Back to Journals » OncoTargets and Therapy » Volume 7
Emerging molecular targets in oncology: clinical potential of MET/hepatocyte growth-factor inhibitors
Authors Smyth E, Sclafani F, Cunningham D
Received 9 January 2014
Accepted for publication 14 February 2014
Published 12 June 2014 Volume 2014:7 Pages 1001—1014
DOI https://doi.org/10.2147/OTT.S44941
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Elizabeth C Smyth, Francesco Sclafani, David Cunningham
Department of Gastrointestinal Oncology, Royal Marsden Hospital, Sutton, UK
Abstract: The MET/hepatocyte growth-factor (HGF) signaling pathway plays a key role in the processes of embryogenesis, wound healing, and organ regeneration. Aberrant activation of MET/HGF occurs through multiple mechanisms including gene amplification, mutation, protein overexpression, and abnormal gene splicing interrupting autocrine and paracrine regulatory feedback mechanisms. In many cancers including non-small-cell lung cancer, colorectal, gastric, renal, and hepatocellular cancer, dysregulation of MET may lead to a more aggressive cancer phenotype and may be a negative prognostic indicator. Successful therapeutic targeting of the MET/HGF pathway has been achieved using monoclonal antibodies against the MET receptor and its ligand HGF in addition to MET-specific and multitargeted small-molecule tyrosine-kinase inhibitors with several drugs in late-phase clinical trials including onartuzumab, rilotumumab, tivantinib, and cabozantinib. MET frequently interacts with other key oncogenic tyrosine kinases including epidermal growth-factor receptor (EGFR) and HER-3 and these interactions may be responsible for resistance to anti-EGFR therapies. Similarly, resistance to MET inhibition may be mediated through EGFR activation, or alternatively by increasing levels of MET amplification or acquisition of novel “gatekeeper” mutations. In order to optimize development of effective inhibitors of the MET/HGF pathway clinical trials must be enriched for patients with demonstrable MET-pathway dysregulation for which robustly standardized and validated assays are required.
Keywords: MET, HGF, colorectal cancer, gastric cancer, NSCLC, renal cancer, hepatocellular cancer, onartuzumab, rilotumumab, cabozantinib
MET signaling pathways and function in healthy tissue
The MET proto-oncogene was first identified in a chemically transformed osteosarcoma-derived cell line in 1984, and its protein product was subsequently found to describe a receptor tyrosine kinase the ligand for which was identified as hepatocyte growth factor (HGF; or scatter factor).1–3 Ligand-dependent activation by binding of HGF to MET leads to receptor dimerization and phosphorylation of three kinase-domain tyrosine residues which then initiate the process of autophosphorylation of tyrosine (Tyr) 1349 and Tyr1356 in the bidentate substrate-binding site, facilitating recruitment of cytoplasmic effector proteins and activating transmembrane signaling.4 Downstream signaling effects are transmitted via mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K)/Akt (protein kinase B), signal transducer and activator of transcription proteins (STAT), and nuclear factor-κB.5–7 The final output of the terminal effector components of these pathways is activation of cytoplasmic and nuclear processes leading to increases in cell proliferation, survival and mobilization, and invasive capacity.8
The MET/HGF signaling pathway plays a key role in hepatocyte and placental formation during embryogenesis, and additionally in voluntary muscle and central nervous system formation.9–12 The effects of MET/HGF are critical for wound healing and organ regeneration; signaling through this pathway encourages proliferation of keratinocytes and their mobilization into de-epithelized zones, and increased levels of HGF produced in response to injury by hepatocytes and renal epithelial cells lead to mitotic and antiapoptotic activity.13–15 These constitutive effects of MET on proliferation, apoptosis, and migration are subverted during the process of tumor growth and metastasis, leading to an aggressive MET-addicted tumor phenotype.
MET activation in cancer
Aberrant MET signaling is a hallmark of multiple cancer types, and may occur through gene amplification or mutation, protein overexpression, or abnormal gene splicing, which interrupt normal autocrine and paracrine regulatory feedback mechanisms.6 Missense mutations of MET have been demonstrated in the germ line of families with a history of hereditary papillary renal cell carcinoma (RCC) and in the tumors of a subset of sporadic papillary renal cancers.16 Production of mouse models with an activating mutation replacing endogenous MET yielded diverse cancers, including carcinomas, lymphomas, and sarcomas providing proof of concept of oncogenic activity for the mutated genotype.17 MET amplification on chromosome 7q31 has been described in gastroesophageal, colorectal, and endometrial carcinoma, medulloblastoma, non-small-cell lung cancer (NSCLC), and glioma.18–23 Overexpression of the protein receptor tyrosine kinase is more common than amplification, and has been demonstrated in all tumor types with gene amplification in addition to breast, cervical, head and neck, renal, hepatocellular, melanoma, thyroid, and mesothelioma cancer types.24
MET also interacts with other key oncogenic signaling pathways, in particular HER2 (human epidermal growth factor receptor 2) superfamily members epidermal growth-factor receptor (EGFR) and HER-3. For example, cells that express EGFR and MET demonstrate ligand-independent MET phosphorylation and activation through EGFR, whereas in EGFR-mutant NSCLC, MET amplification leads to escape from gefitinib sensitivity by HER3-mediated activation of PI3K signaling.25,26 In Kirsten rat sarcoma (KRAS) wild-type colorectal cancer cell lines overexpression of the EGFR ligand TGFα (transforming growth factor-α) leads to MET activation and cetuximab resistance, and MET amplification appears to be a resistance mechanism for colorectal cancer patients treated with anti-EGFR antibody therapy.27,28 The MET pathway also increases the malignant potential of tumors through induction of angiogenesis; MET/HGF is a potent inducer of vascular endothelial growth factor (VEGF)-A production and suppressor of thrombspondin-1, and acts synergistically with the VEGF receptor (VEGFR) through common downstream signaling molecules to increase neovascularization activity.7,29 Finally, there appears to be an emerging role for MET/HGF signaling in maintaining the stem cell niche in cancer; Wnt activity in colorectal cancer stem cells has been described to be supported by myofibroblast-secreted HGF.30 These interconnected and diverse functions underlie the key role of the MET/HGF axis in driving tumor growth and supporting an intercellular milieu that is conducive to the metastatic spread of the primary tumor.
Development of MET-inhibitor therapies
Greater understanding of the structure, function, and role of MET/HGF in cancer has led to the development of multiple compounds targeting this pathway. These include monoclonal antibodies targeting the receptor and ligand, and small-molecule tyrosine-kinase inhibitors (TKIs) functional at an intracellular level. Monoclonal antibodies in clinical trials include onartuzumab (MetMab; Roche, Basel, Switzerland), rilotumumab (Amgen, Thousand Oaks, CA, USA) and ficlatuzumab (Aveo Pharmaceuticals, Cambridge, MA, USA). Onartuzumab, a human immunoglobulin (Ig)-G1 antibody with murine variable domains is a potent MET antagonist that competes with HGF for binding at that site.31 Rilotumumab and ficlatuzumab are fully humanized monoclonal anti-HGF antibodies that block HGF binding to MET.32 Onartuzumab and rilotumumab bind to the Sema and SPH (serine protease-homology) domains of MET and HGF respectively, and the monovalent binding design of onartuzumab has been demonstrated to prevent activation of the receptor induced by dimerization which may occur with bivalent antibodies.33
The majority of small-molecule inhibitors of MET may be classified as one of three subtypes each of which impedes adenosine triphosphate (ATP) binding, but of which type II and type III also occupy other distinct binding sites within the MET receptor.6 Most type I inhibitors (eg, crizotinib) preferentially bind to the inactive form of the enzyme and are therefore ineffective against tumors harboring an activating Tyr1230H mutation. Type I inhibitors are most specific for the MET kinase; however, crizotinib has also demonstrated significant efficacy against NSCLC tumors harboring echinoderm microtubule-associated protein-like 4–anaplastic lymphoma kinase (EML4-ALK) fusion and ALK-positive large-cell lymphomas.34,35 Type II inhibitors are more promiscuous: in addition to MET, foretinib is an inhibitor of AXL, RON (Recepteur d’Origine Nantais), VEGFR2, PDGFR (platelet-derived growth-factor receptor-β), and KIT.36,37 Cabozantinib is a multitargeted TKI with activity against MET, VEGFR2, RET, KIT, AXL, tyrosine kinase with immunoglobulin-like and EGF-like domains (TIE)-2 and FLT, and is active against both the active and inactive conformations of MET.38 Tivantinib, which cannot be accurately classified into any of these subtypes, is a non-ATP competitive MET inhibitor that impedes ligand-dependent and -independent activation.39
MET in lung cancer
In NSCLC overexpression of MET occurs in up to 60% of tumor tissues.40 The most common mechanism of MET activation is protein expression secondary to transcriptional upregulation.41 Although less common, amplification and mutation of the MET gene are alternative mechanisms leading to MET activation; MET amplification has been reported in 1%–21% of cases, whereas mutations of the MET gene occur less frequently and usually lead to activation of the HGF/MET pathway through alternative splice forms deleting the juxtamembrane domain.22,42–45 Although MET activation does not appear to be associated with specific clinicopathological characteristics studies have consistently reported an association of both MET amplification/overexpression and intratumoral/plasma HGF levels with poor prognosis.43,46–51 Moreover, the HGF/MET pathway also plays an important role in mediating resistance to EGFR TKIs through the activation of both PI3K/Akt and extracellular signal-regulated kinase (ERK) signaling.52,53 Preclinical data suggest that combining EGFR TKIs and MET inhibitors is a promising strategy to restore gefitinib sensitivity in cell lines.26,54
In recent years, several inhibitors of the HGF/MET pathway have been investigated in NSCLC, largely in combination with EGFR TKIs. Onartuzumab (the monoclonal antibody that competes with HGF for MET binding) has been examined in a randomized Phase II trial of erlotinib ± onartuzumab in EGFR-unselected, chemorefractory, NSCLC patients (Table 1). A total of 137 patients were enrolled, and no differences in survival outcomes were observed in the non-molecularly selected intention-to-treat population.55 However, in a prespecified subgroup analysis of MET-positive patients (n=66), the combination of onartuzumab plus erlotinib was associated with a significant improvement in both progression-free survival (PFS; hazard ratio [HR] 0.53, P=0.04) and overall survival (OS; HR 0.37, P=0.002) compared to erlotinib alone. Outcomes were inferior for MET-negative patients treated with onartuzumab compared to those treated with erlotinib alone (PFS HR 1.71, P=0.06; OS HR 2.61, P=0.004). A randomized Phase III trial of erlotinib ± onartuzumab in MET-positive patients is ongoing.56 In contrast, a trial of ficlatuzumab, the humanized IgG1 antibody, has provided contrasting results in a similar patient population.57 Despite promising preliminary activity observed in Phase I trials, a randomized Phase II trial of gefitinib ± ficlatuzumab in 188 treatment-naïve Asian patients with high incidence of sensitizing EGFR mutations failed to demonstrate superiority of the combination therapy.58–60 PFS was 5.6 months in the combination arm versus 4.7 months in the gefitinib-alone arm. Interestingly, in contrast to the results reported in the trial with onartuzumab, a subgroup analysis showed that the addition of ficlatuzumab to gefitinib seemed to benefit more those patients with low tumoral MET expression (7.3 versus 2.8 months), and this difference appeared to be more pronounced in the presence of tumors with sensitizing EGFR mutations. However, as the patients in this subset analysis were fewer than 20 it is difficult to draw any firm conclusions from these data.
