")
Back to Journals » Journal of Blood Medicine » Volume 12
Efficacy, Safety and Pharmacokinetic Results of a Phase III, Open-Label, Multicenter Study with a Plasma-Derived Von Willebrand Factor (VWF)/Factor VIII (FVIII) Concentrate in Pediatric Patients <12 Years of Age with Hemophilia A (SWIFTLY-HA Study)
Authors Djambas Khayat C, Iosava G, Romashevskaya I , Stasyshyn O , Lopez MJ, Pompa MT, Rogosch T , Seifert W
Received 6 January 2021
Accepted for publication 29 May 2021
Published 21 June 2021 Volume 2021:12 Pages 483—495
DOI https://doi.org/10.2147/JBM.S299130
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Martin H Bluth
Claudia Djambas Khayat,1 Genadi Iosava,2 Irina Romashevskaya,3 Oleksandra Stasyshyn,4 Marta Julia Lopez,5 Maria Teresa Pompa,6 Tobias Rogosch,7 Wilfried Seifert7
1Hospital Hôtel Dieu de France, Saint Joseph University, Beirut, Lebanon; 2Joint Stock Hematology and Transfusiology Research Institute, Tbilisi, Georgia; 3Republican Research Centre of Radiation Medicine and Human Ecology, Gomel, Belarus; 4Institute of Blood Pathology and Transfusion Medicine, Lviv, Ukraine; 5Guatemala City Hospital Roosevelt, Guatemala, Guatemala; 6Monterrey Nuevo Leon OCA Hospital (MIRC), Monterrey Nuevo Leon, Mexico; 7CSL Behring, Clinical Research and Development, Marburg, Germany
Correspondence: Claudia Djambas Khayat
Hospital Hôtel Dieu de France, Saint Joseph University, Beirut, Lebanon
Tel +9611613027
Email [email protected]
Background: Plasma-derived von Willebrand factor/factor VIII (pdVWF/FVIII; VONCENTO®, CSL Behring) is a high-concentration, low-volume, high-purity concentrate, with a high level of VWF high-molecular-weight multimers and a VWF/FVIII ratio of ∼ 2.4:1.
Methods: This study (NCT01229007) investigated the pharmacokinetics (PK), efficacy and safety of pdVWF/FVIII in 35 previously treated (minimum 20 exposure days [EDs]) pediatric patients (< 12 years) with severe hemophilia A. PK was evaluated with a single 50 IU FVIII/kg dose of pdVWF/FVIII. Efficacy and safety analyses were performed during on-demand treatment (n=17) or prophylaxis (n=18) for up to 100 EDs with a maximum study duration of 12 months.
Results: PK profiles were similar for patients aged < 6 years and those aged 6– 12 years, and, as expected, the youngest patients had an increased clearance. On-demand patients reported 320 non-surgical bleeding (NSB) events and received a median number of 29.0 infusions (median dose 34.2 IU FVIII/kg). Hemostatic efficacy was assessed by the investigator as excellent/good in all cases (24%/76%). The 18 patients in the prophylaxis arm experienced 173 NSB events (97 NSBs [56%] in three patients). Five patients (28%) had no NSB events. Overall, patients received a median number of 92 infusions (median dose 30.6 IU FVIII/kg). The majority of bleeds (92%) were successfully controlled with only one infusion. Hemostatic efficacy was assessed by the investigator as excellent (86%) or good (14%). Inhibitors occurred in three patients of which two were transient (low titer) and one persisted (high titer). These three patients had known risk factors for inhibitor development.
Conclusion: This study demonstrated comparable PK profiles for pediatric patients aged < 6 years and aged 6– 12 years, and an excellent efficacy and safety profile in this population. The adverse events reported were mostly mild to moderate with inhibitor rates within the expected incidence range.
