Back to Journals » Journal of Hepatocellular Carcinoma » Volume 1
Emerging role of insulin-like growth factor-binding protein 7 in hepatocellular carcinoma
Authors Akiel M ![]() , Rajasekaran D, Gredler R, Siddiq A, Srivastava J, Robertson C, Jariwala N, Fisher P, Sarkar D
, Rajasekaran D, Gredler R, Siddiq A, Srivastava J, Robertson C, Jariwala N, Fisher P, Sarkar D
Received 7 January 2014
Accepted for publication 3 February 2014
Published 26 March 2014 Volume 2014:1 Pages 9—19
DOI https://doi.org/10.2147/JHC.S44460
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Maaged Akiel, Devaraja Rajasekaran, Rachel Gredler, Ayesha Siddiq, Jyoti Srivastava, Chadia Robertson, Nidhi Himanshu Jariwala, Paul B Fisher, Devanand Sarkar
Department of Human and Molecular Genetics, Massey Cancer Center, VCU Institute of Molecular Medicine, Virginia Commonwealth University, Richmond, Virginia, USA
Abstract: Hepatocellular carcinoma (HCC) is a vicious and highly vascular cancer with a dismal prognosis. It is a life-threatening illness worldwide that ranks fifth in terms of cancer prevalence and third in cancer deaths. Most patients are diagnosed at an advanced stage by which time conventional therapies are no longer effective. Targeted molecular therapies, such as the multikinase inhibitor sorafenib, provide a modest increase in survival for advanced HCC patients and display significant toxicity. Thus, there is an immense need to identify novel regulators of HCC that might be targeted effectively. The insulin-like growth factor (IGF) axis is commonly abnormal in HCC. Upon activation, the IGF axis controls metabolism, tissue homeostasis, and survival. Insulin-like growth factor-binding protein 7 (IGFBP7) is a secreted protein of a family of low-affinity IGF-binding proteins termed “IGFBP-related proteins” that have been identified as a potential tumor suppressor in HCC. IGFBP7 has been implicated in regulating cellular proliferation, senescence, and angiogenesis. In this review, we provide a comprehensive discussion of the role of IGFBP7 in HCC and the potential use of IGFBP7 as a novel biomarker for drug resistance and as an effective therapeutic strategy.
Keywords: HCC, IGFBP7, gene therapy, angiogenesis, senescence, apoptosis
Introduction
Hepatocellular carcinoma (HCC) is a tumor arising from hepatocytes, the epithelial cells of the liver. Macroscopic pathology is characterized by scattered, large, and multinodular tumors. HCC represents more than 80% of liver cancers. Worldwide, it is a serious life-threatening illness with a poor prognosis. Globally, HCC ranks fifth in terms of cancer prevalence and third in cancer deaths.1,2 HCC is considered the fifth most prevalent cancer in males and the seventh in females, and the likelihood of developing HCC is four to five times higher in males than in females.3 This difference in the occurrence of HCC according to sex is thought to be due to the influence of sex hormones4,5 – while androgen enhances the development of hepatitis B virus (HBV)-triggered HCC in HBV transgenic male mice, estrogen reduces the occurrence of HCC in female mice on exposure to the hepatocarcinogen, diethylnitrosamine.4–7 However, the regulatory action by which androgen and estrogen orchestrate the disparity between the sexes is still not fully understood.
The number of reported new cases of HCC is escalating each year and HCC has a 5-year survival rate of <12%. The American Cancer Society estimates that in 2013 there will be more than 30,000 new cases and more than 21,000 reported deaths in the USA (updated data not yet available to confirm this estimate). Chronic HBV infection constitutes 50% of HCC cases around the world.3,8 In contrast, in the USA, hepatitis C virus (HCV)-related HCC is more common than HBV-related HCC, with the former constituting 48% of all HCC cases in the USA and the latter 16%.8,9 Immigrants from countries where HBV is endemic account for most of the HBV-related HCC cases in the USA.9 The remaining percentage of cases is unequally distributed across various risk factors that modify disease, such as alcohol-related liver disease and metabolic syndromes.8,10 HCV, alcohol-related liver disease, and nonalcoholic fatty liver disease tend to be common risk factors in the USA.3,8
HCC is not a population-specific illness; however, statistical data on the incidence of HCC show that Asians, Pacific Islanders, and Native Americans have the highest rate of incidence, followed by African Americans and Caucasians in the USA.9 The incidence of HCC in the USA has increased from 1.6 to 4.9 cases per 100,000 of US population from 1975 to 2005, an increase of more than 200%.11 As HCV-related HCC has manifested most commonly around the age of 70, the increase of HCV-related HCC in the USA might be linked to the needle sharing involved in drug use at the time when drug abuse was prevalent (more than 30 years ago), as implied by a cohort study of HCV-infected patients.1,12
HCC is a highly heterogeneous cancer, which may be explained by the diversity and increased number of etiological factors.13 The majority of risk factors leads to chronic liver disease that later progresses to HCC.3 Patients with HCC have an extremely low health-related quality of life (a marker considered when assessing HCC prognosis) compared with chronic liver disease patients.14 The American Association for the study of Liver Diseases endorses the use of the Barcelona Clinic Liver Cancer Staging System. The Barcelona Clinic Liver Cancer has implemented a treatment algorithm that tailors a treatment strategy based on the stage of HCC. For example, surgical resection and liver transplantation are offered to patients diagnosed at an early stage resulting in a ≥75% survival rate. In most cases, HCC is diagnosed at an advanced stage at a time when symptoms start to manifest, by which time prognosis is dismal and the aforementioned conventional treatments are no longer efficacious.1–3 The only US Food and Drug Administration-approved drug available for non-resectable advanced HCC is the multikinase inhibitor sorafenib, which provides a survival advantage of only 2.8 months compared with placebo.15 Therefore, the identification of new diagnostic markers is mandatory to diagnose and treat patients at a very early stage when prognosis is highly favorable. Additionally, new regulatory molecules need to be identified that might be targeted to develop effective therapeutic approaches for advanced HCC, thereby providing longstanding benefits to patients.
Insulin-like growth factor (IGF) signaling in HCC
The IGF pathway is an evolutionarily conserved pathway across mammals. The binding of IGF-1 and IGF-2 to their cognate receptors leads to the activation of phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)-AKT and Ras-Raf-mitogen-activated protein kinases (MAPK) signaling through which the IGF axis regulates metabolism, tissue homeostasis, and survival.16,17 The role of IGF in carcinogenesis is becoming apparent, as there is increasing evidence from epidemiological studies associating IGF signaling with malignancy including HCC. Additionally, activation of the IGF axis is associated with resistance to therapeutic drugs.16,18–20 The complexity of IGF signaling resides at the level of regulation conferred by three types of interacting proteins: ligands (IGF-1 and IGF-2), receptors (IGF-1R, IGF-2R) and insulin-like growth factor-binding proteins (IGFBPs).
