXa, activated factor X. * indicates investigational therapies, ¥ indicates agents in preclinical development. |
Standard Half-Life Products
Although the advent of recombinant FVIII and FIX concentrates greatly improved patient QoL, the typical infusion regimen of three times per week in HA (due to a half-life of 8–12 hours) remained burdensome for patients. Factor IX standard half-life (SHL) products have a half-life of 24 hours, with a typical infusion regimen of twice per week in HB.
Extended Half-Life Products
Research has focused on protein modifications to increase factor half-life (Table 1 and Table 2). Modifications to recombinant factor concentrates include conjugation to polyethylene glycol (PEGylation) or fusion to albumin or the Fc region of IgG1, which decreases the clearance of these products by liver-expressed scavenger receptors. Compared with SHL products, EHL-FIX achieves a 4 to 6-fold increase in circulating half-life, while most EHL-FVIII have a 1.3 to 1.7-fold increase in half-life.14 The tight FVIII and von Willebrand factor (VWF) complex stabilizes and safeguards FVIII against degradation and clearance and also creates a “ceiling effect” for FVIII half-life of 15–19 hours.29
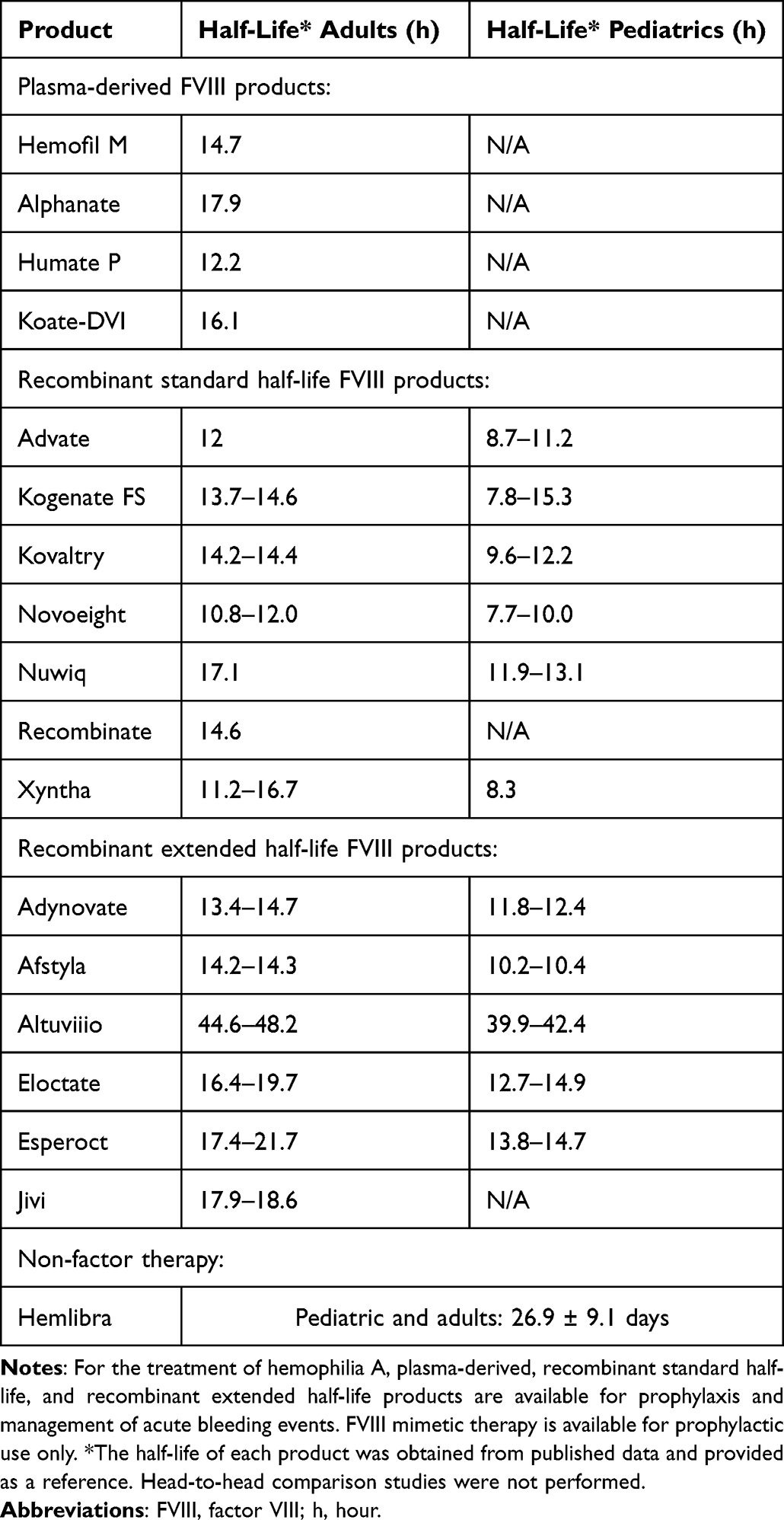 |
Table 1 FDA-Approved Therapeutics for Hemophilia A |
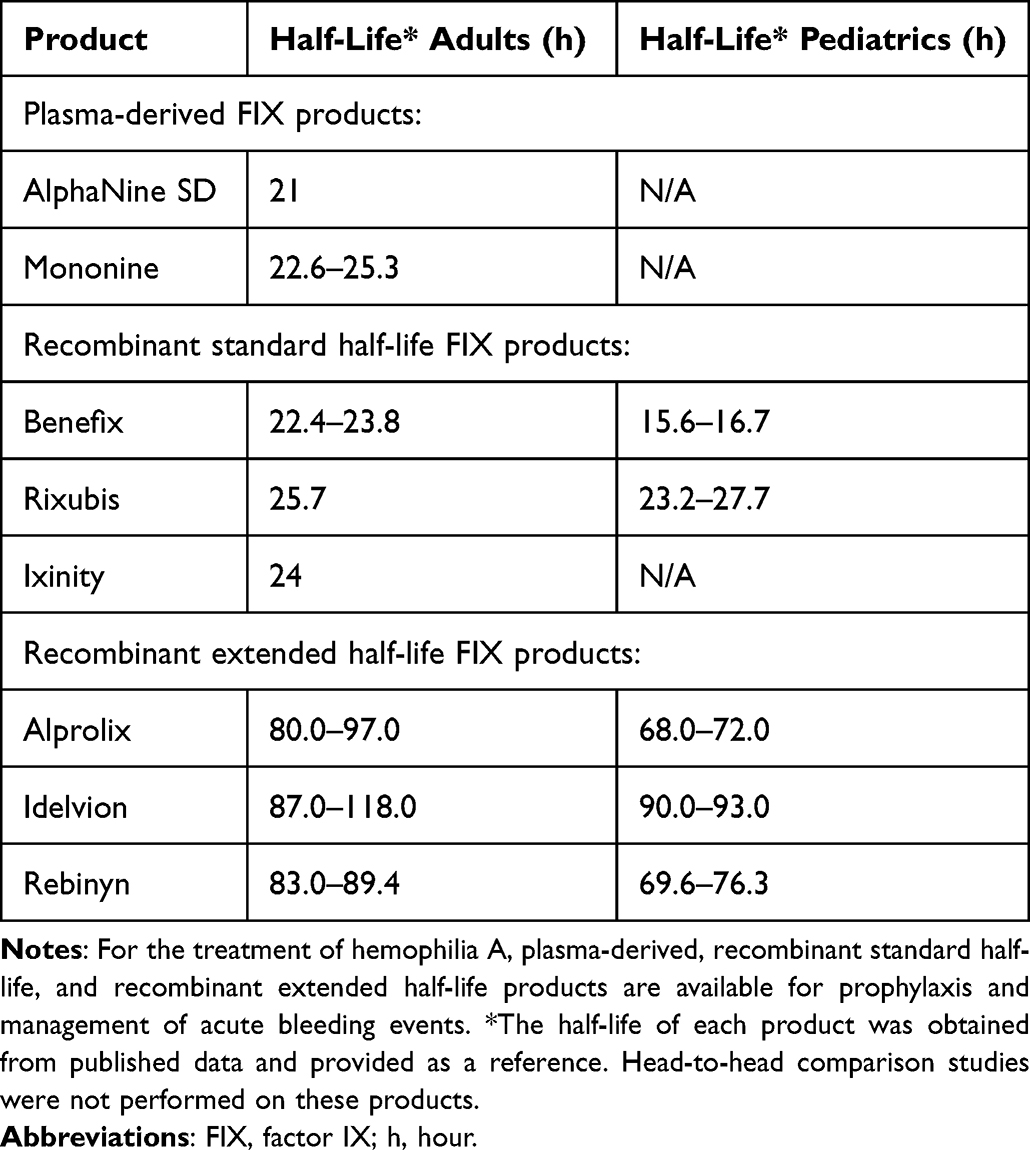 |
Table 2 FDA-Approved Therapeutics for Hemophilia B |
Efanesoctocog Alfa
Approved by the FDA in 2023 for prophylactic and on-demand treatment of adults and children with PwHA, efanesoctocog alfa (Altuviiio®, Sanofi) is the first EHL-FVIII to eliminate the VWF-imposed ceiling on FVIII half-life.22 Efanesoctocog alfa is a B domain-deleted, Fc-fusion recombinant FVIII protein with a VWF-stabilizing D′D3 domain that prevents endogenous VWF binding.30 Efanesoctocog alfa also contains two hydrophilic polypeptides (XTENs) that extend half-life through steric hindrance. The half-life of efanesoctocog alfa is 3- to 4-fold longer than SHL-FVIII, with 40% FVIII activity remaining after 4 days and an expected trough level of 15% 7 days after infusion for adult subjects.31 For PwHA treated with efanesoctocog alfa, a validated one-stage clotting assay should be used to measure FVIII activity, as chromogenic or aPTT-based assays may overestimate FVIII activity by approximately 2.5-fold.32
Non-Factor Therapies: FVIII Mimetics for PwHA
Humanized bispecific antibodies that simultaneously bind activated FIX (FIXa) and factor X (FX) were a transformative advance in care to provide effective prophylaxis for PwHA regardless of inhibitor status. FVIII mimetics are administered by subcutaneous injection and are used only for prophylaxis, with factor concentrates or bypassing agents required for the management of acute bleeding events. The major barrier to access non-factor therapies is cost,33 although for inhibitor patients, prophylaxis with the bispecific antibody emicizumab has been reported to be more cost-effective than bleed control with bypassing agents.34,35 Other barriers to non-factor therapies include adverse reactions such as headaches, arthralgia, pyrexia, and diarrhea,36 aversion to subcutaneous injections in some patients, and concern for thrombosis among inhibitor patients who use aPCC for breakthrough bleed treatment. In addition, FVIII mimetics are not effective for patients with hemophilia B.
