Back to Journals » Therapeutics and Clinical Risk Management » Volume 12
Emerging approaches in Parkinson’s disease – adjunctive role of safinamide
Authors Müller T ![]()
Received 15 May 2016
Accepted for publication 9 July 2016
Published 2 August 2016 Volume 2016:12 Pages 1151—1160
DOI https://doi.org/10.2147/TCRM.S86393
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Garry Walsh
Thomas Müller
Department of Neurology, Alexianer St Joseph Hospital Berlin-Weißensee, Berlin, Germany
Abstract: Ongoing neuronal death in Parkinson’s disease (PD) causes an altered neurotransmission of various biogenic amines, particularly dopamine. As these changes do not follow a distinct pattern, they vary individually, and are differently pronounced. As a result, a heterogeneous onset of motor and nonmotor features occurs in each patient with PD during the whole course of the disease. PD actually describes a set of distinct diseases that manifest themselves in clinical syndromes with certain similarities but also great differences. This clinical picture responds to drugs with a broad spectrum of modes of actions better than to compounds with an exclusive focus on specific receptor subtypes. Therefore, safinamide is an ideal candidate for treatment of patients with PD, since its pharmacological profile includes reversible monoamine oxidase-B inhibition, blockade of voltage-dependent sodium channels, modulation of calcium channels, and inhibition of glutamate release. Safinamide is applied only once daily. Its oral dose ranges from 50 to 100 mg. Safinamide was well tolerated and safe in the clinical development program that demonstrated the amelioration of motor symptoms and OFF phenomena by safinamide when combined with dopamine agonists or levodopa. In the real world of maintenance of patients with PD, effects of safinamide application resemble therapy with classical monoamine oxidase inhibitors or amantadine in combination with other dopamine-substituting drugs. Safinamide is becoming increasingly available in the EU despite complex approval and pricing scenarios.
Keywords: safinamide, MAO-B inhibition, glutamate release inhibition, Parkinson’s disease, dopamine substitution, glutamate
Introduction
The second-most frequent chronic neurodegenerative disease is Parkinson’s disease (PD). The incidence rate of PD ranges from 8 to 18 per 100,000 persons each year. Onset of PD is less under age 50 and considerably rises after age 60.1,2 Earlier epidemiologic studies showed a decline of life expectancy, which was then improved with the introduction of better therapeutic options, particularly l-3,4-dihydroxyphenylalanine (levodopa). This amino acid is looked upon as the most effective drug therapy for PD. A decreasing death rate in the PD patient population was found during the initial 15 years of levodopa prescription. During this interval, levodopa was administered without enzyme blockers, which slow levodopa turnover. High dosing of levodopa was needed owing to rapid degradation of levodopa via dopa decarboxylase to dopamine and, to a lesser extent, via catechol-O-methyltransferase (COMT) to 3-O-methyldopa.3 Then dopa decarboxylase inhibitors (DDIs), such as benserazide or carbidopa, were added to levodopa formulations. DDIs impair peripheral levodopa conversion to dopamine and support the levodopa delivery to the brain, since only levodopa but not dopamine transpasses the blood–brain barrier. To date, this levodopa/DDI combination is the most frequently employed levodopa regimen despite the introduction of COMT inhibitors in the meantime. However, between 1976 and 2011, an increase of 328.7% in the general death rate of PD patients was again noticed, when compared with the first interval of levodopa therapy without DDIs use.3 This phenomenon is not fully understood yet. Nevertheless, PD patients have a life span similar to that of the normal population. PD may considerably limit quality of life of patients and their caregivers. As an example, consequences of falls due to impaired motor behavior or pneumonia as a result of swallowing disturbances may temporarily cause situations with reduced quality of life for PD patients.1,2 The term “PD” actually describes a set of distinct diseases that manifest themselves in clinical syndromes with certain similarities but also great differences. The onset of various combinations of rigidity, akinesia, and resting tremor is still looked upon as the main diagnostic phenomenon for this disease entity. Balance problems mostly appear later in the course of the disease. A wide array of unspecific nonmotor features often precedes an initial temporary onset of motor symptoms. Better awareness of nonmotor signs of PD and initiatives for earlier detection of PD gain more and more interest for a better characterization of this so-called “prodromal” or “premotor” interval of PD.4–6 This research strategy will hopefully ease the development of future disease-modifying treatment strategies or even cure.
