Back to Journals » International Journal of Nanomedicine » Volume 15
Electrospun Gelatin Nanocontainers for Enhanced Biopharmaceutical Performance of Piroxicam: In Vivo and In Vitro Investigations
Authors Zhao L, Mustapha O ![]() , Shafique S, Jamshaid T, Din FU
, Shafique S, Jamshaid T, Din FU ![]() , Mehmood Y
, Mehmood Y ![]() , Anwer K, Yousafi QUA, Hussain T, Khan IU
, Anwer K, Yousafi QUA, Hussain T, Khan IU ![]() , Ghori MU
, Ghori MU ![]() , Shahzad Y
, Shahzad Y ![]() , Yousaf AM
, Yousaf AM ![]()
Received 19 July 2020
Accepted for publication 24 September 2020
Published 10 November 2020 Volume 2020:15 Pages 8819—8828
DOI https://doi.org/10.2147/IJN.S271938
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Lin Zhao, 1 Omer Mustapha, 2 Shumaila Shafique, 2 Talha Jamshaid, 3 Fakhar ud Din, 4 Yasir Mehmood, 5 Khaleeq Anwer, 6 Qurrat ul Ain Yousafi, 7 Talib Hussain, 8 Ikram Ullah Khan, 5 Muhammad Usman Ghori, 9 Yasser Shahzad, 8 Abid Mehmood Yousaf 8
1Department of Rheumatology of Traditional Chinese and Western Medicine, Xinxiang Central Hospital, Xinxiang 453000, People’s Republic of China; 2Faculty of Pharmaceutical Sciences, Dow College of Pharmacy, Dow University of Health Sciences, Karachi 74200, Pakistan; 3Faculty of Pharmacy and Alternative Medicine, Islamia University of Bahawalpur, Bahawalpur 63100, Pakistan; 4Department of Pharmacy, Quaid-i-Azam University, Islamabad 45320, Pakistan; 5Department of Pharmaceutics, Faculty of Pharmaceutical Sciences, Government College University Faisalabad, Faisalabad 38040, Pakistan; 6Office of Chief Executive Officer, District Health Authority, Pakpattan 57400, Pakistan; 7Department of Neurosurgery, District Headquarters Hospital, Rawalpindi 46000, Pakistan; 8Department of Pharmacy, COMSATS University Islamabad, Lahore Campus, Lahore 54000, Pakistan; 9Department of Pharmacy, School of Applied Science, University of Huddersfield, Huddersfield HD1 3DH, UK
Correspondence: Abid Mehmood Yousaf; Talib Hussain Tel +92-300-477-4147; +92-345-722-0536
Email [email protected] Email [email protected]
Background: Piroxicam exhibits low oral bioavailability, due to its meager solubility in water. The intent of this study was to ameliorate the bioavailability of the drug by employing a solubility-enhancing encapsulation technique.
Methods: Seven samples were formulated with piroxicam and gelatin using both solvent evaporation and electrospraying together. Evaluation of solubility and release rate in water and assessment of bioavailability in rats were carried out in comparison with piroxicam plain drug powder (PPDP). Other in vitro explorations were accomplished using powder X-ray diffraction analysis, differential scanning calorimetry, thermogravimetric analysis, scanning electron microscopy, and Fourier-transform infrared spectroscopy.
Results: All piroxicam-loaded gelatinnanocontainers (PLGNs) enhanced solubility and release of the payload in water. In particular, a PLGN formulation consisting of piroxicam and gelatin at a 1:8 (w:w) ratio presented about 600-fold the drug solubility of that shown by PPDP. Moreover, 85.12%± 10.96% of the payload was released from this formulation in 10 minutes which was significantly higher than that dissolved from PPDP in 10 minutes (11.81%± 5.34%). Drug content, drug loading, and encapsulation efficiency of this formulation were 93.41%± 0.56%, 10.45%± 0.06%, and 66.74%± 6.87%, respectively. The drug loaded in PLGNs existed in the amorphous state, as confirmed by X-ray diffraction and differential scanning–calorimetry analyses, and was more stable when analyzed by thermogravimetric analysis. Moreover, Fourier-transform infrared spectroscopy analysis suggested nonexistence of any piroxicam–gelatin interaction in the formulation. In the scanning electron–microscopy image, PLGNs appeared as round, smooth particles, with particle size of < 1,000 nm. Amelioration in bioavailability of piroxicam with the aforementioned PLGN formulation was fourfold that of PPDP.
Conclusion: The PLGN formulation fabricated with piroxicam and gelatin at 1:8 (w:w) might be a promising system for enhanced biopharmaceutical performance of the drug.
