Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Elevated circulating PAI-1 levels are related to lung function decline, systemic inflammation, and small airway obstruction in chronic obstructive pulmonary disease
Authors Wang H, Yang T, Li D, Wu Y, Zhang X, Pang C, Zhang J, Ying B, Wang T ![]() , Wen F
, Wen F
Received 29 February 2016
Accepted for publication 27 April 2016
Published 26 September 2016 Volume 2016:11(1) Pages 2369—2376
DOI https://doi.org/10.2147/COPD.S107409
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Richard Russell
Hao Wang,1,2,* Ting Yang,1,2,* Diandian Li,1,2 Yanqiu Wu,1,2 Xue Zhang,1,2 Caishuang Pang,1,2 Junlong Zhang,3 Binwu Ying,3 Tao Wang,1,2 Fuqiang Wen1,2
1Department of Respiratory and Critical Care Medicine, West China Hospital of Sichuan University, Chengdu, Sichuan, People’s Republic of China; 2Division of Pulmonary Diseases, State Key Laboratory of Biotherapy of China, West China Hospital of Sichuan University, Chengdu, Sichuan, People’s Republic of China; 3Department of Laboratory Medicine, West China Hospital of Sichuan University, Chengdu, Sichuan, People’s Republic of China
*These authors contributed equally to this work
Background: Plasminogen activator inhibitor-1 (PAI-1) and soluble urokinase-type plasminogen activator receptor (suPAR) participate in inflammation and tissue remolding in various diseases, but their roles in chronic obstructive pulmonary disease (COPD) are not yet clear. This study aimed to investigate if PAI-1 and suPAR were involved in systemic inflammation and small airway obstruction (SAO) in COPD.
Methods: Demographic and clinical characteristics, spirometry examination, and blood samples were obtained from 84 COPD patients and 51 healthy volunteers. Serum concentrations of PAI-1, suPAR, tissue inhibitor of metalloproteinase-1 (TIMP-1), Matrix metalloproteinase-9 (MMP-9), and C-reactive protein (CRP) were detected with Magnetic Luminex Screening Assay. Differences between groups were statistically analyzed using one-way analysis of variance or chi-square test. Pearson’s partial correlation test (adjusted for age, sex, body mass index, cigarette status, and passive smoke exposure) and multivariable linear analysis were used to explore the relationships between circulating PAI-1 and indicators of COPD.
Results: First, we found that serum PAI-1 levels but not suPAR levels were significantly increased in COPD patients compared with healthy volunteers (125.56±51.74 ng/mL versus 102.98±36.62 ng/mL, P=0.007). Then, the correlation analysis showed that circulating PAI-1 was inversely correlated with pulmonary function parameters including the ratio of forced expiratory volume in 1 second to forced vital capacity (FEV1/FVC), FEV1/Pre (justified r=-0.308, P<0.001; justified r=-0.295, P=0.001, respectively) and SAO indicators such as FEV3/FVC, MMEF25–75/Pre (justified r=-0.289, P=0.001; justified r=-0.273, P=0.002, respectively), but positively related to the inflammatory marker CRP (justified r=0.351, P<0.001), the small airway remolding biomarker TIMP-1, and MMP-9 (justified r=0.498, P<0.001; justified r=0.267, P=0.002, respectively). Besides, multivariable linear analysis showed that FEV1/FVC, CRP, and TIMP-1 were independent parameters associated with PAI-1.
Conclusion: Our findings first illustrate that elevated serum PAI-1 levels are related to the lung function decline, systemic inflammation, and SAO in COPD, suggesting that PAI-1 may play critical roles in the pathogenesis of COPD.
Keywords: plasminogen activator inhibitor-1 (PAI-1), chronic obstructive pulmonary disease (COPD), systemic inflammation, small airway obstruction (SAO)
Introduction
Chronic obstructive pulmonary disease (COPD) is a worldwide health care problem characterized by progressive airflow limitation, which is rarely reversible.1 The World Health Organization predicted that COPD would become the third leading cause of death and might move up to the fifth leading cause of disability-adjusted life years by 2030.2,3 Aside from airway inflammation and oxidative stress, recent studies suggested that small airway obstruction (SAO) caused by small airway remolding (SAR) along with aberrant extracellular matrix (ECM) deposition also contributed to the pathogenesis of COPD.4 On the other hand, more and more evidences showed that airway inflammation might also “spread” into the circulatory system and cause systemic inflammatory injuries and organ damage.5 However, arguments about how they work in COPD and what factors are involved have never stopped.