Emicizumab
Emicizumab is currently the only FDA-approved humanized bispecific monoclonal antibody; it mimics the scaffold effect of FVIIIa in the tenase complex by bridging FIXa and FX. The FDA licensed emicizumab for prophylactic treatment in PwHA, with or without inhibitors for all age groups and severity levels.18,19 Emicizumab has revolutionized the treatment of PwHA by overcoming the need for intravenous access for administration, maintaining effectiveness even in the presence of inhibitors, and achieving stable hemostatic levels comparable to patients with mild hemophilia. Standard aPTT-based assays cannot assess FVIII activity in people taking emicizumab.37 Monitoring exogenous FVIII administration is performed utilizing a bovine substrate-based chromogenic assay rather than human components.
For hemophilia patients with inhibitors using emicizumab, the concurrent use of aPCC at ≥100 U/kg/day for ≥24 hours to manage acute bleeding events is contraindicated due to the increased risk for thrombosis.38 In a Phase 3 clinical trial, 5/109 patients with hemophilia and inhibitors receiving once weekly emicizumab experienced thrombosis or thrombotic microangiopathy adverse events when aPCC was used for acute bleed control.19 These events have not been reported with the concomitant use of rFVIIa.
An analysis of the HAVEN 1–4 clinical studies demonstrated that 3.5% of participants (14/398) developed antidrug antibodies against emicizumab; of these, 0.75% (3/394) were considered to have antidrug antibodies with neutralizing potential,39 associated with increased emicizumab clearance and risk of breakthrough bleeding events.40 The formation of anti-emicizumab antibodies may render this treatment ineffective for a small subset of patients.
The long-term effects of emicizumab use, in association with decreased exposure to FVIII concentrates, are unknown. In animal models, FVIII has been shown to play a role in maintaining bone mineral density.41 Emicizumab treatment does not impact levels of bone biomarkers,42,43 however, the effects of novel agents such as emicizumab on bone health may require long-term surveillance. In addition, studies are ongoing to determine the impact of delayed exposure to FVIII on the rate of inhibitor development in previously untreated patients.
Patients with severe hemophilia receiving non-factor replacement therapy (eg, emicizumab) have fewer BEs and are converted to a mild bleeding phenotype. Consequently, management problems such as decreased proficiency with self-infusion and difficulty recognizing BEs or injuries that may cause a bleed can result in delayed treatment and more advanced/severe bleeding events. HTCs must now adapt their approaches to improve patient education, facilitate communication using technology or AI, routinely evaluate self-infusion proficiency, and establish backup infusion plans involving external providers or facilities as required.
Next-Generation Mimetics
Two next-generation mimetic therapies are currently in clinical trials. Mim8 (denecimig, Novo Nordisk) exhibits a 15-fold increased potency compared with emicizumab in animal models.44 Preliminary phase 3 trial results involving 254 hemophilia A patients with or without inhibitors using Mim8 prophylaxis administered once-weekly or once-monthly for 26 weeks were compared to episodic treatment or prophylaxis using factor concentrates during a 26-week run-in period.45 For subjects who previously treated on-demand, the mean ABR was 15.75 (95% CI: 10.7–23.2) compared to 0.45 (95% CI: 0.18–1.14) for once-weekly Mim8 and 0.20 (95% CI: 0.06–0.72) for once-monthly Mim8. For subjects who received pre-study factor prophylaxis, the mean ABR was 4.83 (95% CI: 3.59–6.51) compared with 2.51 (95% CI: 1.42–4.42) for once-weekly Mim8, and 3.10 (95% CI: 2.23–4.29) compared to 1.78 (95% CI: 1.17–2.71) for once-monthly Mim8 prophylaxis. There were no reports of anti-drug antibodies or other safety concerns. NXT007 (Hoffman-La Roche), another FVIIIa mimetic engineered and optimized based on emicizumab, is currently being evaluated in Phase 1 trials.46
Nonfactor Therapies: Rebalancing Agents
For hemophilia patients without access to prophylaxis, including hemophilia B patients with inhibitors and patients who do not respond adequately to existing therapies, there is a need for additional prophylactic agents. Novel prophylactic therapies that rebalance hemostasis by decreasing levels of endogenous inhibitors or down-regulators of coagulation can restore thrombin generation in the absence of FVIII or FIX and have been developed to meet these therapeutic gaps (Figure 2). Notably, for some of these new prophylactic therapies, efficacy is evaluated in comparison to usual therapy and/or episodic treatment. Additional research is needed to demonstrate product efficacy relative to the current standard of care with emicizumab or factor concentrate prophylaxis, which would help inform the selection of optimal prophylactic therapy for individual patients.
 |
Figure 2 Anticoagulants and rebalancing agents. For hemophilia patients with inhibitors, rebalancing agents that inhibit the activity of activated protein C, protein S, or tissue factor pathway inhibitor or downregulate antithrombin levels are under investigation or in preclinical development. This figure was reproduced with permission from: Lewandowska M, Nasr S, Shapiro AD. Therapeutic and technological advancements in haemophilia care: Quantum leaps forward. Haemophilia. 2022;28 Suppl 4:77–92. © 2022 John Wiley & Sons Ltd.102 Abbreviations: APC, activated protein C; AT, antithrombin; TFPI, tissue factor pathway inhibitor, FVa, activated factor V; FVIIa, activated factor VII; FVIIIa, activated factor VIII; FIXa, activated factor IX; FXa, activated factor X, FXIa, activated factor XI. * indicates investigational therapies, ¥ indicates agents in preclinical development. |
Monoclonal Antibodies Against Tissue Factor Pathway Inhibitor
Tissue factor pathway inhibitor (TFPI) is a Kunitz-type serine protease inhibitor that binds tissue factor, factor VIIa, and factor X, thereby downregulating the generation of FXa by tissue factor and FVIIa.47 Monoclonal antibodies designed to inhibit TFPI increase tissue factor-dependent activation of FX and subsequent thrombin generation.
Concizumab
Concizumab (Alhemo®, Novo Nordisk) is a high-affinity humanized IgG4 antibody that binds the K2 domain of TFPI; it is administered once daily by subcutaneous injection.48 In 2023, concizumab received regulatory approval in Canada, Australia, and Switzerland for the prophylactic treatment of adolescents and adults with hemophilia A or B and inhibitors,49 with approval in the U.S. following in December 2024. In Japan, concizumab is licensed for prophylaxis in adult and adolescent patients with hemophilia A and B with and without inhibitors. The safety and efficacy of concizumab were evaluated in 133 PwH and inhibitors aged >12 years (81 nonrandomized and 52 randomized).50 The trial was temporarily paused after three nonfatal thromboembolic events occurred in study participants; data analysis led to the development of a risk-mitigation strategy starting with a loading dose of 1.0 mg/kg, followed by 0.2 mg/kg daily which was potentially adjusted either up to 0.25 or downward to 0.15 mg/kg/day based upon the concizumab level at week 4.51 In addition, when other agents are required for concurrent treatment of breakthrough bleeding events, guidelines have been provided for use of the minimal effective dose.50 Since this strategy was implemented, no further thromboembolic events have occurred. For hemophilia patients with inhibitors, the estimated mean ABR was 1.7 (95% CI: 1.0–2.9) with concizumab versus 11.8 (95% CI: 7.0–19.9) in participants using on-demand factor replacement (rate ratio 0.14; 95% CI: 0.07–0.29; p < 0.001).50,52 For hemophilia A patients without inhibitors, the mean ABR during concizumab prophylaxis was 2.7 (inter-quartile ratio (IRQ): 1.6–4.6) compared with 19.3 (IQR 11.3–33.0) for patients not on prophylaxis. For hemophilia B patients without inhibitors, the mean ABR on concizumab prophylaxis was 3.1 (IQR: 1.9–5.0) compared to 14.8 (IQR: 8.1–26.9) for patients not using prophylaxis.49
Marstacimab
Marstacimab (Hympavzi®, Pfizer), a low-affinity human IgG1 antibody that binds the K2 domain of TFPI, is administered weekly by subcutaneous injection. Marstacimab was approved in October 2024 by the FDA and November 2024 in Europe for prophylaxis in adult and adolescent patients (≥12 years of age) with hemophilia A or B without inhibitors.20 A Phase 2 clinical trial involving 20 severe HA and HB patients with and without inhibitors, aged >12 years that evaluated six treatment cohorts demonstrated that ABR decreased with marstacimab treatment (mean ABR range: 0–3.6; SD: 0–7.2; median ABR range: 0–2.0; 0–14.4) compared with pre-treatment ABR (mean ABR range: 14.0–22.0; SD: 1.6–7.9; median ABR: 14.0–20.0; SD: 12.0–42.0).53 Preliminary analysis of a phase 3 clinical trial involving 128 adults and adolescents with HA or HB without inhibitors showed a mean ABR of 38 (95% CI: 31.0–46.5) during the observational phase where BEs were managed on-demand compared to a mean ABR of 3.18 (95% CI: 2.1–4.9) associated with marstacimab treatment (ratio estimate: 0.084; 95% CI: 0.059–0.119; p < 0.001).54 For patients who used prophylaxis during the observational phase, the mean ABR was 7.85 (95% CI: 5.1–10.6) compared with 5.08 (95% CI: 3.4–6.8) associated with marstacimab treatment (estimated difference: –2.8; 95% CI: –5.4, –0.2; p = 0.0376).55 No thromboembolic adverse events have been reported for patients receiving marstacimab.56
RNA-Interfering Therapeutics Targeting Antithrombin
Antithrombin is an endogenous anticoagulant that binds and inhibits the activity of thrombin, factor Xa, and factor IXa.57 For hemophilia management, antithrombin production in the liver has been targeted using antisense oligonucleotide small interfering RNA (siRNA) therapeutic technologies that bind and lead to the degradation of antithrombin messenger RNAs.58 This silences antithrombin SERPINC1 gene expression and prevents the production of the antithrombin protein.
Fitusiran
The siRNA fitusiran (Sanofi) has a short circulating half-life (3–5 hours); however, the mean rate of antithrombin recovery post-treatment is 10–15% per month, resulting in persistent decreased antithrombin expression for several months after discontinuation. The efficacy of fitusiran has been described in a Phase 3 clinical study involving 57 patients >12 years old with severe hemophilia A or B and inhibitors treated with fitusiran once monthly compared to on-demand bypassing agents. Negative binomial model-based mean ABR was significantly lower in the fitusiran prophylaxis group (1.7; 95% CI: 1.0–2.7) than in the on-demand bypassing agents group (18.1; 95% CI: 10.6–30.8), corresponding to a 90.8% (95% CI: 80.8–95.6; p < 0.0001) reduction in ABR in favor of fitusiran prophylaxis.59 These results are consistent with the report that fitusiran reduced the ABR by 90% compared to on-demand factor concentrates in patients with severe hemophilia A and B without inhibitors.60
In 2017, a Phase 2 extension trial of fitusiran was halted after a patient death attributed to cerebral venous sinus thrombosis. The review revealed that the patient had been given clotting factor concentrates at a dose that exceeded guidelines for management of breakthrough bleeding events.61–63 Subsequently, in 2020, clinical development of fitusiran was voluntarily paused by the sponsor following four nonfatal thrombotic events among 259 clinical trial subjects who had received at least 1 dose of fitusiran.64 The study protocol was adjusted to target 15–35% residual antithrombin levels by reducing the initial fitusiran dose and adjusting subsequent doses according to antithrombin levels. Phase 3 clinical studies of these adjusted protocols are currently ongoing.