Keywords: hemophilia A, von Willebrand factor, factor VIII, on-demand therapy, prophylaxis, hemostatic efficacy
Plain Language Summary
Hemophilia A is an inherited bleeding disorder where the main symptom is spontaneous bleeding into the joints or muscles. Repeated bleeding episodes can be acutely painful and lead to long-term damage; therefore, the aim of hemophilia treatment is to prevent and manage these bleeding episodes to improve quality of life. One treatment option to control excessive bleeding for patients with hemophilia A is the use of von Willebrand factor/factor VIII (VWF/FVIII) concentrates derived from human plasma. Our study evaluated the safety and efficacy of a plasma-derived (pd)VWF/FVIII in children (aged <12 years old) with severe hemophilia A who had previously received hemophilia treatment with a FVIII product for a minimum of 20 exposure days. Treatment was either on-demand, to treat bleeding episodes as they arose, or prophylactic at regular intervals, with the aim of preventing bleeding. We also compared how children in different age groups (<6 versus 6–12 years old) responded to a one-off dose of pdVWF/FVIII. Hemostatic efficacy was assessed as excellent or good for both on-demand (24% and 76%, respectively) and prophylactic (86% and 14%, respectively) treatment with pdVWF/FVIII. Three patients developed inhibitors to FVIII, of which two were transient and one persisted; no other safety findings of concern were noted. These findings support those seen in adult/adolescent patients with severe hemophilia A. Comparable pharmacokinetic profiles were observed between the two age groups studied. Overall, these results demonstrate a favorable benefit-risk profile for pdVWF/FVIII, and support its use to treat bleeding events in children with hemophilia A.
Introduction
Hemophilia A is an X-chromosome-linked, congenital bleeding disorder, which is caused by decreased activity of factor VIII (FVIII) in plasma (FVIII:C) due to mutations in the FVIII gene. The incidence is estimated to be 1 in 5000 live male births.1,2 Severe hemophilia A is characterized by a FVIII:C plasma level less than 1% of normal levels.3 Intracranial, muscle and joint bleeding can occur in these patients, even with the minimal activities of daily life. Treatment for hemophilia is aimed at preventing and managing bleeding episodes and their subsequent complications. Prophylaxis with factor replacement therapy has been shown in many studies to prevent or at least reduce the progression of damage to target sites, such as joints.4,5 A systematic review and meta-analysis of six randomized controlled trials demonstrated that prophylaxis started early in childhood preserves joint function as compared with on-demand treatment, due to a reduction in total bleeds and bleeding into joints, resulting in improved quality of life.6
A number of virus-depleted/inactivated plasma-derived FVIII and recombinant FVIII containing replacement products are currently available.2,7–9 FVIII inhibitor development is the most significant treatment complication of these products seen in these patients.9 The development of inhibitors in patients with hemophilia A is correlated with a variety of endogenous and exogenous risk factors.10,11 The advantages and disadvantages of plasma-derived versus recombinant FVIII products are still an area of controversy, especially since the von Willebrand factor (VWF) present in different concentrations in some of the plasma-derived products might reduce inhibitor development.11–19
Plasma-derived VWF/FVIII concentrate (pdVWF/FVIII; Voncento®, CSL Behring, Germany) is a high-concentration, low-volume, high-purity VWF/FVIII concentrate which contains a large proportion of high-molecular-weight VWF multimers and a VWF:FVIII ratio of ~2.4:1.20 The efficacy and safety of pdVWF/FVIII has been previously demonstrated in adults with hemophilia A.20 In this study, the efficacy, safety and pharmacokinetics (PK) of pdVWF/FVIII were investigated in pediatric patients aged 0 to <12 years with severe hemophilia A (FVIII:C <1%) who had received limited previous FVIII treatment for a minimum of 20 exposure days (EDs).