Heterogeneity of etiology, progress, and therapeutic response
Chronic neuronal death causes an individual different and heterogeneous neurotransmission with a deficit of biogenic amines in various brain areas of PD patients. The progress of degeneration takes place in a nonlinear fashion. The etiology of PD is still unknown and probably of multifactorial origin. A genetic cause occurs only in approximately 5% of all cases.7 The nigrostriatal dopamine deficit is mainly responsible for the initial onset of noticeable symptoms of disturbed motor behavior. A rough estimate is that approximately 70% of neuronal dopamine-synthesizing neurons is already gone at that moment. However, it is also unknown how many of these neurons are permanently lost to death or just perform hibernation of function owing to reversible neuronal injury related to intracellular α-synuclein overload. Nevertheless, clinical diagnosis is made relatively late in the course of PD. Upregulated formation of Lewy bodies that contain proteins, ie, ubiquitin and α-synuclein, are major neuropathological hallmarks. This process of wrong folding of proteins may result from the disease process itself or from unspecific defense mechanisms in still healthy neurons, which are able to eliminate these wrongly folded proteins by a packaging procedure. Many reports also describe microglia activation leading to inflammation with concomitant release of a series of pro-inflammatory and neurotoxic molecules in combination with free radical exposure as an essential component of the apoptotic suicide program of neurons. The heterogeneity of neuronal death mechanisms is probably one of many reasons why real groundbreaking, efficacious disease-modifying therapies are not available yet.8
Current treatment of PD
There are effective symptomatic therapies, which mainly balance the nigrostriatal dopamine deficiency. This deficit is predominantly responsible for the onset of akinesia, rigidity, and tremor, to a lesser extent. Disturbed postural reflexes respond only to specific preventive and symptomatic training programs. An improvement of motor behavior also ameliorates the severity of nonmotor features. The response to therapy also individually varies. Generally, treatment possibilities are satisfying for motor symptoms in the early stage, but not when PD progresses to more advanced stages. It is well accepted that brain delivery of dopamine-replacing drugs should be as continuous as possible. This concept ameliorates the reappearance of motor symptoms or so-called “OFF” phenomena, when the effect of the applied dopamine-substituting drug combination starts to vane. It also prevents overstimulation of the dopaminergic system, which results in onset of involuntary movements or so-called dyskinesia. These fluctuations of movements, characterized by OFF states and dyskinesia, are summarized as motor complications. Intervals with good movement behavior are also termed “ON” times. These periods are characterized by the reappearance of motor symptoms. Onset of dyskinesia, which appears mostly during ON intervals and is subdivided into “troublesome” and quality of life limiting and “non troublesome” with slight severity, is more bothersome for the caregivers than the patients. OFF states are more a serious, not well-tolerated problem for patients, whereas carers or family members are only bothered to a lesser extent.9 Severe motor complications respond to pump systems with continuous dopaminergic drug application or deep brain stimulation (DBS) to a certain extent. Both techniques were developed for advanced PD patients. There is some evidence from the biochemical investigations that DBS only enhances the endogenous synthesis and continuous release of biogenic amines. Increased levels of the biogenic amine derivative homovanillic acid were found during chronic DBS.10,11 Thus, hypothetically, DBS effects resemble one of compounds, which improve PD symptoms owing to their broad spectrum of modes of actions. The continuous electric stimulation in certain nuclei of the basal ganglia network shares the effects of pump systems, which provide a continuous stimulation of dopamine receptors by the applied dopamine-substituting drug. Therefore, it is no big surprise that DBS reduces the amount of dopamine-substituting compounds necessary for an adequate therapy of disturbed motor behavior, diminishes the intensity of motor complications, and improves motor symptoms. DBS does not provide benefit for balance problems in PD.8
General issues for established and emerging drug approaches in PD
The focus on motor behavior for drug development in PD in the past 40 years finally came to a standstill. This interval was bridged by the development of sometimes very detailed and bureaucratic treatment guidelines, summarized under the term “evidence-based medicine”. All of them have one common disadvantage. They neglect the heterogeneity of PD in terms of diagnosis, treatment, and progression, and are misinterpreted or even misused by the authorities when they decide on the value of new compounds ignoring the opinions of clinicians dealing with PD patients in daily clinical practice.12 This arbitrary approval procedure with its focus on motor behavior is somehow fatal for future innovative therapies in PD. However, clinicians already realized that this too specific focus on certain dopamine receptor subtypes or on drugs with only one mechanism of action aiming at a certain molecular structure will fail in the real world. Mostly, experimental research demonstrates positive outcomes in certain well-accepted animal models of PD within a standardized study design. However, these results are not of high value in their daily clinical practice for the treatment of PD patients. Maintenance of PD patients is complex and demands an individually adapted, careful and cautious titration, and recurrent adaptation of antiparkinsonian drug regimes to the ongoing disease process.13 To date, nearly all the treatment concepts of PD have only one thing in common: They emphasize that therapy should start once the diagnosis of PD has been established. Moreover, an optimized therapeutic regime prevents adaptation of the human body to features of PD. Certain features of PD, such as bound posture, reduced swinging of arms, walking with small steps, speaking in a low voice, and so on, are at least partially aggravated by an unconscious learning process. As a result, nonpharmacological approaches, so-called activating therapies, such as physiotherapy, occupational therapy, or speech therapy, gain increasing importance and, essentially, supplement drug therapy. Research on standardized training methods to overcome these PD-related deficits of emotional and motor behavior have gained more and more attention in past years. Thus, experimental and clinical research investigated the effects of rehabilitation programs. They demonstrate the beneficial effects of regular performance of exercise, Tai chi, dance therapy, and so on on the motor and nonmotor symptoms associated with PD.14,15 A certain drug portfolio exists for amelioration of motor symptoms. Continuous consideration of the tolerability, safety, and efficacy of the applied drug combinations and of the needs of the patients and their caregivers essentially contributes to successful treatment in the long term. Accordingly, standardized treatment approaches are of limited value in clinical practice, when these heterogeneous forms of PD are treated.13 A certain relationship is well known in terms of onset and severity of nonmotor and motor symptoms in all PD stages.
Drugs for PD
The oldest employed compounds are anticholinergics with their tremorlytic activity. Nowadays, they are only rarely used because of their negative effects on short-term memory dysfunction sooner or later during chronic intake. There are good treatment options for motor symptoms by balancing the nigrostriatal dopamine deficiency.16 Three pharmacologic principles are known. One is administration of the metabolic dopamine precursor levodopa, which traverses the blood–brain barrier in contrast to dopamine itself. Since levodopa has a short half-life in the periphery, fluctuations of levodopa in plasma appear. Following the conversion of levodopa into dopamine in presynaptic neurons, these ups and downs of levodopa contribute to a pulsatile stimulation of postsynaptic dopaminergic receptors with dopamine. In the clinic, fluctuations of nonmotor and motor features sometimes appear within months, particularly during high levodopa dosing.17 Another principle is the direct stimulation of postsynaptic nigrostriatal receptors with dopamine agonists. Important determinants of their effects on motor symptoms are both their affinity to the dopaminergic uptake site and their metabolic half-life. It is well proven that dopamine agonists may delay the appearance of nonmotor and various types of dopamine dysregulation syndromes. Therefore, these compounds are slowly titrated to improve their tolerability. A further essential pharmacologic approach is the continuous, irreversible inhibition of the dopamine metabolizing enzyme monoamine oxidase-B (MAO-B). Thus, they contribute to higher and more stable dopamine concentrations in the synaptic cleft and prolong the stimulation of postsynaptic dopamine receptors.16 More recent findings in PD patients revealed that the commonly administered MAO-B inhibitors selegiline and rasagiline are not specific for MAO-B during chronic intake. Both substances also showed certain MAO-A inhibiting properties in plasma.18,19 Amantadine is a more frequently applied and relatively old drug. Although initially looked upon as an antiviral compound, it was later occasionally discovered by clinicians to have a moderate efficacy in relation to motor symptoms in PD patients. Research on the pharmacological properties of this compound inaugurated the concept of N-methyl-D-aspartate receptor blockade as a therapeutic principle in the treatment of PD. The precise mode of action of amantadine is still not clear. For instance, this compound is also suspected to share some dopamine-mimicking effects. The resurgence of research on amantadine started with the observation of beneficial effects on dyskinesia,20 and since 2015, safinamide has been available in Europe. Pharmacologically, safinamide selectively inhibits MAO-B activity in a reversible fashion only. Blockade of selective sodium channels and modulation of calcium influx with consequent inhibition of excessive glutamate release are further properties. Thus, it resembles a combination of available MAO-B inhibitors and amantadine in one substance. The effects of safinamide on motor symptoms are well proven.21
Objectives and methods
This narrative review aims to describe the role of safinamide in treatment regimes in the “real” world of daily maintenance of PD patients. A PubMed search has been done with the term “safinamide” to date (May 2016). The other cited literature is not based on any systematic queries in databases.