Modified Inhibitors of Activated Protein C
Activated protein C (APC) is a serine-protease that proteolytically inactivates FVa and FVIIIa thereby decreasing FXa and thrombin generation.
SR604
SR604 is a monoclonal antibody in pre-clinical testing that inhibits the binding of APC to FVa and inhibits the anticoagulant function of APC in humanized hemophilic mice without interfering with cytoprotection or endothelial barrier function of protein C.65
SerpinPC
SerpinPC (Centessa Pharmaceuticals) is a recombinant alpha 1 antitrypsin serine protease inhibitor modified to specifically inhibit APC. SerpinPC is administered by subcutaneous injection, with an estimated half-life of 99 hours (personal communication, Centessa Pharmaceuticals, February 12, 2024). Preliminary results from a Phase 2a trial with 20 male participants (16 hA, 4 hB) demonstrated a 96% reduction in median ABR during the pre-specified assessment period (1.0; interquartile range: 1.0–4.5) compared to the pre-exposure observation period of on-demand treatment (35.6; interquartile range: 29.8–40.4). No treatment-related serious adverse events were reported.66 In November 2024, Centessa Pharmaceuticals announced they had discontinued clinical development of SerpinPC.67
Monoclonal Antibody Against Protein S
Protein S functions as a cofactor for APC and TFPI. In mouse models of hemophilia, protein S deficiency increased thrombin generation and protected against bleeding, suggesting that protein S inhibition may have therapeutic efficacy in hemophilia.68
VGA039
VGA039 (Vega Therapeutics) is a monoclonal antibody directed against protein S to inhibit cofactor activity for TFPI and APC. Preclinical in vitro studies performed in human von Willebrand disease (type 1, type 2A, type 2B, type 3), and FVII-, FVIII-, FIX-, FXI-, and FXIII-deficient plasmas showed that VGA039 increased thrombin generation in a dose-dependent manner,69 suggesting it may be effective in treating several inherited bleeding disorders. Currently, VGA039 is in phase 1 clinical trials involving healthy subjects and von Willebrand disease patients.
Recombinant Bypassing Agents
Bypassing agents are required for the management of acute bleeding events in hemophilia inhibitor patients (Table 3). Unlike FVIII and FIX concentrates, the half-life of bypassing agents has not been associated with hemostatic efficacy. Prior to 2020, only two bypassing agents were available, a plasma-derived aPCC (FEIBA®, Takeda), licensed in 1986, and eptacog alfa (NovoSeven®, Novo Nordisk), a rFVIIa licensed in 1999.70 In a single randomized comparative study, the overall efficacy of aPCC and eptacog alfa were found to be similar, despite variable inter- and intra-patient responses.17 For patients who treated with two doses of eptacog alfa (90–120 mcg/kg), efficacy at the 12-hour time point was 84.4%.17 The single-dose efficacy of eptacog alfa (90–270 mcg/kg) has been reported between 10% and 59%.71–73 In a study evaluating different dose ranges of eptacog alpha (<100, 100–150, 150–200 and >200 mcg/kg) the reported bleeding cessation rate was 84% for the three lower dose groups, and 97% for the highest dose group (P < 0.001).74
 |
Table 3 FDA-Approved Bypassing Agents for the Treatment of Bleeding Events in Hemophilia Patients |
Eptacog beta (Sevenfact®, HEMA Biologics, LLC and LFB SA) is a bypassing agent licensed by the FDA in 2020 and EMA, UK, and Mexico in 2022 to treat and control BEs in adolescent and adult PwH and inhibitors. In Phase 3 clinical trials, eptacog beta was used at two initial dose regimens, resulting in a 12-hour primary endpoint efficacy rate of 82% and 91%, depending on the initial dose.75,76 A single 225 mcg/kg dose of eptacog beta was demonstrated to control 84% of BEs, nearing efficacy observed with single doses of factor replacement therapy observed in non-inhibitor patients. Eptacog alfa and eptacog beta are not biosimilars, a designation that requires products to have no clinically meaningful differences in safety, purity, and potency.77
Gene Therapy
To date, one FVIII gene therapy, valoctocogene roxaparvovec (Roctavian®, BioMarin) and two FIX gene therapies, etranacogene dezaparvovec (Hemgenix®, CSL Behring), and fidanacogene elaparvovec (Beqvez®, Pfizer) have been approved in the US and conditionally approved in Europe for the treatment of hemophilia A and B, respectively. The FIX gene therapies etranacogene dezaparvovec and fidanacogene elaparvovec are also approved in Canada. Hemophilia gene therapy offers the promise of a single-dose infusion resulting in enduring factor expression, obviating the need for repeat treatment. However, barriers to access include age, pre-existing immunity to AAV, factor-specific inhibitors, cost, and the requirement for a dedicated setting to administer treatment where personnel and equipment are immediately available to treat potential infusion reactions, and the need for dedicated follow-up post-infusion to ensure that immune-mediated responses do not result in transgene loss.78,79
FDA-Approved Gene Therapies
Valoctocogene roxaparvovec is the first gene therapy product licensed by the FDA for treating adults with severe HA without inhibitors. Valoctocogene roxaparvovec is an AAV serotype 5 (AAV5) vector that contains the B-domain deleted human F8 gene administered as a single infusion of 6 × 1013 vector genomes per kilogram (vg/kg).80 A Phase 3 clinical trial evaluated efficacy and safety in 134 adult males with severe HA. Exclusion criteria included anti-AAV5 capsid antibodies, pre-existing liver fibrosis, excessive alcohol consumption, and the presence of FVIII inhibitors. Clinically meaningful improvements were observed in the mean ABR which decreased from 5.4 (SD: 6.9) with prior prophylaxis to 2.6 (SD: 6.2) after gene therapy (mean difference: −2.8; 95% CI: −4.3, −1.2).78 The mean FVIII activity level at the end of the first year was 43.6%. The durability of the treatment in an extension of the phase 3 study showed that the mean FVIII level decreased to 29.7% at year 3 (n = 132) and a mean of 22% at year 4 (n = 17).81 Overall, 4.5% (6/134) subjects had no response to the study product, while FVIII:C declined to <3% for 17.2% (23/134) subjects during a follow-up period of up to 4.3 years.82 A 7-year extension study of the phase 1/2 cohort found that participants who had received the 6 × 1013 vector infusion had mean FVIII:C levels of 16.2 IU/dL (n = 5), corresponding to mild hemophilia.83 Regression analyses estimated that the rate of change in FVIII activity was −0.001 in the last year. During year 7, the mean ABR was 0.9, a 96% reduction from baseline (mean ABR: 17.6). Two participants resumed prophylaxis in year 7.
Etranacogene dezaparvovec is the first gene therapy product approved by the FDA for the treatment of adult patients with hemophilia B without inhibitors who are receiving FIX prophylaxis or have a history of life-threatening or repeated serious bleeding.84 Etranacogene dezaparvovec is an AAV5 vector containing the Padua variant (FIX-R338L) of the F9 gene.85–88 Padua is a naturally occurring missense mutation that increases the specific activity of FIX up to 8-fold.89 Treatment is administered as a single infusion of 2 × 1013 vg/kg. Efficacy and safety were described in 54 males with FIX <2 IU/dL.79 The trial did not exclude participants with pre-existing antibodies to AAV5. The mean ABR decreased from 4.1 (95% CI: 3.2–5.4) during the 6-month lead-in period of FIX prophylaxis to 1.9 (95% CI: 1.0–3.4) during months 7–18 after treatment, for a rate ratio of 0.46 (95% CI: 0.26–0.81), demonstrating noninferiority of etranacogene dezaparvovec compared with FIX prophylaxis.79 The stability of factor activity was maintained, with a mean FIX activity of 41.5% at 1 year and 38.6% 3 years post-treatment.90 Two of 54 patients had FIX activity <5 IU/dL at month 18 and required prophylaxis.79 One patient received only 10% of the infused gene therapy product dose due to an infusion reaction, and no FIX expression was observed for another patient whose day-of-dosing anti-AAV titer was high (3212).
Fidanacogene elaparvovec was approved by the FDA in 2024 for treating hemophilia B patients who currently use FIX prophylaxis, or have experienced life-threatening bleeding or repeated, serious spontaneous bleeding episodes. Fidanacogene elaparvovec uses a F9 Padua transgene and a bioengineered capsid. Phase 3 trial evaluation of fidanacogene elaparvovec efficacy and safety has been reported. Forty-five adult males with FIX ≤2% were treated with a single low-dose vector infusion (5 × 1011 vg/kg) of fidanacogene elaparvovec.91,92 The mean ABR decreased from 4.4 during the six-month lead-in period of FIX prophylaxis to 1.3 between week 12 and month 15 post-fidanacogene elaparvovec, demonstrating non-inferiority. Mean factor levels 12- and 24 months post-treatment were 25.3% and 26.6% respectively. Durability was assessed in a multi-year follow-up of 14 Phase 1/2a study participants; the mean ABR 6 years post-gene therapy was 0.3 (SD: 0.66) and the mean FIX activity was 27.6%.93 Several additional gene therapy trials are underway (Table 4).
