Back to Journals » Medical Devices: Evidence and Research » Volume 14
Efficient Quality Management in MedTech Start-Ups (Based on ISO 13485)
Authors Kheir O ![]() , Smedts S, Jacoby A, Verwulgen S
, Smedts S, Jacoby A, Verwulgen S
Received 19 May 2021
Accepted for publication 9 July 2021
Published 15 October 2021 Volume 2021:14 Pages 313—319
DOI https://doi.org/10.2147/MDER.S320583
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Omar Kheir, Sam Smedts, Alexis Jacoby, Stijn Verwulgen
Department of Product Development, Faculty of Design Sciences, University of Antwerp, Antwerp, Belgium
Correspondence: Omar Kheir Tel +31657019567
Email [email protected]
Abstract: Start-ups that are involved in the development of medical devices play a key role in innovation, yet among the several limitations they encounter, they suffer a lack of knowledge in quality management. This lack may create a challenge that could hinder or limit their success. This paper seeks to present an overview and act as a guideline for the required documentation to establish a quality management system for start-ups in medical devices development, based on a case-studied start-up, in accordance with ISO 13485. Using this case study along with the actual ISO 13485 standard document, can support medical devices start-ups in their ISO compliance and certification journey.
Keywords: ISO, innovation, medical, start-up, quality, compliance, documentation
Introduction
Review of Medical Devices’ Regulatory Requirements
There is no doubt that the role of medical devices has become pivotal in safeguarding the wellbeing of the progressively health-conscious population. A medical device can include any instrument, apparatus, implement, machine, appliance, implant, reagent for in vitro use, software, material or other similar or related articles, intended by the manufacturer to be used, alone or in combination, for human beings.4 Today, there are an estimated 2 million different kinds of medical devices on the world market, categorized into more than 22, 000 generic devices groups.2 At the same time, medical error is considered among the top leading causes of death in the United States, ranked third after heart disease and cancer and accounting for over 250,000 deaths annually.1 Therefore, medical devices are governed by strict regulations which are set by the different countries in which manufacturers are intending to launch their products. Most importantly are the European Commission (EC) Medical Device Directives and the United States Food Drug Administration regulations.3
In the European Union (EU), medical devices that meet the essential requirements of all relevant European Medical Device Directives are marked with CE (Conformité Européenne) so that they can be sold throughout the European Economic Area (EEA). As a prerequisite to this, medical devices in the EU must undergo a conformity assessment, which may involve an audit by a notified body for the Quality Management System (QMS) of the manufacturer and a possible review of the technical documentation on the performance and safety of the device, to demonstrate that they meet legal requirements and ensure they are safe and perform as intended. Nevertheless, as described by the EC directives, for medical devices that fall under class I, the conformity assessment does not involve a notified body, while for all other classes (IIa, IIb, III) a notified body must be involved. The reference number of the notified body appears on the CE mark when applicable, and each class is susceptible to a relevant set of EC regulations. Figure 1 represents few examples of which medical devices can fall under each class while indicating how risk level increases with devices moving to higher classes, as per The Danish Medicines Agency7.