Materials and Methods
Study Design
The SWIFTLY-HA study (NCT01229007) was conducted in eight centers in Ukraine (n=2), Belarus (n=2), Guatemala, Mexico, Georgia and Lebanon between August 2010 to July 2014. This study was carried out in accordance with the International Conference on Harmonization Good Clinical Practice guidelines, the Declaration of Helsinki (2008), and standard operating procedures for clinical research and development at CSL Behring. Ethics approval, individual written informed consent from patient’s legal guardian (as all patients were <12 years of age), and approval by the Independent Ethics Committee/Institutional Review Board of the participating centers (Ethics Committee of the State Institution “Republican Scientific and Practical Center of Pediatric Oncology and Hematology” and Ethics Committee of State Institution “Republican Scientific and Practical Center of Radiation Medicine and Human Ecology”, Belarus; Joint Stock Company the Institute of Haematology and Transfusiology, Georgia; Hospital Roosevelt Independent Ethics Committee, Guatemala; University of Saint-Joseph Beirut Ethics Committee, Lebanon; OCA Hospital Monterrey International Research Center Ethics & Research Committee, Mexico; Ministry of Health of Ukraine Central Ethics Commission, Ukraine) were obtained prior to enrollment.
Male patients were eligible for enrollment if they were <12 years of age with severe hemophilia A (FVIII:C <1%) and had received previous FVIII treatment for a minimum of 20 EDs. Patients with evidence of vaccination against hepatitis A and B or presence of antibodies against hepatitis A and B were included and provided signed informed consent (children <1 year of age could have been included without evidence of vaccination against hepatitis A); patients with no evidence of previous vaccination and/or no protective antibody titer against hepatitis A and B were vaccinated at screening. Patients were excluded from study participation if they received infusion of any FVIII product, cryoprecipitate, whole blood, plasma, or desmopressin acetate in the 4 days prior to Day 1, or presented with a known history of FVIII inhibitors, or with a FVIII inhibitor level >0.6 Bethesda Units (BU)/mL at screening; the intake of aspirin or other non-steroidal anti-inflammatory drugs within 7 days of study drug administration also resulted in exclusion from study participation.
