Back to Journals » Drug Design, Development and Therapy » Volume 10
Electroporation-delivered transdermal neostigmine in rats: equivalent action to intravenous administration
Authors Berkó S, Szűcs K, Balázs B, Csányi E ![]() , Varju G, Sztojkov-Ivanov A, Budai-Szűcs M, Bóta J, Gaspar R
, Varju G, Sztojkov-Ivanov A, Budai-Szűcs M, Bóta J, Gaspar R ![]()
Received 22 December 2015
Accepted for publication 17 February 2016
Published 19 May 2016 Volume 2016:10 Pages 1695—1701
DOI https://doi.org/10.2147/DDDT.S102959
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Wei Duan
Szilvia Berkó,1,* Kálmán F Szűcs,2,* Boglárka Balázs,1,3 Erzsébet Csányi,1 Gábor Varju,4 Anita Sztojkov-Ivanov,2 Mária Budai-Szűcs,1 Judit Bóta,2 Róbert Gáspár2
1Department of Pharmaceutical Technology, Faculty of Pharmacy, University of Szeged, Szeged, Hungary; 2Department of Pharmacodynamics and Biopharmacy, Faculty of Pharmacy, University of Szeged, Szeged, Hungary; 3Gedeon Richter Plc., Budapest, 4Dr Derm Clinic of Anti-Aging Dermatology, Aesthetic Laser and Plastic Surgery, Budapest, Hungary
*These authors contributed equally to this work
Purpose: Transdermal electroporation has become one of the most promising noninvasive methods for drug administration, with greatly increased transport of macromolecules through the skin. The cecal-contracting effects of repeated transdermal electroporation delivery and intravenous administration of neostigmine were compared in anesthetized rats.
Methods: The cecal contractions were detected with implantable strain gauge sensors, and the plasma levels of neostigmine were followed by high-performance liquid chromatography.
Results: Both intravenously and EP-administered neostigmine (0.2–66.7 µg/kg) increased the cecal contractions in a dose-dependent manner. For both the low doses and the highest dose, the neostigmine plasma concentrations were the same after the two modes of administration, while an insignificantly higher level was observed at a dose of 20 µg/kg after intravenous administration as compared with the electroporation route. The contractile responses did not differ significantly after the two administration routes.
Conclusion: The results suggest that electroporation-delivered neostigmine elicits action equivalent to that observed after intravenous administration as concerning both time and intensity. Electroporation permits the delivery of even lower doses of water-soluble compounds through the skin, which is very promising for clinical practice.
Keywords: transdermal, electroporation, neostigmine, smooth muscle, contraction, plasma level
Introduction
Since the emergence of electroporation (EP) in the early 1980s, it has received appreciable attention in both chemotherapy and gene therapy. With this method, reversible perturbation of the lipid membrane is induced with electrical pulses. The EP could be induced by a large variety of pulses: high-voltage pulses which are typically short, low-voltage pulses which are typically longer, or a combination of high- and low-voltage pulses.1,2 Electrochemotherapy involves EP of intratumorally or intravenously (iv) administered cytotoxic drugs, and serves as an excellent alternative for the therapy of cutaneous and subcutaneous metastases.3–6 Furthermore, the delivery of gene-based drugs and DNA vaccines by EP enhances the transgene expression in the muscle by two to three orders of magnitude.7 EP also seems to be suitable for extended transdermal drug delivery, modifying the multilamellar lipid bilayers of the stratum corneum.8–10
The morphology and the special composition of the stratum corneum make it an impermeable barrier for water-soluble compounds. The stratum corneum consists of keratin-rich enucleate nonviable cells (corneocytes) embedded in an intercellular matrix of multilamellar lipid bilayers. Only a few drugs are able to pass through the skin without the aid of a penetration enhancer.11 Transdermal delivery of drugs by EP was first suggested by Prausnitz et al12 to enhance their bioavailability. Drugs administered transdermally have higher bioavailability than when they are used orally, thanks to their avoidance of the hepatic first-pass metabolism and gastric acid. On the other hand, transdermal administration protects the gastrointestinal system from direct irritation by the drugs.