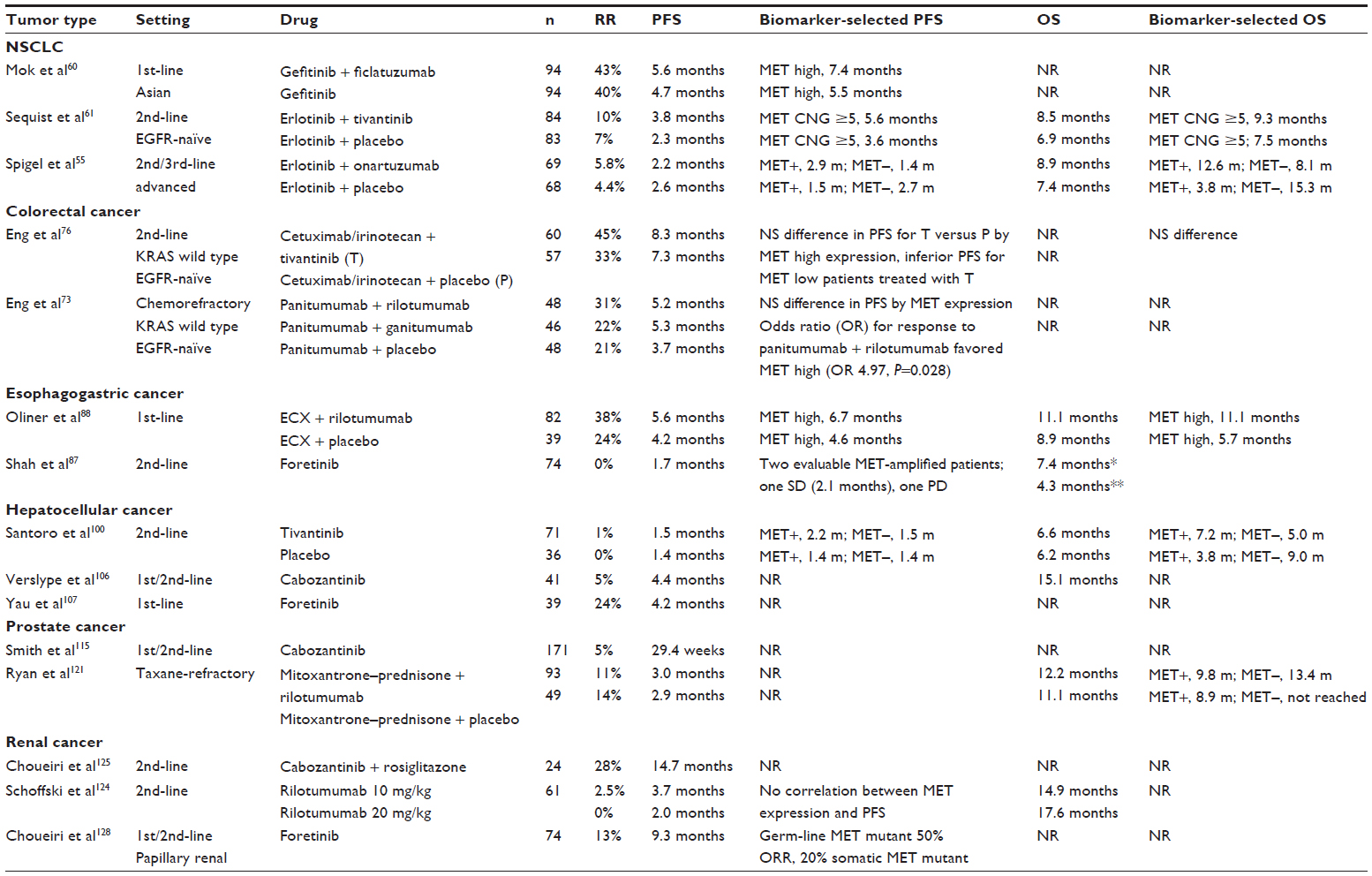 | Table 1 Selected MET-inhibitor clinical trials with biomarker-selected results if available |
Mixed results have also been demonstrated for tivantinib (ARQ 197; ArQule, Woburn, MA, USA), the non-ATP-competitive small-molecule MET inhibitor that exerts its antitumor activity by stabilizing the inactive conformation of MET.39 A randomized Phase II trial of erlotinib with or without tivantinib in 167 previously treated patients with EGFR TKI-naïve NSCLC failed to meet its primary end point of an improvement in PFS in the intention-to-treat non-biomarker-selected patient population.61 In this study, a numerically better but not statistically significant improvement in PFS (3.8 versus 2.3 months, HR 0.81; P=0.24) was reported for the combination therapy. However, in a prespecified proportional hazards model assessing PFS, this difference was found to be statistically significant (HR 0.68, P=0.04). Moreover, in another prespecified exploratory analysis, a significant improvement in PFS was also observed in the group of patients with KRAS-mutant tumors (HR 0.18, P-value for interaction =0.006). Based on the evidence of a trend toward an increased benefit from tivantinib in patients with nonsquamous histology and tumors with high MET gene copy number, a randomized Phase III trial of erlotinib ± tivantinib in pretreated, nonsquamous NSCLC patients was initiated. Although a significant improvement in PFS for the combination treatment was observed, the trial was discontinued early on the advice of the independent data-monitoring committee for futility, as the desired difference in the primary end point of OS would not be reached.24 Other Phase II and III clinical trials investigating tivantinib in combination with erlotinib in NSCLC are ongoing; these include those in the EGFR wild-type62 and KRAS-mutant63 populations.
MET in colorectal cancer
Activation of MET is a common feature in colorectal cancer and upregulation of the HGF/MET pathway, as expressed by MET messenger ribonucleic acid (mRNA) or protein overexpression has been consistently reported in this setting.64–66 In contrast and in keeping with other malignancies, ligand-independent MET activation by amplification or mutation has only been reported in a minority of cases.41,67 In colorectal cancer MET appears to have an important role in tumor progression and has also been reported to be a negative prognostic indicator.23,68–70 Multiple series demonstrate that MET activation in colorectal cancer may provide a selective advantage for the acquisition of an aggressive phenotype that correlates with early stage invasion, liver metastases, and unfavorable clinical outcomes.23,68–71
Preclinical data suggest that HGF-induced MET activation may represent an alternative, RAS-independent mechanism of resistance to cetuximab through the reactivation of the MAPK and Akt pathways. Stimulation with HGF was shown to inhibit the antiproliferative effects of EGFR inhibition, while MET inhibition abrogated this effect.72 These preliminary findings of the importance of MET in resistance to anti-EGFR therapy in colorectal cancer have been confirmed in a recent study where MET amplification emerged as a novel mechanism of both primary and secondary resistance to EGFR-targeted antibodies, possibly accounting for >10% of cetuximab-resistant cases that are wild type for KRAS, BRAF, NRAS (neuroblastoma RAS), PIK3CA (phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha), and HER2.28 Interestingly, MET amplification in patients who progressed on anti-EGFR agents was either a new molecular finding in posttreatment tumor samples or the result of the expansion of a preexisting minor subclonal population of MET-amplified cancer cells under the selective pressure of an EGFR-targeted therapy.