Ligands
The liver produces the majority of IGF-1 and IGF-2, which are 67% identical and share 40% identity with pro-insulin. However, IGF-1 and IGF-2 have additional domains that do not exist in insulin.21,22 IGF-1 is induced in the liver by growth hormone (GH), while IGF-2 seems to be GH independent.23 In animal models, IGF-2 is termed “fetal growth factor”, as IGF-2 expression increases during fetal development and decreases shortly after birth. In contrast, IGF-1 expression is detected in adults and thereby named “adult growth factor.” Both IGF-1 and IGF-2 act in autocrine, paracrine, and endocrine manners.22,24
This developmental difference of expression in IGF-1 and IGF-2 is not the case in humans, as both IGF-1 and IGF-2 are detected in adulthood.24 The IGF-2 gene, however, is different from IGF-1, as it is a maternally imprinted gene in humans and mice.25,26 Through maternal imprinting, heritable epigenetic silencing of genes based on the parent of origin, IGF-2 is monoallelic and expressed by the paternal allele. Loss of this maternal silencing leads to bi-allelic expression, as has been observed in neoplasms such as colorectal cancers and HCC.27–29 The IGF-2 gene is developmentally regulated through highly controlled transcriptional regulation utilizing four distinct promoters (P1 to P4). During fetal development, P2, P3, and P4 are used. In adulthood, the P1 promoter is mostly used and is responsible for approximately 50% of total IGF-2 messenger RNA (mRNA) levels, and the utilization of P2 and P4 is markedly reduced.30 In concert with this tight regulation, a study of a cohort of 104 HCC patients has shown that re-usage of the fetal promoters (P3 and P4) leads to the increased abundance of IGF-2 transcripts.19 In HCC, IGF-2 is more commonly overexpressed than IGF-1, and given the observation that the GH receptor is downregulated in HCC, these studies might explain why the increased abundance of IGF-2, and not IGF-1, is observed in HCC.18,31,32
Receptors
Receptors that mediate IGF signaling are classified as tyrosine kinase receptors. These receptors contain α-subunits (exposed to the extracellular side) and a β-subunit (facing the cytoplasmic side) and include IGF-1R, the insulin receptor, and insulin-related receptors. These receptors are structurally similar and can homodimerize and heterodimerize with each other.16,33 Upon ligand binding, the receptors undergo conformational change resulting in the autophosphorylation of the cytoplasmic β-subunit of the receptors and activation of the insulin-receptor substrate (IRS) 1 and IRS2, and Shc proteins, downstream of the receptors. This process ultimately results in the activation of two major pathways – the PI3K and MAPK – which exert proliferative and survival advantages favoring transformation and cancer development.16
IGF-1R
The expression of IGF-1R in normal hepatocytes is low. The majority of IGF-1R in the liver comes from Kupffer cells, endothelial cells, and stellate cells.34,35 This explains why, in normal physiology, hepatocytes are not sensitive to signaling through IGF-1R and are not the major target of IGF-1.36 However, there is evidence that during the transformation process of hepatocytes into malignant HCC, hepatocytes overexpress IGF-1R.37 IGF-1 and IGF-2 can bind IGF-1R with high affinity.38 Similarly, in animal models of HCC, IGF-1R expression is increased in tumors compared with in preneoplastic lesions.39 Through this overexpression of IGF-1R, HCC cells are able to gain a proliferative advantage and evade apoptosis because IGF-1R blockade exerts an antiproliferative effect and sensitizes cancer cells to apoptosis. These data indicate that excessive signaling through the IGF axis is a critical component in the transformation of preneoplastic cells into malignant HCC.37,40
IGF-2R
IGF-2R, also called mannose-6-phosphate receptor, is a unique single transmembrane protein that binds only to IGF-2 with high affinity. It predominantly binds to mannose-6-phosphate containing proteins and targets them to the lysosomal compartment for degradation.41 Hepatocytes, Kupffer cells, endothelial cells, and stellate cells all express IGF-2R.42 In the liver, IGF-2R binds IGF-2 and facilitates its endocytosis for degradation, thereby maintaining the bioavailability of IGF-2. As a result, a role of IGF-2R as a tumor suppressor has been proposed.40 In HCC, loss of heterozygosity and function mutation in IGF-2R are common events.43–45
IGFBPs
The IGFBP family encompasses six members, IGFBP1–6. Structurally, IGFBPs are clustered together with strong similarity and contain 16–18 conserved cysteines, 10–12 at the N-terminus and six at the C-terminus. At the N-terminus, IGFBPs share a common motif, termed the IGFBP motif (GCGCCXXC). IGFBPs are classified as having a high affinity for binding to IGFs, probably by forming a pocket and both the N-terminus and C-terminus contribute to this high affinity binding.46 Secreted IGFs are generally found in the circulation in a ternary structure bound to IGFBPs and the acid labile subunit (ALS), IGFBP3 being the most abundant IGFBP in the circulation. As a result, the role of IGFBPs is generally thought to limit the bioavailability of IGFs to their cognate receptors. However, some membrane IGFBPs, such as IGFBP5, may enhance the stability of IGFs and their binding to IGF receptors.47 IGFBPs are regulated by proteases that cleave IGFBPs to increase the bioavailability of IGFs.24,46 The role of IGFBP1–6 in HCC is not very well understood; however, there is evidence that IGFBP3 is downregulated by promoter hypermethylation in HCC.48 Moreover, the addition of IGFBP3 mitigates the mitogenic effects of IGF-1 and IGF-2. IGFBPs also have IGF-independent functions; for example, IGFBP3 can still exert antiproliferative effects even after cleavage.46,48–50
IGFBP7
IGFBP7, also known as insulin-like growth factor-binding protein-related protein-1 (IGFBP-rp1), mac25, tumor adhesion factor (TAF), prostacyclin-stimulating factor (PSF), and angiomodulin (AGM), is a secreted protein of a family of low-affinity IGFBPs termed “insulin-like growth-factor binding protein-related proteins” (IGFBP-rp1–10).46,51 The IGFBP7 locus is mapped to chromosome 4q12.52 Mouse IGFBP7 has a 94.4% similarity and 87.5% nucleotide identity to human IGFBP7.53 IGFBP7 shows 40%–45% similarity and 20%–25% identity to other IGFBPs.54 At the N-terminus, IGFBP7 contains the IGFBP motif (GCGCCXXC) in a region of eleven conserved cysteines. IGFBP7 is different from the other IGFBPs in that it lacks the conserved cysteines at the C-terminus and has only one cysteine.54 Moreover, it further differs from the other IGFBPs in that it has 100-times less affinity for binding to IGF-1 and is the only family member that binds insulin with strong affinity, limiting insulin binding to the insulin receptor.55 Additionally, there is evidence of differences in posttranslational modifications of the other IGFBPs compared with IGFBP7. For example, conserved serines (residues 101, 119, and 183), known to enhance growth-factor response to IGF-1 in IGFBP1 via phosphorylation, are not found in IGFBP7. IGFBP7 has 25 serines of which only two are conserved in other IGFBPs. It is worth noting that the ability of IGFBP7 to inhibit insulin binding to its receptor was tested under supraphysiological conditions. The average amount of IGFBP7 in serum is around 33 ng/mL, and insulin binding was analyzed at 100 pmol.54–57 This difference in structure and posttranslational modifications might suggest that IGFBP7 has unique functions that are primarily IGF independent.46