Transdermal EP is currently one of the most promising modes of physical enhancement for the noninvasive transdermal delivery of lipophilic, hydrophilic, and charged or neutral molecules or macromolecules. The transdermal transport of drugs with molecular weight up to 40 kDa across the skin can thereby be increased by several orders of magnitude.8,13 Appropriate transdermal transport necessitates the adjustment of the voltage, pulse duration, and number of series of impulses.14
In the present study, the model drug chosen for EP was neostigmine methylsulfate, a reversible acetylcholinesterase inhibitor and an inducer of gastrointestinal motility. Neostigmine has only poor bioavailability after oral administration; its effective parenteral dose for humans is 0.5 to 2.0 mg, while the equivalent oral dose should be 30 mg or more, but large oral doses may cause toxic effects.15 Neostigmine methylsulfate (molecular weight: 334.39 g/mol, log P: -2.2) is not able to permeate through the skin because of its low penetration through the stratum corneum.
We set out to investigate the gastrointestinal effects of repeated transdermal administration of neostigmine by EP and compare the systemic effects of EP-delivered and iv-administered neostigmine. The plasma neostigmine levels were followed in both cases.
Materials and methods
Housing and handling of the animals
All animals were treated in full accordance with the European Communities Council Directives (86/609/ECC) and the Hungarian Act for the Protection of Animals in Research (XXVIII.tv.32.§). All experiments involving animal subjects were carried out under sterile conditions with the approval of the Hungarian Ethical Committee for Animal Research (registration number: IV/198/2013).
All animal-handling procedures were performed according to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and followed the guidelines of the Animal Welfare Act. Sprague-Dawley rats (Charles-River Laboratories, Budapest, Hungary) were housed at 22°C±3°C in a relative humidity of 30%–70% under a 12-hour light/dark cycle. Pellet food (Charles-River Laboratories) and tap water were provided ad libitum.
Experimental design and contractility measurement
The experimental design is represented in Figure 1. Male Sprague-Dawley rats (6–7 weeks old, body weight: 190–220 g) participated either in repeated iv (n=10) or transdermal EP treatment (n=8) with neostigmine methylsulfate (Teva Pharmaceutical Industries Ltd., Gödöllő, Hungary). Throughout the experiment, the animals were kept under deep anesthesia with a combination of intraperitoneal ketamine and xylazine (36 and 4 mg/kg, respectively). The jugular vein was cannulated for drug administration and the carotid artery for blood collection. The abdominal cavity was opened and an implantable force/displacement transducer or strain gauge (EGG-01 SG; Experimetria, Budapest, Hungary) was sutured onto the surface of the cecum. The animals were then placed onto a 37°C heated operating table (EXP-D-TC/MA-02; Experimetria) in order to maintain the body temperature. The strain gauge converted the cecal mechanical contractions to electrical signals. The amplified signals were recorded and analyzed with an online computer by the S.P.E.L. Advanced ISOSYS Data Acquisition System (Experimetria). The basal contractions were detected for at least 10 minutes before the first dose of neostigmine. The area under the curve of these 10 minutes was used as the control, and the stimulation was calculated as a percentage of this initial period. Following the addition of each concentration of neostigmine, the area under the curve of a 10-minute period was evaluated. The effects were expressed as percentages of the basal activity. Three-parameter, symmetrical sigmoidal concentration–response curves were fitted. The ED50 values, that is, the doses that provoke a response half way between the basal and the maximal response, were calculated and the unpaired t-test was applied for statistical analysis with the Prism 5.0 computer program (GraphPad Software, Inc., La Jolla, CA, USA).
 | Figure 1 The experimental design for the investigation of the effect of neostigmine by EP administration. |
EP protocols
Before EP, the rats were shaved on the abdominal surface and the remaining hair was removed with Veet depilatory cream (Reckit Benckiser, Bristol, UK).