The efficacy and safety of pdVWF/FVIII was assessed in an open-label study comprising an on-demand and a prophylactic treatment arm. The on-demand regimen consisted of either immediate (irregular) treatment with pdVWF/FVIII of a non-surgical spontaneous or traumatic bleeding event, or a preventive (irregular) treatment with pdVWF/FVIII to prevent an anticipated bleeding event (eg, before expected physical activity that was associated with an increased bleeding risk). In the prophylaxis regimen, patients were given regular treatment with pdVWF/FVIII every 2–3 days. In addition, pdVWF/FVIII was given to prevent and treat any surgical bleeding events. The general dosing recommendation for the treatment or prophylaxis of spontaneous or trauma-induced hemorrhages and for surgeries is shown in Table 1. Assignment to the prophylaxis or on-demand arm of the study, as well as each patient’s treatment regimen and individual dose was determined by the investigator, based on the reason for use.
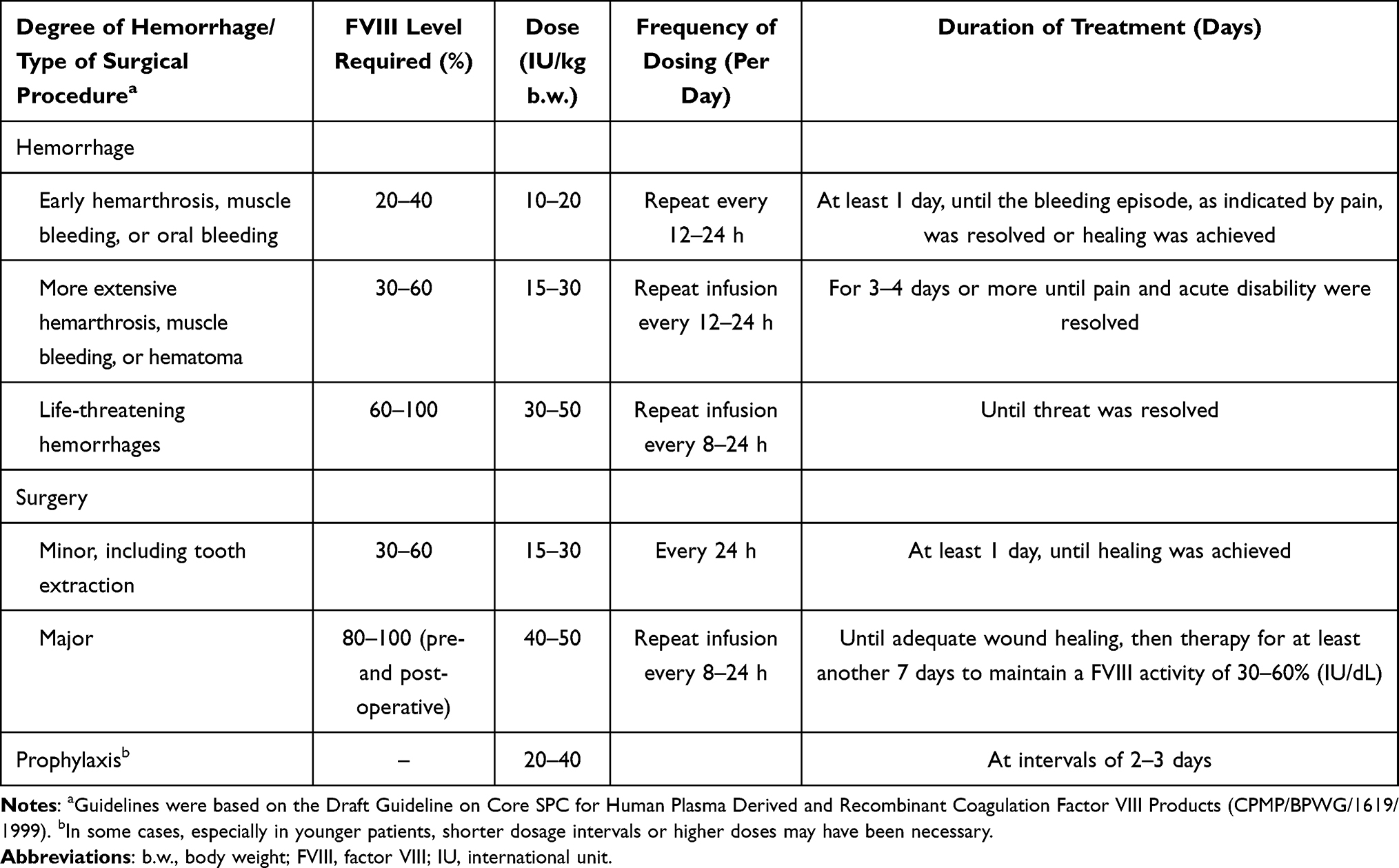 |
Table 1 Guidelines for Dosage During the Efficacy Component77 |
This study consisted of three periods (Figure 1): i) a screening period of up to 35 days; ii) a PK component of up to 3 days consisting of a single dose of pdVWF/FVIII on Day 1 with PK samples collected on Days 1, 2, and 3; iii) an efficacy and safety component of 6 to 12 months (on-demand/preventive therapy or as regular prophylaxis) corresponding to approximately 50 and 100 EDs to pdVWF/FVIII.
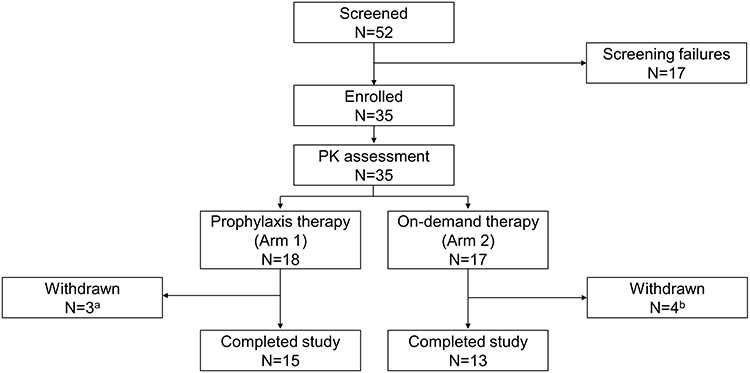 |
Figure 1 Study design. Abbreviations: N, total number of patients; PK, pharmacokinetics. Notes: aDue to site closure (n=2) and non-compliance with prophylaxis regimen (n=1). bDue to FVIII inhibitor development (n=2) and withdrew their consent (n=2). |
The primary objectives were to assess the hemostatic efficacy of pdVWF/FVIII in its usage over ≥50 EDs, and to investigate the pharmacokinetic profile of pdVWF/FVIII. The secondary objective was to assess the safety of pdVWF/FVIII in this patient population.