Physiological expression of IGFBP7
IGFBP7 mRNA is expressed in normal tissues of the brain, liver, heart, small intestine, spleen, kidney, placenta, lung, skeletal muscle, thymus, prostate, testis, ovary pancreas, and colon, as detected by northern blotting.58 IGFBP7 protein expression was analyzed by immunohistochemistry using an antibody directed against the decapeptide on the C-terminus. Peripheral nerves, cilia of the respiratory system, epididymis, and fallopian tubes had strong positive staining for IGFBP7.59 Smooth-muscle cells from gut, bladder, prostate, and endothelial cell walls also showed strong staining. While endothelial cells stained positively for IGFBP7, lymphocytes, plasma cells, and fat cells were negatively stained. In the kidney, the epithelia of distal tubules were more strongly stained than those of proximal tubules. Moreover, cells from the zona reticularis and the zona glomerulosa were more strongly stained than cells from the zona fasciculate. In the liver, using serial analysis of gene expression (SAGE), activated stellate cells were found to be the major contributor to the expression of IGFBP7, as seen by the low expression of IGFBP7 in total liver compared with in isolated activated stellate cells.60 This discrete expression of IGFBP7 in tissue and cell specific manner might suggest that IGFBP7 might have specific functions in those organs.59
Protein–protein interactions and cell-surface interactions of IGFBP7
In addition to the IGFBP family, IGFBP7 also shows a strong homology with follistatin, an activin-binding protein, except that IGFBP7 lacks the C-terminus of follistatin.61 IGFBP7 binds to activin A, thus resembling the function of activated follistatin. As a result, IGFBP7 can modulate the growth-suppressing effects of the transforming growth factor (TGF)-β1 superfamily, as seen in the upregulation of IGFBP7 after treatment with TGF-β1 and retinoic acid. However, the mechanism by which IGFBP7 modulates TGF-β effects is not very well understood.58,61,62 IGFBP7 binds to heparin sulfate on the cell surface, and, on cleavage by matriptase, the binding seems to be affected. Proteolytic cleavage at the N-terminus, containing the heparin binding motif, reduces heparin binding and the occupancy of IGF-1R.63–65 Moreover, IGFBP7 has been shown to bind to type IV collagen on the vascular basement membrane.63,64,66 Additionally, in the high endothelial venules (HEVs), IGFBP7 binds with extracellular matrix components; collagen type IV; heparan sulfate, glycosaminoglycans; secondary lymphoid-tissue chemokine (SLC; also known as chemokine [C-C motif] ligand 21 [CCL21]); interferon (INF)-g-inducible protein 10 (IP-10; also known as C-X-C motif chemokine 10 [CXCL10]); and regulated on activation, normal T cell expressed and secreted (RANTES; also known as chemokine [C-C motif] ligand 5 [CCL5]). Through these interactions at HEVs, IGFBP7 might play a role in controlling lymphocyte trafficking to lymph nodes.67 The functional relevance of these interactions in regulating HCC development, progression, and metastasis has not been reported yet.
The role of IGFBP7 in carcinogenesis
The role of IGFBP7 in carcinogenesis was first documented when it was identified as a differentially expressed gene in normal leptomeninges compared with meningiomas and in primary prostate epithelia cell lines compared with prostate cancer cell lines. Multiple studies have reported IGFBP7 downregulation in breast cancer.52,58,68 IGFBP7 is suggested to be a potential tumor suppressor regulating senescence and cell proliferation. Loss of heterozygosity in the IGFBP7 4q12 locus has been reported in breast cancer in more than 50% of cells isolated from tumor tissue compared with normal tissue. This was in parallel with loss of IGFBP7 in invasive carcinoma cells and was associated with poorer prognosis in estrogen receptor-negative breast cancer.69,70 Similarly, a role of IGFBP7 as a tumor suppressor in colorectal and lung cancer has been proposed.71,72 The downregulation of IGFBP7, probably through aberrant DNA methylation, was associated with poorer prognosis.71–74 Using tumor tissue array sections of invasive breast-cancer samples with known cell-cycle aberration status and other clinicopathological data, decreased expression of IGFBP7 in tumor tissue correlated with poor differentiation, increased cyclin E, and inactivation of retinoblastoma protein and showed an inverse association with proliferation marker Ki-67 in estrogen receptor-negative tumors.70 Gene-expression microarray analysis of ductal carcinomas in situ documented a significant downregulation of IGFBP7 in high-grade compared with low-grade ductal carcinomas in situ.75 Serial transplantation of primary breast tumors in nonobese diabetic (NOD)/severe combined immunodeficiency (SCID) mice resulted in progressive reduction of IGFBP7 expression that correlated with the increased growth and aggressiveness of the serially transplanted tumors.76 Overexpression of IGFBP7 in triple-negative breast-cancer cell line (MDA-MB-468) abrogated their growth and migration via inhibition of phosphorylation of MAPK extracellular signal-regulated kinase (ERK)-1/2. When transplanted into NOD/SCID mice, IGFBP7 overexpressing clones had reduced tumor growth compared with controls.76 Treatment of a triple-negative breast-cancer cell line (MDA-MB-231) with recombinant IGFBP7 protein induced G1 cell-cycle arrest and senescence, as indicated by cell-cycle analysis and β-galactosidase staining, respectively. The IGFBP7-induced cell-cycle arrest and senescence was mediated by induction of the p53–p21 axis and the activation of stress-activated p38 MAPK. Tail-vein injection of recombinant IGFBP7 for 3–4 days in NOD/SCID mice harboring xenografted triple-negative breast-cancer tumors resulted in decreased angiogenesis, as shown by a reduction in cluster of differentiation (CD) 31 and vascular endothelial growth factor (VEGF) expression. Recombinant IGFBP7 was able to induce apoptosis as seen by caspase-3 and poly adenosine diphosphate ribose polymerase cleavages.77 Taken together, these results suggest that IGFBP7 could act as a negative regulator of mitogenesis by suppressing ERK-1/2 and activating p38 MAPK and p53-p21 pathways to curb uncontrolled proliferation, induce senescence, and suppress angiogenesis in breast cancer.
Further studies on breast cancer have demonstrated that IGFBP7 occupies and blocks activation of the IGF-1R receptor, limiting its internalization rather than binding to IGF-1/2 or insulin.65 This blockade was mediated by the N-terminus of IGFBP7 and resulted in reduced phosphorylation of IGF-1R and hence activation of the IGF-1R–AKT–mammalian target of rapamycin (mTOR) axis leading to dephosphorylation of 4E-binding protein-1 and activation of its translation-suppressive actions. However, both intact and truncated (lacking N-terminus) IGFBP7 was able to induce apoptosis.65 These data suggest that IGF-1R–AKT–mTOR–4E-BP1 could be an important axis that IGFBP7 interferes with to decrease protein translation, growth, and survival. Additionally, the data also suggest an IGF-IGFR-independent action of IGFBP7 in inducing apoptosis.