As we know, plasminogen activator inhibitor-1 (PAI-1) was first identified as an important PAI expressed by cultured bovine endothelial cells6 and many other different types of cells in various tissues.7 During the past decades, numerous clinical and laboratory studies have evaluated the various functions of this unique serpin, including regulating fibrinolysis in the process of thrombus formation, promoting ECM, contributing to atherosclerosis,8 bone remolding,9 renal fibrosis,8 myocardial infarction, and diabetic vascular damage.10 Mostly, PAI-1 is found to act in the aforementioned roles by interacting with the urokinase-type activator (uPA) system, which is known for its contribution to the fibrinolysis in blood.11 Meanwhile, recent studies have found that uPA system, including uPA and urokinase-type activator receptor (uPAR), is involved in inflammation and tissue remolding.12 Interestingly, sputum levels of PAI-1 and soluble uPAR (suPAR) were found to be increased in COPD patients.13,14 These findings raised the problem of whether PAI-1 and uPA system is involved in the inflammatory response and tissue remolding in COPD? Unfortunately, there is no definite answer until now.
Therefore, we measured the serum levels of PAI-1 and suPAR in COPD patients and healthy subjects and analyzed if these two circulatory biomarkers were related to lung function decline, systemic inflammation, and SAO so as to explore the potential roles of PAI-1 and suPAR in COPD.
Methods
Study protocol
The study protocol conformed to the principles of the Declaration of Helsinki and was approved by the Ethic Committee of West China Hospital, Sichuan University, People’s Republic of China. All subjects gave written informed consents before the study. Between October 2013 and March 2014, naïve COPD patients and healthy controls without active pulmonary disorders were recruited from the Outpatient Department of West China Hospital. Every subject received a standard lung function test according to the American Thoracic Society and European Respiratory Society guidelines;15 briefly, all the subjects underwent three-time eligible prebronchodilation spirometry tests and postbronchodilation spirometry tests. Then, COPD was diagnosed prospectively for this study on the basis of the Global Initiative for Chronic Obstructive Lung Disease (GOLD) criteria.1 To be included in the study, patients had to show a ratio of forced expiratory volume in the first second to forced vital capacity (FEV1/FVC) below 70% after bronchodilation and an increase in FEV1 below 12% after inhalation of β2-agonist (200 mg salbutamol). Meanwhile, during the final 3 months, they had to be clinically stable. All the enrolled patients and controls had never received standard COPD-related therapies such as inhaled corticosteroids, bronchodilator, etc. Chest X-ray and electrocardiogram were performed to exclude the comorbidities such as pneumonia, pulmonary tuberculosis, lung cancer, cardiomegaly, myocardial ischemia, arrhythmia, and other cardiopulmonary disorders. Patients were also excluded if they had conditions known to affect serum levels of PAI-1, suPAR, or C-reactive protein (CRP), including hemorrhagic and clotting disorders, diabetes, stroke, acute infectious disorders, and so on.
Collection of the basic information
The basic information of enrolled subjects, including age, sex, height, weight, cigarette status, passive cigarette exposure, main complaints, and medical history, was collected by two individual investigators. Body mass index (BMI) was calculated using the formula weight/square of height. To avoid the contrived bias, the third investigator assessed this information and had it confirmed by the subjects, and all the investigators were blind to the spirometry reports of the subjects.
Measurement of PAI-1, suPAR, CRP, TIMP-1, and MMP-9
The COPD patients and healthy controls were requested to fast overnight from 22:00 the night before; the next morning venous blood samples were collected from the median cubital vein, and 1 hour later, serum was separated by centrifugation at 3,000 revolutions per minute for 10 minutes and stored at −80°C until analysis. Serum levels of PAI-1, suPAR, CRP, tissue inhibitor of metalloproteinase-1 (TIMP-1), and Matrix metalloproteinase-9 (MMP-9) were detected using a Magnetic Luminex Screening Assay (R&D Systems, Minneapolis, MN, USA) on the Bio-Plex 200 (Bio-Rad, CA, USA) at the Department of Laboratory Medicine of West China Hospital. The detection sensitivities were 0.67 pg/mL for PAI-1, 43.2 pg/mL for suPAR, 116 pg/mL for CRP, 3.42 pg/mL for TIMP-1, and 13.6 pg/mL for MMP-9. All the measurements were carried out strictly according to the manufacturer’s instructions, and technicians who performed tests were blinded to the clinical details of the subjects.
Statistical analysis
All the data are presented as mean ± standard deviation (SD). Differences between groups were statistically analyzed using one-way analysis of variance or chi-square test when appropriate. Then, correlations between different serum biomarkers and lung functions were tested using Pearson’s partial correlation test adjusting for age, sex, BMI, pack-years, and passive cigarette exposure status. Finally, multivariable linear analysis was conducted to confirm the aforementioned relationships. The threshold of significance was set at two-side 5%. Data were analyzed, and figures were created with GraphPad Prism 6.01 for Windows (GraphPad Software Inc, La Jolla, CA, USA).