Keywords: aqueous solubility, electrospraying, gelatin encapsulation, nanocontainers, oral bioavailability, piroxicam
Introduction
Piroxicam, an anti-inflammatory and analgesic substance, is prescribed to mitigate pain and associated inflammation. In particular, it is useful in the treatment and management of pain accompanied by rheumatoid arthritis and osteoarthritis.1 It impedes the activity of both cyclooxygenase (COX) type 1 and COX type 2 enzymes; therefore, formation of prostaglandins is ceased.2 Prostaglandins are a group of physiologically active lipids that induce pain and agony. Some interactions of piroxicam with other drugs have been reported as well. For example, when administered along with cadmium, it exhibits more toxicity.3 On the other hand, when coadministered with rosuvastatin, its adverse effects, such as peptic ulceration and hepatorenal damage, are mitigated.4
Piroxicam is a member of class II of the Biopharmaceutics Classification System (BCS) of drugs.5 The substances included in this class are either completely insoluble or meagerly soluble in aqueous fluids; however, they possess remarkable ability to traverse cell membranes efficiently.6 Piroxicam is scarcely soluble in aqueous media, as it imparts about 23 µg/mL solubility in water.7 Water is the main component of gastrointestinal (GI) fluid. A BCS class II entity is meagerly dissolved in GI fluid present in the lumen of the GI tract. Only when the drug is in a solution state in GI fluid can it traverse cells.8 Owing to its exiguous solubility in GI fluid, piroxicam is not sufficiently permeated through cells, and hence the drug titer in the systemic circulation is not effective to block the activity of COX type 2 or COX type 2 enzymes well. An alternative way to achieve effective bioavailability is to administer the drug at higher quantities; however, this may induce severe deleterious effects in the GI tract. Therefore, adoption of a suitable solubility-improvement technique is a more sagacious way to cope with this problem.
Solubility of a BCS class II substance can be ameliorated via several techniques, such as fabrication of nanosized drug-loaded hydrophilic particles, dispersion of a drug in hydrophilic polymers, association of a drug with cyclodextrins, wrapping a drug-payload core in hydrophilic wall material, casting of drug-laden porous silica particles, self-emulsifying systems, microemulsions, and cocrystallization.6,9–11 Pharmaceutical nanoencapsulation is a promising technology for resolving poor solubility and bioavailability problems of a BSC class II agent. It is an approach that envelop a solid, liquid, or gaseous substance in diminutive polymeric capsules.12 In this drug-delivery system, the wrapper or wall material protects the core from the surrounding environment, and storage stability of the payload can be extended.13 Gelatin, a heterogeneous mixture of chains of amino acids, is soluble in warm water. It is derived from collagen protein by hydrolysis. It is broadly availed in the pharmaceutical, food, and cosmetic industries, owing to its absolutely atoxic, biodegradable, ecofriendly, biocompatible, and high payload–holding characteristics. Also, it is convenient to administer and is eliminated appropriately in the physiological milieu.14 It is a regularly utilized wrapping or wall material for encapsulation of pharmacologically active substances and nutrients.15 Studies have shown that gelatin can foster the aqueous solubility and oral bioavailability of numerous drugs.9,16 A hydroalcoholic compound solution of a BCS class II drug and gelatin can be transmuted to a dry-particulate form by electrospraying. In this process, water is evaporated a great deal, and the ethanolic solution of the drug (core) is enwrapped or encapsulated by gelatin (shell or wall).12,17 Electrospraying is an excellent technique to obtain drug-loaded spherical gelatin nanocapsules.18 A pharmaceutical nanocapsule is a nanosized (<1 µm) drug-loaded spherical particle with core–shell morphology.19
The aim of this work was to augment the aqueous solubility and release rate of piroxicam using gelatin nanoencapsulation. Piroxicam-loaded gelatin nanocontainers (PLGNs) were prepared by electrospraying. Solubility and release rate of piroxicam in PLGNs were investigated. Crystalline physiognomy was determined using powder X-ray diffraction (PXRD) and differential scanning calorimetry (DSC). Thermogravimetric analysis (TGA) was used for measuring percentage weight loss occurring in a sample due to gradually rising temperature. Fourier-transform infrared (FTIR) spectroscopy analyses were carried out for exploring possible interlinkage between piroxicam and gelatin. Shapes and surface attributes of particles were inspected using scanning electron microscopy (SEM). Oral bioavailability was evaluated in white albino laboratory rats (Sprague Dawley). Area under the curve (AUC), highest drug concentration in plasma (Cmax), and time taken from dose administration to Cmax (Tmax) were used as parameters for assessing bioavailability.
Methods
Materials
Piroxicam (purity ≥98%) was obtained from Sigma-Aldrich (St Louis, MO, USA). Gelatin was procured from Daejung Chemical (Siheung, South Korea). All other chemicals and solvents were of reagent grade.
Preparation of PLGNs
All formulations were prepared by electrospraying. Recipes of samples prepared in this study are shown in Table 1. For each sample, 100 mg piroxicam plain drug powder (PPDP) was dissolved completely in 60 mL ethanol at 40°C (solution A). Gelatin was dissolved separately in 90 mL distilled water at 40°C (solution B). Then, both solutions were mixed together thoroughly to get a final clear solution (solution C). Quantities of solvents in solution A and solution B were taken so that the composition of solvent in solution C was approximately 40% hydroalcoholic mixture. Solution C was sprayed under high voltage using electrospraying (ESR 100; NanoNC Equipment, Seoul, South Korea). The compound solution to be electrospun was put in the barrel of a syringe made of glass (Hamilton, Reno, NV, USA). The solution-loaded syringe was properly attached to the syringe holder. The plunger of the syringe was pushed by the pump to move the solution via a single-lumen nozzle at a constant rate of 1 mL/h. At a voltage of 12.5 kV, a Taylor cone was formed and the jet converted to plume beneath the Taylor cone. Each electrospun sample was collected and kept at 40°C until constant weight, stowed in air-tight microtubes.