In melanocytes, a genome-wide small hairpin RNA screen identified IGFBP7 as an essential gene involved in oncogene-induced senescence (OIS).78 Overexpression of BRAF-V600E, glutamic acid to valine substitution at position 600 (a common mutation in melanomas), initially conferred a proliferative effect on normal melanocytes. However, soon after, activated BRAF induced expression of IGFBP7, which led to OIS. This was further demonstrated in melanocytic nevi, which show increased expression of IGFBP7. Downregulation of IGFBP7 in melanocytic nevi promoted their escape from OIS and facilitated their transformation into melanomas. Treatment of melanomas harboring the BRAF-V600E mutation with recombinant IGFBP7 promoted apoptosis via induction of B-cell lymphoma-2/adenovirus E1B 19 kDa protein-interacting protein 3-like (BNIP3L), a pro-apoptotic B-cell lymphoma-2 family protein. Downregulation of IGFBP7 expression in melanomas was shown to be through increased methylation of the CpG island of the IGFBP7 promoter, a feature also observed in other tumors.78 Intra-tumoral injection of IGFBP7-expressing plasmids inhibited the growth of malignant melanoma in mice via the reduction of VEGF and induction of apoptosis.79 It has also been observed that IGFBP7 might function as a regulator of proliferation in keratinocytes. Knockdown of IGFBP7 using RNA interference in spontaneously immortalized human keratinocytes and primary human keratinocytes increased proliferation and decreased tumor necrosis factor-α-induced apoptosis, and treatment with recombinant IGFBP7 induced apoptosis in keratinocytes.80 Moreover, the downregulation of IGFBP7 was concomitant with the downregulation of genes involved in the calcium-dependent differentiation of keratinocytes.80 These data suggest a potential role of IGFBP7 in regulating cell growth and differentiation.
The role of IGFBP7 in HCC
In our attempt to decipher the function of astrocyte elevated gene-1 (AEG-1), an oncogene overexpressed in >90% of HCC patients, we identified IGFBP7 as one of the most downregulated genes on overexpression of AEG-1. This observation suggested that IGFBP7 downregulation might be required to mediate the tumor-promoting function of AEG-1.81 We documented downregulation of IGFBP7 expression in human HCC patient samples and HCC cell lines compared with normal liver and hepatocytes. Additionally, an inverse correlation between IGFBP7 expression and the grades and stages of HCC was observed (Figure 1A).82 Genomic deletion of IGFBP7 leading to loss of heterozygosity was observed in 26% of patients (Figure 1B). In HCC, downregulation of IGFBP7 expression was associated with hypermethylation of the CpG islands in the IGFBP7 promoter in murine SV40 T/t antigen-induced hepatocarcinogenesis and in human HBV-related HCC.83,84 The HBV-mediated transformation of hepatocytes into HCC is thought to occur at the level of regulation and interaction with a number of genes and signaling pathways that control cell survival and apoptosis.85 For example, hepatitis B virus X (HBx), a viral protein with multifaceted functions, can perturb the expression of a number of genes, including growth-control genes, such as Src tyrosine kinases, Ras, Raf, MAPK, ERK, c-Jun N-terminal kinase, and a number of other genes probably through the inherent capability of HBV to integrate into the genome, which might lead to the upregulation of genes such as oncogenes via strong viral-enhancer elements and the alteration of centrosome replication, leading to genomic instability or the microdeletion of genes such as tumor-suppressor genes.13,86 HBx can bind and nullify the function of p53, a tumor-suppressor gene with multifaceted actions.87 Similarly, HCV can trigger a cascade of events leading to the inactivation of p53 and retinoblastoma, the activation of MAPK, and the increased production of reactive oxygen species. Unlike HBV, HCV does not integrate into the host genome.88,89 Moreover, HBV and HCV have been shown to induce epigenetic changes via the alteration of DNA methylation and histone modifications (reviewed in Herceg and Paliwal).90 The functional link between viral hepatitis and the loss of IGFBP7 has not been established yet. However, it has been observed recently that the frequency of serum IGFBP7-promoter methylation in patients with HBV-induced HCC is significantly increased compared with in chronic hepatitis B patients and healthy controls.84 IGFBP7 is a responsive gene to p53 in colorectal cancer, and it has been shown that p53 binds a p53-responsive element in the intron-1 of the IGFBP7 gene, inducing its expression. This binding is lost in colorectal cancer, probably due to the DNA methylation of CpG island at the promoter region.91 As previously described, IGFBP7 has been shown to interfere with the MAPK-ERK pathway, and its loss was associated with epigenetic alterations or genomic deletions.69,82–84 Thus, it is tempting to hypothesize that HBV or HCV might require the alteration of critical nodes in signaling pathways to facilitate genomic instability and loss of tumor-suppressor genes such as IGFBP7 to facilitate the transformation of hepatocyte and the development of HCC from a chronic disease state.
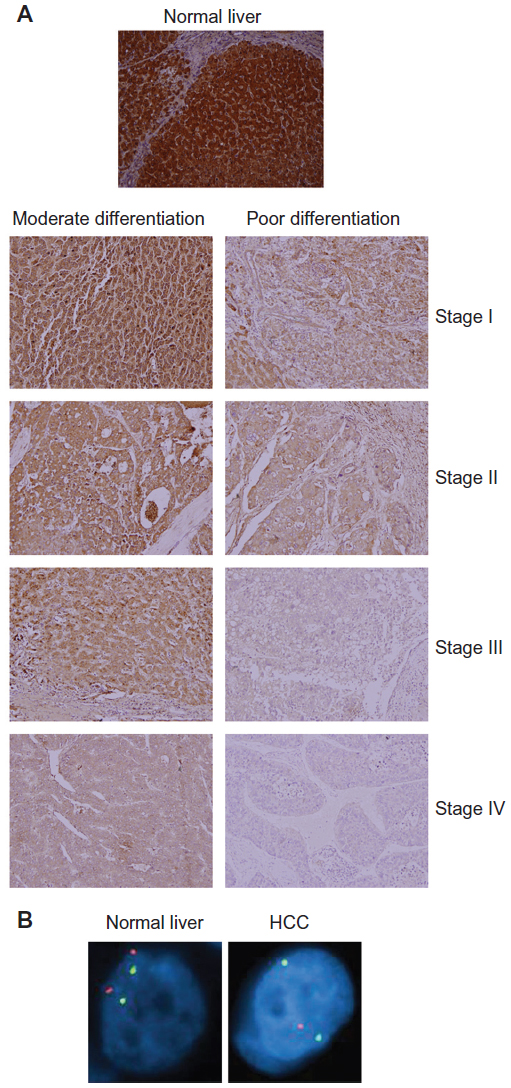 | Figure 1 Insulin-like growth factor-binding protein 7 (IGFBP7) is downregulated in hepatocellular carcinoma (HCC). (A) Tissue microarray using immunohistochemistry examining IGFBP7 expression at/in different stages and grades of HCC. (B) Fluorescent in situ hybridization in normal liver and HCC patient samples showing loss of heterozygosity. Orange, IGFBP7; green, CEP4 (pericentromeric region). Note the loss of one IGFBP7 allele in HCC compared with in normal liver. |
The methylation of IGFBP7 in HCC patients has been associated with vascular invasion.83,84 Recently, it has been shown that reintroducing (SWItch/Sucrose NonFermentable)-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1 (SMARCB1)/SNF, a commonly deleted core subunit of SWI/SNF chromatin remodeling complexes, in malignant rhabdoid tumors increased the expression of IGFBP7.92 SMARCB1/SNF5 directly regulates IGFBP7 and is essential for IGFBP7 expression. Moreover, restoring IGFBP7 expression in SMARCB1/SNF5 null cells sensitized them to apoptosis.92 Whether the downregulation of IGFBP7 by AEG-1 overexpression is mediated by promoter methylation or alteration in chromatin remodeling remains to be determined.