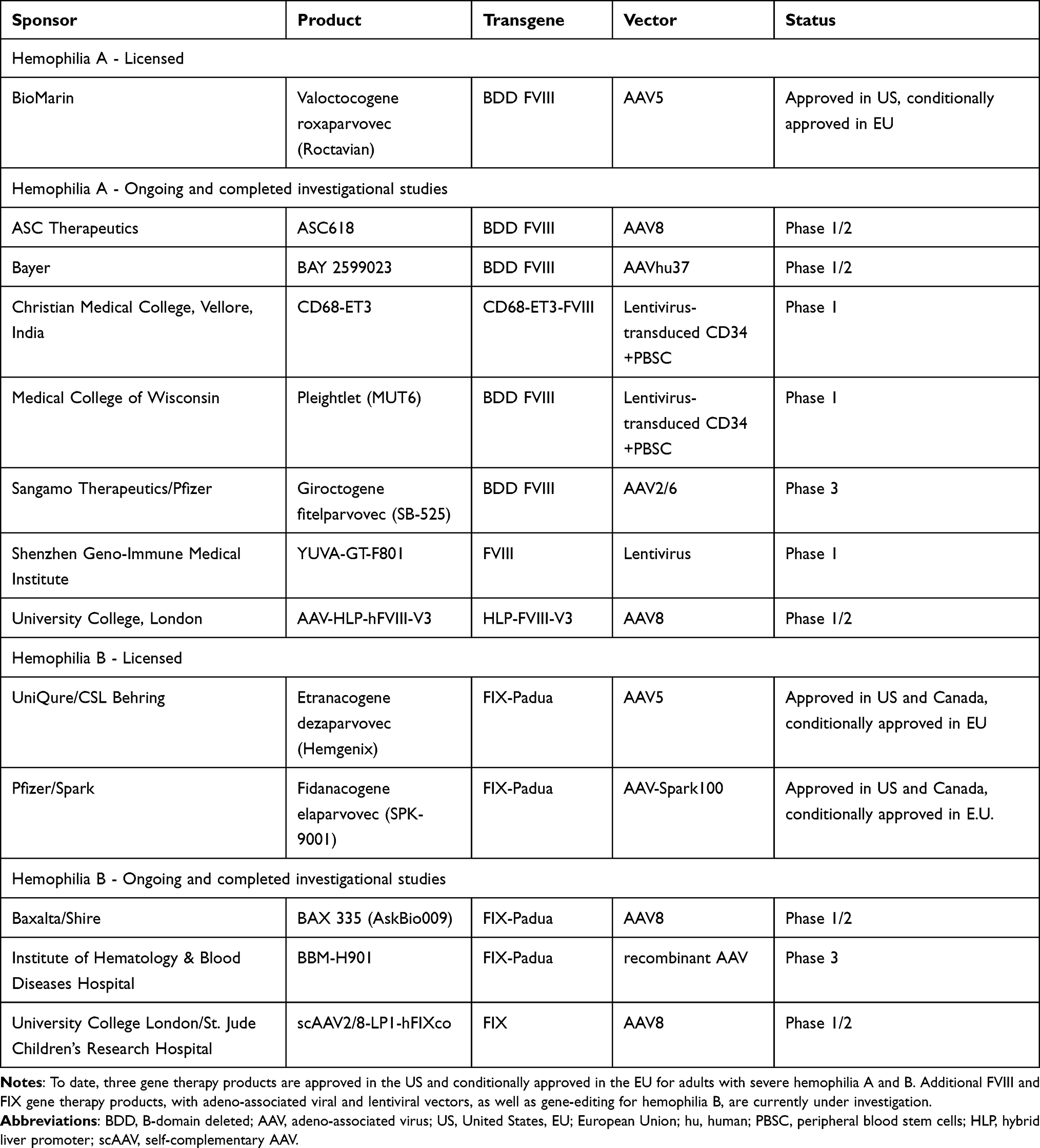 |
Table 4 Approved and Investigational Hemophilia Gene Therapy Products |
Gene Therapy Safety Considerations
There are several safety considerations associated with hemophilia gene therapy. Increased alanine aminotransferase (ALT) levels that often correspond with a reduction or loss of transgene expression have been observed in hemophilia AAV gene therapy 4–12 weeks post-treatment. For valoctocogene roxaparvovec, over 80% of the study participants developed liver function test abnormalities requiring the use of steroids (median treatment duration 35 weeks) and/or other immunosuppressive medications (median treatment duration 26 weeks).78,80,94 16.7% of subjects who received etranacogene dezaparvovec required steroid treatment for abnormal liver function tests for a mean duration of 81.4 days,79 while 53.3% of patients treated with fidanacogene elaparvovec experienced elevated ALT levels, and 62.2% of patients received ≥1 corticosteroid dose, with a mean treatment duration of 107.5 days.92
In addition, the potential genotoxic risk of vector integration into the hepatocyte genome is currently unknown.95 Preclinical studies in AAV-canine FVIII-treated hemophilia A dogs have demonstrated that although the majority of the vector is retained in episomal form, a small proportion may be integrated. Integrated vectors did not preferentially associate with cancer-related genes or dysregulated cancer-associated gene expression, and histopathological evaluation of canine livers more than 10 years post-gene therapy did not show evidence of malignant tumors.96–98 In liver biopsy samples from human subjects 2.6–4.1 years post-treatment with valoctocogene roxaparvovec, no evidence of dysplasia, architectural distortion, fibrosis, chronic inflammation, or endoplasmic reticulum stress was observed in transgene-expressing hepatocytes.99
Gene Therapy Preparedness
Successful and safe treatment of PwH with gene therapy requires multistakeholder participation. The plan proposed by the Medical and Scientific Advisory Council (MASAC) of the National Bleeding Disorders Foundation (NBDF) encompasses education/institutional training, establishing criteria for patient eligibility, insurance authorization and support, procurement and administration, and patient monitoring and follow-up.100 Short- and long-term monitoring is essential to ensure patient safety and durability of transgene expression. Implementing shared decision-making strategies and consideration of individual patient goals will advance the delivery of patient-centered care. To aid in patient education and empower informed decisions, the World Federation of Hemophilia created a digital Shared Decision-Making tool (sdm.wfh.org).101
New Technologies
Technological innovations increasingly contribute to the ability to improve the QoL for PwH (Figure 3). Such advancements include large databases, smartphone applications for remote data collection, telehealth technology to improve access to care, and artificial intelligence (AI) to improve patient and healthcare provider education.102 Adapting patient and clinician practice to incorporate the use of such technological advancements in both clinical and home settings has the potential to improve patient quality of care and overall experience.103
 |
Figure 3 Evolution of treatment for hemophilia. Since the discovery of fresh frozen plasma in the 1950s, hemophilia treatment has progressed from the use of cryoprecipitate in the 1970s and recombinant factor concentrates in the 1990s. Recent developments including extended half-life factor concentrates, FVIII mimetics, rebalancing agents, and gene therapies provide novel options for the management of hemophilia A and B. Implementation of new technologies and a shared decision-making relationship between hemophilia care providers and their patients will enhance the provision of patient-centered care. Abbreviations: PK, pharmacokinetic; POC-MSKUS, point-of-care musculoskeletal ultrasound; AI, artificial intelligence; AAV, adeno-associated virus; T1/2, half-life. |
Many HTCs across North America and Europe have adopted point-of-care musculoskeletal ultrasound (POC-MSKUS). Patient self-imaging of hemophilic joints using mobile ultrasound devices with teleguidance, remote expert-directed real-time feedback, has shown promising results requiring further investigation.104 This technology may expedite the diagnosis and treatment of BEs, decrease the need for travel to an HTC for evaluation, optimize the utilization of costly therapies, and increase patient autonomy.
Challenges in Ensuring Equitable Care
Inequitable access to hemophilia care for underserved populations can result in a higher burden of disease. The promotion of health equity within the hemophilia population requires the identification of barriers to optimal care, and recognizing bias that may influence the quality of care. Inequity in hemophilia care is intersectional, with underserved patients often facing multiple barriers to treatment.
Gender Equity
Women and girls with hemophilia have historically been overlooked due to the X-linked nature of hemophilia inheritance, which is often presumed them to be asymptomatic.105 Updated nomenclature has been devised to highlight clinically relevant bleeding experienced by these patients.2 A study of a surveillance database of 139 US-based HTCs reported that 17.9% of mild, 1.4% of moderate, and 0.5% of severe hemophilia patients are female,28 although these numbers are likely to be underestimated, at least in part due to decreased rates of diagnosis compared with male counterparts. There is also increasing recognition that a subset of hemophilia carriers with factor levels ≥40% may present with a bleeding phenotype.2 Women with hemophilia are often excluded from clinical trials, and therefore the evidence of efficacy and safety of new therapeutics is incomplete in this population. The European Hemophilia Consortium and the European Association for Hemophilia and Allied Disorders (EAHAD) have described the principles of care for women and girls with inherited bleeding disorders, including women and girls with hemophilia, including timely diagnosis, provision of family-centered care, education and care for heavy menstrual bleeding, comprehensive management of pregnancy including pre-conception counseling, prenatal diagnostics, and post-partum management, and inclusion in registries and clinical research.106
Current hemophilia diagnostic guidelines and nomenclature are based on gender-binary categorizations1,2 and do not incorporate language inclusive of transgender, non-binary, and inter-sex patients.107 As the prevalence of individuals who identify as transgender or non-binary is increasing, particularly among children and adolescents, there is also an increased risk of marginalization, misgendering, and stigmatization of these patients, which can contribute to suboptimal care.107 For transgender patients, there is a paucity of data on hemophilia management during gender-affirming hormonal therapy or surgery.105,108 Increased research, awareness, use of gender-neutral language in hemophilia, and expert consensus on diagnostic and management strategies could reduce barriers to care and promote more inclusive, patient-centered treatment.
Race/Ethnicity
Disparities in access to and efficacy of hemophilia treatments have been documented in association with patient race or ethnicity, with Black, Hispanic, and other minority populations experiencing less favorable outcomes. Race and ethnicity have been shown to negatively influence access to home therapy and prophylaxis treatment.109,110 Black and Hispanic hemophilia A patients have a higher incidence of inhibitors than White patients;111 mechanisms underlying these observations are the subject of ongoing investigation. Notably, non-Hispanic Black and Hispanic patients are significantly less likely to receive ITI than White patients.112,113 There are currently conflicting reports on the success rate of ITI between different racial/ethnic groups.114,115 Non-White hemophilia patients have higher chronic pain levels116 and lower QoL than White patients,117 and life expectancy for non-Hispanic Black patients (56 years) is significantly lower than for non-Hispanic White patients (68 years).118 Disparities in hemophilia care outcomes can also be complicated by factors such as delayed diagnosis, socioeconomic status, access to HTCs, insurance, sick leave, as well as educational, and language barriers, all contributing to systemic inequity. Racial and ethnic minority groups are under-represented in hemophilia research and interventional clinical trials, leading to significant knowledge gaps relating to efficacy and safety within differing populations for new therapies.119
Access to HTCs and Hematologists
HTCs provide multidisciplinary integrated care for patients, including individualized treatment planning, specialized laboratory testing, patient education, psychosocial care, and surveillance to identify emerging issues. The care provided by HTCs has been shown to reduce patient mortality by 40% and decrease healthcare resource utilization and cost of care.120 A report from the Indiana Hemophilia Surveillance System found that patients cared for by the IHTC had a higher rate of prophylaxis and self-infusion and a reduction in the use of emergency departments.121 The long-term sustainability of HTCs is essential for optimal patient care.
There is currently a shortage of adult classical hematologists who dedicate their careers to these rare disorders.122 Physicians require increased clinical experience with new agents, and expertise in specialized coagulation laboratory assays and pharmacokinetics to effectively administer and manage current and newly emerging therapeutics. Medical organizations have dedicated significant resources to address these gaps. The American Society of Hematology introduced a Hematology-Focused Fellowship Training Program, to allow physicians to combine comprehensive classical hematology training with career-enhancing education with the goal of creating 50 new adult classical hematologists by 2030. The Partners Physician Academy, a U.S.-based, bleeding disorders-focused educational program delivered by hematology experts, is also working to address this gap. The program’s goal is to support and accelerate the career advancement of physicians in fellowship or those early in their career, for leadership as part of the Hemophilia Treatment Center (HTC) network.