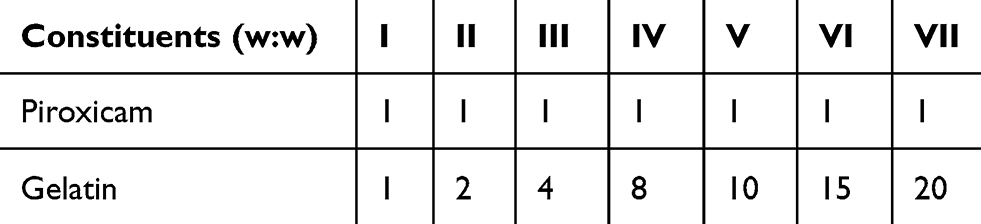 |
Table 1 Composition of piroxicam-loaded gelatin nanocontainers |
Solubility of Piroxicam in PLGNs
For each PLGN formulation, excess dried product was poured into a 2 mL Eppendorf microtube containing 0.5 mL distilled water and mixing done for a minute using a vortexmixer. Then, microtubes were swayed (100 rpm) for 5 days in a waterbath (37°C). After 5 days, samples were centrifuged at 7,000 g for 5 minutes. In another clean microtube, 0.2 mL clear supernatant was poured and diluted with an equal quantity of acetonitrile. The diluted sample (10 µL) was analysed with HPLC (1260 Infinity; Agilent Technologies, Santa Clara, CA, USA) using a Quasar C18 column (4.6 mm ID × 150 mm, 5 μm; PerkinElmer, Shelton, CT, USA) set at 40°C. The mobile phase comprised trifluoroacetic acid and acetonitrile (60:40, v:v), which eluted at a rate of 1 mL/min. The eluent was quantified at 332 nm for the determination of piroxicam concentration.20
Content of Piroxicam in PLGNs
A PLGN formulation equivalent to 5 mg piroxicam was transferred to a 100 mL flask containing 100 mL 40% aqueous ethanol. The lid was tightly closed to minimize evaporation of the solvent. Then, the flask was secured in a waterbath (37°C) for 2 hours. The solution was cleared by magnetic stirring, after which a stock solution of 50 µg/mL drug concentration was obtained. This clear solution was filtered (0.45 μm) and aptly diluted with acetonitrile. The dilution was analyzed in accordance with HPLC for determining actual drug concentration. Piroxicam content in each PLGN sample was computed:
PC = PA/PT × 100
where PC stands for piroxicam amount in the sample, PA the actual quantity of piroxicam in the diluted sample, and PT the theoretical quantity of the dilution. For each composition, the test was performed in triplicate (n=3).
Drug Loading
The percentage of piroxicam loading was computed:
PL = PW/PN × 100
where, PL is the percentage of piroxicam loading in PLGN, PW the weight of piroxicam (g) present in PN of PLGN, and PN the quantity of PLGN (g).
Encapsulation Efficiency
The optimized PLGN formulation, equivalent to 5 mg drug, was placed in a 2 mL Amicon Ultra centrifugal filter (UFC200324, MWCO 3,000 Da; Merck Millipore, Billerica, MA, USA) and rinsed thrice with absolute alcohol, each time with 1 mL volume. The filtrate was discarded each time. Ultracentrifugal filtration was accomplished with 7,000 g for 10 minutes. Then, the residue was dissolved gradually with 40% hydroalcoholic solution that had been preheated to 50°C. Each time, 1 mL was used until complete dissolution. The filtrate was preserved in a 10 mL measuring flask each time. Then, the volume was raised to 100 mL with ethanol. The theoretical concentration of this solution was 50 µg/mL. The actual concentration of piroxicam in this solution was determined by HPLC. The encapsulation efficiency of PLGN was calculated:
EE = PE/PT ×100
where EE is encapsulation efficiency, PE the actual quantity (g) of encapsulated piroxicam, and PT total weight (g) of piroxicam present in PLGN in accordance with the drug content.
Release Rate of Piroxicam from PLGNs
The release rate of piroxicam from a PLGN formulation was determined using a basket apparatus (Vision Classic 6; Hanson Research, Los Angeles, CA, USA). A PLGN formulation corresponding to 40 mg piroxicam was sealed in a dialysis pouch (12–14kDa). Then, the sample-loaded pouch was encaged in the basket by fitting the basket with the shaft. In order to sink the basket in 900 mL 2% (w:v) aqueous solution of Tween 80 filled in the vessel below, the rotating shaft (100 rpm) was pulled down. The temperature of the release medium had already been maintained at 37°C±0.5°C. After each predetermined time interval, 1 mL release medium was sampled using a syringe and filtered into an HPLC vial using a syringe filter (0.22 μm). Piroxicam concentration was quantified with HPLC. For each PLGN formulation, experiments was done in sextuplicate (n=6).
X-Ray Diffraction
X-ray diffraction (D-Max 2500PC; Rigaku, Tokyo, Japan) using a CuKα1 monochromatic emission source was used for determining the degree of crystallinity or amorphousness of the samples. Analyses were carried out in the range of 5°-50° at a scanning speed of 10°/min with a stepsize of 0.02°/s and scanning mode set at 2θ. Current flow and voltage supply were regulated at100 mA and 100 kV, respectively.