Further studies from our laboratory have confirmed a potential tumor-suppressor role of IGFBP7 in HCC. Stable overexpression of IGFBP7 in AEG-1-overexpressing HCC cell lines abrogated proliferation and colony formation, and induced senescence.82 The effect was more pronounced in vivo in a nude mouse xenograft model, as shown by a marked reduction of tumor growth and Ki-67 staining in IGFBP7-overexpressing clones compared with in controls. The profound inhibitory effect of IGFBP7 in vivo was explained by the inhibition of angiogenesis, as evidenced by a reduction in CD31 staining, decreased blood vessel formation in chick chorioallantoic membrane assays, and abrogated differentiation and tube formation of human umbilical vein endothelial cells.82 An anti-angiogenic effect of IGFBP7 might be conferred by a reduction in phosphorylation of IGF-1R and mitogen-activated protein kinase kinase/ERK-1/2, leading to decreased VEGF expression in human umbilical vein endothelial cells and tumor cells.79,93 A separate study showed decreased IGFBP7 staining in 35.6% of HCC patients (104 patients studied), and this decrease was statistically associated with large tumor size, increased vascular invasion, and poor overall survival and disease-free survival rates.94 Thus, IGFBP7 downregulation might be used as a marker to assess HCC progression.
Potential role of IGFBP7 in cancer therapy
We tested the efficacy of IGFBP7 gene delivery using a replication-incompetent adenovirus (the Ad.5/3 virus) as a potential therapeutic approach for HCC.95 Three different HCC cell lines, differing in their p53 and Rb status, were used in this study – namely, Huh7 (mutated p53 and wildtype Rb), Hep3B (p53 and Rb null), and HepG3 cells (wildtype p53 and Rb). Infection with Ad.IGFBP7 in all cell lines abrogated proliferation and induced apoptosis but not senescence, which might be explained by the accumulation of a large amount of IGFBP7 on Ad.IGFBP7 infection, shifting the balance from senescence to apoptosis. Ad.IGFBP7 infection resulted in the generation of reactive oxygen species; the induction of a DNA damage response, as evidenced by the phosphorylation of Ataxia telangiectasia mutated, Ataxia telangiectasia and Rad3 related, checkpoint kinase-1, checkpoint kinase-2, and p53; and the activation of the p38 MAPK pathway. Treatment with an antioxidant or the inhibition of p38 MAPK provided significant protection against IGFBP7-mediated apoptosis.
The growth-inhibiting effect and apoptosis induction by Ad.IGFBP7 was also demonstrated in vivo. Human HCC xenografts were established orthotopically in nude mouse livers. Intravenous injections of Ad.IGFBP7 at a dose of 1×109 viral particles/injection for a total of five injections over a 2-week period significantly inhibited tumor size and growth and reduced levels of alpha-fetoprotein (Figure 2A). Ad.IGFBP7 did not adversely affect the adjacent normal liver, only the xeno-transplanted tumor that showed hemorrhagic necrosis. Similar effects were also observed when subcutaneous xenografts were implanted in both flanks of nude mice (Figure 2B). Ad.IGFBP7 was injected only on one side and the growth-inhibiting effect and apoptosis induction were evident in both sides, as evidenced by a reduction in Ki-67 and CD31 staining. Moreover, conditioned media from Ad.IGFBP7-infected cells could induce apoptosis in freshly plated human HCC cells.95 These findings suggest that IGFBP7 protein might enter the circulation from the initial site of injection and inhibit the growth of distal tumors (illustrative of metastasis). Thus, Ad.IGFBP7 might be an effective therapy for both primary and metastatic HCC.
 | Figure 2 Ad.IGFBP7 is an effective therapy for hepatocellular carcinoma (HCC). (A) Bioluminescence imaging of nude mice with established orthotopic tumors in the liver using human HCC cells before (left) and after (right) treatment with Ad.IGFBP7. (B) Photograph of nude mice bearing subcutaneous xenografts from human HCC cells on both flanks treated with control Ad.vec (top) or Ad.IGFBP7 (bottom). Only the left-sided tumors were injected with Ad. Note the almost complete disappearance of both left- and right-sided tumors in Ad.IGFBP7-treated mice. |
Targeted molecular therapy has ushered in strategies for the treatment of advanced HCC. For example, sorafenib, a multikinase inhibitor, increased the survival of advanced HCC patients by 2.8 months.3,15,96 Unfortunately, systemic therapy is limited in its efficacy, reinforcing the need for the characterization of additional molecular mechanisms driving the progression of HCC that could be targeted in combination with systemic therapy. A recent study of HCC demonstrated that IGFBP7 mediated sensitivity to IFN-α treatment in HCC.97 IFN-α-resistant HCC cell lines were established by repetitive exposure to IFN-α. Microarray analysis of the IFN-α-resistant cells showed a significant downregulation of IGFBP7. Knocking down IGFBP7 using RNA interference further augmented resistance to IFN-α. In contrast, IGFBP7 overexpression restored sensitivity to IFN-α in resistant cells. The mechanism by which IGFBP7 enhances sensitivity to IFN-α-resistant cells might be via the modulation of apoptosis, although this aspect has not been well studied.97
IGFBP7 is downregulated in hypopharynx cancer cells by low-dose treatment with paclitaxel.98 This finding suggests that a reduction in IGFBP7 might be essential in acquiring resistance to paclitaxel. However, it is worth noting that, in that study, the authors did not establish resistant clones, and the role of IGFBP7 in acquired resistance to paclitaxel requires further investigation.98 In an experimental mouse model of metastatic melanoma, tail-vein injection of recombinant IGFBP7 in athymic nude mice suppressed the growth of metastatic melanoma cells and increased the survival rate of IGFBP7-injected animals.99 These findings further strengthen the notion of using IGFBP7 as a biomarker to predict drug resistance and as a potential target for cancer therapy.
Conclusion
Although the tumor-suppressor function of IGFBP7 is established in diverse cancers, the molecular mechanism by which IGFBP7 exerts its effect remains to be clarified. Is IGF-1 inhibition the primary mechanism of IGFBP7 action? How does IGFBP7 mediate its IGF-independent functions? Is there a cell-surface receptor for IGFBP7 similar to that for IGFBP3?100 What is the consequence of insulin inhibition by IGFBP7? These questions might be addressed by analyzing an IGFBP7 knockout mouse. The efficacy of IGFBP7 as a therapy needs to be stringently evaluated in transgenic mouse models of cancer, either alone or in combination with conventional therapy. The studies so far on IGFBP7 are very promising and future in-depth analysis will establish the utility of this molecule as a biomarker and therapeutic agent.