Several MET inhibitors have been tested in colorectal cancer or are currently under investigation; however, most of the available data relate to the monoclonal antibody rilotumumab and the selective, non-ATP-competitive MET TKI tivantinib. In a three-arm, randomized Phase IB/II trial (n=142) of panitumumab (Vectibix®; Amgen) in combination with rilotumumab (anti-HGF antibody), ganitumumab (IGF-1R [insulin-like growth-factor receptor-1 inhibitor]) or placebo, the use of the combination treatment including rilotumumab showed promising activity (overall response rate 31% versus 21%; PFS 5.2 versus 3.7 months) compared to single-agent panitumumab in patients with chemorefractory tumors.73 In a biomarker analysis of 91 of 96 patients allocated to panitumumab ± rilotumumab a correlation between MET expression and activity of rilotumumab was found only when MET-staining intensity (≥2+ versus ≤1+) rather than percentage of MET-positive cells (>50% versus ≤50%) was considered. The anti-MET monoclonal antibody onartuzumab (MetMab) is currently being investigated in a randomized, double-blind, placebo-controlled, Phase II study in conjunction with bevacizumab plus mFOLFOX-6 in chemonaïve metastatic colorectal cancer patients;74 recruitment has completed to this study and results are currently pending.75
As a result of promising signals of activity of the anti-MET TKI tivantinib in a Phase IB study in colorectal cancer where four of nine patients had an objective response, a combination study of irinotecan–cetuximab (Erbitux, Merk-Serono) with or without this drug was investigated in a randomized, Phase II trial in a population of KRAS wild-type metastatic colorectal cancer patients (n=122) who had progressed on or after one line of systemic therapy.76,77 Tivantinib in combination with standard treatment was associated with a higher response rate (45% versus 33.3%) and a slight improvement in PFS (8.3 versus 7.3 months, respectively); however this was not statistically significant (PFS HR 0.85, P=0.38). Surprisingly, but in line with what has been observed with ficlatuzumab in NSCLC, a subgroup analysis conducted in a small number of study patients showed a statistically significant improvement in PFS with tivantinib in patients with low-MET-expressing tumors; however this subgroup contained only 23 patients and requires validation in a larger patient cohort. Tivantinib is currently under investigation in conjunction with cetuximab in a Phase II study in advanced colorectal cancer patients who are refractory to anti-EGFR therapy and who demonstrate high MET (immunohistochemistry [IHC] 2+ or IHC 3+) expression.74
Esophagogastric cancer
The MET pathway is frequently aberrantly activated in gastric carcinoma with evidence of both overexpression and amplification demonstrated in multiple series. Overexpression of MET appears to be more common than amplification, and may be membranous or cytoplasmic. In a recent series of 495 Korean patients membranous and cytoplasmic overexpression of MET was demonstrated in 22% of cases and membranous expression alone in 8%. Simultaneous overexpression correlated well with increased MET mRNA expression, copy-number gain, and amplification, and was prognostic with respect to OS and disease-free survival; however, membranous MET expression alone did not significantly correlate with these end points.78 In another large Asian series (n=438), Lee et al demonstrated a 24% prevalence of overexpression (IHC 2+ and 3+) combined and a 3.4% rate of copy-number gain using silver in situ hybridization. Almost all (>90%) patients with MET IHC 3+ overexpression demonstrated gene amplification, with the remainder demonstrating high gene polysomy.79 Furthermore, all patients with gene amplification demonstrated either IHC 2+ or 3+ overexpression. Therefore it appears that in comparison to overexpression MET amplification in gastric cancer is a relatively infrequent event with rates ranging from 2% to 10%.78–81 Of note, earlier studies examining levels of amplification are likely to have overestimated the incidence of this event due to methodological considerations as these studies frequently used Southern blot or quantitative polymerase chain-reaction techniques which are unable to distinguish between true gene amplification and polysomy, whereas this poses less difficulty with current in situ hybridization techniques.18,19,82
There is particular difficulty in assessing the consistency of measurement of MET overexpression across studies as different investigators have used either varying percentage of cell staining alone as a measure, whereas others have used both percentage of cells and intensity of staining and various composites of these to yield an H-score.83 This is also true of amplification as the level of copy-number gain which has been designated as MET high is between two and five times the level of a reference gene depending on the series.79,80 A recent meta-analysis referencing 15 studies and 2,210 patients attempted to synthesize the prognostic effects of both overexpression and amplification across multiple studies. Both overexpression and amplification were demonstrated to be associated with inferior OS, with HR =2.66 and HR =1.66, respectively.83 This was true for Western and Asian populations, and the prognostic effect of MET was also independent of stage. An additional important consideration when applying these data to potential clinical trial design is the fact that the pattern of MET copy-number alteration in gastric cancer (using high-resolution single-nucleotide polymorphism arrays) appears to be predominantly mutually exclusive of amplification of other relevant receptor tyrosine-kinase genes (FGFR, ERBB2, KRAS, and EGFR).84
Abrogation of MET-pathway signaling in gastric cancer has been successful using both small-molecule TKIs and monoclonal antibody therapy. In the initial Phase I study of tivantinib (the orally available tyrosine kinase MET inhibitor) in a non-molecularly selected population minor regression was noted in a patient with gastric cancer with stable disease for 15 weeks duration.85 Early reports of efficacy of crizotinib in a MET-amplified patient cohort were described by Lennerz et al who reported responses in two of four patients treated with crizotinib in a Phase I trial enriched for MET-amplified patients.81 Additionally, a case report detailing a complete and durable response in a female gastric cancer patient with high MET polysomy and MET overexpression was reported during the Phase I trial of onartuzumab.86 This patient was treated with single-agent onartuzumab at a dose of 20 mg/kg every 3 weeks with a complete response demonstrated following four doses. Unsurprisingly, results of MET inhibition have been less promising in unselected patient populations. Foretinib, a multitargeted TKI targeting MET, RON, AXL, TIE-2, and VEGFR2 failed to demonstrate activity in a largely non-MET-amplified gastric cancer patient population previously treated with chemotherapy.87 In this Phase II study, 69 evaluable patients were treated with foretinib either on an intermittent (240 mg/day for 5 consecutive days every 2 weeks) or daily dosing (80 mg/day during each 2-week cycle) schedule until progression. No patient in either cohort demonstrated a complete or partial response and 23% and 20% of patients in the intermittent and daily dosing cohorts respectively had a best response of stable disease. Three patients in this study were MET-amplified by FISH (fluorescence in situ hybridization): one was unevaluable due to toxicity, one had progressive disease, and one had stable disease of short duration (2.1 months). A Phase II study evaluating the addition of the anti-HGF monoclonal antibody rilotumumab to epirubicin + cisplatin + capecitabine (Xeloda®, Roche) (ECX) chemotherapy in a non-MET-selected population has been reported in abstract form. A total of 121 patients with treatment-naïve advanced gastroesophageal cancer were randomized to ECX chemotherapy plus either placebo or rilotumumab at two dose levels (7.5 mg/kg or 15 mg/kg). In the 90 patients with evaluable MET expression, patients with MET-high tumors (>50% cells with MET expression) had superior survival when treated with rilotumumab than those with MET-low tumors (OS 11.1 versus 5.7 months, HR 0.29; P=0.012). Conversely, patients with MET-low tumors treated with chemotherapy plus rilotumumab had a trend toward worse survival.88 An exposure–response analysis presented at the same meeting demonstrated that increased exposure to rilotumumab in MET-high patients was associated with improvements in PFS and OS in that patient group.89 Both onartuzumab and rilotumumab are currently in global Phase III randomized trials in advanced esophagogastric cancer with MET overexpression: onartuzumab in conjunction with FOLFOX chemotherapy90 and rilotumumab with ECX.91 Multiple MET-targeting TKIs are also currently under evaluation in clinical trials in this setting.