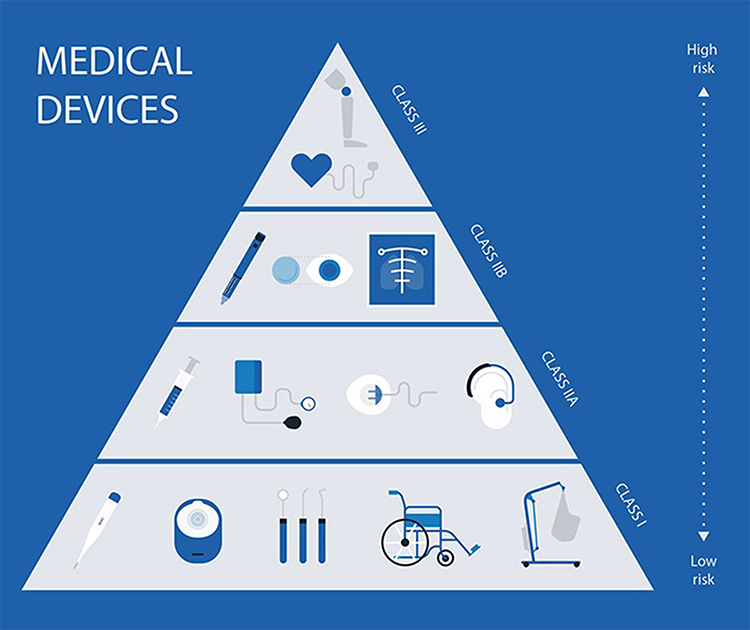 |
Figure 1 Medical devices classes as per EC regulations. Reproduced from Danish Medicines Agency. Medical devices; 2020. Available from: https://laegemiddelstyrelsen.dk/en/devices/#.7 |
Role of ISO 13485
Standardization and/or certification implementation processes have been receiving notable attention from manufacturing companies over the last few years.6 This is mostly because compliance to standards assures the alignment with the safety and quality standards throughout the entire device production line.5 For the medical devices development (MDD) field, ISO 13485 was founded to align the quality management processes of medical devices’ developers to best practice and to the increasingly stringent regulatory requirements that are highly concerned with every step of a product’s life cycle, including service and delivery. This internationally agreed standard was initially introduced in 2003 and its latest version was published in 2016. Also, the newly released Medical Device Regulation (MDR) based on the EU regulation 2017/745, which replaces the Council Directive 93/42/EEC of 14th June 1993 concerning medical devices, obliges to have a QMS with relevant requirements that can be mapped to certain clauses of ISO 13485.9
The role of the ISO 13485 standard is to set the general requirements for a QMS that can be used by an organization for the design, the development, the production, the installation, and the maintenance of medical devices, as well as the design, the development, and the provision of relative services.10 In addition to the organizations that are involved in MDD, other stakeholders such as certification institutions use this ISO standard in order to assess the ability of a MDD organization to meet customers’ and regulatory requirements. The requirements of this QMS are complementary to the technical conditions for the products that are necessary to meet customers’ and applicable regulatory requirements for safety and performance.4 Also, being certified to ISO 13485 does not fulfil the requirements of the FDA or the EC directives but it aligns and serves as a framework for the organization’s management system towards complying with the regulatory requirements, and which is the ultimate objective of the developers of medical devices in order to sell their products.
Establishing the ISO 13485 Documentation
Similar to any ISO certification, organizations in the MDD field that are intending to obtain the ISO 13485 certification should first educate themselves on the requirements of this standard and then implement a customized management system in line with these requirements. The reference document for this management system should be the Quality Manual (QM), in which the organization states its goals, processes, procedures and forms with respect to its design and production process in compliance with this standard’s requirements. This can be developed internally by the organization’s team or externally through a hired consultant with expertise in ISO 13485. This second option may constitute an obstacle to start-ups who usually suffer a structural lack of tangible and intangible resources,11 even though they have a key role in the innovation process since they have direct access to the latest technological and scientific advancements, emerging from academic activities. Once all the processes and procedures are established and referenced in the QM, the organization can seek a certification body for external audit and actual certification if the requirements’ implementation were satisfied. Figure 2 illustrates the hierarchy for the documentation required by the QMS where the QM is the primary document (Level A) cascading down to all remaining needed documents which are usually way more detailed and can be divided into two other levels, documented procedures (Level B), and work instructions or records (Level C). A form is a kind of document yet once it is filled out it becomes a record; hence it is an example of a (Level C) document.
 |
Figure 2 Typical QMS documentation hierarchy. Adapted from Food Control, 18(9), Aggelogiannopoulos D, Drosinos EH, Athanasopoulos P. Implementation of a quality management system (QMS) according to the ISO 9000 family in a Greek small-sized winery: a case study. 1077–1085s, Copyright (2007), with permission from Elsevier.12 |
The extent of documentation of the management quality system can differ in each organization. Nevertheless, as per the ISO 13485:2016 standard, it should not fail to include:
a) Documented statements of a quality policy and quality objectives.
b) A quality manual.
c) Documented procedures and records required by this international standard.
d) Documents, including records, determined by the organization that are necessary to ensure the effective planning, operation, and control of its processes.
e) Other documentation specified by applicable regulatory requirements.