Safinamide
Safinamide (S)-(+)-2-[[4-[(3-fluorobenzyl)oxy]benzyl] amino]propanamide methanesulfonate is a small, water-soluble, stable molecule with low central nervous system toxicity. Safinamide is well absorbed without nutrition interference with linear metabolism and a long half-life. A combination of the two mechanisms of reversible MAO-B inhibition and glutamate release inhibition is considered the essential mode of action for the improvement of motor impairment in PD.22,23
Efficacy, safety, and tolerability of safinamide in humans
Safinamide was safe and well tolerated in healthy volunteers, who were exposed to safinamide with doses ranging from 25 to 10,000 μg/mL. A linear pharmacokinetic behavior occurred proportionally in relation to the applied dosage. The clearance of safinamide from the body was t1/2 22 hours with no clinical relevant accumulation. Tolerability was good in healthy controls.24,25
Efficacy, safety, and tolerability of safinamide in PD patients
The authorities in Europe (European Medicines Agency) approved safinamide as an add-on treatment for levodopa-treated PD patients. In the USA, approval was recently delayed because of concerns about missing data on a potential dependency risk of safinamide. In this review, only the most important safinamide trials are described, having been selected according to the personal opinion of the author. Only in abstract form published trials were considered when they were very important.
Pilot study
A small pilot trial demonstrated clinical benefit in dopamine agonist- and levodopa-treated PD patients, who received safinamide (once a day [OID]) in increasing dosages every 2 weeks.23 Despite the small number of participants, this trial gave positive signals for the efficacy of safinamide on impaired motor behavior in PD patients. This demonstrated that the combination of a dopamine agonist with a MAO-B inhibitor, such as safinamide, may exert a certain positive effect in PD patients such as a decrease in MAO-B activity in platelets (additional results are provided in Table 1). Interestingly, the area under the curve value of dopamine in serum went up ~30%. This was not significant, and may be interpreted as an effect due to MAO-B inhibition in the periphery. Area under the curve of serum levodopa levels was also increased proportionally to the dose of safinamide added to treatment, reaching 88% compared with baseline, when no safinamide was added.
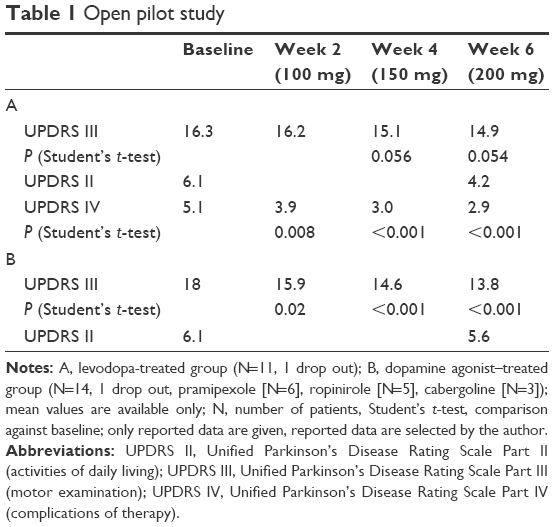 | Table 1 Open pilot study |
PD patients in early stages
The promising findings of the pilot trial led to further clinical trials in dopamine agonist-treated PD patients. In the 009 study, 172 PD patients participated, two withdrew consent, and two did not meet inclusion criteria.26 They were not treated or did not take a dopamine agonist only. Forty-nine study subjects completed the placebo therapy, and 52 finished the lower (0.5 mg/kg, equivalent to ~40 mg/d) and 49 the higher (1 mg/kg equivalent to ~90 mg/d) safinamide dosing. Initially, 56 patients were randomized to each arm. There were no significant differences among study groups at baseline. One hundred and one patients were on dopamine agonists (apomorphine, 1; bromocriptine, 9; cabergoline, 8; pergolide, 31; piribedil, 4; pramipexole, 32; ropinirole, 16). The withdrawal rate did not differ between groups. A positive response to safinamide was looked upon as a ≥30% improvement in the scores on the Unified Parkinson’s Disease Rating Scale, motor examination (UPDRS III) scores between baseline and end of the trial. Responders at the end of the study (intention-to-treat cohort) rose. There were 12 responders (21.4%) in the placebo group, 17 responders (30.9%) in the 0.5 mg/kg group, and 21 responders (37.5%) in the 1 mg/kg arm. The responder rate went up from 20.6% (placebo) to 36.4% in the 0.5 mg/kg arm, and then to 47.1% in the 1 mg/kg dose group in patients on a dopamine agonist regimen (additional results are provide in Table 2).26
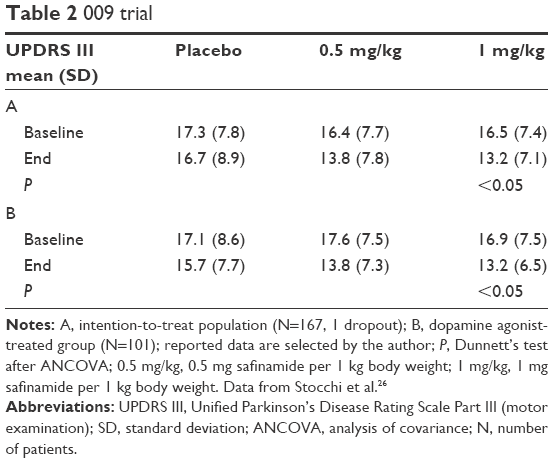 | Table 2 009 trial |