Pharmacokinetics
The actual PK dose was calculated using the volume of each administered batch multiplied by the assigned potency of the specific batch, as determined at the batch release testing. PK parameters for FVIII:C were calculated by a non-compartmental infusion model from plasma concentration values collected after an initial single bolus intravenous infusion (administered at a maximum infusion speed of 6 mL/min) of 50 IU FVIII/kg body weight on Day 1. PK samples were drawn prior to the first dose of pdVWF/FVIII and then at 0.5, 4, 8, 24, and 48 h after the end of the first dose according to the European Medicines Agency guideline for FVIII products.21 Chromogenic FVIII:C assay was performed in a central laboratory (Medilys Laborgesellschaft – Asklepios Institut, Hamburg, Germany).
Efficacy Assessments
The clinical efficacy parameters assessed in the study were hemostasis assessment (for each non-surgical bleeding [NSB] event and surgical event by the investigator and patient/caregiver who filled in a home therapy diary), study product usage, blood product transfusion requirements, and surgeon’s assessment of blood loss during a surgical procedure (“less”, “equivalent”, or “more” compared with the expected blood loss from a patient without a bleeding disorder undergoing the same procedure). Clinical assessments of hemostatic efficacy were based on a four-point grading scale: “excellent” if hemostasis was achieved/cessation of bleeding occurred; “good” if slight oozing or partial but adequate control of bleeding occurred and no additional product was required for unplanned treatment; “moderate” if moderate bleeding or moderate control of bleeding occurred and additional product was required for unplanned treatment; “none” in cases of severe uncontrolled bleeding. The severity of NSB events was assessed as major or minor by the investigator, according to guidance provided. If the patient was unable to self-administer or required more than two doses of pdVWF/FVIII to control an NSB event, they were to be treated at the study center and the investigator conducted daily hemostatic efficacy assessments. The patient visited the study site every 3 months and the investigator made a retrospective assessment of the patient’s response to pdVWF/FVIII for each NSB event as documented in the home diary; ratings were based on the four-point efficacy grading scale described above. For each surgical procedure, the investigator was required to provide a daily assessment of patient response to pdVWF/FVIII during the inpatient period, with an overall investigator assessment performed at patient discharge, using the four-point efficacy grading scale. For prophylactic treatment, the annualized spontaneous bleeding rate (AsBR) was calculated.
Safety
All patients who received at least one dose of pdVWF/FVIII were included in the safety analysis. Safety assessments included the reporting of adverse drug reactions (an adverse event at least possibly related to pdVWF/FVIII), serious adverse events (SAEs, an adverse event that is life-threatening, requires/prolongs hospitalization, results in persistent/significant disability/incapacity, is a congenital anomaly/birth defect, or is considered medically significant), the presence of FVIII inhibitors, laboratory parameters (such as biochemistry, hematology, and urinalysis), and a physical examination and vital signs assessment. The extent of exposure to the study drug included administrations of pdVWF/FVIII during the PK and efficacy analyses. All medications taken 30 days prior to screening and during the entire study duration were recorded.
FVIII:C, the presence of FVIII inhibitors, and the investigation of seroconversion for virus markers (indicative of hepatitis A, B, and C virus infection) were assessed/conducted in a central laboratory (Medilys Laborgesellschaft – Asklepios Institut, Hamburg, Germany). Presence of FVIII inhibitors was analyzed using the Bethesda method (Nijmegen modification). Virology reference samples were collected at Day 1 and at the final visit, but were not analyzed unless deemed necessary.
Statistical Analyses
Descriptive statistics were used to summarize continuous variables (mean and standard deviation, and/or median and range). Categorical variables are presented as numbers and percentages in frequency tables. Summaries are based on observed data (missing data were not replaced). Formal statistical tests were not performed. Descriptive statistics for PK parameters were additionally stratified by age group (<6 years, 6 to <12 years).