Inhibitor Status
Hemophilia inhibitor patients have an increased risk of orthopedic complications and mortality as well as reduced QoL of life compared to non-inhibitor patients.123–125 To date, ITI is the only successful method for inhibitor eradication; however, this process is costly and burdensome to the patient, requiring frequent repeated exposures to replacement products. In FVIII deficiency, ITI outcomes show variable average success rates (63–91%) depending on the protocol used.126 Inhibitor patients have access to fewer therapies for prophylaxis and for acute bleed management than non-inhibitor patients, and the cost of these treatments may be up to 3-fold higher than for non-inhibitor patients.123 For hemophilia A inhibitor patients who have not undergone successful ITI, emicizumab is an effective option for prophylaxis.19 In 2023, concizumab was approved in Canada, Australia, and Japan for prophylactic treatment of adolescents and adults >12 years with hemophilia B and inhibitors.127 FDA approval of non-factor therapies is eagerly awaited for this patient population. Hemophilia B patients with anaphylaxis to FIX are limited to rFVIIa use only, as aPCC contains trace amounts of plasma-derived FIX. Clinical development of additional bypassing rFVIIa agents has been hampered by issues related to product efficacy and immunogenicity.71,128,129
Mild and Moderate Hemophilia
While hemophilia severity is currently classified based on factor levels, there is phenotypic heterogeneity where some mild or moderate patients present with spontaneous bleeding events similar to those observed in severe hemophilia.130,131 Patients with moderate hemophilia report a substantial disease burden including joint impairment, chronic pain, and disability-related unemployment, which may limit lifestyle choices such as participation in sports and other physical activities.132 Prophylaxis is currently the standard of care for severe hemophilia to reduce bleed frequency and prevent arthropathy, however, patients with mild and moderate hemophilia, who may benefit from prophylaxis, are less likely to be offered this treatment. Mild/moderate hemophilia patients are also less likely to be included in interventional clinical trials. A focus group report has described a set of parameters for identifying mild and moderate patients who may be eligible for prophylaxis based on early onset of spontaneous bleeding, ABR, and evidence of early joint disease.130 Incorporating the clinical assessment of patient bleeding phenotype could strengthen the current disease severity classification system and improve outcomes for these patients, including increasing their ability to engage in physical activity.130
Age
As a result of advancements in hemophilia care, PwH now enjoy a near-normal life expectancy.21 However, as hemophilia patients age they are subject to chronic pain and disability associated with suboptimal treatment in early life including joint arthropathy, osteoporosis, and viral illness, as well as age-related comorbidities such as cancer and cardiovascular disease.133 The incidence of cardiovascular disease among aging PwH is rising, affecting up to 15% of patients in the U.S.,134 presenting management challenges to clinicians who must navigate a delicate balance to ensure that patients receive adequate hemostatic control while reducing the risk of thrombotic conditions such as venous thromboembolism, arterial thrombosis, and atrial fibrillation. There is limited research on the optimal management of this growing population. A 2023 international expert-opinion-based guidance document on managing PwH requiring antithrombotic therapy addresses the complex management of these patients.135
Resource-Limited Countries
There is a high cost to hemophilia therapies, recombinant clotting factor concentrates, bypassing agents, non-factor therapies, and gene therapy.123,136 Approximately 15% of hemophilia patients, located in mostly high-income countries, have access to effective treatment, while 10–15% of patients located in high-middle-income regions have access to treatments that are inadequate for high-quality care.137 The remaining 70–75% of hemophilia patients, located in low-to-middle-income regions, have very limited or no access to diagnosis and treatment,25 resulting in high rates of morbidity and mortality. For example, in 2020, only 12.3% of FVIII concentrates were allocated to treating hemophilia patients residing in low- and lower-middle-income countries, which represent 63% of the global hemophilia population.138,139 Transfusion of blood and blood products, including fresh frozen plasma and cryoprecipitate, remain the mainstay of bleed management for many of these patients.140 Transfusion-acquired infections are high due to the incidence of hepatitis B and C, HIV, malaria, and syphilis in these populations, largely as a result of inadequate bloodborne pathogen screening and limited use of viral inactivation technologies.140 Additional challenges include the lack of healthcare infrastructure such as diagnostic laboratories; only 6.3% of patients in low-and-middle-income countries are estimated to have received a hemophilia diagnosis.21,25,141 Patients in resource-limited nations may also lack awareness of bleeding disorders, access to clinicians trained in hemophilia management, HTCs, and laboratories for coagulation monitoring.141
The WFH, which includes 147 national member organizations, has a mandate to ensure treatment for patients worldwide.142 The WFH World Bleeding Disorder Registry collects standardized clinical data on patients with hemophilia,143 allowing for improved research and advocacy on treatment needs in resource-limited areas. In addition, the WFH Twinning program creates partnerships between emerging and established HTCs to provide training and improve diagnosis and clinical management. In 1996, the WFH began the Humanitarian Aid Program (HAP) to provide hemophilia products donated by pharmaceutical manufacturers for the management of life-threatening bleeding in patients from resource-limited regions.137,144 Donations of EHL factor concentrates and emicizumab from industry partners have enabled WFH HAP to provide prophylactic care.139,144 More recently, donations were made by the manufacturer of eptacog beta to provide access to rFVIIa therapy to patients in low-income countries, a long-standing goal of WFH HAP (personal communication with Program Director). Future commitments from the sponsors of novel hemophilia therapies are essential for the maintenance and expansion of these programs. Ultimately, WFH HAP aims to enable recipient countries to independently and sustainably manage inherited bleeding disorders, including hemophilia.137
Conclusions
A new treatment paradigm for the management of hemophilia has been described by a panel of hemophilia care providers, patient advocates, and health economists.145 This set of milestones links outcomes with factor deficiency correction to sustain life, minimize joint impairment, prevent spontaneous bleeding, achieve normal mobility, sustain minor and major surgery or trauma, and achieve normal hemostasis or functional cure. Attainment of such goals will ensure improved QoL, allowing patients to participate in activities such as work and family life without restriction or dependence on specialized care. The ultimate goal in care, as described by Krumb and Hermans, is a “hemophilia-free mind” where patients are free of the psychological burden and constant thoughts of their disease.146
Emerging non-factor hemophilia treatments may reduce the frequency or burden of dosing, allowing for subcutaneous administration with decreased peaks and troughs between doses. In addition, gene therapies may provide sustained factor expression after a single dose. Together with new technologies that enhance and optimize treatment for acute bleeding events, these advancements represent significant progress toward achieving a “hemophilia-free mind”.146 However, similar to existing products, new and emerging therapies and technologies will likely be inaccessible to most hemophilia patients worldwide. Several measures can be implemented to ensure that all patients have an equitable opportunity for optimal care.
As the landscape of hemophilia treatment evolves, the art of medicine and the patient-physician partnership must adapt. Provider education on the current and historical inequity of hemophilia treatment, and improved recognition of unconscious or implicit bias that may lead to a negative evaluation of patients may improve care and begin to address health disparities.109,147 Ensuring diverse racial and ethnic representation among healthcare workers and hemophilia advocacy organizations will improve patient-clinician communication, healthcare outcomes, and patient experiences.109,148 Patients must be educated on their treatment options, and a shared decision making partnership with their hemophilia care team should guide the selection of treatments and regimens that best control disease and meet the patient’s health goals.
Hemophilia stakeholders, including patients, healthcare providers, and funders of treatment (government and insurance companies) must advocate for equitable access to these new therapies. Mahlangu et al describe the stakeholder responsibilities in ensuring the availability, acceptability, and affordability of these treatments for all patients, including education, fundraising, and prioritizing policies aimed at price reduction and equitable distribution of treatment.141 Stakeholders can also guide future research directed at improving equitable access to care. A multi-stakeholder consultation process involving the US inherited bleeding disorder community, recently described by the National Bleeding Disorders Foundation, identified key future research priorities including health services research to optimize efficiency and effectiveness of healthcare delivery, diversity and inclusion research to improve care for underserved populations, and implementation sciences to better translate research findings into real-world practice.149,150
Industry partners can help improve equitable access to novel therapies. Post-marketing studies and future clinical trials must consider equity and diversity in their design. Implementation of variable pricing strategies to improve affordability in low-and-middle-income regions,146 and participation in humanitarian aid programs, such as WFH HAP, will help ensure the distribution of these products to patients otherwise unable to access treatment. Industry sponsors can also support patient and clinician educational initiatives, and research aimed at improving care for underserved populations.
This is an inspirational time, as decades of research and development in hemophilia care culminate in advancements that fulfill the promise of reducing treatment burden and bleeding events. Ensuring accessibility of these treatments is an important step towards health equity for all hemophilia patients, enabling the attainment of physical and mental well-being free from the burden of their disease and its treatment.
Acknowledgments
The authors thank Angela Jansen, PhD, MHS, and Laura Swystun, PhD of GLOVAL LLC for assisting with manuscript preparation.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
No financial support was received from the industry for the development of this manuscript.
Disclosure
M.L. reports compensation paid directly to the Indiana Hemophilia & Thrombosis Center for participating advisory boards, consultation, or speaker’s bureau for: Alnylam, BPL, Agios, Pfizer, and Roche/Genentech. S.N. has received consultancy fees from HEMA Biologics and Kedrion Biopharma, unrelated to this work. A.D.S. reports compensation paid directly to the Indiana Hemophilia & Thrombosis Center for participating in advisory boards and/or consultation for Novo Nordisk, Pfizer, Genentech/Roche, Sanofi-Genzyme/Bioverativ, HEMA Biologics, Be Biopharma, BioMarin, Kedrion Biopharma; research from Novo Nordisk, Pfizer, Genentech/Roche, Kedrion Biopharma, Sanofi-Genzyme/Bioverativ, Regeneron, Takeda Pharmaceuticals, Pharmacosmos A/S, and Centessa Pharmaceuticals/ApcinteX Ltd; speaker’s bureau from: Genentech/Roche, Kedrion Biopharma, Sanofi-Genzyme/Bioverativ; and sits on the board of the Novo Nordisk Haemophilia Foundation. The authors report no other conflicts of interest in this work.
References
1. Blanchette VS, Key NS, Ljung LR, Manco-Johnson MJ, van den Berg HM, Srivastava A. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12(11):1935–1939.
2. van Galen KPM, d’Oiron R, James P, et al. A new hemophilia carrier nomenclature to define hemophilia in women and girls: communication from the SSC of the ISTH. J Thromb Haemost. 2021;19(8):1883–1887. doi:10.1111/jth.15397
3. Srivastava A, Santagostino E, Dougall A, et al. WFH guidelines for the management of hemophilia, 3rd edition. Haemophilia. 2020;26 Suppl 6:1–158. doi:10.1111/hae.14046
4. Pool JG, Shannon AE. Production of high-potency concentrates of antihemophilic globulin in a closed-bag system. N Engl J Med. 1965;273(27):1443–1447. doi:10.1056/NEJM196512302732701
5. Woods GM, Dunn AL. Coagulation factor concentrates for inherited bleeding disorders. Rossi’s Principles Transf Med. 2016;2016:328–343.
6. Britannica, The Editors of Encyclopaedia. “Ryan White”. Encyclopedia Britannica. Availale from: https://www.britannica.com/biography/Ryan-White.
7. Di Paolantonio T, Mariani G, Ghirardini A, et al. Low risk of transmission of the human immunodeficiency virus by a solvent-detergent-treated commercial factor VIII concentrate. J Med Virol. 1992;36(2):71–74. doi:10.1002/jmv.1890360202
8. Gitschier J, Wood WI, Goralka TM, et al. Characterization of the human factor VIII gene. 1984. Biotechnology. 1992;24:288–292.