As for the quality manual, it should outline the structure of the documentation used in the QMS. As per the ISO 13485:2016 standard, it should include:
a) The scope of the QMS, including details or justifications for any exclusion or non-application.
b) The documented procedures for the QMS, or reference to them.
c) A description of the interaction between the processes of the QMS.
This paper describes the documentation to be established for the QMS of an ISO 13485 certified start-up by highlighting the established processes, procedures, and forms to fulfil these ISO standard requirements. The study by Klute-Wenig and Refflinghaus8 confirmed that the processes in start-ups and micro-enterprises, which may constitute of few developers, are rather informal or not yet specified, even though these companies are often in a phase of strong growth. According to the same study, these small companies face the difficulty to decide if and how a QMS should be implemented, how the corresponding requirements can be fulfilled, and which requirements are relevant. Accordingly, the content and the conclusion of this paper can assist start-ups specifically in their ISO 13485 certification track as it will explain the major steps, along with the needed documentation, that should be established in line with the standard’s requirements and registered into the QM.
Materials and Methods
The following case study covers a MDD start-up based in Belgium, specialized in the development and sales of orthopedic braces, supports, and post-operative bandages. Their products are being sold through distributors in most European countries and even overseas. They provide in-house management for all their processes in order to support their flexible operations and to easily respond to specific needs in the market. This is demonstrated through the growing number of products and services they have been offering successively. Even though the company commenced its commercial activities with CE marked devices in 2009, it has achieved the ISO 13485 certification in 2016 to further align the internal processes to international standards and to stimulate the quality assurance process. This was successfully implemented internally by hired quality personnel in charge and the direct involvement of the top management.
To pursue this study, two comprehensive meetings were held with the design specialist of this MDD start-up who has shared and discussed with the researcher the adopted design and QMS processes. In both meetings, the researcher and the design specialist discussed the formation of the QMS documentation and used printouts to link and map the documentation involved. In the upcoming section, their adopted QM is sketched and discussed along with references to other quality documents which the case-studied start-up have established to reach ISO 13485 compliance and certification.
Results
Table 1 describes the content of the QM with respect to the case-studied start-up and maps the components against the referenced quality documents. The adopted QM is divided into 5 sections and is titled ”General Design Procedure (GDP)” in which the start-up lists all the documentation required by ISO 13485 in relation to the utilized design process. The manual starts with a “scope” which entails the scale of applicability and the boundaries of this document: “This document describes the general design process for the R&D department that must work according to ISO 13485ʹ. The scope needs to be extensively discussed and agreed upon with the certification body, as it will be stated on the ISO 13485 certificate. The second section of the manual describes the “purpose” which entails the objective(s) from this document:
The aim for this document is to provide the R&D method structure. The working method is based on the ISO 13485 standards and forms. This makes internal communication better and the work can be traced and assessed. Errors are detected preventively during the process. In addition, the document is intended to integrate the ISO standards in a comprehensible and intuitive way into the work process.
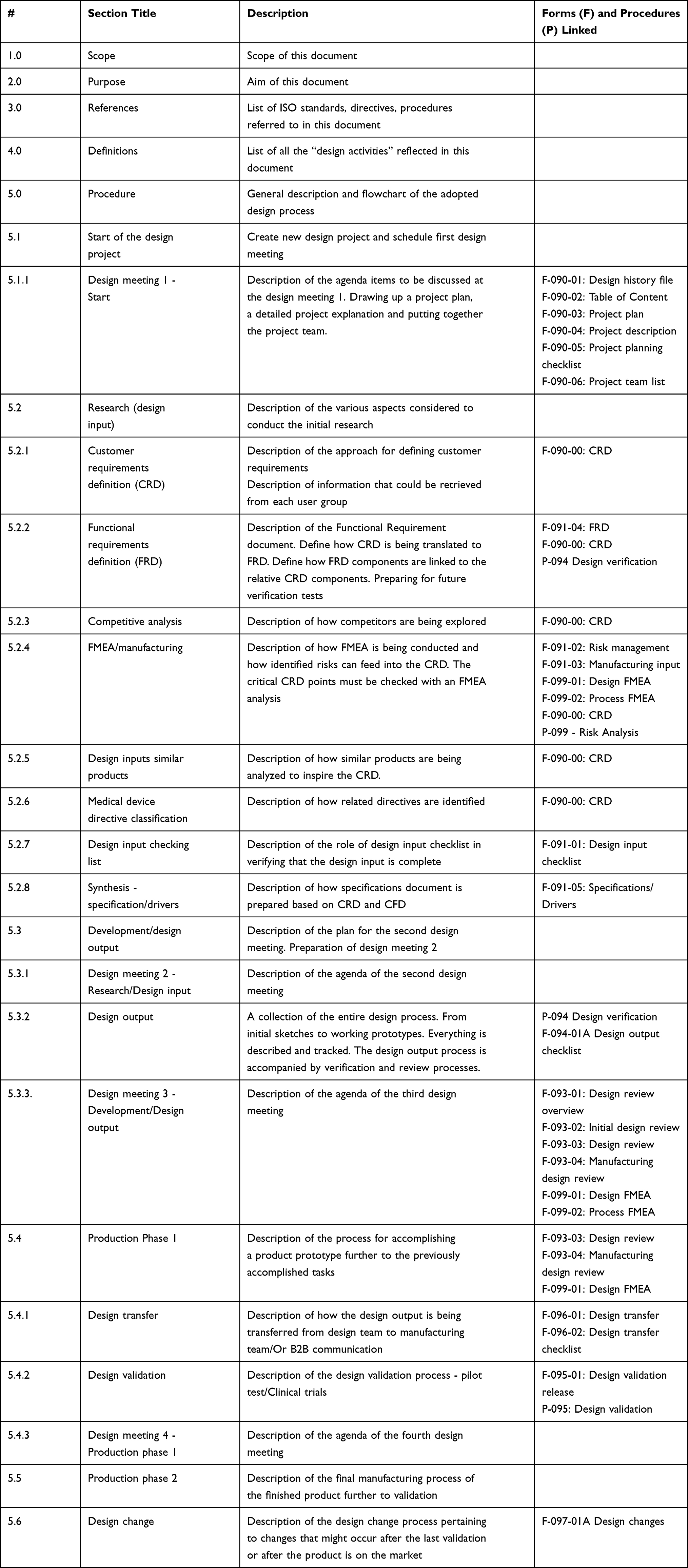 |
Table 1 Description of Content of the QM in the Case-Studied Start-Up |
The purpose section in this document is important to identify the intention of the start-up behind this document and the way it will help in meeting the ISO 13485 requirements. As described in the scope statement, the case-studied start-up has chosen to combine between the adopted general design process with the requirements of the ISO standard because this will ease the communication flow and will integrate the ISO requirements with the various steps of the design process to ensure comprehensive requirements’ implementation.
The rest of the document breaks down the design and development procedure into steps while each step fulfils the relevant requirements of the standard and refers, when applicable, to a separate procedure or process that should be executed, or a form that should be completed. The manual’s content map in Table 1 lists the 3 procedures/processes and 23 forms that were established to comply with the ISO requirements, while being reflected into the design process. Start-ups should not be bound to any number of documents as long as the requirements are implemented and can be proven whenever needed to the notified bodies. In Table 1, each required step of the design process has been briefly described in the second column while referring to the related QMS procedures and forms in the third column.
The main purpose of the QM is to provide written evidence that the start-up is committed to meet all the requirement of the standard. Some requirements may require a different documentation as is the case in our case-studied start-up where different requirements were branched out into separate documents. Nevertheless, it is most crucial that the manual shows how does all the requirements interact with each other. Having a flowchart or a design process, as in the case of the start-up where all the requirements are linked to the design process, can provide a better perception, both internally for the staff and externally for other stakeholders such as consultants and notified bodies. Table 1 also shows the interconnection between all the documentation while each document is fulfilling one more requirement. As an example, the Customer Requirements Definition (CRD) document is being referenced to several steps in the design process to fulfil required action(s) for each of the steps which it has been referenced to. On the other hand, some steps do not require separate documentation to fulfil them such as the step of “production Phase 2”. Therefore, the QM is a living document that should be strictly and continuously updated, whereby any changes with respect to the implementation of this standard’s requirements shall be approved and then disclosed.