Results
Clinical characteristics of subjects
We enrolled 43 COPD smokers, 41 COPD nonsmokers, 18 healthy smokers, and 33 healthy nonsmokers. According to the GOLD standard, among these 84 COPD patients, there were 44 GOLD 1, 35 GOLD 2, 4 GOLD 3, and 1 GOLD 4 patients. Table 1 lists the demographic and clinical characteristics including age, sex, BMI, smoking status, pack-years, passive smoke exposure, lung function data, and serum levels of PAI-1, suPAR, and CRP. No significant difference in age, BMI, and passive smoke exposure was observed among these four groups. However, irrespective of COPD group or the healthy group, almost all the smokers were males. Lung function data illustrated that COPD patients were suffering from more serious SAO as their maximum midexpiratory flow (MMEF)/Pre and FEV3/FVC were largely decreased when compared with healthy controls.
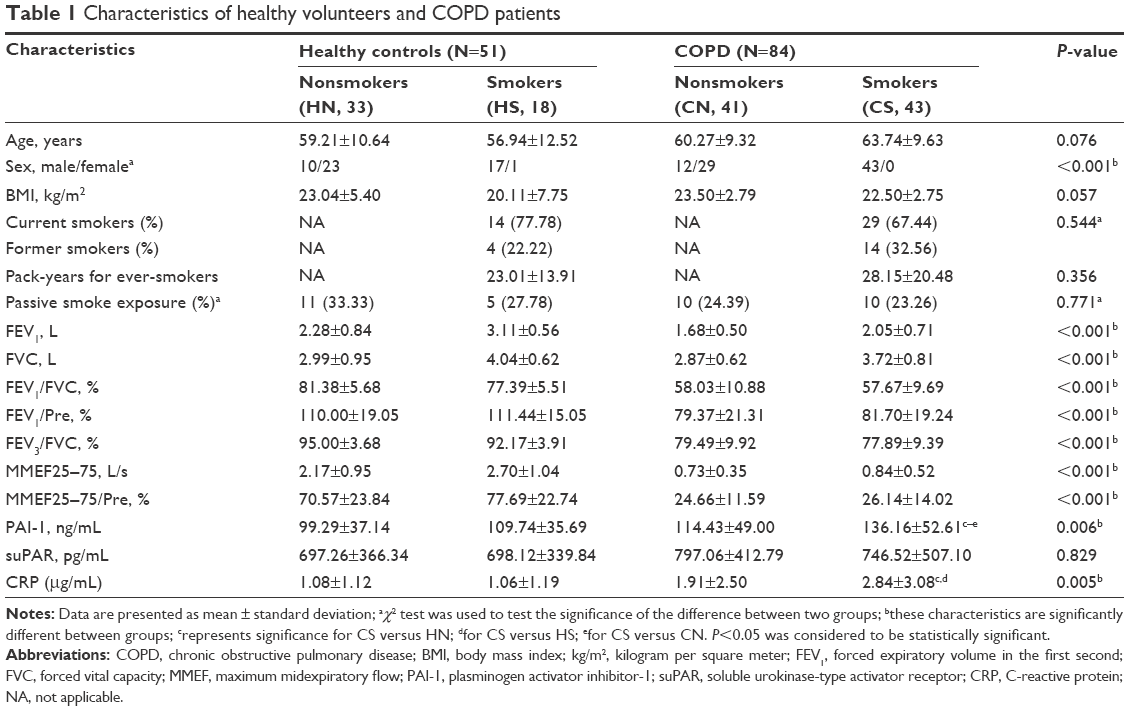 | Table 1 Characteristics of healthy volunteers and COPD patients |
Circulatory PAI-1 but not suPAR was upregulated in COPD patients
First, we compared PAI-1 and suPAR levels between COPD patients and healthy subjects. As listed in Table 1, serum PAI-1 levels in COPD patients was significantly increased in comparison with healthy subjects (125.56±51.74 ng/mL versus 102.98±36.62 ng/mL, P=0.007); more specifically, COPD smokers had higher levels of PAI-1 compared with healthy nonsmokers, healthy smokers, and COPD nonsmokers (136.16±52.61 ng/mL versus 99.29±37.14 ng/mL, 109.74±35.69 ng/mL, and 114.43±49.00 ng/mL, P<0.05, respectively, Figure 1A). Similarly, serum levels of CRP in COPD smokers were the highest among these four groups (2.84±3.08 μg/mL, P=0.005). Moreover, PAI-1 levels increased in patients with GOLD 2–4 COPD compared with healthy subjects and GOLD 1 COPD patients (138.85±55.59 ng/mL versus 102.98±36.62 ng/mL and 113.47±45.28 ng/mL, P<0.05, respectively, Figure 1B). However, serum suPAR levels showed no difference among these different patients and healthy subjects (P=0.829).