9. Wood WI, Capon DJ, Simonsen CC, et al. Expression of active human factor VIII from recombinant DNA clones. Nature. 1984;312(5992):330–337. doi:10.1038/312330a0
10. Choo KH, Gould KG, Rees DJ, Brownlee GG. Molecular cloning of the gene for human anti-haemophilic factor IX. Nature. 1982;299(5879):178–180. doi:10.1038/299178a0
11. Anson DS, Austen DE, Brownlee GG. Expression of active human clotting factor IX from recombinant DNA clones in mammalian cells. Nature. 1985;315(6021):683–685. doi:10.1038/315683a0
12. Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535–544. doi:10.1056/NEJMoa067659
13. Hazendonk H, van Moort I, Mathot RAA, et al. Setting the stage for individualized therapy in hemophilia: what role can pharmacokinetics play? Blood Rev. 2018;32(4):265–271. doi:10.1016/j.blre.2018.01.001
14. Mancuso ME, Santagostino E. Outcome of clinical trials with new extended half-life FVIII/IX concentrates. J Clin Med. 2017;6(4):39. doi:10.3390/jcm6040039
15. Male C, Andersson NG, Rafowicz A, et al. Inhibitor incidence in an unselected cohort of previously untreated patients with severe haemophilia B: a PedNet study. Haematologica. 2021;106(1):123–129. doi:10.3324/haematol.2019.239160
16. Gouw SC, van den Berg HM, Fischer K, et al. Intensity of factor VIII treatment and inhibitor development in children with severe hemophilia A: the RODIN study. Blood. 2013;121(20):4046–4055. doi:10.1182/blood-2012-09-457036
17. Astermark J, Donfield SM, DiMichele DM, et al. A randomized comparison of bypassing agents in hemophilia complicated by an inhibitor: the FEIBA NovoSeven comparative (FENOC) study. Blood. 2007;109(2):546–551.
18. Mahlangu J, Oldenburg J, Paz-Priel I, et al. Emicizumab prophylaxis in patients who have hemophilia A without inhibitors. N Engl J Med. 2018;379(9):811–822. doi:10.1056/NEJMoa1803550
19. Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N Engl J Med. 2017;377(9):809–818. doi:10.1056/NEJMoa1703068
20. U.S. FDA approves Pfizer’s HYMPAVZI™ (marstacimab-hncq) for the treatment of adults and adolescents with hemophilia A or B without inhibitors. Business Wire. Available from: https://finance.yahoo.com/news/u-fda-approves-pfizer-hympavzi-172300267.html.
21. Iorio A, Stonebraker JS, Chambost H, et al. Establishing the prevalence and prevalence at birth of hemophilia in males: a meta-analytic approach using national registries. Ann Intern Med. 2019;171(8):540–546. doi:10.7326/M19-1208
22. von Drygalski A, Chowdary P, Kulkarni R, et al. Efanesoctocog alfa prophylaxis for patients with severe hemophilia A. N Engl J Med. 2023;388(4):310–318.
23. von Mackensen S, Shah J, Seifert W, Kenet G. Health-related quality of life in paediatric haemophilia B patients treated with rIX-FP. Haemophilia. 2019;25(1):45–53. doi:10.1111/hae.13624
24. Oldenburg J, Kulkarni R, Srivastava A, et al. Improved joint health in subjects with severe haemophilia A treated prophylactically with recombinant factor VIII Fc fusion protein. Haemophilia. 2018;24(1):77–84. doi:10.1111/hae.13353
25. World Federation of Hemophilia. Report on the Annual Global Survey. 2022.
26. O’Mahony B, Black C. Expanding hemophilia care in developing countries. Semin Thromb Hemost. 2005;31(5):561–568. doi:10.1055/s-2005-922228
27. Hacker MR, Geraghty S, Manco-Johnson M. Barriers to compliance with prophylaxis therapy in haemophilia. Haemophilia. 2001;7(4):392–396. doi:10.1111/j.1365-2516.2001.00534.x
28. Miller CH, Soucie JM, Byams VR, et al. Women and girls with haemophilia receiving care at specialized haemophilia treatment centres in the United States. Haemophilia. 2021;27(6):1037–1044. doi:10.1111/hae.14403
29. Mannucci PM. Hemophilia therapy: the future has begun. Haematologica. 2020;105(3):545–553. doi:10.3324/haematol.2019.232132
30. Seth Chhabra E, Liu T, Kulman J, et al. BIVV001, a new class of factor VIII replacement for hemophilia A that is independent of von Willebrand factor in primates and mice. Blood. 2020;135(17):1484–1496. doi:10.1182/blood.2019001292
31. Efanesoctocog alfa (Altuviiio) [package insert]. Waltham, MA: Bioverativ Therapetuics Inc; 2024.
32. Konkle BA, Shapiro AD, Quon DV, et al. BIVV001 fusion protein as factor VIII replacement therapy for hemophilia A. N Engl J Med. 2020;383(11):1018–1027. doi:10.1056/NEJMoa2002699
33. Mahlangu J, Iorio A, Kenet G. Emicizumab state-of-the-art update. Haemophilia. 2022;28 Suppl 4(Suppl 4):103–110. doi:10.1111/hae.14524
34. Lee H, Cho H, Han JW, et al. Cost-utility analysis of emicizumab prophylaxis in haemophilia A patients with factor VIII inhibitors in Korea. Haemophilia. 2021;27(1):e12–e21. doi:10.1111/hae.14143
35. Cortesi PA, Castaman G, Trifiro G, et al. Cost-effectiveness and budget impact of emicizumab prophylaxis in haemophilia A patients with inhibitors. Thromb Haemost. 2020;120(2):216–228. doi:10.1055/s-0039-3401822
36. Gelbenegger G, Schoergenhofer C, Knoebl P, Jilma B. Bridging the missing link with emicizumab: a bispecific antibody for treatment of hemophilia A. Thromb Haemost. 2020;120(10):1357–1370. doi:10.1055/s-0040-1714279
37. Emicizumab (Hemlibra) [package insert]. Basel, Switzerland: Roche; November 2017.
38. Langer AL, Etra A, Aledort L. Evaluating the safety of emicizumab in patients with hemophilia A. Expert Opin Drug Saf. 2018;17(12):1233–1237. doi:10.1080/14740338.2019.1551356
39. Paz-Priel I, Chang T, Asikanius E, et al. Immunogenicity of emicizumab in people with hemophilia A (PwHA): results from the HAVEN1-4 studies. Blood. 2018;132(Suppl_1):633. doi:10.1182/blood-2018-99-118492
40. Kizilocak H, Guerrera MF, Young G. Neutralizing antidrug antibody to emicizumab in patients with severe hemophilia A: case report of a first noninhibitor patient and review of the literature. Res Pract Thromb Haemost. 2023;7(6):102194. doi:10.1016/j.rpth.2023.102194
41. Taves S, Sun J, Livingston EW, et al. Hemophilia A and B mice, but not VWF(-/-)mice, display bone defects in congenital development and remodeling after injury. Sci Rep. 2019;9(1):14428. doi:10.1038/s41598-019-50787-9
42. Manco-Johnson M, Briones N, Tran A, et al. Emicizumab: will it suffice for bone metabolism? Res Pract Thromb Haemost. 2021;5(S2):104–105. doi:10.1002/rth2.12459
43. Kiialainen A, Niggli M, Kempton CL, et al. Effect of emicizumab prophylaxis on bone and joint health markers in people with haemophilia A without factor VIII inhibitors in the HAVEN 3 study. Haemophilia. 2022;28(6):1033–1043. doi:10.1111/hae.14642
44. Østergaard H, Lund J, Greisen PJ, et al. A factor VIIIa-mimetic bispecific antibody, Mim8, ameliorates bleeding upon severe vascular challenge in hemophilia A mice. Blood. 2021;138(14):1258–1268. doi:10.1182/blood.2020010331
45. Mancuso M, Matsushita T, Chowdary P, et al. Efficacy and safety of Mim8 prophylaxis in adults and adolescents with hemophilia A with or without inhibitors: phase 3, open-label, randomized, controlled FRONTIER2 study. Res Pract Thromb Haemost. 2024;2024:1.
46. Teranishi-Ikawa Y, Soeda T, Koga H, et al. A bispecific antibody NXT007 exerts a hemostatic activity in hemophilia A monkeys enough to keep a nonhemophilic state. J Thromb Haemost. 2023;22:430–440. doi:10.1016/j.jtha.2023.09.034
47. Mast AE. Tissue factor pathway inhibitor: multiple anticoagulant activities for a single protein. Arterioscler Thromb Vasc Biol. 2016;36(1):9–14. doi:10.1161/ATVBAHA.115.305996
48. Pasca S. Concizumab as a subcutaneous prophylactic treatment option for patients with hemophilia A or B: a review of the evidence and patient’s perspectives. J Blood Med. 2022;13:191–199. doi:10.2147/JBM.S242219
49. Chowdary P, Angchaisuksiri P, Apte S, et al. Concizumab prophylaxis in people with haemophilia A or haemophilia B without inhibitors (explorer8): a prospective, multicentre, open-label, randomised, phase 3a trial. Lancet Haematol. 2024;11(12):e891–e904. doi:10.1016/S2352-3026(24)00307-7
50. Matsushita T, Shapiro A, Abraham A, et al. Phase 3 trial of concizumab in hemophilia with inhibitors. N Engl J Med. 2023;389(9):783–794.
51. Ck SSV, Rasmussen JS, Rose TH, Tamer S, Porstmann T, Haaning J. Risk mitigation strategy for concizumab clinical trials after pause due to non-fatal thrombotic events. Blood. 2020;136(Supplement 1):40.
52. Shapiro AD, Angchaisuksiri P, Astermark J, et al. Subcutaneous concizumab prophylaxis in hemophilia A and hemophilia A/B with inhibitors: phase 2 trial results. Blood. 2019;134(22):1973–1982. doi:10.1182/blood.2019001542
53. Mahlangu J, Luis Lamas J, Cristobal Morales J, et al. Long-term safety and efficacy of the anti-tissue factor pathway inhibitor marstacimab in participants with severe haemophilia: Phase II study results. Br J Haematol. 2023;200(2):240–248. doi:10.1111/bjh.18495
54. As MD, Palladino A, Hwang E, McDonald R, Taylor CT, Teeter J. Efficacy and safety of the anti-tissue factor pathway inhibitor marstacimab in participants with severe hemophilia without inhibitors: results from the phase 3 basis trial. Blood. 2023;142(Supplement 1):285. doi:10.1182/blood-2023-181263
55. Jiménez-Yuste V, Palladino A, McDonald R, et al. Dose escalation of the anti-tissue factor pathway inhibitor marstacimab in participants with severe hemophilia without inhibitors: results from the phase 3 BASIS and long-term extension trials.