Acknowledgments
The review was supported in part by the James S McDonnell Foundation and National Cancer Institute Grant number R01 CA138540 (to DS) and developmental funds from the VCU Massey Cancer Center (to DS and PBF).
Disclosure
PBF holds the Thelma Newmeyer Corman Chair in Cancer Research. DS is the Harrison Endowed Scholar in Cancer Research and a Blick Scholar. The authors declare no other conflicts of interest in this work.
References
El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–2576. | |
Carr BI, Pujol L. Pain at presentation and survival in hepatocellular carcinoma. J Pain. 2010;11(10):988–993. | |
El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365(12):1118–1127. | |
Naugler WE, Sakurai T, Kim S, et al. Gender disparity in liver cancer due to sex differences in MyD88-dependent IL-6 production. Science. 2007;317(5834):121–124. | |
Li Z, Tuteja G, Schug J, Kaestner KH. Foxa1 and Foxa2 are essential for sexual dimorphism in liver cancer. Cell. 2012;148(1–2):72–83. | |
Wu MH, Ma WL, Hsu CL, et al. Androgen receptor promotes hepatitis B virus-induced hepatocarcinogenesis through modulation of hepatitis B virus RNA transcription. Sci Transl Med. 2010;2(32):32ra35. | |
Yu MW, Chen CJ. Elevated serum testosterone levels and risk of hepatocellular carcinoma. Cancer Res. 1993;53(4):790–794. | |
American Cancer Society. Cancer Facts and Figures 2012. Atlanta, GA: American Cancer Society; 2012. Available from: http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-031941.pdf. Accessed February 14, 2013. | |
El-Serag HB. Hepatocellular carcinoma and hepatitis C in the United States. Hepatology. 2002;36(5 Suppl 1):S74–S83. | |
Welzel TM, Graubard BI, Zeuzem S, El-Serag HB, Davila JA, McGlynn KA. Metabolic syndrome increases the risk of primary liver cancer in the United States: a study in the SEER-Medicare database. Hepatology. 2011;54(2):463–471. | |
Yang JD, Roberts LR. Hepatocellular carcinoma: A global view. Nat Rev Gastroenterol Hepatol. 2010;7(8):448–458. | |
Armstrong GL, Wasley A, Simard EP, McQuillan GM, Kuhnert WL, Alter MJ. The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med. 2006;144(10):705–714. | |
Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6(9):674–687. | |
Steel JL, Chopra K, Olek MC, Carr BI. Health-related quality of life: Hepatocellular carcinoma, chronic liver disease, and the general population. Qual Life Res. 2007;16(2):203–215. | |
Llovet JM, Ricci S, Mazzaferro V, et al; SHARP Investigators Study Group. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. | |
Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nature Rev Cancer. 2012;12(3):159–169. | |
Steiner DF, Chan SJ, Welsh JM, Kwok SC. Structure and evolution of the insulin gene. Annu Rev Genet. 1985;19:463–484. | |
Scharf JG, Braulke T. The role of the IGF axis in hepatocarcinogenesis. Horm Metab Res. 2003;35(11–12):685–693. | |
Tovar V, Alsinet C, Villanueva A, et al. IGF activation in a molecular subclass of hepatocellular carcinoma and pre-clinical efficacy of IGF-1R blockage. J Hepatol. 2010;52(4):550–559. | |
Lu Y, Zi X, Pollak M. Molecular mechanisms underlying IGF-I-induced attenuation of the growth-inhibitory activity of trastuzumab (Herceptin) on SKBR3 breast cancer cells. Int J Cancer. 2004; 108(3):334–341. | |
Kuemmerle JF. Insulin-like growth factors in the gastrointestinal tract and liver. Endocrinol Metab Clin North Am. 2012;41(2):409–423, vii. | |
Daughaday WH, Rotwein P. Insulin-like growth factors I and II. Peptide, messenger ribonucleic acid and gene structures, serum, and tissue concentrations. Endocr Rev. 1989;10(1):68–91. | |
Goya L, de la Puente A, Ramos S, Martín MA, Escrivá F, Pascual-Leone AM. Regulation of insulin-like growth factor-I and -II by glucose in primary cultures of fetal rat hepatocytes. J Biol Chem. 1999;274(35):24633–24640. | |
LeRoith D, Roberts CT Jr. The insulin-like growth factor system and cancer. Cancer Lett. 2003;195(2):127–137. | |
Giannoukakis N, Deal C, Paquette J, Goodyer CG, Polychronakos C. Parental genomic imprinting of the human IGF2 gene. Nature Genet. 1993;4(1):98–101. | |
Hu JF, Nguyen PH, Pham NV, Vu TH, Hoffman AR. Modulation of Igf2 genomic imprinting in mice induced by 5-azacytidine, an inhibitor of DNA methylation. Mol Endocrinol. 1997;11(13):1891–1898. | |
Cui H, Cruz-Correa M, Giardiello FM, et al. Loss of IGF2 imprinting: a potential marker of colorectal cancer risk. Science. 2003;299(5613):1753–1755. | |
Rainier S, Johnson LA, Dobry CJ, Ping AJ, Grundy PE, Feinberg AP. Relaxation of imprinted genes in human cancer. Nature. 1993;362(6422):747–749. | |
Poirier K, Chalas C, Tissier F, et al. Loss of parental-specific methylation at the IGF2 locus in human hepatocellular carcinoma. J Pathol. 2003;201(3):473–479. | |
Li X, Cui H, Sandstedt B, Nordlinder H, Larsson E, Ekstrom TJ. Expression levels of the insulin-like growth factor-II gene (IGF2) in the human liver: developmental relationships of the four promoters. J Endocrinol. 1996;149(1):117–124. | |
Breuhahn K, Longerich T, Schirmacher P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene. 2006;25(27):3787–3800. | |
Su TS, Liu WY, Han SH, et al. Transcripts of the insulin-like growth factors I and II in human hepatoma. Cancer Res. 1989;49(7):1773–1777. | |
Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocrine Rev. 2009;30(6):586–623. | |
Zindy F, Lamas E, Schmidt S, Kirn A, Brechot C. Expression of insulin-like growth factor II (IGF-II) and IGF-II, IGF-I and insulin receptors mRNAs in isolated non-parenchymal rat liver cells. J Hepatol. 1992;14(1):30–34. | |
Brenzel A, Gressner AM. Characterization of insulin-like growth factor (IGF)-I-receptor binding sites during in vitro transformation of rat hepatic stellate cells to myofibroblasts. Eur J Clin Chem Clin Biochem. 1996;34(5):401–409. | |
Caro JF, Poulos J, Ittoop O, Pories WJ, Flickinger EG, Sinha MK. Insulin-like growth factor I binding in hepatocytes from human liver, human hepatoma, and normal, regenerating, and fetal rat liver. J Clin Invest. 1988;81(4):976–981. | |
Yao WF, Liu JW, Sheng GL, Huang DS. Blockade of IGF-IR exerts anticancer effects in hepatocellular carcinoma. Mol Med Rep. 2011;4(4):719–722. | |