Hepatocellular carcinoma
The MET/HGF pathway has been attributed an important role in the genesis and maintenance of hepatocellular carcinoma, and has emerged as an attractive therapeutic target for this disease. In hepatocellular carcinoma MET overexpression has been reported in 20%–48% of cases.92–94 This phenomenon has not been consistently associated with gene amplification, suggesting that for hepatocellular carcinoma alternative mechanisms including autocrine or paracrine HGF-induced activation or epigenetic regulation of expression may account for a significant number of MET-overexpressing tumors.95,96 In studies investigating the correlation between MET expression and clinicopathological features or clinical outcome in hepatocellular carcinoma MET has been largely shown to correlate with aggressive tumor phenotype and poor survival in both the early stage and advanced setting.97–100 A possible association of MET overexpression with favorable clinical characteristics as suggested by other studies, is likely to be due to the small number of patients analyzed, heterogeneity of the patient populations, or differences in study methodology.96,101
In vitro and in vivo studies demonstrate that MET overexpression is associated with the development of hepatocellular carcinoma, while knockdown of MET leads to the inhibition of tumor growth and regression of advanced tumors.102–104 The promising results observed with MET inhibition in preclinical studies of hepatocellular carcinoma raised interest in assessment of MET as a therapeutic target in the clinical setting, in particular because effective systemic treatment options are limited for patients with this disease.39,103,104 Several selective MET inhibitors are under development and being tested in early stage clinical trials; however tivantinib (ARQ197; Aveo) is the agent with the majority of clinical data available. In a randomized, double-blind, placebo-controlled, crossover Phase II trial, 74 patients with advanced, Child–Pugh A hepatocellular carcinoma previously treated with one systemic therapy were randomly allocated in a 2:1 ratio to receive oral tivantinib or placebo.100 Although clinically marginal, a statistically significant improvement in median time to progression (1.6 versus 1.4 months, HR 0.64; P=0.04) was observed in favor of tivantinib. Importantly, a prespecified subgroup analysis indicated that MET overexpression may represent a potential predictive biomarker for tivantinib benefit as the most clinically and statistically significant tivantinib effects in terms of tumor stabilization (50% versus 20%), time to progression (2.7 versus 1.4 months, HR 0.43; P=0.03) and OS (7.2 versus 3.8 months, HR 0.38; P=0.01) were observed in the group of patients with MET-overexpressing tumors. However, given the modest activity of the drug in the unselected population and the small numbers of patients assessed for MET expression in the subgroup analysis (n=22), confirmatory evidence of clinical benefit will be sought in a Phase III randomized trial comparing tivantinib with placebo in pretreated patients with MET-overexpressing tumors.105
Other multitargeted TKIs with activity against MET have also recently been investigated in hepatocellular carcinoma.106–108 In particular, in a Phase II randomized discontinuation trial cabozantinib (an oral inhibitor of MET and VEGFR2), was investigated in 41 patients with hepatocellular carcinoma half of whom had been previously treated with sorafenib.106 Although only 5% of patients demonstrated a partial response at 12 weeks prior to the randomization, the overall disease-control rate (partial response + stable disease) at this time point was 68%, and 38% of patients with serial α-fetoprotein measurements demonstrated a decline of >50% from baseline. These encouraging results which may in part have been driven also by the antiangiogenic properties of this drug, have led to the development of a large Phase III controlled trial of cabozantinib versus placebo in hepatocellular carcinoma patients previously treated with sorafenib.109 The monoclonal antibody onartuzumab is also being investigated in conjunction with sorafenib in the first line setting for patients with hepatocellular carcinoma.110
Prostate cancer
MET expression in prostate cancer is associated with high-grade tumors and the presence of metastases, in particular bone metastases, and in prostate cancer cell lines MET expression is inversely correlated with expression of the androgen receptor.111,112 The androgen receptor has been demonstrated to be a negative regulator of MET, and accordingly the effect of small-molecule MET inhibitors has been demonstrated to be more potent in androgen-insensitive prostate cancer cells.113,114 Cabozantinib, an inhibitor of MET, VEGFR, and multiple other tyrosine kinases, was investigated in a randomized discontinuation study in advanced castration-resistant prostate cancer at a dose of 100 mg daily; patients with stable disease by response-evaluation criteria in solid tumors (RECIST) at 12 weeks were randomized to cabozantinib or placebo.115 Recruitment was halted following enrollment of 171 patients due to efficacy in the experimental arm of the trial. Although the overall response rate at 12 weeks was 5%, an additional 75% of patients had stable disease, of whom 31 were randomized at week 12. PFS was 23.9 weeks for men treated with cabozantinib, and 5.9 weeks for those receiving placebo (HR 0.12, P<0.001). Bone pain and narcotic use were also significantly decreased in the majority of patients. Dose reductions were frequent (51% at 12 weeks) in this initial study and a subsequent dose-ranging study demonstrated superior tolerability and comparable efficacy for a 40 mg daily dose which was recommended for subsequent randomized clinical trials.115,116
Significant resolution of bone lesions on bone scan has been a notable effect of cabozantinib in prostate cancer trials; it has recently been demonstrated that in addition to direct cytotoxic effects on prostate cancer cells, cabozantinib has an inhibitory effect on osteoclast production and a biphasic dose-dependent effect on osteoblast activity both mediated through MET and VEGFR signaling.117 Therefore, the effects of cabozantinib on bone scintigraphy are as a result of cytotoxicity in addition to direct effects on bone remodeling.
Cabozantinib is currently under investigation in several large randomized studies in metastatic castration-resistant prostate cancer in previously treated patients118,119 and in combination with abiraterone in patients who are treatment-naïve.120 However, the addition of rilotumumab to mitoxantrone and prednisolone therapy in metastatic castration-resistant prostate cancer patients previously treated with docetaxel did not result in any improvements in PFS or OS when compared to standard therapy (PFS 3.0 versus 2.9 months, OS 12.2 versus 11.1 months, respectively), including in MET-high (n=38) patients.121 Therefore it was not recommended that rilotumumab proceed to a Phase III trial in this setting.
Renal cell carcinoma
The MET pathway is activated through at least two separate mechanisms in RCC of distinct histological subtypes. In clear-cell RCC inactivation of the VHL gene is common, and preclinical data suggest that this may induce constitutive phosphorylation of MET leading to enhanced cell mobilization and invasive capacity.122 MET expression is common in RCC and associated with a negative prognosis; in a recent study examining MET expression on 330 RCC cores, expression was highest in papillary and sarcomatoid subtypes and those with a higher Fuhrman grade but was also present on clear-cell RCC, and in an analysis limited to clear-cell subtypes remained a negative prognostic marker.123 In MET-activated clear-cell RCC cell lines treatment with tivantinib led to inhibition of cell proliferation providing a clinical rationale for targeting MET-activated clear-cell RCC with these agents.
A Phase II study with the anti-HGF monoclonal antibody rilotumumab was conducted in 61 patients with metastatic RCC of varying histologies (clear-cell 75.4%, papillary 11.5%), the majority of whom had previously received antiangiogenic therapy.124 Although one partial response was maintained for 2.5 years no other responses were seen, median PFS was 3.7 months at 10 mg/kg and 2.0 months at 20 mg/kg rilotumumab doses and tumoral MET expression was not associated with response or survival outcomes. As a result, further development of rilotumumab has not been pursued in this disease.
The antiangiogenic properties of the TKI cabozantinib make this an attractive agent for treatment of RCC. Promising results in clear-cell RCC patients were seen in a drug–drug interaction study examining the effects of rosiglitazone on cabozantinib pharmacokinetics; of 25 patients treated with a median of two prior treatments, 24% had a confirmed partial response by RECIST, and 86% experienced some tumor regression.125 These encouraging results have led to the development of multiple clinical trials investigating cabozantinib in clear-cell RCC: in comparison to everolimus in a Phase II randomized study for patients who have previously progressed following TKI therapy,126 and in comparison to sunitinib in previously untreated patients.127
A second mechanism of MET activation is seen in the papillary subtype of renal cancer, with activating mutations of MET found in the germ line of families with hereditary papillary RCC and in a proportion of sporadic noninherited cases. In a nonrandomized study assessing the effect of the nonselective MET inhibitor foretinib 74 patients with papillary RCC were recruited, eleven of whom had germ-line and five of whom had somatic MET mutations.128 Two patients demonstrated MET amplification with no mutation. Median PFS was 9.3 months and 1-year survival was 70% with median OS not reached. Of the ten patients with a germ-line mutation, half had a partial response and half had stable disease, whereas only one of five patients with a somatic mutation had a response and no MET amplified patient did. Although the trial failed to meet its primary end point of a response rate of >25% the response rate in germ-line-mutant patients is noteworthy, and MET inhibition would appear to be worthwhile in this patient group.