56. Md AS, Mahlangu J, Taylor CT, et al. Marstacimab, an anti-tissue factor pathway inhibitor, in participants with hemophilia Α or B, with and without inhibitors: an integrated analysis of safety. Blood. 2023;142(Supplement 1):3980. doi:10.1182/blood-2023-174682
57. Rezaie AR, Giri H. Anticoagulant and signaling functions of antithrombin. J Thromb Haemost. 2020;18(12):3142–3153. doi:10.1111/jth.15052
58. Young G, Lenting PJ, Croteau SE, Nolan B, Srivastava A. Antithrombin lowering in hemophilia: a closer look at fitusiran. Res Pract Thromb Haemost. 2023;7(4):100179. doi:10.1016/j.rpth.2023.100179
59. Young G, Srivastava A, Kavakli K, et al. Efficacy and safety of fitusiran prophylaxis in people with haemophilia A or haemophilia B with inhibitors (ATLAS-INH): a multicentre, open-label, randomised phase 3 trial. Lancet. 2023;401(10386):1427–1437. doi:10.1016/S0140-6736(23)00284-2
60. Srivastava A, Brewer AK, Mauser-Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19(1):e1–47. doi:10.1111/j.1365-2516.2012.02909.x
61. Delgado CD. Fitusiran dosing in trials reduced to lower blood-clot risk. Available from: https://hemophilianewstoday.com/2021/03/01/fitusiran-dosing-phase-3-trials-reduced-to-lower-risk-of-blood-clots.
62. Pasi KJ, Lissitchkov T, Mamonov V, et al. Targeting of antithrombin in hemophilia A or B with investigational siRNA therapeutic fitusiran-Results of the phase 1 inhibitor cohort. J Thromb Haemost. 2021;19(6):1436–1446. doi:10.1111/jth.15270
63. Pasi KJ, Rangarajan S, Georgiev P, et al. Targeting of antithrombin in hemophilia A or B with RNAi therapy. N Engl J Med. 2017;377(9):819–828. doi:10.1056/NEJMoa1616569
64. Peyvandi F, Garagiola I, Abbattista M. Fitusiran in haemophilia: a breakthrough drug with many unknowns. Lancet. 2023;401(10386):1400–1401. doi:10.1016/S0140-6736(23)00514-7
65. Jiang M, Yang F, Jiang Y, et al. Safety and efficacy of an anti-human APC antibody for prophylaxis of congenital factor deficiencies in preclinical models. Blood. 2023;142(12):1071–1081. doi:10.1182/blood.2023020005
66. Hj BT, Koch A, Mocanu I, Makhaldiani L. Serpin-PC in persons with severe hemophilia (PwH): updated results from a multicenter multi-part,
67. Lobo A. Centessa discontinues development of SerpinPC for hemophilia. Hemophilia News Today. Available from: https://hemophilianewstoday.com/news/development-serpinpc-treatment-hemophilia-halted-centessa/.
68. Prince R, Bologna L, Manetti M, et al. Targeting anticoagulant protein S to improve hemostasis in hemophilia. Blood. 2018;131(12):1360–1371. doi:10.1182/blood-2017-09-800326
69. Leong L, Byun T, Kim B, et al. Pre-clinical characterization of VGA039, an anti-protein S monoclonal antibody being developed as a universal hemostatic agent for various bleeding disorders. Blood. 2022;140(S1):1666–1667. doi:10.1182/blood-2022-170245
70. NOVOSEVEN [package insert]. Novo Nordisk, Bagsvaerd, Denmark, 2014.
71. Lentz SR, Ehrenforth S, Karim FA, et al. Recombinant factor VIIa analog in the management of hemophilia with inhibitors: results from a multicenter, randomized, controlled trial of vatreptacog alfa. J Thromb Haemost. 2014;12(8):1244–1253. doi:10.1111/jth.12634
72. Santagostino E, Mancuso ME, Rocino A, Mancuso G, Scaraggi F, Mannucci PM. A prospective randomized trial of high and standard dosages of recombinant factor VIIa for treatment of hemarthroses in hemophiliacs with inhibitors. J Thromb Haemost. 2006;4(2):367–371. doi:10.1111/j.1538-7836.2006.01772.x
73. Almagro D, Agramonte O, Castillo D, Zamora Y, Ballester JM. Experience with a single dose of recombinant activated factor VII for the management of mild-to-moderate bleeds in haemophilia. Haemophilia. 2011;17(2):322–323. doi:10.1111/j.1365-2516.2010.02403.x
74. Parameswaran R, Shapiro AD, Gill JC, Kessler CM, Investigators HR. Dose effect and efficacy of rFVIIa in the treatment of haemophilia patients with inhibitors: analysis from the hemophilia and thrombosis research society registry. Haemophilia. 2005;11(2):100–106. doi:10.1111/j.1365-2516.2005.01075.x
75. SEVENFACT [package insert]. Puteaux, France: Laboratoire Francais du Fractionnement et des Biotechnologies S.A.; 2022.
76. Wang M, Lawrence JB, Quon DV, et al. PERSEPT 1: a phase 3 trial of activated eptacog beta for on-demand treatment of haemophilia inhibitor-related bleeding. Haemophilia. 2017;23(6):832–843. doi:10.1111/hae.13301
77. Biosimilars. U.S. Food & Drug Adminstration. https://www.fda.gov/drugs/therapeutic-biologics-applications-bla/biosimilars.
78. ROCTAVIAN [Package Insert]. Novato, California: Biomarin Pharmaceutical, Inc.; 2023.
79. HEMGENIX (etranacogene dezaparvovec-drlb) [package insert]. King of Prussia, United States: CSL Behring LLC; 2022.
80. Ozelo MC, Mahlangu J, Pasi KJ, et al. Valoctocogene Roxaparvovec Gene Therapy for Hemophilia A. N Engl J Med. 2022;386(11):1013–1025.
81. Ozelo MC, Mahlangu J, Madan B, et al. Bleeding, FVIII activity, and safety 3 years after gene transfer with valoctocogene roxaparvovec: results from GENER8-1. Hematol Transf Cell Ther. 2023;45:S451–S452. doi:10.1016/j.htct.2023.09.842
82. Valoctocogene roxaparvovec (Roctavian) [package insert]. Novato CA USA: BioMarin Pharmaceutical Inc; 2024.
83. Symington E, Rangarajan S, Lester W, et al. Valoctocogene roxaparvovec gene therapy provides durable haemostatic control for up to 7 years for haemophilia A. Haemophilia. 2024;30(5):1138–1147. doi:10.1111/hae.15071
84. George LA, Sullivan SK, Giermasz A, et al. Hemophilia B gene therapy with a high-specific-activity Factor IX variant. N Engl J Med. 2017;377(23):2215–2227. doi:10.1056/NEJMoa1708538
85. Miesbach W, Meijer K, Coppens M, et al. Gene therapy with adeno-associated virus vector 5-human factor IX in adults with hemophilia B. Blood. 2018;131(9):1022–1031. doi:10.1182/blood-2017-09-804419
86. Pipe SW, Leebeek FWG, Recht M, et al. Gene therapy with etranacogene dezaparvovec for hemophilia B. N Engl J Med. 2023;388(8):706–718. doi:10.1056/NEJMoa2211644
87. Von Drygalski A, Giermasz A, Castaman G, et al. Etranacogene dezaparvovec (AMT-061 phase 2b): normal/near normal FIX activity and bleed cessation in hemophilia B. Blood Adv. 2019;3(21):3241–3247. doi:10.1182/bloodadvances.2019000811
88. von Drygalski A, Gomez E, Giermasz A, et al. Stable and durable factor IX levels in patients with hemophilia B over 3 years after etranacogene dezaparvovec gene therapy. Blood Adv. 2023;7(19):5671–5679. doi:10.1182/bloodadvances.2022008886
89. Arruda VS-JB, Factor IXP. From biochemistry to gene therapy. Blood. 2016;128(22):SCI–9. doi:10.1182/blood.V128.22.SCI-9.SCI-9
90. Pipe S, Pvd V, Verhamme P, et al. Long-term bleeding protection, sustained FIX activity, reduction of FIX consumption and safety of hemophilia B gene therapy: results from the HOPE-B trial 3 years after administration of a single dose of etranacogene dezaparvovec in adult patients with severe or moderately severe hemophilia B. Blood. 2023;142(S1):1055.
91. Cuker A, ALzahrani H, Astermark J, et al. Efficacy and safety of fidanacogene elaparvovec in adults with moderately severe or severe hemophilia B: results from the phase 3 BENEGENE-2 gene therapy trial. Res Pract Thrombosis Haemostasis. 2023;7(S2):124–125. doi:10.1016/j.rpth.2023.100452
92. Klamroth R, Kalac M, Fuiman J, et al. Efficacy and safety of fidanacogene elaparvovec in adults with moderately severe to severe hemophilia B: updated results from the phase 3 BENEGENE-2 gene therapy trial.
93. Samelson-Jones B, George L, Rasko J, et al. A multi-year follow-up study of fidanacogene elaparvovec gene therapy for hemophilia B.
94. Mahlangu J, Kaczmarek R, von Drygalski A, et al. Two-year outcomes of valoctocogene roxaparvovec therapy for hemophilia A. N Engl J Med. 2023;388(8):694–705. doi:10.1056/NEJMoa2211075
95. Batty P, Lillicrap D. Hemophilia gene therapy: approaching the first licensed product. Hemasphere. 2021;5(3):e540.
96. Batty P, Mo AM, Hurlbut D, et al. Long-term follow-up of liver-directed, adeno-associated vector-mediated gene therapy in the canine model of hemophilia A. Blood. 2022;140(25):2672–2683. doi:10.1182/blood.2021014735
97. Batty P, Fong S, Franco M, et al. Vector integration and fate in the hemophilia dog liver multiple years after AAV-FVIII gene transfer. Blood. 2024;143(23):2373–2385. doi:10.1182/blood.2023022589
98. Nguyen GN, Everett JK, Kafle S, et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat Biotechnol. 2021;39(1):47–55. doi:10.1038/s41587-020-0741-7
99. Fong S, Yates B, Sihn CR, et al. Interindividual variability in transgene mRNA and protein production following adeno-associated virus gene therapy for hemophilia A. Nat Med. 2022;28(4):789–797. doi:10.1038/s41591-022-01751-0
100. National Hemophilia Foundation. MASAC Recommendations on Hemophilia Treatment Center Preparedness for Delivering Gene Therapy for Hemophilia. MASAC Document 282. Available from: https://www.hemophilia.org/healthcare-professionals/guidelines-on-care/masac-documents/masac-document-282-masac-recommendations-on-hemophilia-treatment-center-preparedness-for-delivering-gene-therapy-for-hemophilia.