A further similarly designed study was the 015 trial, which lasted 6 months.27 It was performed in a randomized, double-blind, placebo-controlled fashion. The addition of safinamide once daily to a stable dosing of a single dopamine agonist was investigated in 270 early patients suffering from PD <5 years. Exclusion criteria were motor complications, intake of more than one dopamine agonist or any other PD drug in the 4 weeks before screening, dementia, or cognitive dysfunction (cutoff: Mini Mental State Examination score <24), or a score of 3 on Item I of the UPDRS. Finally, patients with serious medical, mental, or physical conditions that would preclude collection of safety or efficacy data were also not allowed. Participants were allocated to one of the three study groups. They received either safinamide 100 mg OID (90 patients) or safinamide 200 mg OID (89 patients) or matching placebo tablets (90 patients) as an add-on therapy to a dopamine agonist treatment. In each safinamide arm, safinamide was titrated up. Patients assigned to 100 mg/d first took 50 mg/d for 2 weeks, before receiving the target dose of 100 mg/d. Patients of the 200 mg/d arm were initially put on 100 mg safinamide per day in addition to their stable PD drug regimen. After 7 days, their dose was elevated to 150 mg, and then following Day 14, the target dose of daily 200 mg safinamide was applied. A hierarchical statistical approach for the analysis of covariance comparisons (covariates: treatment, visit, and treatment visit-by-visit interaction as fixed effects, baseline UPDRS III as covariate, and country as random effect). If any of the primary comparisons (first, 200 mg safinamide vs placebo; second, 100 mg safinamide vs placebo) were not significant, then subsequent tests were considered exploratory.
Safinamide 100 mg/d improved the UPDRS III score in comparison with the effect of dopamine agonist application only plus placebo (results are provided in Table 3). Application of safinamide 100 mg/day also declined UPDRS activities of daily living (II) score in comparison with placebo and dopamine agonist therapy alone (difference between end of study and baseline of –2.2±3.8 in the safinamide-treated group versus –1.2±3.5 in the placebo group; P=0.02). Safinamide 200 mg/day showed no significant outcome (Table 3).27 Generally, however, higher safinamide dosing caused an elevated rate of early terminations in comparison with the low-dose and placebo groups.28–30
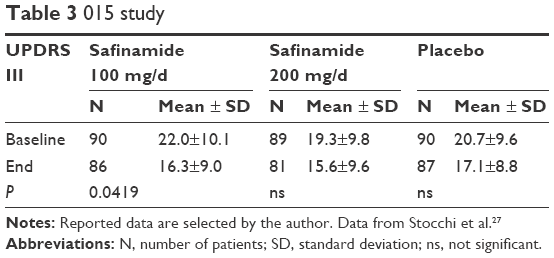 | Table 3 015 study |
The 015 trial was extended for 52 weeks in a blinded fashion in the 017 study. Primary endpoints were the interval to necessary treatment interventions. They were defined as an increase in dopamine agonist dosing, addition of a further dopamine agonist, introduction of levodopa or another PD drug, or discontinuation due to lack of efficacy. Pooled data from both safinamide-treated groups did not reach statistical significance for this primary endpoint in particular. The most probable reason was the lack of response observed in patients of the 200 mg safinamide arm. However, a post hoc analysis revealed that patients taking safinamide 100 mg/d experienced a lower rate of interventions compared with patients on a dopamine agonist only (25% [safinamide] vs 51% [placebo]; P=0.04). A secondary endpoint during this extension interval was the mean change of UPDRS III. Only safinamide 100 mg/d improved UPDRS II and III scores over the 18-month treatment phase (Table 4).31
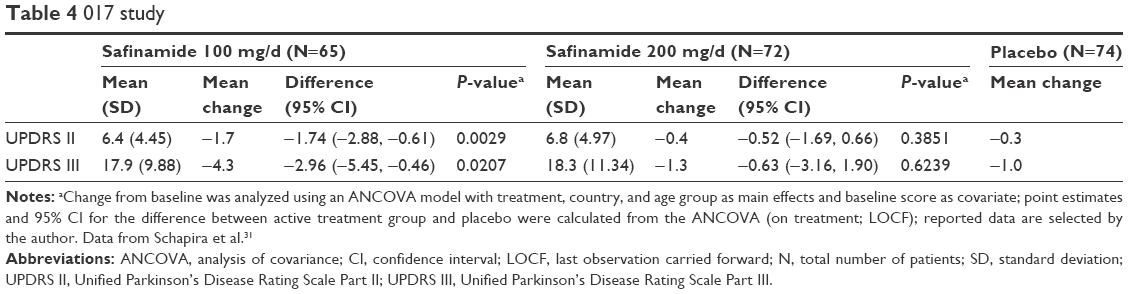 | Table 4 017 study |
The “MOTION” trial
The MOTION study (only abstract available) also showed the safety and tolerability of a combination of 50 or 100 mg safinamide with one dopamine agonist.32 Of 679 randomized patients, 607 completed the 24-week treatment period. In patients on monotherapy with a single dopamine agonist (666 patients), safinamide 100 mg OID significantly improved UPDRS III scores in comparison with placebo. The comparisons between PD patients on safinamide 50 mg OID with placebo arm outcomes were not significant (Table 5).