Toxicity of MET inhibition
The extracellular inhibitors of the MET pathway (onartuzumab, rilotumumab, and ficlatuzumab) appear to be well tolerated, with relatively few treatment-related serious adverse events reported in clinical trials to date. In the Phase I studies for both onartuzumab and rilotumumab, the maximum tolerated dose was not reached.129 Peripheral edema appears to be a class effect of these compounds, and increased rates of neutropenia have been demonstrated when rilotumumab is used in conjunction with chemotherapy.88 Activation of the MET pathway has been associated with dysregulation of the clotting cascade in preclinical models; however, with the caveat of relatively small control groups treated to date, significant differences in the incidence of thromboembolic disease have not been noted with these drugs.130 Class-effect toxicities associated with nonselective tyrosine kinase inhibition (fatigue, gastrointestinal upset, hepatotoxicity) are frequent but usually mild.87,115 However, awareness of toxicity related to off-target effects, such as those on VEGFR (hypertension, hemorrhage, perforation), is also necessary, as these may be significant.115 Additionally, tivantinib appears to have cytotoxic effects that are independent of its MET-inhibitory activity, and significant rates of neutropenia and neutropenia-related deaths have been documented with the use of this compound.100,131
Resistance to MET inhibition
Acquisition of novel mutations, redundancy in intracellular signaling pathways, and downregulation of inhibitory feedback mechanisms have been demonstrated to be responsible for de novo and acquired resistance to other TKIs, such as those inhibiting EGFR, BRAF and mitogen-activated protein-kinase kinase (MEK). The mechanisms by which resistance to MET inhibition may occur have recently begun to emerge, and preeminent among these is the interplay between the MET and the EGFR pathways. In MET-amplified gastric cancer lines treated with the MET inhibitor PHA-665752 EGF and heregulin-dependent activation of EGFR and HER3 led to downstream effects on the MAPK and PI3K pathways and abrogation of the effects of MET inhibition.132 However, combined blockade of MET and EGFR using gefitinib or with MEK and Akt inhibitors led to reversal of MET resistance. In a separate experiment, resistance to MET therapy in SNU6838 cells was mediated through TGFα expression and EGFR activation.133 Similarly, activation of the EGFR pathway has been demonstrated to be responsible for acquired resistance to the MET inhibitor PF2341066 in MET-amplified NSCLC lines and while combination therapy with PF2341066 and the EGFR inhibitor erlotinib did not result in decreased cell proliferation, it did suppress emergence of MET resistance.134
Alternative escape mechanisms from MET inhibition include increased amplification of MET, acquisition of mutations affecting binding-site conformation, and upregulation of non-EGFR-signaling pathways. In MET-amplified gastric (GTL16) and NSCLC (EBC-1) cell lines when initially sensitive cells were treated with either of two MET inhibitors (PHA-665752 or JNJ38877605), the MET gene acquired further amplification with subsequent increased levels of protein expression leading to adequate levels of phosphorylation which successfully maintained enzymatic activity.135 However, at higher levels of drug which overcame the increased MET amplification, amplification and overexpression of KRAS emerged and this remained sensitive to downstream inhibition of MAPK elements using U0126 and PD0325901. An additional pathway by which MET amplified gastric cancer cell line GTL16-acquired resistance to MET inhibition with PF04217903 is the emergence of a novel SND1 (staphylococcal nuclease domain 1)–BRAF fusion protein that uses activated BRAF to escape MET suppression.136 Again, the activity of this resistance mechanism could be suppressed through combined MET and BRAF or MEK inhibition. Further evidence of the efficacy of combination therapy in overcoming resistance is demonstrated by NSCLC cell lines resistant to erlotinib and the MET inhibitor SU11274, which display upregulation of both mTOR (mammalian target of rapamycin) and Wnt pathway components and restoration of sensitivity to EGFR/MET inhibition by the addition of everolimus.137
A final mechanism of resistance in gastric cancer cell lines has been demonstrated when MET-amplified SNU6838 gastric cancer cell lines were treated with the MET inhibitors PHA-665752 and PF2341066; a novel mutation occurred in the activation loop of MET, causing a conformal change that blocked inhibitor binding analogous to the gatekeeper mutations seen in EGFR (T790M) following erlotinib treatment and in ABL (T315I) following imatinib.133 Although the MET Y1230H mutation renders cancers insensitive to type I MET inhibitors, conformal differences between these and type II compounds may allow treatment of MET Y1230H mutant cancers or prevent the emergence of resistance due to the mutation.138,139
Conclusion and future directions
The ubiquity of MET-pathway activation in cancer and the malignant phenotype that it confers on MET-mutated, -amplified, or -overexpressed tumors ensure that this is an attractive therapeutic target for many cancers. Pharmacological inhibition of this pathway has clear benefits in terms of response and survival, albeit in limited numbers to date. It is clear that to optimize these benefits clinical trials must be enriched for patients with demonstrable MET-pathway dysregulation; what is less clear is the best means by which to achieve this. Robust standardization and validation of assay methodology for MET expression is essential in order to confidently address the benefit of MET inhibition across distinct patient populations, and assessment of the correlation between gene amplification, protein expression, and treatment efficacy is also mandated. With respect to clinical trial development, treatment with anti-MET/HGF antibodies and chemotherapy and/or other antibodies appears to be an attractive option given the lack of significant additive toxicities seen for combination regimens, whereas the small-molecule TKIs may potentially be combined with other similar drugs targeting other relevant pathways. These combinatorial approaches may be designed in order to delay or prevent the emergence of resistance to MET inhibition via intimately connected pathways, such as EGFR, HER3, and RAS. Ultimately, collaborative clinical trials and serial tissue collection will be required in order to fully evaluate the impact of inhibition of this promising target on oncology outcomes.
Acknowledgment
We acknowledge support from the National Institute for Health Research Royal Marsden/Institute for Cancer Research Biomedical Research Centre.
Disclosure
Dr Smyth and Dr Sclafani declare no relevant conflicts. Professor Cunningham has received research funding from Roche, Amgen, Celgene, Sanofi, Merck Serono, Novartis, and Astra Zeneca.
References
Cooper CS, Park M, Blair DG, et al. Molecular cloning of a new transforming gene from a chemically transformed human cell line. Nature. 1984;311(5981):29–33. | |
Giordano S, Ponzetto C, Di Renzo MF, Cooper CS, Comoglio PM. Tyrosine kinase receptor indistinguishable from the c-met protein. Nature. 1989;339(6220):155–156. | |
Naldini L, Vigna E, Narsimhan RP, et al. Hepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the proto-oncogene MET. Oncogene. 1991;6(4):501–504. | |
Ponzetto C, Bardelli A, Zhen Z, et al. A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell. 1994;77(2):261–271. | |
Sipeki S, Bander E, Buday L, et al. Phosphatidylinositol 3-kinase contributes to Erk1/Erk2 MAP kinase activation associated with hepatocyte growth factor-induced cell scattering. Cell Signal. 1999;11(12):885–890. | |
Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12(2):89–103. | |
Zhang YW, Wang LM, Jove R, Vande Woude GF. Requirement of Stat3 signaling for HGF/SF-Met mediated tumorigenesis. Oncogene. 2002;21(2):217–226. | |
Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11(12):834–848. | |
Schmidt C, Bladt F, Goedecke S, et al. Scatter factor/hepatocyte growth factor is essential for liver development. Nature. 1995;373(6516):699–702. | |
Uehara Y, Minowa O, Mori C, et al. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature. 1995;373(6516):702–705. | |
Maina F, Hilton MC, Ponzetto C, Davies AM, Klein R. Met receptor signaling is required for sensory nerve development and HGF promotes axonal growth and survival of sensory neurons. Genes Dev. 1997;11(24):3341–3350. | |