Results
Patients
A total of 35 patients were enrolled, 18 patients were treated in the prophylaxis arm and 17 patients were treated in the on-demand arm (Figure 1). Patients participated in the study for a median (range) of 300 (1–388) days. A total of 28 patients completed the study (15 in the prophylaxis arm and 13 in the on-demand arm). In the prophylaxis arm, two patients who had developed a low titer FVIII inhibitor were withdrawn due to site closure and one patient who was non-compliant to treatment was withdrawn. In the on-demand arm, one patient was withdrawn due to a pre-existing FVIII inhibitor, one patient because he developed a high titer FVIII inhibitor and two patients withdrew their consent.
Baseline patient characteristics are shown in Table 2. In the on-demand arm, 11 patients (65%) had less than 50 EDs to previous FVIII treatment. In the prophylaxis arm, 2 patients (11%) had less than 50 EDs. The proportion of patients aged <6 years was lower in the prophylaxis arm (22.2%) than in the on-demand arm (70.6%), resulting in a higher mean age in the prophylaxis arm (7.3 years) than in the on-demand arm (4.9 years).
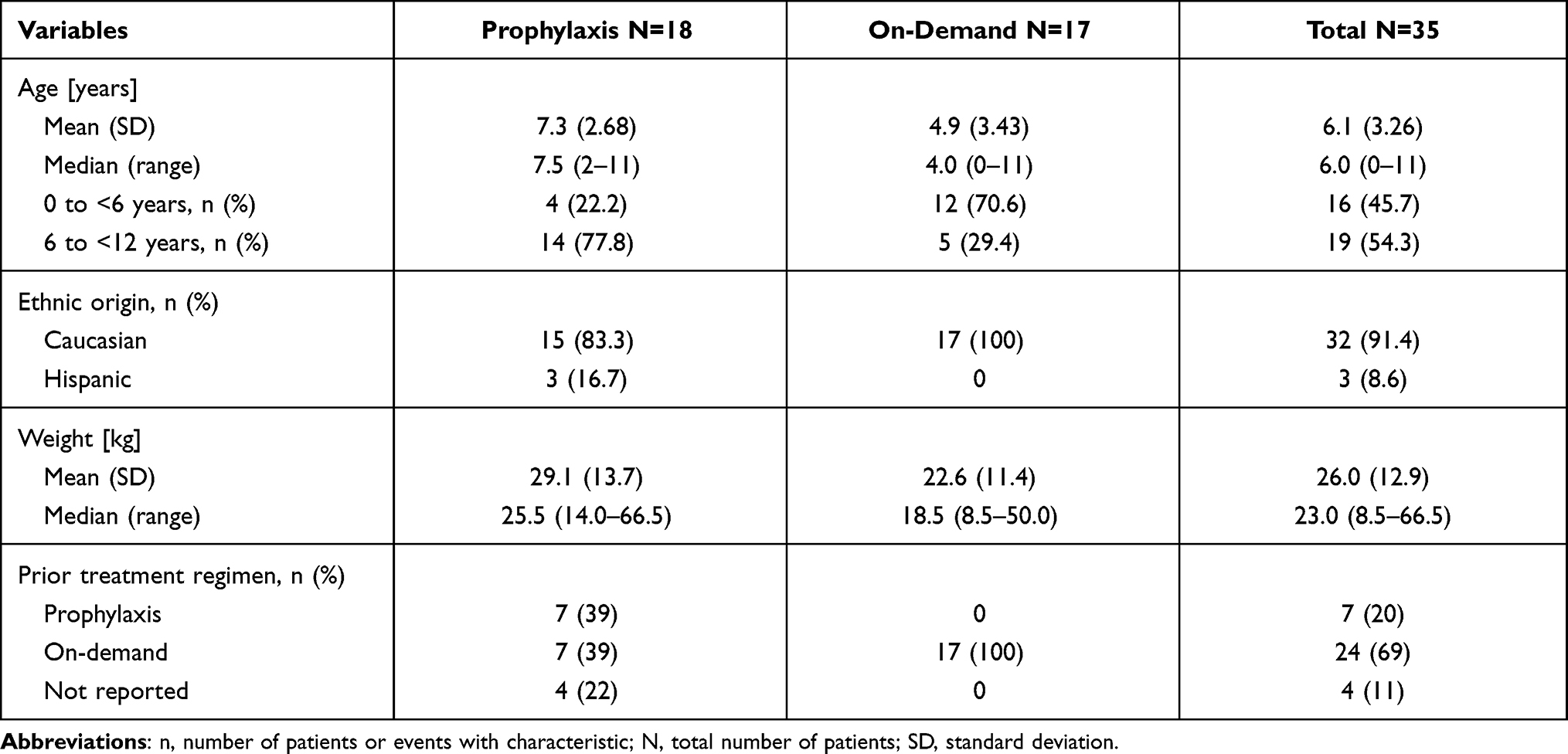 |
Table 2 Patient Characteristics |
Pharmacokinetics