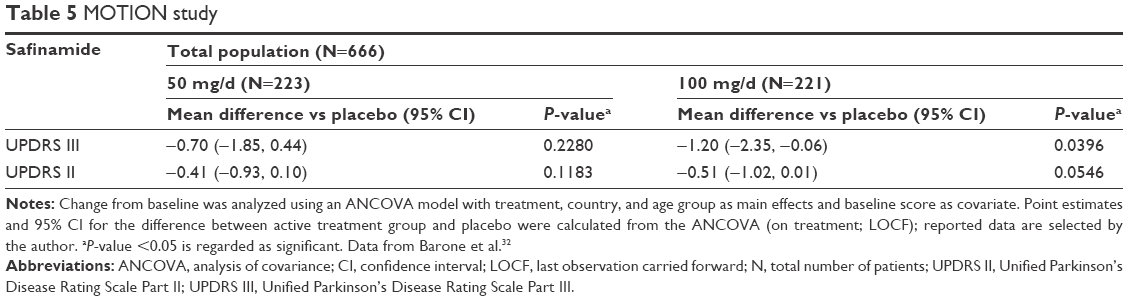 | Table 5 MOTION study |
More severe PD patients on a chronic levodopa regimen
As a result of the initial findings from the pilot study, the efficacy of safinamide was also investigated in combination with levodopa therapy. The first one was the so-called 016 trial, which lasted 6 months. This randomized, double-blind, placebo-controlled global multicenter study included 669 mid- to late-stage idiopathic PD patients with more than 3 years of disease duration. The levodopa regimen had to be stable. Study participants had to suffer from motor fluctuations. The daily duration of “OFF” intervals should be at least 1.5 hours. Concomitant therapy with stable doses of a dopamine agonist and/or an anticholinergic drug was allowed. First, dosing of levodopa was stabilized, then participants were allocated to one of the three groups of the trial (1:1:1). They received either one of two different doses of safinamide (50 or 100 mg once daily: 223 and 224 patients, respectively) or placebo (222 patients), as an adjunctive treatment to their levodopa therapy. The primary efficacy endpoint was the increase in mean daily ON time (ON time without dyskinesia plus ON time with minor dyskinesia) during an 18-hour period assessed by patients’ recordings on a diary. Both safinamide doses prolonged the daily total ON time (mean difference to baseline: 1.3 hours) compared with placebo (0.7 hours) significantly (P=0.022, safinamide 50 mg; P=0.013, safinamide 100 mg). There was no significant prolongation of the daily ON time with troublesome dyskinesia. Of 669 study patients, 89% of safinamide-treated subjects completed the trial (91%: 50 mg; 87%: 100 mg) in comparison with 89% in the placebo group. Secondary efficacy endpoints of this study were also positive; eg, there was a decrease in daily OFF time and in mean OFF time following first levodopa dosing in the morning, and an improvement in the UPDRS III score during ON time. There was a possibility for participants to prolong safinamide intake in the extension of 016, the study 018. Over 90% of the 016 patients entered this 78-week trial, which continued in a placebo-controlled and double-blind fashion.33,34 The objectives were to assess the effect on dyskinesias determined with the dyskinesia rating scale as a primary endpoint. The result was negative. Post hoc analyses revealed that patients with moderate-to-severe dyskinesia at baseline (ie, dyskinesia rating scale total score >4) improved when they took safinamide 100 mg/d (P=0.032). Moreover, all the secondary and tertiary endpoints of the 6-month study were met in the safinamide 100 mg/d arm (Table 6).35
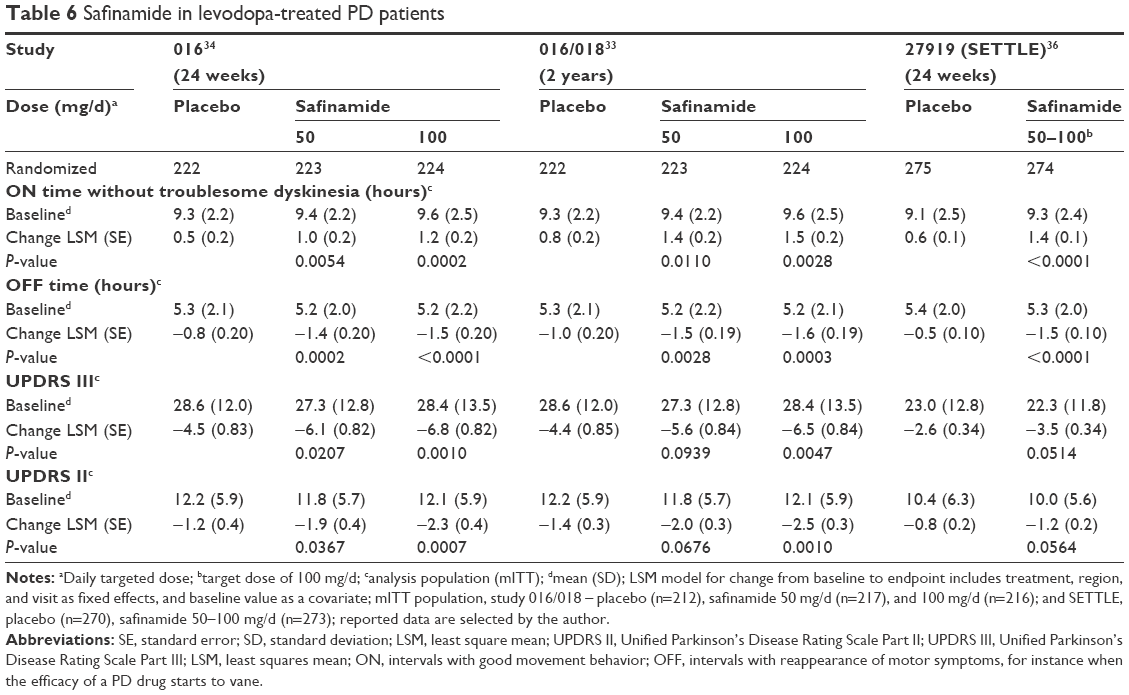 | Table 6 Safinamide in levodopa-treated PD patients |
The SETTLE study is published only in abstract form.36 In this Phase III trial, the study participants received 50 and 100 mg safinamide, respectively, in comparison with the placebo-treated group. This global study lasted 24 weeks and included 549 patients, 484 of whom finished the trial. Safinamide showed significantly more reduced OFF time, more ON time, and better UPDRS scores in the safinamide-treated study population compared with placebo. Before randomization, the dopamine-substituting drug regime was stabilized and optimized in at least levodopa-treated patients with motor fluctuations (Table 6).