Bladt F, Riethmacher D, Isenmann S, Aguzzi A, Birchmeier C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature. 1995;376(6543):768–771. | |
Chmielowiec J, Borowiak M, Morkel M, et al. c-Met is essential for wound healing in the skin. J Cell Biol. 2007;177(1):151–162. | |
Huh CG, Factor VM, Sánchez A, Uchida K, Conner EA, Thorgeirsson SS. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci U S A. 2004;101(13):4477–4482. | |
Liu Y. Hepatocyte growth factor in kidney fibrosis: therapeutic potential and mechanisms of action. Am J Physiol Renal Physiol. 2004;287(1):F7–F16. | |
Schmidt L, Duh FM, Chen F, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997;16(1):68–73. | |
Graveel CR, London CA, Vande Woude GF. A mouse model of activating Met mutations. Cell Cycle. 2005;4(4):518–520. | |
Nakajima M, Sawada H, Yamada Y, et al. The prognostic significance of amplification and overexpression of c-met and c-erb B-2 in human gastric carcinomas. Cancer. 1999;85(9):1894–1902. | |
Kuniyasu H, Yasui W, Kitadai Y, Yokozaki H, Ito H, Tahara E. Frequent amplification of the c-met gene in scirrhous type stomach cancer. Biochem Biophys Res Commun. 1992;189(1):227–232. | |
Fischer U, Müller HW, Sattler HP, Feiden K, Zang KD, Meese E. Amplification of the MET gene in glioma. Genes Chromosomes Cancer. 1995;12(1):63–65. | |
Samuelson E, Levan K, Adamovic T, Levan G, Horvath G. Recurrent gene amplifications in human type I endometrial adenocarcinoma detected by fluorescence in situ hybridization. Cancer Genet Cytogenet. 2008;181(1):25–30. | |
Beau-Faller M, Ruppert AM, Voegeli AC, et al. MET gene copy number in non-small cell lung cancer: molecular analysis in a targeted tyrosine kinase inhibitor naïve cohort. J Thorac Oncol. 2008;3(4):331–339. | |
Zeng ZS, Weiser MR, Kuntz E, et al. c-Met gene amplification is associated with advanced stage colorectal cancer and liver metastases. Cancer Lett. 2008;265(2):258–269. | |
Scagliotti GV, Novello S, von Pawel J. The emerging role of MET/HGF inhibitors in oncology. Cancer Treat Rev. 2013;39(7):793–801. | |
Dulak AM, Gubish CT, Stabile LP, Henry C, Siegfried JM. HGF-independent potentiation of EGFR action by c-Met. Oncogene. 2011; 30(33):3625–3635. | |
Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043. | |
Troiani T, Martinelli E, Napolitano S, et al. Increased TGF-α as a mechanism of acquired resistance to the anti-EGFR inhibitor cetuximab through EGFR–MET interaction and activation of MET signaling in colon cancer cells. Clin Cancer Res. 2013;19(24):6751–6765. | |
Bardelli A, Corso S, Bertotti A, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 2013;3(6):658–673. | |
Sulpice E, Ding S, Muscatelli-Groux B, et al. Cross-talk between the VEGF-A and HGF signalling pathways in endothelial cells. Biol Cell. 2009;101(9):525–539. | |
Vermeulen L, De Sousa EMF, van der Heijden M, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Bioly. 2010;12(5):468–476. | |
Jin H, Yang R, Zheng Z, et al. MetMAb, the one-armed 5D5 anti-c-Met antibody, inhibits orthotopic pancreatic tumor growth and improves survival. Can Res. 2008;68(11):4360–4368. | |
Burgess TL, Sun J, Meyer S, et al. Biochemical characterization of AMG 102: a neutralizing, fully human monoclonal antibody to human and nonhuman primate hepatocyte growth factor. Mol Cancer Ther. 2010;9(2):400–409. | |
Merchant M, Ma X, Maun HR, et al. Monovalent antibody design and mechanism of action of onartuzumab, a MET antagonist with anti-tumor activity as a therapeutic agent. Proc Natl Acad Sci U S A. 2013;110(32):E2987–E2996. | |
Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013; 368(25):2385–2394. | |
Mosse YP, Lim MS, Voss SD, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013;14(6):472–480. | |
Qian F, Engst S, Yamaguchi K, et al. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res. 2009;69(20):8009–8016. | |
Eder JP, Shapiro GI, Appleman LJ, et al. A phase I study of foretinib, a multi-targeted inhibitor of c-Met and vascular endothelial growth factor receptor 2. Clin Cancer Res. 2010;16(13):3507–3516. | |
Yakes FM, Chen J, Tan J, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. 2011;10(12):2298–2308. | |
Munshi N, Jeay S, Li Y, et al. ARQ 197, a novel and selective inhibitor of the human c-Met receptor tyrosine kinase with antitumor activity. Mol Cancer Ther. 2010;9(6):1544–1553. | |
Ichimura E, Maeshima A, Nakajima T, Nakamura T. Expression of c-met/HGF receptor in human non-small cell lung carcinomas in vitro and in vivo and its prognostic significance. Jpn J Cancer Res. 1996;87(10):1063–1069. | |
Smolen GA, Sordella R, Muir B, et al. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc Natl Acad Sci U S A. 2006;103(7):2316–2321. | |
Onozato R, Kosaka T, Kuwano H, Sekido Y, Yatabe Y, Mitsudomi T. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J Thorac Oncol. 2009;4(1):5–11. | |
Cappuzzo F, Janne PA, Skokan M, et al. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann Oncol. 2009;20(2):298–304. | |
Ma PC, Kijima T, Maulik G, et al. MET mutational analysis in small cell lung cancer: novel juxtamembrane domain mutations regulating cytoskeletal functions. Cancer Res. 2003;63(19):6272–6281. | |
Boland JM, Jang JS, Li J, et al. MET and EGFR mutations identified in ALK-rearranged pulmonary adenocarcinoma: molecular analysis of 25 ALK-positive cases. J Thorac Oncol. 2013;8(5):574–581. | |
Okuda K, Sasaki H, Yukiue H, Yano M, Fujii Y. Met gene copy number predicts the prognosis for completely resected non-small cell lung cancer. Cancer Sci. 2008;99(11):2280–2285. | |
Lim EH, Zhang SL, Li JL, et al. Using whole genome amplification (WGA) of low-volume biopsies to assess the prognostic role of EGFR, KRAS, p53, and CMET mutations in advanced-stage non-small cell lung cancer (NSCLC). J Thorac Oncol. 2009;4(1):12–21. | |
Siegfried JM, Weissfeld LA, Singh-Kaw P, Weyant RJ, Testa JR, Landreneau RJ. Association of immunoreactive hepatocyte growth factor with poor survival in resectable non-small cell lung cancer. Cancer Res. 1997;57(3):433–439. | |
Onitsuka T, Uramoto H, Ono K, et al. Comprehensive molecular analyses of lung adenocarcinoma with regard to the epidermal growth factor receptor, K-ras, MET, and hepatocyte growth factor status. J Thorac Oncol. 2010;5(5):591–596. | |
Hosoda H, Izumi H, Tukada Y, et al. Plasma hepatocyte growth factor elevation may be associated with early metastatic disease in primary lung cancer patients. Ann Thorac Cardiovasc Surg. 2012;18(1):1–7. | |
Ujiie H, Tomida M, Akiyama H, et al. Serum hepatocyte growth factor and interleukin-6 are effective prognostic markers for non-small cell lung cancer. Anticancer Res. 2012;32(8):3251–3258. | |
Turke AB, Zejnullahu K, Wu YL, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17(1):77–88. | |
Yano S, Yamada T, Takeuchi S, et al. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a Japanese cohort. J Thorac Oncol. 2011;6(12):2011–2017. | |
Zucali PA, Ruiz MG, Giovannetti E, et al. Role of cMET expression in non-small-cell lung cancer patients treated with EGFR tyrosine kinase inhibitors. Ann Oncol. 2008;19(9):1605–1612. | |
Spigel DR, Ervin TJ, Ramlau RA, et al. Randomized phase II trial of onartuzumab in combination with erlotinib in patients with advanced non-small-cell lung cancer. J Clin Oncol. November 10, 2013;31(32):4105–4114. | |
Genentech. A Study of Onartuzumab (MetMAb) in Combination With Tarceva (Erlotinib) in Patients With Met Diagnostic-Positive Non-Small Cell Lung Cancer Who Have Received Chemotherapy For Advanced or Metastatic Disease (MetLung). Available from: http://clinicaltrials.gov/show/NCT01456325. NLM identifier: NCT01456325. Accessed March 5, 2014. | |
Elez E, Tabernero J, Prudkin L. Pharmacodynamic (PD)–pharmacokinetic (PK) study of ficlatuzumab (F), a monoclonal antibody (MAB) directed to the hepatocyte growth factor (HGF), in patients (Pts) with advanced solid tumors who have live metastases (Mets). Ann Oncol. 2012; (23 Suppl 9):443PD. | |
Meetze K, Boudrow A, Connoly K. Anti-tumor activity of SCH 900105 (AV299), an anti-HGF antibody, in non-small cell lung cancer models. Mol Cancer Ther. 2009;8 Suppl 12:C173. | |
Patnaik A, Weiss GJ, Papadopoulos K, et al. Phase I study of SCH 900105 (SC), an anti-hepatocyte growth factor (HGF) monoclonal antibody (MAb), as a single agent and in combination with erlotinib (E) in patients (pts) with advanced solid tumors. J Clin Oncol. 2010; 28 Suppl 15:2525. | |