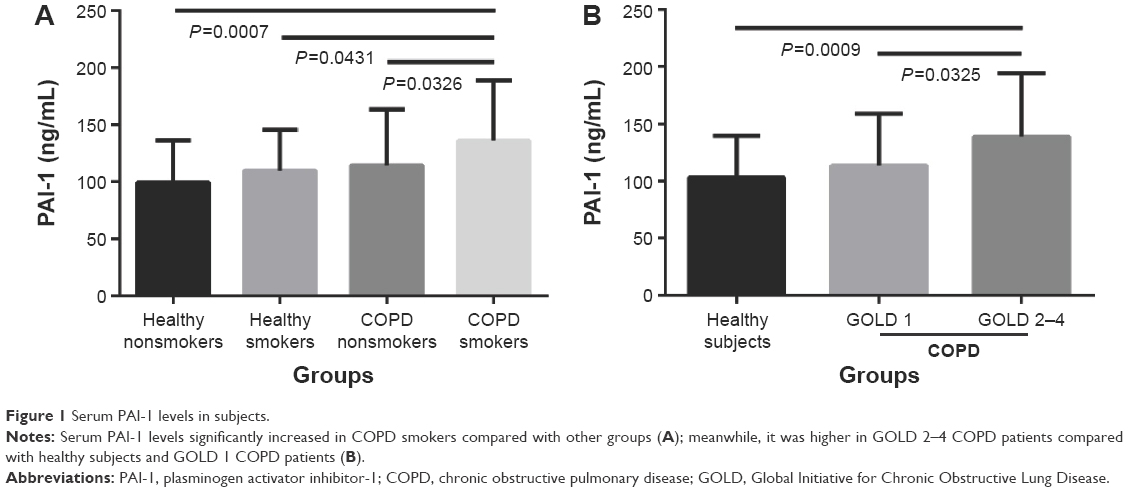 | Figure 1 Serum PAI-1 levels in subjects. |
PAI-1 levels were correlated with lung function and systemic inflammation
Next, we studied the relationship between PAI-1 and COPD-related parameters (Table 2). As can be seen from Table 2, serum PAI-1 levels were inversely related to FEV1/FVC (justified r=−0.308, P<0.001, Figure 2A) and FEV1/Pre (justified r=−0.295, P=0.001, Figure 2B) after adjusting for age, sex, BMI, pack-years, and passive cigarette exposure status. Meanwhile, PAI-1 levels showed a positive relationship with CRP levels (justified r=−0.351, P<0.001, Figure 3A).
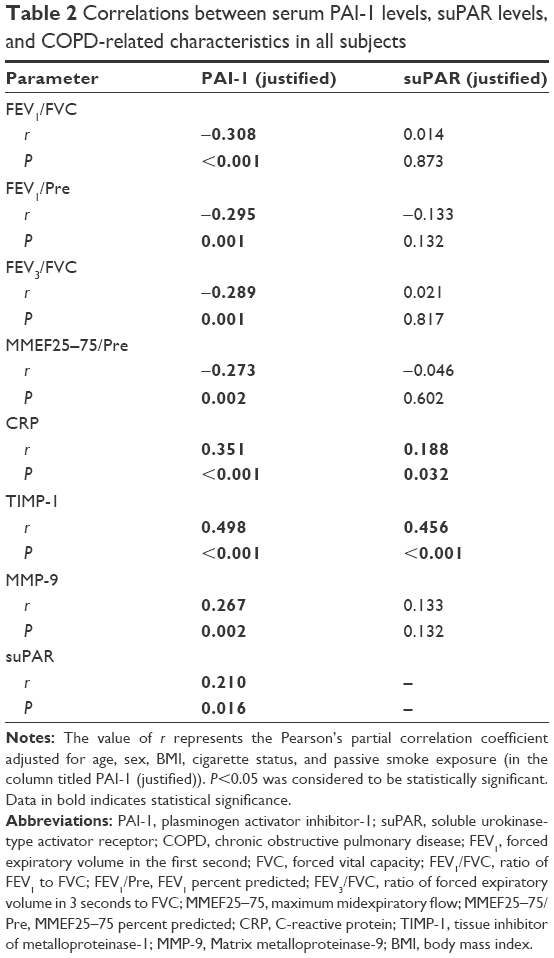 | Table 2 Correlations between serum PAI-1 levels, suPAR levels, and COPD-related characteristics in all subjects |