Neostigmine was administered in cumulative doses of 0.2, 0.67, 2.0, 6.7, 20.0, and 66.7 μg/kg. For EP, neostigmine methylsulfate was dissolved in a 2.5% hydroxyethylcellulose-containing hydrogel (Natrosol™ HEC, Ashland Inc., Covington, KY, USA). The final volume of the neostigmine-containing hydrogel was 25 μL and the pH of the hydrogel was 6.95.
The Mezoforte Duo EP device (Serial Number Mez 120905-D), produced by Dr Derm Equipment Ltd. (Budapest, Hungary), was provided by the Derm Clinic of Anti-Aging Dermatology, Aesthetic Laser and Plastic Surgery (Budapest, Hungary). The device operates on the basis of a pulsed electromagnetic field. The polypropylene-covered treating headpiece contains a plate electrode 25 mm in diameter indirectly contacting with the treated surface. Modulation was achieved with 1,800 Vpp (peak-to-peak voltage) pulses with a duration of 5 milliseconds, followed by a 20 milliseconds break. This pulse treatment was utilized on the abdominal surface for 2 minutes (2,200 periods/min), during which period the applied neostigmine-containing hydrogel was absorbed through the skin, with only a dry film layer remaining on the surface of the skin. A 10-minute waiting period ensued before the next EP-delivered neostigmine dose.
For iv administration, a neostigmine methylsulfate injection (Stigmosan™, Pharmamagist, Budapest, Hungary) was diluted in physiological saline solution. The cumulative neostigmine doses and recording protocol were the same as in the EP protocol. The period for iv administration was ~20–30 seconds, and the recording of contractions was started 2 minutes after the start of the neostigmine administration.
HPLC analysis
Chemicals and reagents for HPLC
Analytical grade sodium dihydrogenphosphate and disodium hydrogenphosphate dodecahydrate were obtained from Sigma-Aldrich (Budapest, Hungary). High-performance liquid chromatography (HPLC) grade glacial acetic acid, acetonitrile, and methanol were purchased from Merck (Darmstadt, Germany). Benzyltri-n-propylammonium chloride (internal standard) and neostigmine methylsulfate were from Sigma-Aldrich. The water used was purified and deionized with the Milli-Q system (Millipore, Milford, MA, USA).
HPLC system and conditions
The HPLC apparatus consisted of a Shimadzu system (Shimadzu Corporation, Kyoto, Japan) equipped with a solvent delivery system (LC-20AD), a DGU-20A3 on-line degasser, a SIL 20A HT autoinjector, a CTO-20A column oven, an SPD-M20A photodiode-array detector, and a CBM-20A system controller. The chromatographic system was equipped with a Rheodyne Model 7125 injector (Rheodyne Corp., Cotati, CA, USA) with a 20 μL loop. The system control and data acquisition were performed with Shimadzu LC solution software (Shimadzu Corporation).
The chromatographic separations were performed on a PhenoSphere SCX (5 μm, 150×4.6 mm) analytical column (Phenomenex Inc., Budapest, Hungary), protected by a 5 μm guard column. The column temperature was maintained constant at 35°C. Separations were performed in isocratic mode. The mobile phase used for the separation consisted of 0.5 M NaH2PO4 buffer (pH =3.5):acetonitrile =45:55 (v/v) pumped at a flow rate of 1 mL/min. The mobile phase was filtered by a Millipore vacuum filtration system equipped with a 0.45 μm pore size filter and degassed by ultrasonication. The detection wavelength was 200 nm.
Preparation of standard solutions and calibration samples
A stock solution of neostigmine methylsulfate (1 mg/mL) was prepared in the HPLC mobile phase and diluted with mobile phase to obtain a series of working standard solutions with concentrations ranging from 3 to 50 μg/mL. The internal standard stock solution (1 mg/mL in methanol) was further diluted to 50 μg/mL with water. Stock solutions were stored at −20°C, and working solutions were kept refrigerated at 2°C–6°C. The working solutions were used to prepare seven calibration samples in blank plasma (0.3, 1, 3, 5, 10, 30, and 50 μg/mL).