Differential Scanning Calorimetry
DSC (DSC-Q20; TA Instruments, New Castle, DE, USA) was employed for confirming the crystalline or amorphous status and other thermal physiognomy of the samples. About 5 mg of the sample to be tested was placed and sealed in a Tzero aluminum crucible (TA Instruments). The sealed sample was placed in the calorimeter, which had a constant supply of nitrogen (30 cm3/min). Scanning was performed by heating the sample in the range of 30°–300°C at a rate of 10°C/min.
Thermogravimetric Analysis
The decrease in weight of a sample occurring with gradual raise in temperature was determined by TGA (Q50; TA Instruments). About 5 mg quantity of a sample put into the hangable platinum crucible was lowered into the furnace by the automatic moving arm. The sample enclosed in the furnace was gradually heated in the range of 50–550°C at 10°C/min with a constant supply of nitrogen (25 cm3/min). The decrease in weight (%) of the sample was noted.
Scanning Electron Microscopy
SEM (S-4800; Hitachi, Tokyo, Japan) was employed for studying the shape, size, and surface quality of the particles. Samples to be inspected were clung on the exposed surface of a double-side adhesive tape affixed to a brass stub. Then, samples were coated with platinum (Emitech K-575-K ion-sputter coater) under 8×10−3 mbar vacuum. The current supply and turbo speed settings were adjusted at 20 mA and 90%, respectively. Coated samples were scanned with SEM and images recorded.
Fourier-Transform Infrared Spectroscopy
FTIR spectra of the samples were recorded in the range of 4,000–400 cm−1 at a resolution of 2 cm−1 using a Nicolet6700 (Thermo Fisher Scientific, Waltham, MA, USA).
Bioavailability Assessment in Rats
Twelve white albino laboratory rats (Sprague Dawley), each of 260–300 g body weight, were divided into two equal groups. They were kept in cages under a controlled environment of 21°C–28°C temperature and 45%–65% relative humidity. Before giving a dose of PPDP or PLGN formulation via the oral route, each rat was fasted for 1.0–1.5 days. Access to drinking water was not discontinued. All steps involved in the use of animals as experimental organisms in research were conducted strictly following the Guiding Principles in the Use of Animals in Toxicology adopted by the Society of Toxicology and NIH policy regarding animal welfare. All protocols were also reviewed by the Institutional Animal Care and Use Committee Government College University Faisalabad prior to granting approval for the study (GCUF/ERC/2138).
Each fasted animal was anesthetized by injecting a mixture of tiletamine and xylazine (2:1, v:v) intramuscularly. Cannulation of a polyethylene tube into the right carotid artery was accomplished surgically. The tube was passed through the hollow spring attached to an infusion harness, in order to protect it from likely damage by the animal. Each harnessed rat was kept in a separate adequately sized cage, where it was able to move freely.A dose of PPDP or optimized PLGN formulation corresponding to 7 mg/kg weight was suspended in 700 μL water and promptly given to harnessed rats through the oral route using an oral gavage.
A 200 μL aliquot of blood was withdrawn from the carotid artery of cannulated rats with the help of a 1 mL heparinized syringe at 1, 2, 3, 4, 5, 6, 12, 24, 36, and 48 hours. Plasma was separated swiftly by centrifugation at 5,000 g for 10 minutes. Subsequently, plasma samples were preserved at −20°C until further experimentation.Plasma (50 µL) was poured into a 2 mL microtube and an aliquot (20µg/mL) of naproxen sodium added as an internal standard. Subsequently, 700 µL acetonitrile was added to it as a menstruum for liquid–liquid extraction. After thorough mixing, centrifugation was performed at 5,000 g for 3 minutes. Then, the clear supernatant was carefully shifted to a small-volume HPLC vial and analyzed.
Pharmacokinetic parameters, such as area under the curve (AUC; h·µg/mL), highest piroxicam concentration in plasma (Cmax; µg/mL) and time taken from dose administration to Cmax (Tmax; hours), were computed employing noncompartmental analysis (PK Solver version 2.0; Pharsight, Sunnyvale, CA, USA) following a linear trapezoidal method.21 Corresponding to each time point, the level of significance (p-value) between two means of piroxicam concentrations was determined using Student’s t-test. In general, a p<0.05 suggests presence of a significant difference. On the other hand, p>0.05 represents a nonsignificant difference.
Results and Discussion
Gelatin is a hydrophilic polypeptide that has been used extensively in encapsulation of BCS class II drugs to enhance their solubility and oral bioavailability.12,22 Polymeric nanoparticulated drug-delivery systems also improve solubility, dissolution rate, and oral bioavailability of such drugs.9,23 A drug-laden entity measuring <1 µm is considered a pharmaceutical nanoparticle.19 Electrospraying is an awesome way to obtain drug-loaded polymeric nanoparticles,8,23 so in this research PLGNs were prepared using electrospraying. The recipe of each nanocontainer sample is given in Table 1.