Safinamide in clinical practice
Safinamide has been available for more than 1 year in Germany. The effect of safinamide on reduced OFF times and an improvement of motor symptoms is nearly similar to the one provided by the peripherally acting COMT inhibitor entacapone and irreversible MAO-B inhibitors such as selegiline or rasagiline.37,38 Peripherally acting COMT inhibitors prolong the effects of levodopa owing to inhibition of its metabolism. All these drugs have a better tolerability profile than dopamine agonists.
Future potentially interesting research with safinamide in the real world
It would be interesting to compare a so-called “triple concept” of initial chronic levodopa/DDI application with immediate concomitant COMT and MAO-B inhibition to initial dopamine against monotherapy of PD. The objective should be the general quality of life of patients and caregivers and delayed onset of motor complications in the long term.39 In comparison with dopamine agonist therapy, this kind of triple combination seems to provide a better safety and tolerability, both of which depend on an individual exposure to the patient. However, there may be two essential considerations that favor this concept of levodopa implementation in the therapy of PD patients.
First, COMT inhibition provides a more continuous levodopa brain delivery based on fewer fluctuations of levodopa in plasma during COMT inhibition and a more stable dopamine concentration in the synaptic cleft due to MAO-B inhibition, which also contributes to levodopa sparing in the long term.40 Thus, both pharmacologic principles complement each other in providing a continuous dopaminergic stimulation of postsynaptic dopaminergic receptors in the nigrostriatal system. To date, there are no observational data from the real world yet available for a detailed long-term evaluation of such a concept.
Second, it is well known that dopamine agonists often cause edema, particularly when administered in high dosages as levodopa-sparing treatment alternative.41 Further interesting side effects related more to dopamine agonists than to levodopa are frequent onset of nausea, dizziness, and low blood pressure. In other words, one may assume that safinamide use, in particular, provides some benefit in patients with a certain disposition toward dopamine agonist-induced nausea, which can easily be improved with domperidone, available only in Europe, or a pronounced fall in blood pressure. In clinical practice, this is a problem and may cause falls because of syncope.16 There are only a few therapeutic options for this drug-induced autonomic dysregulation syndrome. Blood pressure-elevating compounds like midodrine show only limited benefit.16 Therefore, prevention of this side effect with a combination of safinamide and a dopamine agonist, which is efficacious and supports dopamine agonist sparing, may be beneficial.
Is safinamide an antidyskinetic drug?
Some attempts are afoot to provide additional proof for a potential antidyskinetic efficacy of safinamide. But is this so important for the daily maintenance of PD patients? Pharmacologically, this concept is based on the glutamate release-inhibiting properties of safinamide, which generally resemble the glutamate receptor antagonizing properties of amantadine. A possible study design would be to replace a certain percentage of a patient’s levodopa with safinamide with a view to reducing OFF time. The study objective would be to assess whether the patient’s dyskinesia improves, worsens, or stays about the same? This would be a crucial outcome to test in a randomized trial of safinamide. Generally, onset of dyskinesia results more from levodopa than from dopamine agonists. The presentation of dyskinesia varies from day to day and is influenced considerably not only by fluctuations of levodopa in plasma and dopamine in the brain but also by environmental and psychological stress. All these components of dyskinesia interfere with the assessment in trials. Therefore, an antidyskinetic effect is probably measurable only in small, well-designed studies performed in a few experienced study centers with a standardized application of levodopa formulations. It is well known, from pharmacokinetic investigations, that levodopa may accumulate over the day in relation to the intervals between each intake and the applied dose.42,43 A concomitant observation of onset, frequency, and severity of dyskinesia over an interval of several hours with standardized objective documentation and assessment may also work as an additional assessment procedure in a clinical trial.8 In summary, such an approach is complex, time consuming, sensible to placebo effects, and expensive.44,45 It will also be questionable whether a putative positive study outcome would convince authorities, payers, the prescribing office neurologists, and patients in the “real” world. A variable expression of slight dyskinesia is often better accepted by the patients themselves in contrast to the OFF states, whereas the caregivers are more tangled by the dyskinesia and to a lesser extent by the OFF intervals.46 Instead, an alternative prospective study program could investigate whether early versus late safinamide introduction together with initiation of traditional levodopa treatment may attenuate the ensuing basal ganglia plasticity that underlies levodopa-induced dyskinesia. However, such a study is risky and would demand a longer duration with this preventive approach, which resembles the delayed start design. Moreover, one must consider that a symptomatic drug treatment alternative for severe dyskinesia is on the horizon. Retarded release formulations of amantadine showed a distinct positive impact on dyskinesia.47
Conclusion and future perspectives
The outcomes of the clinical study program led to the approval of the use of safinamide as an adjunct to levodopa therapy in the EU. Safinamide ameliorates motor impairment in PD patients. To date, these clinical trials have focused mainly on motor behavior. There are a lot of options for drug therapy for this indication, especially for the attenuation of OFF phenomena in patients with fluctuating, advanced PD. Safinamide was particularly efficacious in combination with levodopa. Levodopa has a complex, peripheral gastrointestinal absorption, pharmacokinetic behavior, and brain delivery, all of which differs from patient to patient. These features of levodopa therapy contribute to its long-term limitations, such as motor complications. The future of safinamide may also be an approaching change of treatment paradigms with again earlier use of retarded release resembling levodopa formulations, such as IPX066, in combination with dopamine agonist-sparing treatment alternatives. The clinical handling, safety, and side effect profile of safinamide is better than that of levodopa and of the dopamine agonists, and similar to those of other monoamine oxidase inhibitors, such as rasagiline, with their limited efficacy in regard to motor symptoms.20,39 From the clinical point of view, a future advantage of safinamide use may be the reduction of levodopa and dopamine agonist dosing in the treatment of PD patients in view of the better safety and tolerability of safinamide.