Foulstone E, Prince S, Zaccheo O, et al. Insulin-like growth factor ligands, receptors, and binding proteins in cancer. J Pathol. 2005;205(2):145–153. | |
Scharf JG, Ramadori G, Dombrowski F. Analysis of the IGF axis in preneoplastic hepatic foci and hepatocellular neoplasms developing after low-number pancreatic islet transplantation into the livers of streptozotocin diabetic rats. Lab Invest. 2000;80(9):1399–1411. | |
Kasprzak A, Adamek A. The insulin-like growth factor (IGF) signaling axis and hepatitis C virus-associated carcinogenesis (review). Int J Oncol. 2012;41(6):1919–1931. | |
Massoner P, Ladurner-Rennau M, Eder IE, Klocker H. Insulin-like growth factors and insulin control a multifunctional signalling network of significant importance in cancer. Br J Cancer. 2010;103(10):1479–1484. | |
Novosyadlyy R, Tron K, Dudas J, Ramadori G, Scharf JG. Expression and regulation of the insulin-like growth factor axis components in rat liver myofibroblasts. J Cell Physiol. 2004;199(3):388–398. | |
De Souza AT, Hankins GR, Washington MK, Orton TC, Jirtle RL. M6P/IGF2R gene is mutated in human hepatocellular carcinomas with loss of heterozygosity. Nat Genet. 1995;11(4):447–449. | |
Oka Y, Waterland RA, Killian JK, et al. M6P/IGF2R tumor suppressor gene mutated in hepatocellular carcinomas in Japan. Hepatology. 2002;35(5):1153–1163. | |
Kishnani PS, Chuang TP, Bali D, et al. Chromosomal and genetic alterations in human hepatocellular adenomas associated with type Ia glycogen storage disease. Hum Mol Genet. 2009;18(24):4781–4790. | |
Hwa V, Oh Y, Rosenfeld RG. The insulin-like growth factor-binding protein (IGFBP) superfamily. Endocrine Rev. 1999;20(6):761–787. | |
Firth SM, Baxter RC. Cellular actions of the insulin-like growth factor binding proteins. Endocrine Rev. 2002;23(6):824–854. | |
Hanafusa T, Yumoto Y, Nouso K, et al. Reduced expression of insulin-like growth factor binding protein-3 and its promoter hypermethylation in human hepatocellular carcinoma. Cancer Lett. 2002;176(2):149–158. | |
Aishima S, Basaki Y, Oda Y, et al. High expression of insulin-like growth factor binding protein-3 is correlated with lower portal invasion and better prognosis in human hepatocellular carcinoma. Cancer Sci. 2006;97(11):1182–1190. | |
Jogie-Brahim S, Feldman D, Oh Y. Unraveling insulin-like growth factor binding protein-3 actions in human disease. Endocrine Rev. 2009;30(5):417–437. | |
Shibata Y, Tsukazaki T, Hirata K, Xin C, Yamaguchi A. Role of a new member of IGFBP superfamily, IGFBP-rP10, in proliferation and differentiation of osteoblastic cells. Biochem Biophys Res Commun. 2004;325(4):1194–1200. | |
Swisshelm K, Ryan K, Tsuchiya K, Sager R. Enhanced expression of an insulin growth factor-like binding protein (mac25) in senescent human mammary epithelial cells and induced expression with retinoic acid. Proc Natl Acad Sci U S A. 1995;92(10):4472–4476. | |
Kato MV, Sato H, Tsukada T, Ikawa Y, Aizawa S, Nagayoshi M. A follistatin-like gene, mac25, may act as a growth suppressor of osteosarcoma cells. Oncogene. 1996;12(6):1361–1364. | |
Oh Y, Nagalla SR, Yamanaka Y, Kim HS, Wilson E, Rosenfeld RG. Synthesis and characterization of insulin-like growth factor-binding protein (IGFBP)-7. Recombinant human mac25 protein specifically binds IGF-I and -II. J Biol Chem. 1996;271(48):30322–30325. | |
Yamanaka Y, Wilson EM, Rosenfeld RG, Oh Y. Inhibition of insulin receptor activation by insulin-like growth factor binding proteins. J Biol Chem. 1997;272(49):30729–30734. | |
Vorwerk P, Hohmann B, Oh Y, Rosenfeld RG, Shymko RM. Binding properties of insulin-like growth factor binding protein-3 (IGFBP-3), IGFBP-3 N- and C-terminal fragments, and structurally related proteins mac25 and connective tissue growth factor measured using a biosensor. Endocrinology. 2002;143(5):1677–1685. | |
Kutsukake M, Ishihara R, Momose K, et al. Circulating IGF-binding protein 7 (IGFBP7) levels are elevated in patients with endometriosis or undergoing diabetic hemodialysis. Reprod Biol Endocrinol. 2008;6:54. | |
Hwa V, Tomasini-Sprenger C, Bermejo AL, Rosenfeld RG, Plymate SR. Characterization of insulin-like growth factor-binding protein-related protein-1 in prostate cells. J Clin Endocrinol Metab. 1998;83(12):4355–4362. | |
Degeorges A, Wang F, Frierson HF Jr, Seth A, Sikes RA. Distribution of IGFBP-rP1 in normal human tissues. J Histochem Cytochem. 2000; 48(6):747–754. | |
Boers W, Aarrass S, Linthorst C, Pinzani M, Elferink RO, Bosma P. Transcriptional profiling reveals novel markers of liver fibrogenesis: gremlin and insulin-like growth factor-binding proteins. J Biol Chem. 2006;281(24):16289–16295. | |
Kato MV. A secreted tumor-suppressor, mac25, with activin-binding activity. Mol Med. 2000;6(2):126–135. | |
Matsumoto T, Hess S, Kajiyama H, et al. Proteomic analysis identifies insulin-like growth factor-binding protein-related protein-1 as a podocyte product. Am J Physiol Renal Physiol. 2010;299(4):F776–F784. | |
Sato J, Hasegawa S, Akaogi K, et al. Identification of cell-binding site of angiomodulin (AGM/TAF/Mac25) that interacts with heparan sulfates on cell surface. J Cell Biochem. 1999;75(2):187–195. | |
Ahmed S, Jin X, Yagi M, et al. Identification of membrane-bound serine proteinase matriptase as processing enzyme of insulin-like growth factor binding protein-related protein-1 (IGFBP-rP1/angiomodulin/mac25). FEBS J. 2006;273(3):615–627. | |
Evdokimova V, Tognon CE, Benatar T, et al. IGFBP7 binds to the IGF-1 receptor and blocks its activation by insulin-like growth factors. Science Signal. 2012;5(255):ra92. | |
Ahmed S, Yamamoto K, Sato Y, et al. Proteolytic processing of IGFBP-related protein-1 (TAF/angiomodulin/mac25) modulates its biological activity. Biochem Biophys Res Commun. 2003;310(2):612–618. | |
Nagakubo D, Murai T, Tanaka T, et al. A high endothelial venule secretory protein, mac25/angiomodulin, interacts with multiple high endothelial venule-associated molecules including chemokines. J Immunol. 2003;171(2):553–561. | |
Murphy M, Pykett MJ, Harnish P, Zang KD, George DL. Identification and characterization of genes differentially expressed in meningiomas. Cell Growth Differ. 1993;4(9):715–722. | |