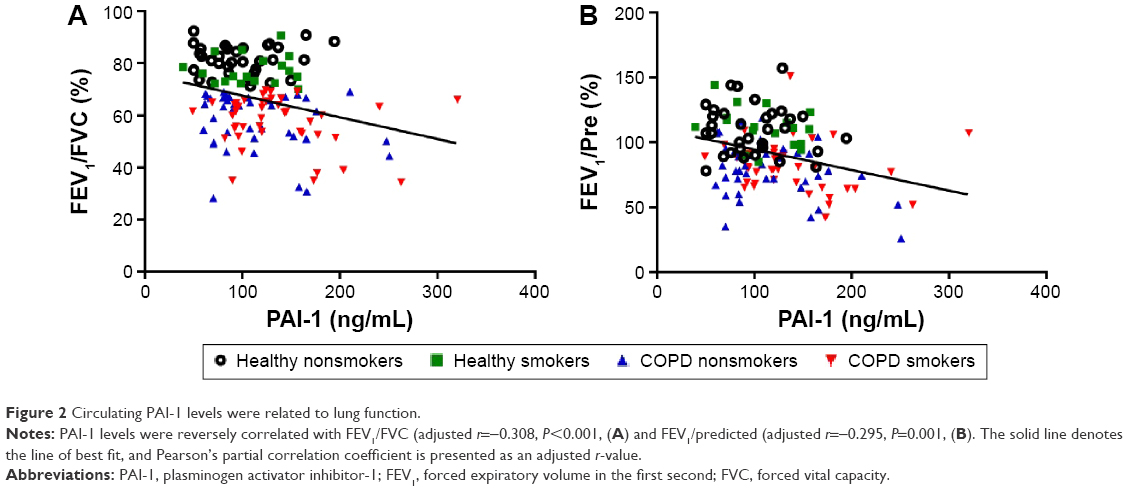 | Figure 2 Circulating PAI-1 levels were related to lung function. |
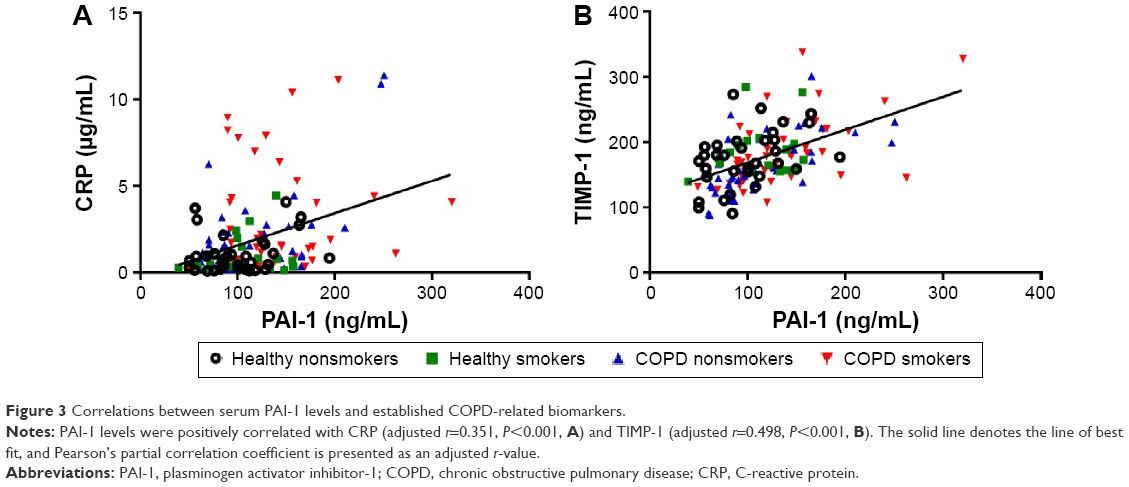 | Figure 3 Correlations between serum PAI-1 levels and established COPD-related biomarkers. |
Serum PAI-1 levels reflected SAO and SAR
Also, from the data given in Table 2, we observe inverse correlations between PAI-1 levels and spirometry index of SAO, including the ratio of FEV3/FVC (justified r=−0.289, P=0.001) and the ratio of MMEF25–75/Pre (justified r=−0.273, P=0.002). Furthermore, PAI-1 levels were positively related to TIMP-1 levels (justified r=0.498, P<0.001, Figure 3B) and MMP-9 levels (justified r=0.267, P=0.002). Besides, a positive relationship was observed between PAI-1 levels and suPAR levels (justified r=0.210, P=0.016).