Sample extraction procedure
The blood samples were collected via the previously inserted polyethylene cannula 2 minutes after each EP or iv neostigmine treatment. The samples were centrifuged at 5,000× g for 10 minutes to obtain serum. Serum samples were stored at −70°C until HPLC analysis.
An aliquot of plasma (200 μL) was combined with 20 μL of internal standard working solution. The mixture was loaded into a Strata-X-CW 33u solid-phase extraction tube (Phenomenex Inc.) previously conditioned with 0.5 mL 5% glacial acetic acid in methanol, 0.5 mL methanol, and 0.5 mL 0.2 M Na2HPO4 (pH =9). The cartridge was washed with 1 mL of a 0.2 M Na2HPO4 (pH =9):methanol =90:10 mixture (v/v). The neostigmine methylsulfate retained in the cartridge was eluted with 1 mL 5% glacial acetic acid in methanol into a glass tube. The eluate was evaporated to dryness under a stream of nitrogen at 40°C and reconstituted with 100 μL mobile phase, and 20 μL was then injected into the HPLC system.
Calibration curve
The seven-point calibration curves, prepared in triplicate, exhibited good linearity (R2=0.9984) in the concentration range 0.3–50 μg/mL for neostigmine methylsulfate. The recovery of neostigmine methylsulfate from the plasma samples was 79.84%±3.52%. The limits of quantification and detection were 0.05 and 0.1 μg/mL, respectively.
Results
Neostigmine increased the cecal contractions after both iv and transdermal EP administration (Figure 2A). The calculated ED50 value for neostigmine was approximately twice as high after iv administration as compared with the EP route. However, this difference is statistically not significant. The calculated maximum increase in contractions was only 20% lower after EP administration than that after iv treatment (Table 1). The cecal contractile responses to iv- or EP-administered neostigmine did not differ significantly in the investigated dose interval (0.2–66.7 μg/kg). The cecal smooth muscle did not show any response to EP stimulation without neostigmine (Figure 2B).
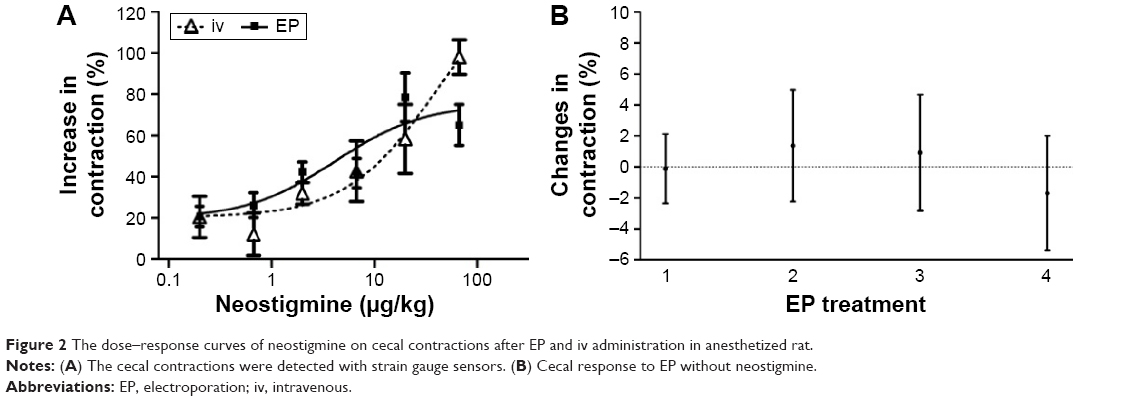 | Figure 2 The dose–response curves of neostigmine on cecal contractions after EP and iv administration in anesthetized rat. |
 | Table 1 The doses for the ED50 and Emax effects of neostigmine on cecal contractions after EP and iv administration in rat |
A representative chromatogram for neostigmine plasma determination is presented in Figure 3. The chromatogram of blank rat plasma spiked with 10 μg/mL neostigmine gave a clear neostigmine peak with a retention time of 7.73±0.31 minutes (Figure 3A). In neostigmine-treated rats, a smaller but identifiable peak was found at the same retention time as that of the neostigmine-spiked blank rat plasma (Figure 3B). The magnitudes of the peak of the internal standard were the same in both cases, indicating the reliability of the method. The HPLC analysis of the neostigmine plasma levels revealed that the two routes of administration resulted in very similar drug concentrations. The neostigmine plasma concentrations were the same after both iv and EP administrations; statistically there was no difference between the two plasma curves (Figure 4).