Burger AM, Zhang X, Li H, et al. Down-regulation of T1A12/mac25, a novel insulin-like growth factor binding protein related gene, is associated with disease progression in breast carcinomas. Oncogene. 1998;16(19):2459–2467. | |
Landberg G, Ostlund H, Nielsen NH, et al. Downregulation of the potential suppressor gene IGFBP-rP1 in human breast cancer is associated with inactivation of the retinoblastoma protein, cyclin E overexpression and increased proliferation in estrogen receptor negative tumors. Oncogene. 2001;20(27):3497–3505. | |
Chen Y, Pacyna-Gengelbach M, Ye F, et al. Insulin-like growth factor binding protein-related protein 1 (IGFBP-rP1) has potential tumour-suppressive activity in human lung cancer. J Pathol. 2007;211(4):431–438. | |
Ruan W, Xu E, Xu F, et al. IGFBP7 plays a potential tumor suppressor role in colorectal carcinogenesis. Cancer Biol Ther. 2007;6(3):354–359. | |
Lin J, Lai M, Huang Q, Ma Y, Cui J, Ruan W. Methylation patterns of IGFBP7 in colon cancer cell lines are associated with levels of gene expression. J Pathol. 2007;212(1):83–90. | |
Lin J, Lai M, Huang Q, Ruan W, Ma Y, Cui J. Reactivation of IGFBP7 by DNA demethylation inhibits human colon cancer cell growth in vitro. Cancer Biol Ther. 2008;7(12):1896–1900. | |
Seth A, Kitching R, Landberg G, Xu J, Zubovits J, Burger AM. Gene expression profiling of ductal carcinomas in situ and invasive breast tumors. Anticancer Res. 2003;23(3A):2043–2051. | |
Amemiya Y, Yang W, Benatar T, et al. Insulin like growth factor binding protein-7 reduces growth of human breast cancer cells and xenografted tumors. Breast Cancer Res Treat. 2011;126(2):373–384. | |
Benatar T, Yang W, Amemiya Y, et al. IGFBP7 reduces breast tumor growth by induction of senescence and apoptosis pathways. Breast Cancer Res Treat. 2012;133(2):563–573. | |
Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132(3):363–374. | |
Chen RY, Chen HX, Jian P, et al. Intratumoral injection of pEGFC1-IGFBP7 inhibits malignant melanoma growth in C57BL/6J mice by inducing apoptosis and down-regulating VEGF expression. Oncology Rep. 2010;23(4):981–988. | |
Nousbeck J, Sarig O, Avidan N, et al. Insulin-like growth factor-binding protein 7 regulates keratinocyte proliferation, differentiation and apoptosis. J Invest Dermatol. 2010;130(2):378–387. | |
Yoo BK, Emdad L, Su ZZ, et al. Astrocyte elevated gene-1 regulates hepatocellular carcinoma development and progression. J Clin Invest. 2009;119(3):465–477. | |
Chen D, Yoo BK, Santhekadur PK, et al. Insulin-like growth factor-binding protein-7 functions as a potential tumor suppressor in hepatocellular carcinoma. Clin Cancer Res. 2011;17(21):6693–6701. | |
Komatsu S, Okazaki Y, Tateno M, et al. Methylation and downregulated expression of mac25/insulin-like growth factor binding protein-7 is associated with liver tumorigenesis in SV40T/t antigen transgenic mice, screened by restriction landmark genomic scanning for methylation (RLGS-M). Biochem Biophys Res Commun. 2000;267(1):109–117. | |
Li F, Fan YC, Gao S, Sun FK, Yang Y, Wang K. Methylation of serum insulin-like growth factor-binding protein 7 promoter in hepatitis B virus-associated hepatocellular carcinoma. Genes Chromosomes Cancer. 2014;53(1):90–97. | |
Tang H, Oishi N, Kaneko S, Murakami S. Molecular functions and biological roles of hepatitis B virus x protein. Cancer Sci. 2006;97(10):977–983. | |
Matsuda Y, Ichida T. Impact of hepatitis B virus X protein on the DNA damage response during hepatocarcinogenesis. Medical Mol Morphol. 2009;42(3):138–142. | |
Ueda H, Ullrich SJ, Gangemi JD, et al. Functional inactivation but not structural mutation of p53 causes liver cancer. Nature Genet. 1995;9(1):41–47. | |
Levrero M. Viral hepatitis and liver cancer: the case of hepatitis C. Oncogene. 2006;25(27):3834–3847. | |
Machida K, Cheng KT, Lai CK, Jeng KS, Sung VM, Lai MM. Hepatitis C virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and STAT3 activation. J Virol. 2006;80(14):7199–7207. | |
Herceg Z, Paliwal A. Epigenetic mechanisms in hepatocellular carcinoma: how environmental factors influence the epigenome. Mutation Res. 2011;727(3):55–61. | |
Suzuki H, Igarashi S, Nojima M, et al. IGFBP7 is a p53-responsive gene specifically silenced in colorectal cancer with CpG island methylator phenotype. Carcinogenesis. 2010;31(3):342–349. | |
Darr J, Klochendler A, Isaac S, Eden A. Loss of IGFBP7 expression and persistent AKT activation contribute to SMARCB1/Snf5-mediated tumorigenesis. Oncogene. Epub July 15, 2013. | |
Tamura K, Hashimoto K, Suzuki K, Yoshie M, Kutsukake M, Sakurai T. Insulin-like growth factor binding protein-7 (IGFBP7) blocks vascular endothelial cell growth factor (VEGF)-induced angiogenesis in human vascular endothelial cells. Eur J Pharmacol. 2009;610(1–3):61–67. | |
Tomimaru Y, Eguchi H, Wada H, et al. IGFBP7 downregulation is associated with tumor progression and clinical outcome in hepatocellular carcinoma. Int J Cancer. 2012;130(2):319–327. | |
Chen D, Siddiq A, Emdad L, et al. Insulin-like growth factor-binding protein-7 (IGFBP7): a promising gene therapeutic for hepatocellular carcinoma (HCC). Mol Ther. 2013;21(4):758–766. | |
Cervello M, McCubrey JA, Cusimano A, Lampiasi N, Azzolina A, Montalto G. Targeted therapy for hepatocellular carcinoma: novel agents on the horizon. Oncotarget. 2012;3(3):236–260. | |
Tomimaru Y, Eguchi H, Wada H, et al. Insulin-like growth factor-binding protein 7 alters the sensitivity to interferon-based anticancer therapy in hepatocellular carcinoma cells. Br J Cancer. 2010;102(10):1483–1490. | |
Xu CZ, Shi RJ, Chen D, et al. Potential biomarkers for paclitaxel sensitivity in hypopharynx cancer cell. Int J Clin Exp Pathol. 2013;6(12):2745–2756. | |
Wajapeyee N, Kapoor V, Mahalingam M, Green MR. Efficacy of IGFBP7 for treatment of metastatic melanoma and other cancers in mouse models and human cell lines. Mol Cancer Ther. 2009;8(11):3009–3014. | |
Ingermann AR, Yang YF, Han J, et al. Identification of a novel cell death receptor mediating IGFBP-3-induced anti-tumor effects in breast and prostate cancer. J Biol Chem. 2010;285(39):30233–30246. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.