Serum suPAR were related to CRP and TIMP-1
On the other hand, serum suPAR levels showed positive correlations with CRP levels (adjusted r=0.188, P=0.032) and TIMP-1 levels (adjusted r=0.456, P<0.001). However, it showed no relationship with lung function parameters and MMP-9 levels.
Multivariable linear analysis
Finally, we performed a multivariable linear analysis for circulating PAI-1 levels and found that age, FEV1/FVC, CRP, and TIMP-1 were the independent parameters associated with PAI-1, as listed in Table 3.
 | Table 3 Multivariable linear analysis for circulating PAI-1 levels |
Discussion
Our main goal was to explore the role of PAI-1 and suPAR in COPD. First, we measured and compared the serum PAI-1 and suPAR levels in healthy nonsmokers, healthy smokers, COPD nonsmokers, and COPD smokers. Then, we analyzed the correlations between serum PAI-1, suPAR levels, and indicators of lung function decline, systemic inflammation, and SAO. Finally, we found that serum PAI-1 levels were significantly increased in COPD patients, especially COPD smokers, and serum PAI-1 levels were related to lung function parameters such as FEV1/Pre and FEV1/FVC, systemic inflammation indicators represented by serum CRP, and SAO parameters including FEV3/FVC, MMEF25–75/Pre, serum TIMP, and serum MMP-9. On the other hand, however, serum suPAR levels only showed association with CRP and TIMP-1 levels. These findings implied that PAI-1 might be involved in the inflammation responses and airway remolding process in COPD.
First, we found that serum PAI-1 level, but not suPAR, was increased significantly in COPD patients, especially COPD smokers compared with other subjects, and negatively related to FEV1/Pre and FEV1/FVC. As a serpin with multiple functions, PAI-1 was first identified as an inhibitor of plasminogen activation system.16 By interacting with uPAR, PAI-1 showed profibrogenic properties and acted as a meaningful serum biomarker in various pulmonary diseases such as pulmonary fibrosis17 and asthma.18 Previous studies illustrated that cigarette smoke exposure could apparently increase the expression of PAI-1 in mice,19 and sputum PAI-1 levels are significantly increased in COPD patients.13 Thus, our finding confirmed that PAI-1 might be related to cigarette smoke exposure in COPD. Besides, we found that serum PAI-1 was positively related to suPAR, which was consistent with the result of the previous study, which indicated that plasminogen activation system (especially uPA system) was invoked in COPD.11
Then, our data showed a positive relationship between serum PAI-1 and serum CRP. Recently, it has been widely accepted that COPD-related inflammation was not only limited to the local airway, but could also “spread” to the whole body, which was termed systemic inflammation.20 As the most widely investigated biomarker of systemic inflammation, CRP was found to relate to airflow obstruction21 and disease severity of COPD.22 This was confirmed in our study by demonstrating that serum CRP was increased in COPD patients. Meanwhile, basic research indicated that PAI-1 regulated lipopolysaccharide-induced inflammation via targeting on the TLR4/NF-κB signaling pathway in alveolar macrophages,23 and elevated PAI-1 expression promoted alveolar epithelial cell apoptosis and exacerbated lung inflammation induced by influenza A virus.19 On the other hand, Chen et al24 found that CRP could increase the expression of PAI-1 via mediating oxidative stress and MAPK signal pathway in human coronary endothelial cells. Therefore, our findings suggested for the first time that PAI-1 may be associated with systemic inflammatory responses in COPD.
In addition, we observed that serum PAI-1 was related to SAO parameters, including MMEF25–75/Pre and FEV3/FVC, and SAR indicators such as TIMP-1 and MMP-9. As we know, SAR affecting bronchioles, especially respiratory bronchioles that were the transition zones between the airway and alveolar spaces, was the leading cause of SAO, which induces the persistent airflow limitation in COPD.25 More specifically, as the main mechanism of SAR, epithelial–mesenchymal transition (EMT) mainly involving MMP and TIMP families played vital roles in the pathogenesis of COPD.26 Previous studies have identified MMPs as a family of enzymes that were capable of degrading all components of the ECM involved in the EMT process27 and found that members of the MMP family were selectively inhibited by specific individuals of TIMPs. Particularly, TIMP-1 has been shown to bind to both the active and latent forms of MMP-9 and inhibit its enzymatic activity. Overall, TIMP-1 and MMP-9 regulated the balance between degradation and deposition of ECM, which were involved in airway remolding.28 On the other hand, clinical investigations showed that serum TIMP-1 and MMP-9 levels were markedly increased and significantly negatively related to FEV1/FVC or FEV1% predicted in COPD patients.29,30 These studies identified TIMP-1 and MMP-9 as serum biomarkers of SAR and SAO in COPD. Therefore, the positive relationship between serum TIMP-1, MMP-9 and PAI-1 found in our study indicated that serum PAI-1 might be involved in the EMT and SAR process in COPD.