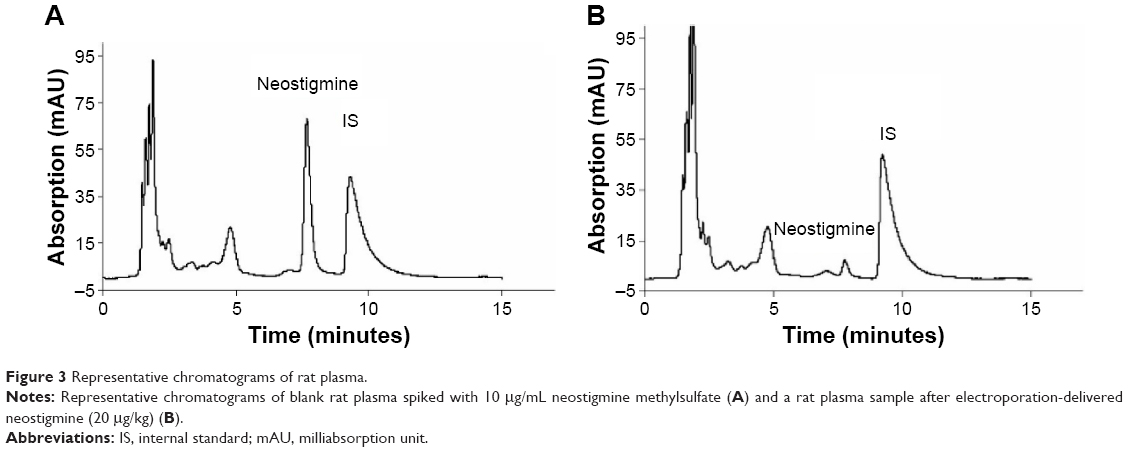 | Figure 3 Representative chromatograms of rat plasma. |
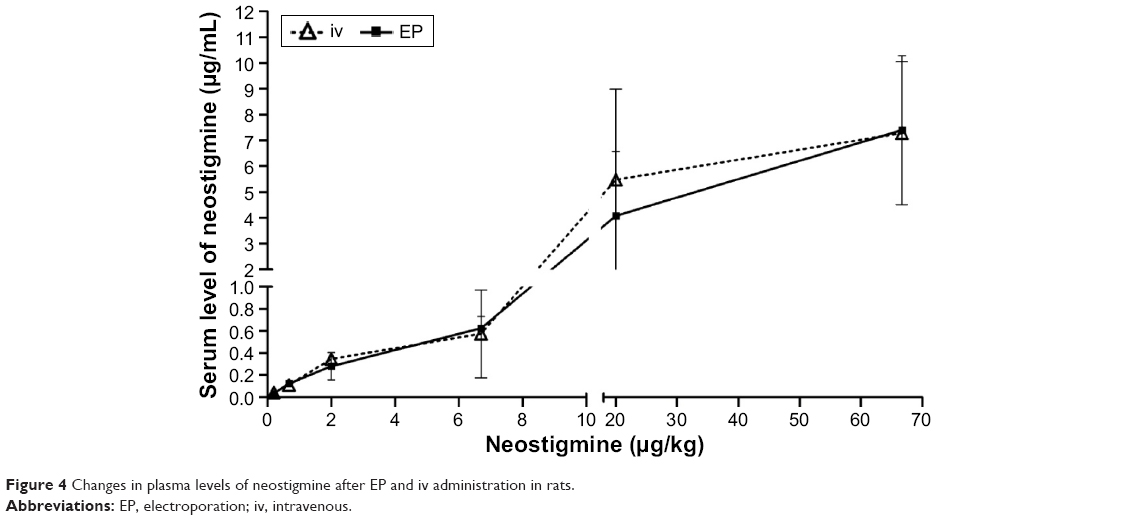 | Figure 4 Changes in plasma levels of neostigmine after EP and iv administration in rats. |
When the cecal contractions were represented as a function of the plasma concentration of neostigmine, the concentration–response curves were similar to the dose–response curves (Figure 5).