101. WFH shared decision making tool. Available from: https://sdm.wfh.org/Accessed%20October%2020.
102. Lewandowska M, Nasr S, Shapiro AD. Therapeutic and technological advancements in haemophilia care: quantum leaps forward. Haemophilia. 2022;28 Suppl 4:77–92. doi:10.1111/hae.14531
103. Gallastegui N, Steiner B, Aguero P, et al. The role of point-of-care musculoskeletal ultrasound for routine joint evaluation and management in the hemophilia clinic - A real world experience. BMC Musculoskelet Disord. 2022;23(1):1111. doi:10.1186/s12891-022-06042-w
104. Aguero P, Barnes RF, Flores A, von Drygalski A. Teleguidance for patient self-imaging of hemophilic joints using mobile ultrasound devices: a pilot study. J Ultrasound Med. 2023;42(3):701–712. doi:10.1002/jum.16084
105. Gualtierotti R, Garagiola I, Mortarino M, Spena S, Romero-Lux O, Peyvandi F. Gender equity in hemophilia: need for healthcare, familial, and societal advocacy. Front Med Lausanne. 2024;11:1345496. doi:10.3389/fmed.2024.1345496
106. van Galen K, Lavin M, Skouw-Rasmussen N, et al. European principles of care for women and girls with inherited bleeding disorders. Haemophilia. 2021;27(5):837–847. doi:10.1111/hae.14379
107. Bercovitz RS. “A new hemophilia carrier nomenclature to define hemophilia in women and girls: communication from the SSC of the ISTH”: comment. J Thromb Haemost. 2022;20(7):1744–1745. doi:10.1111/jth.15727
108. Valk C. A transgender person with haemophilia. J Haem Pract. 2018;5(1):147–151. doi:10.17225/jhp00128
109. Merz LE, Weyand AC. Bad blood: inequity in hemophilia care. Res Pract Thromb Haemost. 2024;8(1):102290. doi:10.1016/j.rpth.2023.102290
110. Baker JR, Riske B, Voutsis M, Cutter S, Presley R. Insurance, home therapy, and prophylaxis in U.S. youth with severe hemophilia. Am J Prev Med. 2011;41(6 Suppl 4):S338–45. doi:10.1016/j.amepre.2011.09.002
111. Miller CH, Benson J, Ellingsen D, et al. F8 and F9 mutations in US haemophilia patients: correlation with history of inhibitor and race/ethnicity. Haemophilia. 2012;18(3):375–382. doi:10.1111/j.1365-2516.2011.02700.x
112. Kempton CL, Payne AB, Fedewa SA. Race, ethnicity, and immune tolerance induction in hemophilia A in the United States. Res Pract Thromb Haemost. 2023;7(8):102251. doi:10.1016/j.rpth.2023.102251
113. Ahmed AE, Pratt KP. Race, ethnicity, F8 variants, and inhibitor risk: analysis of the “My Life Our Future” hemophilia A database. J Thromb Haemost. 2023;21(4):800–813. doi:10.1016/j.jtha.2022.12.017
114. Fedewa SA, Kempton CL. Race and ethnicity and the success of immune tolerance induction among people with severe haemophilia A in the United States. Haemophilia. 2024;30(3):628–637. doi:10.1111/hae.14980
115. Callaghan MU, Rajpurkar M, Chitlur M, Warrier I, Lusher J. Immune tolerance induction in 31 children with haemophilia A: is ITI less successful in African Americans? Haemophilia. 2011;17(3):483–489. doi:10.1111/j.1365-2516.2010.02429.x
116. McLaughlin JM, Lambing A, Witkop ML, Anderson TL, Munn J, Tortella B. Racial differences in chronic pain and quality of life among adolescents and young adults with moderate or severe hemophilia. J Racial Ethn Health Disparities. 2016;3(1):11–20. doi:10.1007/s40615-015-0107-x
117. Fedewa SA, Buckner TW, Parks SG, et al. Racial and ethnic differences in distress, depression, and quality of life in people with hemophilia. J Racial Ethn Health Disparities. 2024;11(3):1394–1404. doi:10.1007/s40615-023-01616-3
118. Fedewa SA, Payne AB, Tran D, Cafuir L, Antun A, Kempton CL. Racial and ethnic differences in reported haemophilia death rates in the United States. Haemophilia. 2023;29(6):1410–1418. doi:10.1111/hae.14859
119. Fedewa SA, Valentino LA, Koo A, et al. Race and ethnicity reporting and representation in hemophilia clinical trials. Blood Adv. 2024;8(10):2351–2360. doi:10.1182/bloodadvances.2024012862
120. Soucie JM, Nuss R, Evatt B, et al. Mortality among males with hemophilia: relations with source of medical care. the hemophilia surveillance system project investigators. Blood. 2000;96(2):437–442.
121. Okolo AI, Soucie JM, Grosse SD, et al. Population-based surveillance of haemophilia and patient outcomes in Indiana using multiple data sources. Haemophilia. 2019;25(3):456–462. doi:10.1111/hae.13734
122. Sharma D, Wallace N, Levinsohn EA, et al. Trends and factors affecting the US adult hematology workforce: a mixed methods study. Blood Adv. 2019;3(22):3550–3561. doi:10.1182/bloodadvances.2019000307
123. D’Angiolella LS, Cortesi PA, Rocino A, et al. The socioeconomic burden of patients affected by hemophilia with inhibitors. Eur J Haematol. 2018;101(4):435–456. doi:10.1111/ejh.13108
124. Walsh CE, Soucie JM, Miller CH. Impact of inhibitors on hemophilia A mortality in the United States. Am J Hematol. 2015;90(5):400–405. doi:10.1002/ajh.23957
125. Morfini M, Haya S, Tagariello G, et al. European study on orthopaedic status of haemophilia patients with inhibitors. Haemophilia. 2007;13(5):606–612. doi:10.1111/j.1365-2516.2007.01518.x
126. Nakar C, Shapiro A. Hemophilia A with inhibitor: immune tolerance induction (ITI) in the mirror of time. Transfus Apher Sci. 2019;58(5):578–589. doi:10.1016/j.transci.2019.08.008
127. Alhemo [package insert]. Novo Nordisk Canada Inc. July 26; 2023.
128. Mahlangu J, Paz P, Hardtke M, Aswad F, Schroeder J. TRUST trial: BAY 86-6150 use in haemophilia with inhibitors and assessment for immunogenicity. Haemophilia. 2016;22(6):873–879. doi:10.1111/hae.12994
129. Ljung R, Karim FA, Saxena K, et al. 40K glycoPEGylated, recombinant FVIIa: 3-month, double-blind, randomized trial of safety, pharmacokinetics and preliminary efficacy in hemophilia patients with inhibitors. J Thromb Haemost. 2013;11(7):1260–1268. doi:10.1111/jth.12237
130. Castaman G, Peyvandi F, De Cristofaro R, Pollio B, Di Minno DMN. Mild and moderate hemophilia A: neglected conditions, still with unmet needs. J Clin Med. 2023;12(4):1368. doi:10.3390/jcm12041368
131. Di Minno MN, Ambrosino P, Franchini M, Coppola A, Di Minno G. Arthropathy in patients with moderate hemophilia a: a systematic review of the literature. Semin Thromb Hemost. 2013;39(7):723–731. doi:10.1055/s-0033-1354422
132. den Uijl IE, Fischer K, Van Der Bom JG, Grobbee DE, Rosendaal FR, Plug I. Clinical outcome of moderate haemophilia compared with severe and mild haemophilia. Haemophilia. 2009;15(1):83–90. doi:10.1111/j.1365-2516.2008.01837.x
133. Hodroj MH, El Hasbani G, Al-Shamsi HO, Samaha H, Musallam KM, Taher AT. Clinical burden of hemophilia in older adults: beyond bleeding risk. Blood Rev. 2022;53:100912. doi:10.1016/j.blre.2021.100912
134. Sood SL, Cheng D, Ragni M, et al. A cross-sectional analysis of cardiovascular disease in the hemophilia population. Blood Adv. 2018;2(11):1325–1333. doi:10.1182/bloodadvances.2018018226
135. Schutgens REG, Jimenez-Yuste V, Escobar M, et al. Antithrombotic treatment in patients with hemophilia: an EHA-ISTH-EAHAD-ESO clinical practice guidance. Hemasphere. 2023;7(6):e900. doi:10.1097/HS9.0000000000000900
136. Zhou ZY, Koerper MA, Johnson KA, et al. Burden of illness: direct and indirect costs among persons with hemophilia A in the United States. J Med Econ. 2015;18(6):457–465. doi:10.3111/13696998.2015.1016228
137. Pierce GF, Adediran M, Diop S, et al. Achieving access to haemophilia care in low-income and lower-middle-income countries: expanded humanitarian aid program of the world federation of hemophilia after 5 years. Lancet Haematol. 2022;9(9):e689–e697. doi:10.1016/S2352-3026(22)00209-5
138. World Federation of Hemophilia. Report on the Annual Global Survey. 2020.
139. Ozelo MC, Yamaguti-Hayakawa GG. Impact of novel hemophilia therapies around the world. Res Pract Thromb Haemost. 2022;6(3):e12695. doi:10.1002/rth2.12695
140. Ndoumba-Mintya A, Diallo YL, Tayou TC, Mbanya DN. Optimizing haemophilia care in resource-limited countries: current challenges and future prospects. J Blood Med. 2023;14:141–146. doi:10.2147/JBM.S291536
141. Mahlangu J, Diop S, Lavin M. Diagnosis and treatment challenges in lower resource countries: state-of-the-art. Haemophilia. 2024;30 Suppl 3:78–85. doi:10.1111/hae.14956
142. Skinner MW. WFH: closing the global gap--achieving optimal care. Haemophilia. 2012;18 Suppl 4:1–12. doi:10.1111/j.1365-2516.2012.02822.x
143. Coffin D, Gouider E, Konkle B, et al. The world federation of hemophilia world bleeding disorders registry: insights from the first 10,000 patients. Res Pract Thromb Haemost. 2023;7(8):102264. doi:10.1016/j.rpth.2023.102264
144. World Federation of Hemophilia Humanitarian Aid Program - Impact Report. 2022.
145. Skinner MW, Nugent D, Wilton P, et al. Achieving the unimaginable: health equity in haemophilia. Haemophilia. 2020;26(1):17–24. doi:10.1111/hae.13862
146. Krumb E, Hermans C. Living with a “hemophilia-free mind” - The new ambition of hemophilia care? Res Pract Thromb Haemost. 2021;5(5):e12567. doi:10.1002/rth2.12567
147. FitzGerald C, Hurst S. Implicit bias in healthcare professionals: a systematic review. BMC Med Ethics. 2017;18(1):19. doi:10.1186/s12910-017-0179-8
148. Wilbur K, Snyder C, Essary A, Reddy S, Will K, Saxon M. Developing workforce diversity in the health professions: a social justice perspective. Health Professions Educ. 2020;6(222–229):222–229. doi:10.1016/j.hpe.2020.01.002
149. Valentino LA, Witkop ML, Santaella ME, DiMichele D, Recht M. Building the blueprint: formulating a community-generated national plan for future research in inherited bleeding disorders. Haemophilia. 2022;28(5):760–768. doi:10.1111/hae.14588
150. Byams VR, Baker JR, Bailey C, et al. Building the foundation for a community-generated national research blueprint for inherited bleeding disorders: research priorities in health services; diversity, equity, and inclusion; and implementation science. Expert Rev Hematol. 2023;16(sup1):87–106. doi:10.1080/17474086.2023.2183836
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Detection Rate of Diabetic Retinopathy Before and After Implementation of Autonomous AI-based Fundus Photograph Analysis in a Resource-Limited Area in Belize
Esmaeilkhanian H, Gutierrez KG, Myung D, Fisher AC
Clinical Ophthalmology 2025, 19:993-1006
Published Date: 21 March 2025
The AI Health Arms Race: A Critical Perspective on Big Tech and the Widening Global Health Equity Gap
Ahmed MM, Othman ZK
Journal of Multidisciplinary Healthcare 2026, 19:610208
Published Date: 21 April 2026