Aqueous solubility outcomes for piroxicam in PLGNs are shown in Figure 1. Solubility in samples I–IV was augmented as the drug:gelatin ratio increased to 1:8 (w:w). This was due to enhanced wettability of the drug by hydrophilic gelatin. As the drug:gelatin ratio increased in samples V–VII — from 1:10 (w:w) to 1:20 (w:w) — an apparent reduction in solubility was noticed. Even so, solubility outcomes were not significantly different (p>0.05) among samples. The presence of a relatively higher proportion of gelatin against the drug in the formulation eventuated in supersaturation more quickly such that no further formulation was dissolved. The drug was enveloped or encapsulated in the gelatin shell, and thus no further drug was dissolved either. For that reason, samples V, VI, and VII, containing relatively larger quantities of gelatin, showed decreased piroxicam solubility. Formulation IV exhibited the highest solubility of piroxicam: 600-fold that of PPDP (94.24±25.65 µg/mL vs 0.16±0.21 µg/mL). Piroxicam solubility afforded by the corresponding physical mixture (PM) was 29.53±24.34 µg/mL, less than that of the optimized formulation. The PM was prepared by simply triturating piroxicam and gelatin at a 1:8 (w:w) ratio using a pestle and mortar.
 |
Figure 1 Effect of composition on aqueous solubility of piroxicam in piroxicam-loaded gelatinnanocontainers). Solubility of piroxicam plain drug powder (PPDP) and physical mixture (PM) in water are also shown. Each value shows the mean ± SD (n=3). |
Piroxicam content in the optimized PLGN formulation was 93.41%±0.56%. The high drug content was due to the solvent-evaporation method used. In this approach, all the constituents of a formulation are first entirely dissolved in a solvent system to get a transparent homogeneous mixture and then subjected to drying, and thus the drug:excipient ratio is not affected, even if some of the solution is spilt accidentally. This method also ensures high content uniformity, as molecular-level mixing is achieved when a clear solution is formed. Drug loading in the optimized formulation was 10.45%±0.06%.Encapsulation efficiency calculated was 66.74%±6.87%. The optimized PLGN formulation was rinsed thrice with absolute ethanol, because the free drug is soluble in absolute ethanol, while gelatin is insoluble in absolute ethanol. As such, free drug was percolated with filtrate, and the encapsulated drug remained preserved inside the gelatin shell. Furthermore, 40% hydroalcoholic solution at 50°C was used for dissolving the residue and leaching of encapsulated drug, because gelatin is soluble in 40% aqueous ethanol at 35°C–40°C.12
The release rate of piroxicam from PLGNs is shown in Figure 2. All formulations showed higher dissolution of the payload than PPDP. The improved release of the drug from the formulations might be credited to enhanced wetting and conversion of piroxicam to the amorphous counterpart by gelatin. Gelatin hinders recrystallization of the drug from solution. The amorphous drug has more surface area exposed to surrounding medium, consequently fostering aiding release . Among all the trialed formulations in this study, PLGN formulation IV, consisting of drug:gelatin (1:8, w:w) exhibited the most expedited release rate. The release rate was about sevenfold that of PPDP (85.12%±10.96% vs 11.81%±5.34%) and threefold that of PM (85.12%±10.96% vs 29.56%±6.52%) in 10 minutes. The release profile of piroxicam pertaining to PM was erratic. The release rate of the drug gradually decreased with increasing drug:gelatin ratios from 1:10 (w:w) to 1:20 (w:w) in formulations V–VII, respectively. This might be ascribed to the existence of a relatively thicker gelatin shell around the core delaying drug release a little. Based on the highest apparent solubility and release rate, PLGN sample IV was selected for further characterization.
 |
Figure 2 Effect of composition on release rate of piroxicam from piroxicam-loaded gelatinnanocontainers. Dissolution with piroxicam plain drug powder (PPDP) and physical mixture (PM) is also shown. Each value shows the mean ± SD (n=6). |
The degree of crystallinity was scrutinized using PXRD and further confirmed by DSC. The PXRD pattern of PPDP represented its particular crystalline nature (Figure 3A). The distinguishing spikes of piroxicam appeared at 8.58°, 11.73°, 14.54°, 16.69°, 17.62°, 18.78°, 21.76°, 22.48°, and 27.40°. The absence of any sharp-pointed projection in the pattern of gelatin confirmed its amorphous nature (Figure 3B). The distinctive barbs of piroxicam were also noticed in the pattern of PM (Figure 3C), which suggested that the drug retained its crystalline disposition in PM. On the other hand, the spikes corresponding to piroxicam vanished in the pattern of PLGN formulation IV (Figure 3D). This suggested that piroxicam lost its inherent crystalline property during the preparation of PLGNs and embraced an amorphous one after loading. The conclusions obtained by DSC analyses were in accord with those on PXRD analyses. A sharp endotherm emulating the melting point of piroxicam was seen at 200°C in the thermogram of PPDP (Figure 4A) which