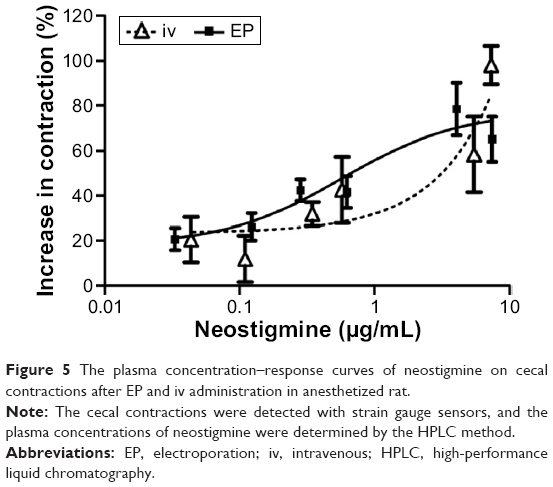 | Figure 5 The plasma concentration–response curves of neostigmine on cecal contractions after EP and iv administration in anesthetized rat. |
Discussion
There is an increasing need for new and noninvasive routes of drug administration in clinical practice. Moreover, there is a demand for fast and safe drug administration for acute care and even chronic treatment. The increasing numbers of peptide drugs have also given rise to a requirement for noninvasive therapy.16 Novel biological therapies involve peptide compounds, mainly monoclonal antibodies, and the antidiabetic or ovulation induction therapies also apply peptide hormones. These peptide drugs are not applicable via oral administration because of their sensitivity to peptidases in the gastrointestinal tract. The most frequent route of administration of such compounds is subcutaneous administration, but many patients fear the needles, especially in self-treatment.
EP is a method of noninvasive drug administration which is widely used for cosmetic interventions,17,18 delivery of genes into cells19 and experimental animals,20,21 or the delivery of anticancer agents into tumor cells.22 Attempts have been made to apply EP for transdermal drug delivery in systemic therapies.9 A number of papers have described the local action of EP-delivered drugs,23 but only a few reports can be found concerning the systemic applicability of this method for drug delivery.9,10,12
The electrodes and the electric pulse parameters are the key information in EP applications. Another way to apply electric pulses for drug delivery is iontophoresis. Iontophoresis applies a low intensity (<0.5 mA/cm2) electric field for a period of minutes or hours with low voltage (5–10 V or less). Direct current is usually used, but pulsed direct current can enhance the efficacy of iontophoresis if the pulse frequency is not too high.24 EP makes use of short (<1 second) and high-voltage pulses (>50 V), creating an aqueous pathway for transport through lipid bilayers of the skin.25 In our EP method, an amplitude-modulated sine wave radiofrequency current was utilized as the source of pulses for EP. The high voltage (1,800 Vpp) and the high pulse frequency (2,200/min) applied exclude the possibility of neostigmine being delivered by iontophoretic processes in our system.
We detected the systemic action of EP-delivered neostigmine methylsulfate with plasma drug level control. In anesthetized rats, no adverse effects were experienced by EP. Neostigmine methylsulfate is a highly water-soluble compound that is unable to penetrate through the barriers of the dermal lipid layers. Since neostigmine exerts a well expressed contracting effect on the gastrointestinal smooth muscle, we selected a part of the large intestine (the cecum) as the target organ of our measurements, and the contraction-increasing effects and plasma levels of EP-delivered neostigmine were compared with its effects after iv administration. For the detection of plasma levels of neostigmine, an HPLC setup was employed. Although the plasma concentration of neostigmine was relatively low, the sensitivity of the method was sufficient for qualitative determination.