Besides, serum suPAR only showed positive correlations with serum TIMP-1 and CRP in our study, which is consistent with previous findings that increased uPAR expression in the small airway epithelium is involved in the EMT process in COPD patients.31 These findings implied that suPAR might act synergistically with PAI-1 in SAR and systemic inflammation in COPD.
Finally, we performed multivariable linear analysis for PAI-1 and found that FEV1/FVC, CRP, and TIMP-1 were the independent parameters associated with PAI-1, which confirmed the relationship between PAI-1 and lung function decline, systemic inflammation, and SAO in COPD.
In this study, we only enrolled 135 subjects because of the rigorous inclusion and exclusion criteria; this would limit the P-value in the correlation analysis. Meanwhile, we evaluated the relationship between PAI-1 levels and SAR only based on the correlation analysis with four indirect indicators TIMP-1, MMP-9, FEV3/FVC, and MMEF25–75/Pre. To avoid these limitations, future studies should enroll more COPD and healthy subjects and collect more evidences of SAR, such as high-resolution computed tomography, biopsy, or other direct indicators.
Conclusion
In conclusion, our findings first revealed the hypothesis that PAI-1 might be associated with lung function decline, systemic inflammation, and SAO in COPD. A previous study showed that airway inflammation and chronic airway remolding induced by ovalbumin were attenuated in PAI-1 gene knockout mice and wide-type mice receiving intratracheal administrations of small interfering RNA against PAI-1.32 In combination with this study, we suggested that PAI-1 might be introduced into clinical practice as a therapeutic target of COPD in the future. However, larger, in-depth clinical and laboratory studies are needed to confirm our findings and explore how elevated PAI-1 play roles in COPD.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (NSFC, Grants 81470236, 81230001, and 31171103) and National key Technology Research and Development Program of the Ministry of Science and Technology of China during of the “12th Five-Year Plan” (Grants 2012BAI05B02 and 2014BAI08B04) to Prof Fuqiang Wen, NSFC Grant 81300011 to Dr Ting Yang, and NSFC Grant 81200031 to Dr Lei Chen.
Author contributions
Hao Wang and Fuqiang Wen designed this research, and all authors contributed toward subjects recruit, data collection, statistical analysis, drafting, and critically revising the paper, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors declare no conflicts of interests in this work.
References
GOLD. Global Strategy for the Diagnosis, Management, and Prevention of COPD (Updated 2015). Global Initiative for Chronic Obstructive Lung Disease; 2015. Available from: http://goldcopd.org/. Accessed September 14, 2016. | ||
WHO. The Global Burden of Disease (2004 Update). Geneva: World Health Organization; 2008. | ||
WHO. Global Health Estimates Summary Tables: Deaths by Cause, Age and Sex by Various Regional Grouping. Geneva: World Health Organization; 2013. | ||
Kranenburg AR, Willems-Widyastuti A, Moori WJ, et al. Enhanced bronchial expression of extracellular matrix proteins in chronic obstructive pulmonary disease. Am J Clin Pathol. 2006;126(5):725–735. | ||
Agusti A, Edwards LD, Rennard SI, et al. Persistent systemic inflammation is associated with poor clinical outcomes in COPD: a novel phenotype. PloS One. 2012;7(5):e37483. | ||
Loskutoff DJ, van Mourik JA, Erickson LA, Lawrence D. Detection of an unusually stable fibrinolytic inhibitor produced by bovine endothelial cells. Proc Natl Acad Sci U S A. 1983;80(10):2956–2960. | ||
Simpson AJ, Booth NA, Moore NR, Bennett B. Distribution of plasminogen activator inhibitor (PAI-1) in tissues. J Clin Pathol. 1991;44(2):139–143. | ||
Ha H, Oh EY, Lee HB. The role of plasminogen activator inhibitor 1 in renal and cardiovascular diseases. Nat Rev Nephrol. 2009;5(4):203–211. | ||
Daci E, Verstuyf A, Moermans K, Bouillon R, Carmeliet G. Mice lacking the plasminogen activator inhibitor 1 are protected from trabecular bone loss induced by estrogen deficiency. J Bone Miner Res. 2000;15(8):1510–1516. | ||