confirmed its particular crystalline disposition. The thermogram of gelatin was devoid of any sharp endotherm (Figure 4B), yet a wide curve sloping downward was seen in the range of 30°C–180°C owing to consumption of heat for vaporizing moisture present in the sample. A sharp endotherm emulating the melting point of the drug at 200°C and the broad curve sloping downward in the range of 30°C–180°C were seen in the thermogram of PM (Figure 4C). This confirmed that piroxicam did not transmute its crystalline disposition in PM. On the other hand, neither of these endothermic behaviours was present in the thermogram of PLGN sample IV (Figure 4D). This suggested that the encapsulated drug was in an amorphous state and the formulation prepared completely dry. Conversion to the amorphous state might be ascribed to the influence of gelatin in the formulation and the preparation method.24
 |
Figure 3 PXRD patterns: (A) piroxicam plain drug powder, (B) gelatin, (C) physical mixture, and (D) piroxicam-loaded gelatinnanocontainers. |
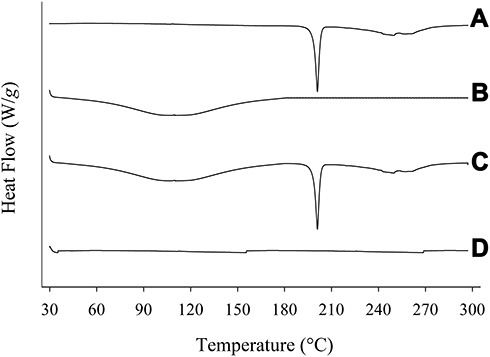 |
Figure 4 DSC: (A) piroxicam plain drug powder, (B) gelatin, (C) physical mixture, and (D) piroxicam-loaded gelatinnanocontainers. |
TGA thermograms recorded in the range of 50°C–550°C are revealed in Figure 5. There was only a 10% loss in weight up to 230°C, due to escape of physically attached moisture. Approximately 3% degradation of PPDP occurred in the range of 50°C–250°C (Figure 5A). Then, 71% further sudden degradation of PPDP was observed in the range of 250°C–300°C. Overall, 88% PPDP was degraded in the range of 50°C–550°C. In the case of gelatin, 7% weight loss was observed in the range of 50°C–200°C (Figure 5B). About 44% further degradation of gelatin was seen in the range of 200°C–300°C. In sum, 67% degradation of gelatin took place in the range of 50°C–550°C. The PM showed 6% weight loss in the range of 50°C–180°C, followed by 56% further weight loss in the range of 180°C–350°C, while total weight loss was about 69% in the range of 50°C–550°C (Figure 5C). In the case of PLGN, only 1% weight loss occurred in the range of 50°C–180°C, the next 40% weight loss took place in the range of 180°C–350°C, and 48% total weight loss was noticed in the range of 50°C–550°C (Figure 5D). Comparison of the TGA thermogram of PM with that of PLGN suggested that the encapsulated drug was thermally more stable. Gelatin (wall material) acts as a protective shield for the encapsulated products (core content) and thereby prolongs the shelflife of the products.13
 |
Figure 5 TGA: (A) piroxicam plain drug powder, (B) gelatin, (C) physical mixture, and (D) piroxicam-loaded gelatinnanocontainers. |
SEM of PPDP (Figure 6A) and gelatin (Figure 6B) showed particles with irregular contours and surface features. Each spherical particle of PLGN formulation IV was bearing a dimple on one side and exhibited an unwrinkled surface (Figure 6C). A pharmaceutical nanocapsule is a nanosized (≤1,000 nm) drug-loaded spherical particle with a core–shell morphology. In Figure 6C, it can be observed that the size of all the particles shown was <1,000 nm. Therefore, particles of electrosprayed PLGN formulation IV were nanocontainers. Smooth of PLGN particles and core–shell morphology is shown in Figure 6D.
 |
Figure 6 SEM: (A) piroxicam plain drug powder (5,000×), (B) gelatin (300×), (C) piroxicam-loaded gelatinnanocontainers (PLGNs) (15,000×), and (D) core–shell morphology of PLGNs. |
FTIR spectra are shown in Figure 7. The spectrum of PPDP revealed sharp distinctive spikes, mainly in the fingerprint region, at 452 cm−1, 527 cm−1, 560 cm−1, 581 cm−1, 611 cm−1, 686 cm−1, 733 cm−1, 758 cm−1, 825 cm−1, 876 cm−1, 936 cm−1, 1,145 cm−1, 1,180 cm−1, 1,345 cm−1, 1,435 cm−1, 1,529 cm−1, and 3,338 cm−1. Other peaks appeared at 1575 cm−1 and 1630 cm−1 (Figure 7A). The absorption band appearing at 3,338 cm−1 was due to valence vibrations of hydroxyl groups (–OH) and secondary amide groups (–NH).25,26 Absorption bands corresponding to conjugating benzene and pyridine rings are situated in the range of 1,650–1,550 cm−1.27 The band appearing at 1,630 cm−1 was due to valence vibrations of the carbonyl group (C=O) of the secondary amides, while a high intensity band at 1,529 cm−1 was because of deformation vibrations of amide groups (–NH). Bands located at 1,575 cm−1 and 14,35 cm−1 were due to valence vibrations (C=C) of benzene and pyridine rings. The presence of sulphur dioxide in piroxicam was confirmed by the bands at 1,345 cm−1 and 1,180 cm−1 arising from the asymmetric and symmetric valence vibrations, respectively. The spikes appearing at 876 cm−1, 825 cm−1, and 758 cm−1 were characteristic peaks of deformation vibrations in the plane (C–H) of aromatic rings present in piroxicam. The FTIR spectrum of gelatin is shown in Figure 7B. All the distinctive spikes of piroxicam mentioned were also witnessed at the same positions in both PM (Figure 7C) and PLGN formulation IV (Figure 7D) spectra. Moreover, the PM spectrum was identical to that of formulation IV; therefore, this suggested nonexistence of interactions between piroxicam and gelatin.