Somewhat surprisingly, no difference in cecal-contracting effect or plasma level of neostigmine was observed after 2 minutes of EP and iv administration. EP may therefore be considered to be equivalent to iv administration for systemic drug delivery. Moreover, the action of neostigmine could be adjusted through the use of relatively small doses, which suggests that distinguished dosages can be applied via EP.
We also proved that the plasma concentrations of neostigmine correlate with the increases in cecal contractions after both iv and EP administrations. This correlation is further evidence that EP-administered neostigmine reaches the circulation and is solely responsible for the measured physiological response.
EP can induce muscle contractions as an adverse effect. However, in our system, the EP did not have any direct contracting effect on the smooth muscle, and thus all the registered changes in the cecal contractions were consequences of the drug action.
There are some limitations of our study. The period for iv administration was ~20–30 seconds, and the recording of contractions was therefore started 2 minutes after the start of the injection in order to make the time frame similar to that of EP. The plasma concentration of an iv-administered drug is highest immediately after the injection, but the blood sample was collected after 2 minutes of iv delivery. This means that we missed the highest plasma peaks after iv administration, which may explain why the plasma curves are similar after iv and EP drug delivery. For the contractions, the neostigmine should be distributed in the large intestine tissue; this is not an immediate action, and we therefore suppose that any difference between the maximum effects of iv and EP drug delivery is not significant, or the distribution process equilibrates the difference. Another limitation is that the impulse of the applied EP parameters was constant; we did not test the effects of different voltages or pulse durations on the bowel action and plasma level of neostigmine. Further studies are necessary to reveal the threshold voltage needed to induce the systemic absorption of the drugs. However, with regard to the earlier and present findings, we have a good reason to suspect that EP works on the principle of all or nothing, opening all the biological barriers in the tissues or cell membranes at once.26 Additionally, we do not know what size of plate electrode should be used in humans. A diameter proportional to that used in rats would certainly be too large. On the other hand, the drug concentration of the hydrogel and the area of the treated surface can be modified easily to find the optimal setup for human therapy. Drugs with different chemical properties should also be tested with this delivery system in order to assess the applicability of this method.
The potential clinical application of EP may involve the rapid therapy of a life-threatening condition to replace iv treatment (eg, epileptic seizure or a cardiac arrest) and may make protein drug delivery (eg, insulin or low molecular weight heparin) easy and painless during self-administration by the patients.
Conclusion
We conclude that transdermal EP-delivered neostigmine elicits action equivalent to that of intravenous administration both in time and intensity. Through EP, even low doses of water-soluble compounds can be delivered through the skin, which is promising for clinical practice. Although our findings are potentially promising for the clinical application of EP for drug delivery, a number of questions remain to be clarified before human therapy can be initiated.
Acknowledgments
The technical support of Zoltánné Csiszár is highly appreciated. Furthermore, we wish to thank Dr Derm Equipment Ltd. for providing the Mezoforte Duo Mez 120905-D device. Special thanks to Veronika Verseghy for taking photos.
Disclosure
The authors report no conflicts of interest in this work.