Lyon CJ, Hsueh WA. Effect of plasminogen activator inhibitor-1 in diabetes mellitus and cardiovascular disease. Am J Med. 2003;115 (Suppl 8A):62s–68s. | ||
Schuliga M, Westall G, Xia Y, Stewart AG. The plasminogen activation system: new targets in lung inflammation and remodeling. Curr Opin Pharmacol. 2013;13(3):386–393. | ||
Del Rosso M, Margheri F, Serrati S, Chilla A, Laurenzana A, Fibbi G. The urokinase receptor system, a key regulator at the intersection between inflammation, immunity, and coagulation. Curr Pharm Des. 2011;17(19):1924–1943. | ||
To M, Takagi D, Akashi K, et al. Sputum plasminogen activator inhibitor-1 elevation by oxidative stress-dependent nuclear factor-kappaB activation in COPD. Chest. 2013;144(2):515–521. | ||
Xiao W, Hsu YP, Ishizaka A, Kirikae T, Moss RB. Sputum cathelicidin, urokinase plasminogen activation system components, and cytokines discriminate cystic fibrosis, COPD, and asthma inflammation. Chest. 2005;128(4):2316–2326. | ||
Miller MR, Hankinson J, Brusasco V, et al. Standardisation of spirometry. Eur Respir J. 2005;26(2):319–338. | ||
Declerck PJ, Gils A. Three decades of research on plasminogen activator inhibitor-1: a multifaceted serpin. Semin Thromb Hemost. 2013;39(4):356–364. | ||
Kotani I, Sato A, Hayakawa H, Urano T, Takada Y, Takada A. Increased procoagulant and antifibrinolytic activities in the lungs with idiopathic pulmonary fibrosis. Thromb Res. 1995;77(6):493–504. | ||
Cho S, Kang J, Lyttle C, et al. Association of elevated plasminogen activator inhibitor 1 levels with diminished lung function in patients with asthma. Ann Allergy Asthma Immunol. 2011;106(5):371–377. | ||
Bhandary YP, Shetty SK, Marudamuthu AS, et al. Plasminogen activator inhibitor-1 in cigarette smoke exposure and influenza A virus infection-induced lung injury. PloS One. 2015;10(5):e0123187. | ||
Agusti AG, Noguera A, Sauleda J, Sala E, Pons J, Busquets X. Systemic effects of chronic obstructive pulmonary disease. Eur Respir J. 2003;21(2):347–360. | ||
Eickhoff P, Valipour A, Kiss D, et al. Determinants of systemic vascular function in patients with stable chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2008;178(12):1211–1218. | ||
de Torres JP, Cordoba-Lanus E, Lopez-Aguilar C, et al. C-reactive protein levels and clinically important predictive outcomes in stable COPD patients. Eur Respir J. 2006;27(5):902–907. | ||
Ren W, Wang Z, Hua F, Zhu L. Plasminogen activator inhibitor-1 regulates LPS-induced TLR4/MD-2 pathway activation and inflammation in alveolar macrophages. Inflammation. 2015;38(1):384–393. | ||
Chen C, Nan B, Lin P, Yao Q. C-reactive protein increases plasminogen activator inhibitor-1 expression in human endothelial cells. Thromb Res. 2008;122(1):125–133. | ||
Hogg JC, Chu F, Utokaparch S, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350(26):2645–2653. | ||
Pain M, Bermudez O, Lacoste P, et al. Tissue remodelling in chronic bronchial diseases: from the epithelial to mesenchymal phenotype. Eur Respir Rev. 2014;23(131):118–130. | ||
Woessner JF Jr. Matrix metalloproteinases and their inhibitors in connective tissue remodeling. FASEB J. 1991;5(8):2145–2154. | ||
Baratelli FE, Heuze-Vourc’h N, Krysan K, et al. Prostaglandin E2-dependent enhancement of tissue inhibitors of metalloproteinases-1 production limits dendritic cell migration through extracellular matrix. J Immunol. 2004;173(9):5458–5466. | ||
Higashimoto Y, Yamagata Y, Iwata T, et al. Increased serum concentrations of tissue inhibitor of metalloproteinase-1 in COPD patients. Eur Respir J. 2005;25(5):885–890. | ||
Linder R, Ronmark E, Pourazar J, Behndig A, Blomberg A, Lindberg A. Serum metalloproteinase-9 is related to COPD severity and symptoms – cross-sectional data from a population based cohort-study. Respir Res. 2015;16(1):28. | ||
Wang Q, Wang Y, Zhang Y, Zhang Y, Xiao W. The role of uPAR in epithelial-mesenchymal transition in small airway epithelium of patients with chronic obstructive pulmonary disease. Respir Res. 2013;14:67. | ||
Miyamoto S, Hattori N, Senoo T, et al. Intra-airway administration of small interfering RNA targeting plasminogen activator inhibitor-1 attenuates allergic asthma in mice. Am J Physiol Lung Cell Mol Physiol. 2011;301(6):L908–L916. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.