 |
Figure 7 FTIR spectra: (A) piroxicam plain drug powder, (B) gelatin, (C) physical mixture, and (D) piroxicam-loaded gelatinnanocontainers. |
Figure 7 showss the mean piroxicam concentration–time graphs constructed with the data on oral administration of PPDP (Figure 8A) and PLGN formulation IV (Figure 8B). The formulation resulted in higher mean plasma concentrations of piroxicam at all time intervals during 1–48 hours (t-test, p<0.05) than PPDP. Pharmacokinetic attributes, such as AUC, Cmax, and Tmax, are displayed in Table 2. AUC and Cmax with formulation IV were significantly ameliorated compared to those obtained with PPDP (t-test, p<0.05); however, Tmax did not alter significantly (t-test, p>0.05). Compared to PPDP, bioavailability was ~fourfold with PLGN formulation IV. The better bioavailability can be credited to augmented solubility and dissolution of piroxicam, due to improved wetting by gelatin, nanosizing, and transmutation of the inherent crystalline structure of the drug to the amorphous one. As such, this formulation of piroxicam–gelatin (1:8, w:w) might be an important bioavailability-enhancing system for delivery of piroxicam orally.
 |
Table 2 Pharmacokinetic parameters obtained after oral administration |
 |
Figure 8 Piroxicam mean titer in plasma versus time: (A) piroxicam plain drug powder and (B) piroxicam-loaded gelatinnanocontainers. *P<0.05 vs PPDP. |
Conclusion
PLGN formulation IV, consisting of piroxicam and gelatin at a ratio of 1:8 (w:w), provided the most boosted solubility (94.24±25.65 µg/mL) and release rate (85.12±10.96% in 10 minutes). The solubility was 600-fold that of PPDP and threefold that of PM, while release rates was sevenfold and threefold, respectively. Drug content, drug loading, and encapsulation efficiency of this formulation were 93.41%±0.56%, 10.45%±0.06% and 66.74%±6.87%, respectively. The encapsulated amorphous piroxicam exhibited more stability and had no covalent linkage with gelatin. The reinforcement in solubility and dissolution of piroxicam in water could have been due to improved wetting of piroxicam due to the presence of gelatin and conversion of piroxicam from its crystalline structure to the amorphous one in PLGN. The amelioration in dissolution rate and solubility of piroxicam in PLGN led to greater bioavailability of piroxicam. Compared to PPDP, enhancement in bioavailability with PLGN formulation IV was fourfold. Therefore, this formulation is a prospective bioavailability-enhancing drug-delivery system for administration of piroxicam via the oral route.
Funding
The present research was not supported by any external funding.
Disclosure
The authors report no conflicts of interest.
References
1. Chaudhary H, Rohilla A, Rathee P, Kumar V. Optimization and formulation design of carbopol loaded Piroxicam gel using novel penetration enhancers. Int J Biol Macromol. 2013;55:246–253. doi:10.1016/j.ijbiomac.2013.01.015
2. Campione E, Paternò EJ, Candi E, et al. The relevance of piroxicam for the prevention and treatment of nonmelanoma skin cancer and its precursors. Drug Design Dev Ther. 2015;9:5843–5850. doi:10.2147/DDDT.S84849
3. Abdeen A, Abou-Zaid OA, Abdel-Maksoud HA, et al. Cadmium overload modulates piroxicam-regulated oxidative damage and apoptotic pathways. Environ Sci Pollut Res. 2019;26(24):25167–25177. doi:10.1007/s11356-019-05783-x
4. Abdeen A, Aboubakr M, Elgazzar D, et al. Rosuvastatin attenuates piroxicam-mediated gastric ulceration and hepato-renal toxicity in rats. Biomed Pharmacother. 2019;110:895–905. doi:10.1016/j.biopha.2018.11.004
5. Shohin IE, Kulinich JI, Ramenskaya GV, et al. Biowaiver monographs for immediate release solid oral dosage forms: piroxicam. J Pharm Sci. 2014;103(2):367–377. doi:10.1002/jps.23799
6. Penkina A, Semjonov K, Hakola M, et al. Towards improved solubility of poorly water-soluble drugs: cryogenic co-grinding of piroxicam with carrier polymers. Drug Dev Ind Pharm. 2016;42(3):378–388. doi:10.3109/03639045.2015.1054400
7. Tantishaiyakul V, Permkam P, Suknuntha K. Use of DRIFTS and PLS for the determination of polymorphs of piroxicam alone and in combination with pharmaceutical excipients: a technical note. AAPS PharmSciTech. 2008;9(1):95–99. doi:10.1208/s12249-007-9003-y
8. Yousaf AM, Malik UR, Shahzad Y, et al. Silymarin-laden PVP-nanocontainers prepared via the electrospraying technique for improved aqueous solubility and dissolution rate. Braz Arch Biol Technol. 2019;62. doi:10.1590/1678-4324-2019170754
9. Yousaf AM, Kim DW, Oh Y-K, Yong CS, Kim JO, Choi H-G. Enhanced oral bioavailability of fenofibrate using polymeric nanoparticulated systems: physicochemical characterization and in vivo investigation. Int J Nanomedicine. 2015;10:1819–1830.