References
Neumann E, Kakorin S, Toensing K. Principles of membrane electroporation and transport of macromolecules. Methods Mol Med. 2000;37:1–35. | ||
Miklavcic D, Pucihar G, Lebar A, Krmelj J, Towhidi L. Advanced Electroporation Techniques in Biology and Medicine. Boca Raton, FL: CRC Press; 2010. | ||
Marty M, Sersa G, Garbay JR, et al. Electrochemotherapy – an easy, highly effective and safe treatment of cutaneous and subcutaneous metastases: results of ESOPE (European Standard Operating Procedures of Electrochemotherapy) study. Ejc Suppl. 2006;4(11):3–13. | ||
Sersa G, Cemazar M, Snoj M. Electrochemotherapy of tumours. Curr Oncol. 2009;16(2):34–35. | ||
Campana LG, Testori A, Mozzillo N, Rossi CR. Treatment of metastatic melanoma with electrochemotherapy. J Surg Oncol. 2014;109(4):301–307. | ||
Spratt DE, Gordon Spratt EA, Wu S, et al. Efficacy of skin-directed therapy for cutaneous metastases from advanced cancer: a meta-analysis. J Clin Oncol. 2014;32(28):3144–3155. | ||
Mir LM, Bureau MF, Rangara R, Schwartz B, Scherman D. Long-term, high level in vivo gene expression after electric pulse-mediated gene transfer into skeletal muscle. C R Acad Sci III. 1998;321(11):893–899. | ||
Denet AR, Vanbever R, Preat V. Skin electroporation for transdermal and topical delivery. Adv Drug Deliv Rev. 2004;56(5):659–674. | ||
Blagus T, Markelc B, Cemazar M, et al. In vivo real-time monitoring system of electroporation mediated control of transdermal and topical drug delivery. J Control Release. 2013;172(3):862–871. | ||
Denet AR, Preat V. Transdermal delivery of timolol by electroporation through human skin. J Control Release. 2003;88(2):253–262. | ||
Prausnitz MR, Elias PM, Franz TJ. Skin barrier and transdermal drug delivery. In: Bolognia JL, Jorizzo JL, Schaffer JV, editors. Dermatology. 3rd ed. Philadelphia: Elsevier Health Sciences; 2012:2065–2073. | ||
Prausnitz MR, Bose VG, Langer R, Weaver JC. Electroporation of mammalian skin: a mechanism to enhance transdermal drug delivery. Proc Natl Acad Sci U S A. 1993;90(22):10504–10508. | ||
Lombry C, Dujardin N, Preat V. Transdermal delivery of macromolecules using skin electroporation. Pharm Res. 2000;17(1):32–37. | ||
Wong TW. Electrical, magnetic, photomechanical and cavitational waves to overcome skin barrier for transdermal drug delivery. J Control Release. 2014;193:257–269. | ||
Mann DE. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 6th ed. New York: Macmillan; 1981. | ||
Lewis AL, Richard J. Challenges in the delivery of peptide drugs: an industry perspective. Ther Deliv. 2015;6(2):149–163. | ||
Zhang L, Lerner S, Rustrum WV, Hofmann GA. Electroporation-mediated topical delivery of vitamin C for cosmetic applications. Bioelectrochem Bioenerg. 1999;48(2):453–461. | ||
Redziniak G. [Liposomes and skin: past, present, future]. Pathol Biol. 2003;51(5):279–281. | ||
Young JL, Dean DA. Electroporation-mediated gene delivery. Adv Genet. 2015;89:49–88. | ||
Gothelf A, Eriksen J, Hojman P, Gehl J. Duration and level of transgene expression after gene electrotransfer to skin in mice. Gene Ther. 2010;17(7):839–845. | ||
Gothelf A, Hojman P, Gehl J. Therapeutic levels of erythropoietin (EPO) achieved after gene electrotransfer to skin in mice. Gene Ther. 2010;17(9):1077–1084. | ||
Kulbacka J, Daczewska M, Dubinska-Magiera M, et al. Doxorubicin delivery enhanced by electroporation to gastrointestinal adenocarcinoma cells with P-gp overexpression. Bioelectrochemistry. 2014;100:96–104. | ||
Mir-Bonafe JM, Vilalta A, Alarcon I, et al. Electrochemotherapy in the treatment of melanoma skin metastases: a report on 31 cases. Actas Dermosifiliogr. 2015;106(4):285–291. | ||
Rawat S, Vengurlekar S, Rakesh B, Jain S, Srikarti G. Transdermal delivery by iontophoresis. Indian J Pharm Sci. 2008;70(1):5–10. | ||
Jadoul A, Bouwstra J, Preat VV. Effects of iontophoresis and electroporation on the stratum corneum. Review of the biophysical studies. Adv Drug Deliv Rev. 1999;35(1):89–105. | ||
Luft C, Ketteler R. Electroporation knows no boundaries: the use of electrostimulation for siRNA delivery in cells and tissues. J Biomol Screen. 2015;20(8):932–942. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.