Mok T, Park K, Geater SL. A randomized phase (Ph) 2 study with exploratory biomarker analysis of ficlatuzumab (F) a humanized hepatocyte growth factor (HGF) inhibitory MAB in combination with gefitinib (G) versus G in Asian patients (pts) with lung adenocarcinoma (LA). Ann Oncol. 2012;23 Suppl 9:1198P. | |
Sequist LV, von Pawel J, Garmey EG, et al. Randomized phase II study of erlotinib plus tivantinib versus erlotinib plus placebo in previously treated non-small-cell lung cancer. J Clin Oncol. 2011;29(24):3307–3315. | |
Kyowa Hakko Kirin Company, Limited. A Phase 3, Randomized, Double-Blinded, Placebo-Controlled Study of ARQ 197 Plus Erlotinib Versus Placebo Plus Erlotinib (ATTENTION). Available from: http://clinicaltrials.gov/ct2/show/NCT01377376. NLM identifier: NCT01377376. Accessed March 5, 2014. | |
ArQule. Erlotinib Plus Tivantinib (ARQ 197) Versus Single Agent Chemotherapy in Locally Advanced or Metastatic Non-Small Cell Lung Cancer. Available from: http://clinicaltrials.gov/ct2/show/NCT01395758. NLM identifier: NCT01395758. Accessed March 5, 2014. | |
Liu C, Park M, Tsao MS. Overexpression of c-met proto-oncogene but not epidermal growth factor receptor or c-erbB-2 in primary human colorectal carcinomas. Oncogene. 1992;7(1):181–185. | |
Fujita S, Sugano K. Expression of c-met proto-oncogene in primary colorectal cancer and liver metastases. Jpn J Clin Oncol. 1997;27(6):378–383. | |
Hiscox SE, Hallett MB, Puntis MC, Nakamura T, Jiang WG. Expression of the HGF/SF receptor, c-met, and its ligand in human colorectal cancers. Cancer Invest. 1997;15(6):513–521. | |
Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–337. | |
Fazekas K, Csuka O, Köves I, Rásó E, Tímár J. Experimental and clinicopathologic studies on the function of the HGF receptor in human colon cancer metastasis. Clin Exp Metastasis. 2000;18(8):639–649. | |
Takeuchi H, Bilchik A, Saha S, et al. MET expression level in primary colon cancer: a predictor of tumor invasion and lymph node metastases. Clin Cancer Res. 2003;9(4):1480–1488. | |
Kammula US, Kuntz EJ, Francone TD, et al. Molecular co-expression of the c-Met oncogene and hepatocyte growth factor in primary colon cancer predicts tumor stage and clinical outcome. Cancer Lett. 2007;248(2):219–228. | |
Lee CT, Chow NH, Su PF, Lin SC, Lin PC, Lee JC. The prognostic significance of RON and MET receptor coexpression in patients with colorectal cancer. Dis Colon Rectum. 2008;51(8):1268–1274. | |
Liska D, Chen CT, Bachleitner-Hofmann T, Christensen JG, Weiser MR. HGF rescues colorectal cancer cells from EGFR inhibition via MET activation. Clin Cancer Res. 2011;17(3):472–482. | |
Eng C, Van Cutsem E, Nowara E, et al. A randomized, phase Ib/II trial of rilotumumab (AMG 102; ril) or ganitumab (AMG 479; gan) with panitumumab (pmab) versus pmab alone in patients (pts) with wild-type (WT) KRAS metastatic colorectal cancer (mCRC): Primary and biomarker analyses. J Clin Oncol. 2011;29 Suppl:3500. | |
Genentech. FOLFOX/Bevacizumab With Onartuzumab (MetMAb) Versus Placebo as First-Line Treatment in Patients With Metastatic Colorectal Cancer. Available from: http://clinicaltrials.gov/ct2/show/NCT01418222. NLM identifier: NCT01418222. Accessed March 5, 2014. | |
Bendell JC, Ervin TJ, Gallinson D, et al. Treatment rationale and study design for a randomized, double-blind, placebo-controlled phase II study evaluating onartuzumab (MetMAb) in combination with bevacizumab plus mFOLFOX-6 in patients with previously untreated metastatic colorectal cancer. Clin Colorectal Cancer. 2013;12(3):218–222. | |
Eng C, Hart LL, Severtsev A, et al. A randomized, placebo-controlled, phase I/II study of tivantinib (ARQ 197) in combination with cetuximab and irinotecan in patients (pts) with KRAS wild-type (WT) metastatic colorectal cancer (CRC) who had received previous front-line systemic therapy. J Clin Oncol. 2013;Suppl 31:3508. | |
Eng C, Bessudo A, Gabrail N. Phase I/II study of tivantinib (ARQ197), irinotecan, and cetuximab in patients with KRAS wild type, previously treated, metastatic colorectal cancer patients. Ann Oncol. 2012;23 Suppl 4:PD-0018. | |
Ha SY, Lee J, Kang SY, et al. MET overexpression assessed by new interpretation method predicts gene amplification and poor survival in advanced gastric carcinomas. Mod Pathol. 2013;26(12):1632–1641. | |
Lee HE, Kim MA, Lee HS, et al. MET in gastric carcinomas: comparison between protein expression and gene copy number and impact on clinical outcome. Br J Cancer. 2012;107(2):325–333. | |
Graziano F, Galluccio N, Lorenzini P, et al. Genetic activation of the MET pathway and prognosis of patients with high-risk, radically resected gastric cancer. J Clin Oncol. 2011;29(36):4789–4795. | |
Lennerz JK, Kwak EL, Ackerman A, et al. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J Clin Oncol. 2011;29(36):4803–4810. | |
Tsujimoto H, Sugihara H, Hagiwara A, Hattori T. Amplification of growth factor receptor genes and DNA ploidy pattern in the progression of gastric cancer. Virchows Arch. 1997;431(6):383–389. | |
Yu S, Yu Y, Zhao N, Cui J, Li W, Liu T. c-Met as a prognostic marker in gastric cancer: a systematic review and meta-analysis. PloS One. 2013;8(11):e79137. | |
Deng N, Goh LK, Wang H, et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut. 2012;61(5):673–684. | |
Yap TA, Olmos D, Brunetto AT, et al. Phase I trial of a selective MET inhibitor ARQ 197 incorporating proof of mechanism pharmacodynamic studies. J Clin Oncol. 2011;29(10):1271–1279. | |
Catenacci DV, Henderson L, Xiao SY, et al. Durable complete response of metastatic gastric cancer with anti-Met therapy followed by resistance at recurrence. Cancer Discov. 2011;1(7):573–579. | |
Shah MA, Wainberg ZA, Catenacci DV, et al. Phase II study evaluating 2 dosing schedules of oral foretinib (GSK1363089), cMET/VEGFR2 inhibitor, in patients with metastatic gastric cancer. PloS One. 2013;8(3):e54014. | |
Oliner KS, Tang R, Anderson A, et al. Evaluation of MET pathway biomarkers in a phase II study of rilotumumab (R, AMG 102) or placebo (P) in combination with epirubicin, cisplatin, and capecitabine (ECX) in patients (pts) with locally advanced or metastatic gastric (G) or esophagogastric junction (EGJ) cancer. J Clin Oncol. 2012;30 Suppl:4005. | |
Zhu M, Tang R, Doshi S, et al. Exposure-response (E-R) analysis of rilotumumab (R, AMG 102) plus epirubicin/cisplatin/capecitabine (ECX) in patients (pts) with locally advanced or metastatic gastric or esophagogastric junction (G/EGJ) cancer. J Clin Oncol. 2012; 30 Suppl:2535. | |
Hoffmann-La Roche. A Study of Onartuzumab (MetMAb) in Combination With mFOLFOX6 in Patients With Metastatic HER2-Negative And Met-Positive Gastroesophageal Cancer (MetGastric). Available from: http://clinicaltrials.gov/ct2/show/NCT01662869. NLM identifier: NCT01662869. Accessed March 5, 2014. | |
Amgen. First-Line Treatment for Locally Advanced or Metastatic Mesenchymal Epithelial Transition Factor (MET) - Positive Gastric, Lower Esophageal, or Gastroesophageal Junction (GEJ) Adenocarcinoma (RILOMET-1). Available from: http://clinicaltrials.gov/ct2/show/NCT01697072. NLM identifier: NCT01697072. Accessed March 5, 2014. | |
Kiss A, Wang NJ, Xie JP, Thorgeirsson SS. Analysis of transforming growth factor (TGF)-alpha/epidermal growth factor receptor, hepatocyte growth factor/c-met, TGF-beta receptor type II, and p53 expression in human hepatocellular carcinomas. Clin Cancer Res. 1997;3(7):1059–1066. | |
Ljubimova JY, Petrovic LM, Wilson SE, Geller SA, Demetriou AA. Expression of HGF, its receptor c-met, c-myc, and albumin in cirrhotic and neoplastic human liver tissue. J Histochem Cytochem. 1997;45(1):79–87. | |
Okano J, Shiota G, Kawasaki H. Expression of hepatocyte growth factor (HGF) and HGF receptor (c-met) proteins in liver diseases: an immunohistochemical study. Liver. 1999;19(2):151–159. | |
Kondo S, Ojima H, Tsuda H, et al. Clinical impact of c-Met expression and its gene amplification in hepatocellular carcinoma. Int J Clin Oncol. 2013;18(2):207–213. | |
Lee SJ, Lee J, Sohn I, et al. A Survey of MET expression and amplification in 287 patients with hepatocellular carcinoma. Anticancer Res. 2013;33(11):5179–5186. | |
Osada S, Kanematsu M, Imai H, Goshima S. Clinical significance of serum HGF and c-Met expression in tumor tissue for evaluation of properties and treatment of hepatocellular carcinoma. Hepatogastroenterology. 2008;55(82–83):544–549. | |
Ke AW, Shi GM, Zhou J, et al. Role of overexpression of CD151 and/or c-Met in predicting prognosis of hepatocellular carcinoma. Hepatology. 2009;49(2):491–503. | |
Wang ZL, Liang P, Dong BW, Yu XL, Yu de J. Prognostic factors and recurrence of small hepatocellular carcinoma after hepatic resection or microwave ablation: a retrospective study. J Gastrointest Surg. 2008;12(2):327–337. | |