Currently, safinamide is increasingly being launched all over the EU. However, the introduction of this drug also faces new hurdles, for instance in terms of complex pricing procedures in Germany or approval in the USA. The conditions for drug approval in PD nowadays focus too much on motor behavior, for instance, reduction of OFF times or amelioration of motor symptoms. In this regard, one even discusses that a certain degree of improved motor behavior should be achieved with the applied rating scales before this benefit is looked upon as clinically relevant.48 This approach neglects, for instance, the heterogeneity of PD with its complex, distinct mixture of motor and nonmotor symptoms.8 The true value of an innovative compound, such as safinamide, will be realized only in the real world of patient maintenance and not in the artificial study world, which is increasingly influenced by the approving authorities and not by clinicians.8 Failures of drug development may also result from the overrestrictive conditions for drug approval or misconceptions of the studied disease per se.49 Generally, innovation in medicine presents a complex scenario nowadays. Health politicians, in collaboration with administrative authorities, try to slow the introduction of new developments by increasing the hurdles for approval, particularly in Europe. The situation in the USA was recently simplified and improved, since a manufacturer can ask for a breakthrough status. Nevertheless, political institutions and approving authorities try to guide and control creativity in research. This is a wrong approach for new ideas or emerging therapies in general, not just those for PD alone. Good science needs creative scientists, time, and freedom, and all these three pillars are shrinking nowadays.
Disclosure
The author participated in the trials on safinamide as principal investigator and currently advises safinamide-developing pharmaceutical companies (Newron, Merck Serono, Zambon). The author reports no other conflicts of interest in this work.
References
de Rijk MC, Launer LJ, Berger K, et al. Prevalence of Parkinson’s disease in Europe: a collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology. 2000;54(11 Suppl 5):S21–S23. | ||
Rajput AH, Birdi S. Epidemiology of Parkinson’s disease. Parkinsonism Relat Disord. 1997;3(4):175–186. | ||
Hinz M, Stein A, Cole T. The Parkinson’s disease death rate: carbidopa and vitamin B6. Clin Pharmacol. 2014;6:161–169. | ||
Berg D, Postuma RB, Adler CH, et al. MDS research criteria for prodromal Parkinson’s disease. Mov Disord. 2015;30(12):1600–1611. | ||
Przuntek H, Müller T, Riederer P. Diagnostic staging of Parkinson’s disease: conceptual aspects. J Neural Transm (Vienna). 2004;111(2):201–216. | ||
Schapira AH, McDermott MP, Barone P, et al. Pramipexole in patients with early Parkinson’s disease (PROUD): a randomised delayed-start trial. Lancet Neurol. 2013;12(8):747–755. | ||
Berg D, Lang AE, Postuma RB, et al. Changing the research criteria for the diagnosis of Parkinson’s disease: obstacles and opportunities. Lancet Neurol. 2013;12(5):514–524. | ||
Müller T, Foley P. Clinical drug research in chronic central neurodegenerative disorders. Expert Rev Neurother. 2016;16(5):497–504. | ||
Politis M, Wu K, Molloy S, Bain G, Chaudhuri KR, Piccini P. Parkinson’s disease symptoms: the patient’s perspective. Mov Disord. 2010;25(11):1646–1651. | ||
Figee M, de KP, Klaassen S, et al. Deep brain stimulation induces striatal dopamine release in obsessive-compulsive disorder. Biol Psychiatry. 2014;75(8):647–652. | ||
Meissner W, Reum T, Paul G, et al. Striatal dopaminergic metabolism is increased by deep brain stimulation of the subthalamic nucleus in 6-hydroxydopamine lesioned rats. Neurosci Lett. 2001;303(3): 165–168. | ||
Weiner WJ. There is no Parkinson disease. Arch Neurol. 2008;65(6):705–708. | ||
Weiner WJ. What do clinical trials tell us about treating patients. Parkinsonism Relat Disord. 2009;15(Suppl 3):S34–S37. | ||
Tomlinson CL, Herd CP, Clarke CE, et al. Physiotherapy for Parkinson’s disease: a comparison of techniques. Cochrane Database Syst Rev. 2014;6:CD002815. | ||
Uhrbrand A, Stenager E, Pedersen MS, Dalgas U. Parkinson’s disease and intensive exercise therapy – a systematic review and meta-analysis of randomized controlled trials. J Neurol Sci. 2015;353(1–2): 9–19. | ||
Müller T. Drug therapy in patients with Parkinson’s disease. Transl Neurodegener. 2012;1(1):1–10. | ||
Stocchi F, Rascol O, Kieburtz K, et al. Initiating levodopa/carbidopa therapy with and without entacapone in early Parkinson disease: the STRIDE-PD study. Ann Neurol. 2010;68(1):18–27. | ||
Bartl J, Müller T, Grunblatt E, Gerlach M, Riederer P. Chronic monoamine oxidase-B inhibitor treatment blocks monoamine oxidase-A enzyme activity. J Neural Transm (Vienna). 2014;121(4):379–383. | ||
Riederer P, Lachenmayer L, Laux G. Clinical applications of MAO-inhibitors. Curr Med Chem. 2004;11(15):2033–2043. | ||
Müller T. Current status of safinamide for the drug portfolio of Parkinson’s disease therapy. Expert Rev Neurother. 2013;13(9):969–977. | ||
Deeks ED. Safinamide: first global approval. Drugs. 2015;75(6):705–711. | ||