10. Park JH, Kim DS, Mustapha O, et al. Comparison of a revaprazan-loaded solid dispersion, solid SNEDDS and inclusion compound: physicochemical characterisation and pharmacokinetics. Colloids Surf B Biointerfaces. 2018;162:420–426. doi:10.1016/j.colsurfb.2017.12.017
11. Tahir H, Shahzad Y, Waters LJ, et al. Impact of processing methods on the dissolution of artemether from two non-ordered mesoporous silicas. Eur J Pharm Sci. 2018;112:139–145. doi:10.1016/j.ejps.2017.11.016
12. Yousaf AM, Kim DW, Kim JK, Kim JO, Yong CS, Choi H-G. Novel fenofibrate-loaded gelatin microcapsules with enhanced solubility and excellent flowability: preparation and physicochemical characterization. Powder Technol. 2015;275:257–262. doi:10.1016/j.powtec.2015.02.004
13. Li L, Wang H, Chen M, et al. Butylated hydroxyanisole encapsulated in gelatin fiber mats: volatile release kinetics, functional effectiveness and application to strawberry preservation. Food Chem. 2018;269:142–149. doi:10.1016/j.foodchem.2018.06.150
14. Ahmed S, Ikram S. Chitosan and gelatin based biodegradable packaging films with UV-light protection. J Photochem Photobiol B. 2016;163:115–124. doi:10.1016/j.jphotobiol.2016.08.023
15. Foox M, Zilberman M. Drug delivery from gelatin-based systems. Expert Opin Drug Deliv. 2015;12(9):1547–1563. doi:10.1517/17425247.2015.1037272
16. Kim JS, Park JH, Jeong SC, et al. Novel revaprazan-loaded gelatin microsphere with enhanced drug solubility and oral bioavailability. J Microencapsul. 2018;35(5):421–427. doi:10.1080/02652048.2018.1515997
17. Atay E, Fabra MJ, Martínez-Sanz M, Gomez-Mascaraque LG, Altan A, Lopez-Rubio A. Development and characterization of chitosan/gelatin electrosprayed microparticles as food grade delivery vehicles for anthocyanin extracts. Food Hydrocoll. 2018;77:699–710. doi:10.1016/j.foodhyd.2017.11.011
18. Hani N, Azarian MH, Torkamani AE, Kamil Mahmood WA. Characterisation of gelatin nanoparticles encapsulated with Moringa oleifera bioactive extract. Int J Food Sci Technol. 2016;51(11):2327–2337. doi:10.1111/ijfs.13211
19. Reis CP, Neufeld RJ, Ribeiro AJ, Veiga F. Nanoencapsulation I. Methods for preparation of drug-loaded polymeric nanoparticles. Nanomed Nanotechnol. 2006;2(1):8–21. doi:10.1016/j.nano.2005.12.003
20. Dragomiroiu G, Cimpoiesu A, Ginghina O, et al. The development and validation of a rapid HPLC method for determination of piroxicam. Farmacia. 2015;63(1):123–131.
21. Zhang Y, Huo M, Zhou J, Xie S. PKSolver: an add-in program for pharmacokinetic and pharmacodynamic data analysis in microsoft excel. Comput Methods Programs Biomed. 2010;99(3):306–314. doi:10.1016/j.cmpb.2010.01.007
22. Kumar R, Nagarwal RC, Dhanawat M, Pandit JK. In-vitro and in-vivo study of indomethacin loaded gelatin nanoparticles. J Biomed Nanotechnol. 2011;7(3):325–333. doi:10.1166/jbn.2011.1290
23. Sun R, Shen C, Shafique S, et al. Electrosprayed polymeric nanospheres for enhanced solubility, dissolution rate, oral bioavailability and antihyperlipidemic activity of bezafibrate. Int J Nanomedicine. 2020;15:705–715. doi:10.2147/IJN.S235146
24. Yousaf AM, Mustapha O, Kim DW, et al. Novel electrosprayed nanospherules for enhanced aqueous solubility and oral bioavailability of poorly water-soluble fenofibrate. Int J Nanomedicine. 2016;11:213–221.
25. Cavallari C, Abertini B, González-Rodrı́guez ML, Rodriguez L. Improved dissolution behaviour of steam-granulated piroxicam. Eur J Pharm Biopharm. 2002;54(1):65–73. doi:10.1016/S0939-6411(02)00021-8
26. Lyn LY, Sze HW, Rajendran A, Adinarayana G, Dua K, Garg S. Crystal modifications and dissolution rate of piroxicam. Acta Pharm. 2011;61(4):391–402. doi:10.2478/v10007-011-0037-z
27. Zhang X, Wu D, Lai J, Lu Y, Yin Z, Wu W. Piroxicam/2-hydroxypropyl-β-cyclodextrin inclusion complex prepared by a new fluid-bed coating technique. J Pharm Sci. 2009;98(2):665–675. doi:10.1002/jps.21453
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.