Santoro A, Rimassa L, Borbath I, et al. Tivantinib for second-line treatment of advanced hepatocellular carcinoma: a randomised, placebo-controlled phase 2 study. Lancet Oncol. 2013;14(1):55–63. | |
Ang CS, Sun MY, Huitzil-Melendez DF, et al. MET and HGF mRNA expression in hepatocellular carcinoma: correlation with clinicopathological features and survival. Anticancer Res. 2013;33(8):3241–3245. | |
Horiguchi N, Takayama H, Toyoda M, et al. Hepatocyte growth factor promotes hepatocarcinogenesis through c-Met autocrine activation and enhanced angiogenesis in transgenic mice treated with diethylnitrosamine. Oncogene. 2002;21(12):1791–1799. | |
Zhang SZ, Pan FY, Xu JF, et al. Knockdown of c-Met by adenovirus-delivered small interfering RNA inhibits hepatocellular carcinoma growth in vitro and in vivo. Mol Cancer Ther. 2005;4(10):1577–1584. | |
You H, Ding W, Dang H, Jiang Y, Rountree CB. c-Met represents a potential therapeutic target for personalized treatment in hepatocellular carcinoma. Hepatology. 2011;54(3):879–889. | |
Daiichi Sankyo Inc. Study of Tivantinib in Subjects With Inoperable Hepatocellular Carcinoma Who Have Been Treated With One Prior Therapy (METIV-HCC). Available from: http://clinicaltrials.gov/ct2/show/NCT01755767. NLM identifier: NCT01755767. Accessed March 5, 2014. | |
Verslype C, Cohn AL, Kelley RK, et al. Activity of cabozantinib (XL184) in hepatocellular carcinoma: results from a phase II randomized discontinuation trial (RDT). J Clin Oncol. 2012;30 Suppl:4007. | |
Yau TC, Sukeepaisarnjaroen W, Chao Y, et al. A phase I/II study of foretinib, an oral multikinase inhibitor targeting MET, RON, AXL, TIE-2, and VEGFR in advanced hepatocellular carcinoma (HCC). J Clin Oncol. 2012;30 Suppl:4108. | |
O’Neil BH, Bendell JC, Modiano MR, et al. Phase I/II study of E7050 (golvantinib) in combination with sorafenib in patients (pts) with advanced hepatocellular carcinoma (HCC): Phase I results. J Clin Oncol. 2012;30 Suppl 34:294. | |
Exelixis. Study of Cabozantinib (XL184) vs Placebo in Subjects With Hepatocellular Carcinoma Who Have Received Prior Sorafenib (CELESTIAL). Available from: http://clinicaltrials.gov/ct2/show/NCT01908426. NLM identifier: NCT01908426. Accessed March 5, 2014. | |
Hoffmann-La Roche. A Study of Onartuzumab as Single Agent and in Combination With Sorafenib in Patients With Advanced Hepatocellular Carcinoma. Available from: http://clinicaltrials.gov/ct2/show/NCT01897038. NLM identifier: NCT01897038. Accessed March 5, 2014. | |
Knudsen BS, Gmyrek GA, Inra J, et al. High expression of the Met receptor in prostate cancer metastasis to bone. Urology. 2002;60(6):1113–1117. | |
Pisters LL, Troncoso P, Zhau HE, Li W, von Eschenbach AC, Chung LW. C-met proto-oncogene expression in benign and malignant human prostate tissues. J Urol. 1995;154(1):293–298. | |
Tu WH, Zhu C, Clark C, Christensen JG, Sun Z. Efficacy of c-Met inhibitor for advanced prostate cancer. BMC Cancer. 2010;10:556. | |
Verras M, Lee J, Xue H, Li TH, Wang Y, Sun Z. The androgen receptor negatively regulates the expression of c-Met: implications for a novel mechanism of prostate cancer progression. Cancer Res. 2007;67(3):967–975. | |
Smith DC, Smith MR, Sweeney C, et al. Cabozantinib in patients with advanced prostate cancer: results of a phase II randomized discontinuation trial. J Clin Oncol. 2013;31(4):412–419. | |
Lee RJ, Saylor PJ, Michaelson MD, et al. A dose-ranging study of cabozantinib in men with castration-resistant prostate cancer and bone metastases. Clin Cancer Res. 2013;19(11):3088–3094. | |
Dai J, Zhang H, Karatsinides A, et al. Cabozantinib inhibits prostate cancer growth and prevents tumor-induced bone lesions. Clin Cancer Res. 2014;20(3):617–630. | |
Exelixis. Study of Cabozantinib (XL184) Versus Prednisone in Men With Metastatic Castration-resistant Prostate Cancer Previously Treated With Docetaxel and Abiraterone or MDV3100 (COMET-1). Available from: http://clinicaltrials.gov/ct2/show/NCT01605227. NLM identifier: NCT01605227. Accessed March 5, 2014. | |
Exelixis. Study of Cabozantinib (XL184) Versus Mitoxantrone Plus Prednisone in Men With Previously Treated Symptomatic Castration-resistant Prostate Cancer (COMET-2). Available from: http://clinicaltrials.gov/ct2/show/NCT01522443. NLM identifier: NCT01522443. Accessed March 5, 2014. | |
Exelixis. Study of Cabozantinib in Combination With Abiraterone in Chemotherapy-Naïve Subjects With Bone-Metastatic Castration-Resistant Prostate Cancer. Available from: http://clinicaltrials.gov/ct2/show/NCT01995058. NLM identifier: NCT01995058. Accessed March 5, 2014. | |
Ryan CJ, Rosenthal M, Ng S, et al. Targeted MET inhibition in castration-resistant prostate cancer: a randomized phase II study and biomarker analysis with rilotumumab plus mitoxantrone and prednisone. Clin Cancer Res. 2013;19(1):215–224. | |
Nakaigawa N, Yao M, Baba M, et al. Inactivation of von Hippel-Lindau gene induces constitutive phosphorylation of MET protein in clear cell renal carcinoma. Cancer Res. 2006;66(7):3699–3705. | |
Gibney GT, Aziz SA, Camp RL, et al. c-Met is a prognostic marker and potential therapeutic target in clear cell renal cell carcinoma. Ann Oncol. 2013;24(2):343–349. | |
Schoffski P, Garcia JA, Stadler WM, et al. A phase II study of the efficacy and safety of AMG 102 in patients with metastatic renal cell carcinoma. BJU Int. 2011;108(5):679–686. | |
Choueiri TK, Pal SK, McDermott DF, et al. Activity of cabozantinib (XL184) in patients (pts) with metastatic, refractory renal cell carcinoma (RCC). J Clin Oncol. 2012;30 Suppl 5:364. | |
Exelixis. A Study of Cabozantinib (XL184) vs Everolimus in Subjects With Metastatic Renal Cell Carcinoma (METEOR). Available from: http://clinicaltrials.gov/ct2/show/NCT01865747. NLM identifier: NCT01865747. Accessed March 5, 2014. | |
National Cancer Institute (NCI). Cabozantinib-s-malate or Sunitinib Malate in Treating Patients With Previously Untreated Locally Advanced or Metastatic Kidney Cancer. Available from: http://clinicaltrials.gov/ct2/show/NCT01835158. NLM identifier: NCT01835158. Accessed March 5, 2014. | |
Choueiri TK, Vaishampayan U, Rosenberg JE, et al. Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J Clin Oncol. 2013;31(2):181–186. | |
Gordon MS, Sweeney CJ, Mendelson DS, et al. Safety, pharmacokinetics, and pharmacodynamics of AMG 102, a Fully human hepatocyte growth factor-neutralizing monoclonal antibody, in a first-in-human study of patients with advanced solid tumors. Clin Cancer Res. 2010;16(2):699–710. | |
Boccaccio C, Sabatino G, Medico E, et al. The MET oncogene drives a genetic programme linking cancer to haemostasis. Nature. 2005;434(7031):396–400. | |
Basilico C, Pennacchietti S, Vigna E, et al. Tivantinib (ARQ197) displays cytotoxic activity that is independent of its ability to bind MET. Clin Cancer Res. 2013;19(9):2381–2392. | |
Bachleitner-Hofmann T, Sun MY, Chen CT, et al. HER kinase activation confers resistance to MET tyrosine kinase inhibition in MET oncogene-addicted gastric cancer cells. Mol Cancer Ther. 2008;7(11):3499–3508. | |
Qi J, McTigue MA, Rogers A, et al. Multiple mutations and bypass mechanisms can contribute to development of acquired resistance to MET inhibitors. Cancer Res. 2011;71(3):1081–1091. | |
McDermott U, Pusapati RV, Christensen JG, Gray NS, Settleman J. Acquired resistance of non-small cell lung cancer cells to MET kinase inhibition is mediated by a switch to epidermal growth factor receptor dependency. Cancer Res. 2010;70(4):1625–1634. | |
Cepero V, Sierra JR, Corso S, et al. MET and KRAS gene amplification mediates acquired resistance to MET tyrosine kinase inhibitors. Cancer Res. 2010;70(19):7580–7590. | |
Lee NV, Lira ME, Pavlicek A, et al. A novel SND1-BRAF fusion confers resistance to c-Met inhibitor PF-04217903 in GTL16 cells through [corrected] MAPK activation. PloS One. 2012;7(6):e39653. | |
Fong JT, Jacobs RJ, Moravec DN, et al. Alternative signaling pathways as potential therapeutic targets for overcoming EGFR and c-Met inhibitor resistance in non-small cell lung cancer. PloS One. 2013;8(11):e78398. | |
Pan B-S, Chan GKY, Chenard M, et al. MK-2461, a novel multitargeted kinase inhibitor, preferentially inhibits the activated c-Met receptor. Cancer Res. 2010;70(4):1524–1533. | |
Bellon SF, Kaplan-Lefko P, Yang Y, et al. c-Met inhibitors with novel binding mode show activity against several hereditary papillary renal cell carcinoma-related mutations. J Biol Chem. 2008;283(5):2675–2683. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.