Fariello RG. Safinamide. Neurotherapeutics. 2007;4(1):110–116. | ||
Stocchi F, Vacca L, Grassini P, et al. Symptom relief in Parkinson disease by safinamide: biochemical and clinical evidence of efficacy beyond MAO-B inhibition. Neurology. 2006;67(7 Suppl 2):S24–S29. | ||
Park KD, Yang XF, Dustrude ET, et al. Chimeric agents derived from the functionalized amino acid, lacosamide, and the alpha-aminoamide, safinamide: evaluation of their inhibitory actions on voltage-gated sodium channels, and antiseizure and antinociception activities and comparison with lacosamide and safinamide. ACS Chem Neurosci. 2015;6(2):316–330. | ||
Perez-Lloret S, Rascol O. The safety and efficacy of safinamide mesylate for the treatment of Parkinson’s disease. Expert Rev Neurother. 2016; 16(3):245–258. | ||
Stocchi F, Arnold G, Onofrj M, et al. Improvement of motor function in early Parkinson disease by safinamide. Neurology. 2004;63(4): 746–748. | ||
Stocchi F, Borgohain R, Onofrj M, et al. A randomized, double-blind, placebo-controlled trial of safinamide as add-on therapy in early Parkinson’s disease patients. Mov Disord. 2012;27(1):106–112. | ||
Onofrj M, Bonanni L, Thomas A. An expert opinion on safinamide in Parkinson’s disease. Expert Opin Investig Drugs. 2008;17(7):1115–1125. | ||
Müller T. Safinamide for symptoms of Parkinson’s disease. Drugs Today (Barc). 2015;51(11):653–659. | ||
Schapira AH. Safinamide in the treatment of Parkinson’s disease. Expert Opin Pharmacother. 2010;11(13):2261–2268. | ||
Schapira AH, Stocchi F, Borgohain R, et al. Long-term efficacy and safety of safinamide as add-on therapy in early Parkinson’s disease. Eur J Neurol. 2013;20(2):271–280. | ||
Barone P, Fernandez HH, Ferreira J, et al. Safinamide as an add-on therapy to a stable dose of a single dopamine agonist: results from a randomized, placebo-controlled, 24-week multicenter trial in early idiopathic Parkinson disease (PD) patients (MOTION Study). Neurology. 2013;80(Meeting Abstracts 1):P01.061. | ||
Borgohain R, Szasz J, Stanzione P, et al. Two-year, randomized, controlled study of safinamide as add-on to levodopa in mid to late Parkinson’s disease. Mov Disord. 2014;29(10):1273–1280. | ||
Borgohain R, Szasz J, Stanzione P, et al. Randomized trial of safinamide add-on to levodopa in Parkinson’s disease with motor fluctuations. Mov Disord. 2014;29(2):229–237. | ||
Cattaneo C, Ferla RL, Bonizzoni E, Sardina M. Long-term effects of safinamide on dyskinesia in mid- to late-stage Parkinson’s disease: a post-hoc analysis. J Parkinsons Dis. 2015;5(3):475–481. | ||
Schapira AH, Fox SH, Hauser RA, et al. Safinamide add on to L-dopa: a randomized, placebo-controlled, 24-week global trial in patients with Parkinson’s disease (PD) and motor fluctuations (SETTLE). Neurology. 2013;80(Meeting Abstracts 1):P01.062. | ||
Hauser RA, Silver D, Choudhry A, Eyal E, Isaacson S. Randomized, controlled trial of rasagiline as an add-on to dopamine agonists in Parkinson’s disease. Mov Disord. 2014;29(8):1028–1034. | ||
Schnitker J, Müller T. Meta-analysis of placebo-controlled clinical trials of safinamide and entacapone as add-on therapy to levodopa in the treatment of Parkinson’s disease. Eur Neurol Rev. 2015;10(1):15–22. | ||
Müller T. Catechol-O-methyltransferase inhibitors in Parkinson’s disease. Drugs. 2015;75(2):157–174. | ||
Przuntek H, Conrad B, Dichgans J, et al. SELEDO: a 5-year long-term trial on the effect of selegiline in early Parkinsonian patients treated with levodopa. Eur J Neurol. 1999;6(2):141–150. | ||
Whone AL, Watts RL, Stoessl AJ, et al. Slower progression of Parkinson’s disease with ropinirole versus levodopa: the REAL-PET study. Ann Neurol. 2003;54(1):93–101. | ||
Muhlack S, Herrmann L, Salmen S, Müller T. Fewer fluctuations, higher maximum concentration and better motor response of levodopa with catechol-O-methyltransferase inhibition. J Neural Transm (Vienna). 2014;121(11):1357–1366. | ||
Müller T. Pharmacokinetic considerations for the use of levodopa in the treatment of Parkinson disease: focus on levodopa/carbidopa/entacapone for treatment of levodopa-associated motor complications. Clin Neuropharmacol. 2013;36(3):84–91. | ||
Goetz CG, Laska E, Hicking C, et al. Placebo influences on dyskinesia in Parkinson’s disease. Mov Disord. 2008;23(5):700–707. | ||
Goetz CG, Damier P, Hicking C, et al. Sarizotan as a treatment for dyskinesias in Parkinson’s disease: a double-blind placebo-controlled trial. Mov Disord. 2007;22(2):179–186. | ||
Müller T, Russ H. Levodopa, motor fluctuations and dyskinesia in Parkinson’s disease. Expert Opin Pharmacother. 2006;7(13): 1715–1730. | ||
Pahwa R, Tanner CM, Hauser RA, et al. Amantadine extended release for levodopa-induced dyskinesia in Parkinson’s disease (EASED Study). Mov Disord. 2015;30(6):788–795. | ||
Hauser RA, Gordon MF, Mizuno Y, et al. Minimal clinically important difference in Parkinson’s disease as assessed in pivotal trials of pramipexole extended release. Parkinsons Dis. 2014;2014:467131. | ||
Hauser RA, Stocchi F, Rascol O, et al. Preladenant as an adjunctive therapy with levodopa in Parkinson disease: two randomized clinical trials and lessons learned. JAMA Neurol. 2015;72(12):1491–1500. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.