On the other hand, top management plays a key role in ensuring that the QMS is being applied as planned, as witnessed in this in this case-studied start-up. According to the study of Aggelogiannopoulos et al12, it is confirmed that the QMS is a management’s responsibility in which their commitment plays a key role for a comprehensive implementation. Accordingly, the amount, the quality, and the details of the QMS documentation can vary among start-ups. The approach and the content exclusion depend largely on the way management finds feasible and can justify respectively.
Conclusion
Start-ups have a key role in the innovation process. Nevertheless, most of the critical problems they encounter concerning the QMS process and its related documentation are owed to their lack of ISO requirements’ knowledge and experience, the lack of time and resources, the lack of commitment of top management and personnel, and the absence of a system of ownership.13 This paper provides a sample of the main QMS documentation which has been implemented by an ISO certified MDD start-up in line with the ISO 13485:2016 requirements. It emphasizes the specifics of the ISO 13485 core document, referred as the Quality Manual, which is a comprehensive document that details the design and development process, and connects its various phases with the established processes, procedures, and forms that are needed to accomplish this standard’s requirements. Even more, this manual shows the dependency and interaction among these processes, procedures and forms while orchestrating the entire design process. The presented QM map in this paper branches out all the required ISO documentation from the adopted design process to ensure the fulfillment of these ISO requirements. Having the design process leading the quality manual is not required by ISO, yet it may assist developers with covering all the needed quality steps and related documentation throughout the entire design stages. The design process can be even updated with hyperlinks to support even more the alignment of the design phases with the ISO requirements. With the help of the actual standard document along the visible support of the management, and after assessing and deciding on the applicability of the relevant clauses from the standard, start-ups can start building their own QM, procedures, policies, and forms in an effort to reach compliance and certification if desired.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Makary M, Daniel M. Medical error: the third leading cause of death in the US. BMJ. 2016;353:i2139. doi:10.1136/bmj.i2139
2. World Health Organization. Medical devices; 2019. Available from: https://www.who.int/health-topics/medical-devices#tab=tab_1.
3. Money A, Barnett J, Kujis J, Craven M, Martin J, Young T. The role of the user within the medical device design and development process: medical device manufacturers’ perspective’. BMC Med Inf Decis Makin. 2011;11(1):1–2. doi:10.1186/1472-6947-11-15
4. ISO/TR 10013. Guidelines for quality management system documentation; 2001.
5. Ullmann B, Angrisani N, Reifenrath J, et al. The effects of handling and storage on magnesium based implants — first results. Mater Sci Eng C. 2013;33(5):3010–3017. doi:10.1016/j.msec.2013.03.034
6. Gadotti Martins E, Pinheiro de Lima E, Gouvea da Costa SE. Developing a quality management system implementation process for a medical device manufacturer. J Manuf Technol Manage. 2015;26(7):955–979. doi:10.1108/jmtm-12-2012-0119
7. The Danish Medicines Agency. Medical devices; 2020. Available from: https://laegemiddelstyrelsen.dk/en/devices/#.
8. Klute-Wenig S, Refflinghaus R. Quality management for microenterprises and start-ups–is the ISO 9001 suitable? Int J Qual Serv Sci. 2020;12(4):435–446. doi:10.1108/ijqss-01-2018-0003
9. Green JIJ. Medical device regulation: requirements for dental professionals who prescribe and manufacture custom-made devices. Prim Dent J. 2021;10(1):64–88. PMID: 33722134. doi:10.1177/2050168420980980.
10. Kim CY, Ko SS, Han YH. The impact of ISO 13485 on the performance of Korean medical device manufacturers. J Soc Korea Ind Syst Eng. 2018;41(1):11–23. doi:10.11627/jkise.2018.41.1.011
11. Spender J-C, Corvello V, Grimaldi M, Rippa P. Start-ups & open innovation: a review of the literature. Eur J Innov Manage. 2017;20(1):4–30. doi:10.1108/EJIM-12-2015-0131
12. Aggelogiannopoulos D, Drosinos EH, Athanasopoulos P. Implementation of a quality management system (QMS) according to the ISO 9000 family in a Greek small-sized winery: a case study. Food Control. 2007;18(9):1077–1085. doi:10.1016/j.foodcont.2006.07.010
13. Barnes D. The manufacturing strategy formation process in small and medium‐sized enterprises. Journal of Small Business and Enterprise Development. 2002;9(2):130–149. doi:10.1108/14626000210427384
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.