The PK population comprised 31 patients, excluding four patients who had received a lower dose than planned (n=2), no PK concentrations were available (n=1) or the patient was excluded due to a pre-existing FVIII inhibitor (n=1). Concentration-time curves showed similar profiles for the overall PK population and the two age groups (Figure 2). In the overall PK population, FVIII:C PK parameters were raised immediately following pdVWF/FVIII infusion, reaching a median Cmax of 0.76 IU/mL at the first sampling point 30 min post-dose, and then declined over time with a median t1/2 of 9.78 h (Table 3). Median IR was 0.016 (IU/mL)/(IU/kg), median CL was 5.44 mL/h/kg, and median Vss was 73.7 mL/kg. Median values for PK parameters were slightly higher in the 6 to <12 years age group compared to the 0 to <6 years age group (with the exception of CL and Vss, Table 3).
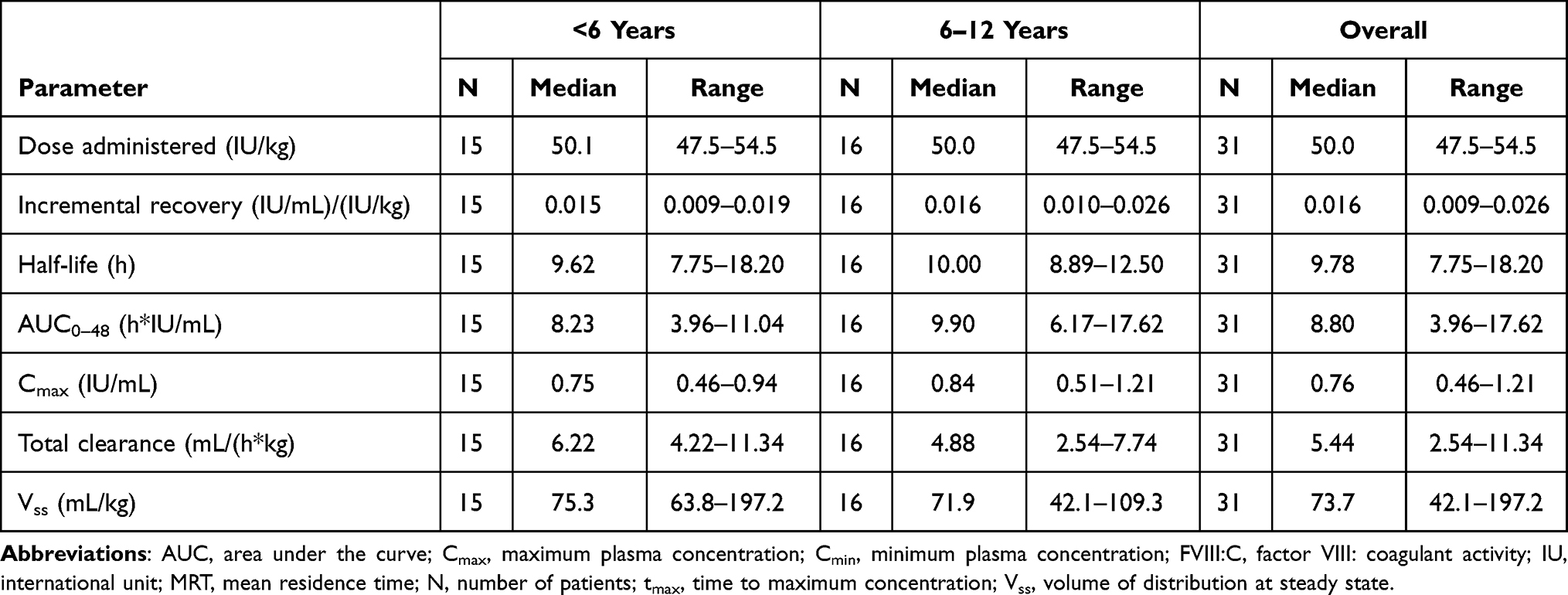 |
Table 3 PK Parameters of FVIII:C in Patients <6 (N=15) and 6–12 Years of Age (N=16) |
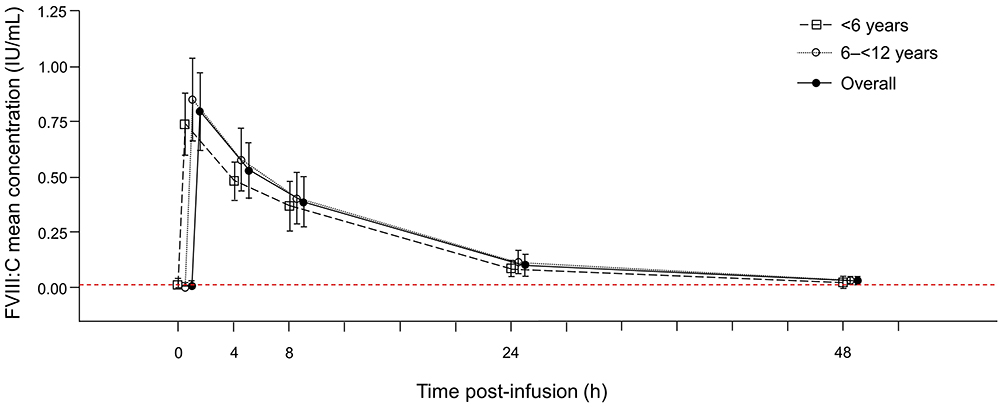 |
Figure 2 Mean (SD) concentration profiles (IU/mL) of FVIII:C by age group and overall. Abbreviations: FVIII:C, factor VIII: coagulant activity; IU, international unit; PK, pharmacokinetics; SD, standard deviation. Notes: Solid line with circles: overall population; black dashed line with squares: 0 to <6 years; black dashed line with circles: 6 to <12 years; red dashed line: lower limit of quantitation (0.008 IU/mL). |
Hemostatic Efficacy
On-demand arm: The 17 patients who were treated on-demand reported 320 NSB events and received a median number of 29.0 infusions (median dose of 34.2 IU FVIII/kg [range: 22.7–49.7 IU/kg]). Two of the NSB events did not require treatment, and are not included in the efficacy evaluation. A total of 75 (23.4%) NSB events required >1 infusion of pdVWF/FVIII, up to a maximum of seven infusions; all remaining NSB events were treated with 1 infusion. Hemostatic efficacy was assessed by the investigator for all 318 events that were treated with pdVWF/FVIII (Table 4). The hemostatic efficacy was reported by the investigator as either excellent (77 events [24.2%]) or good (241 [75.8%]) in all cases. A similar distribution of hemostatic efficacy outcomes was also seen within bleeding event categories for type, severity, or location, with the exception of the 98 major bleeding events, where the hemostatic efficacy of a relatively higher proportion of events was assessed as good (95.9%). There were no relevant differences in the evaluation of hemostatic efficacy between patients aged <6 years and 6–<12 years.
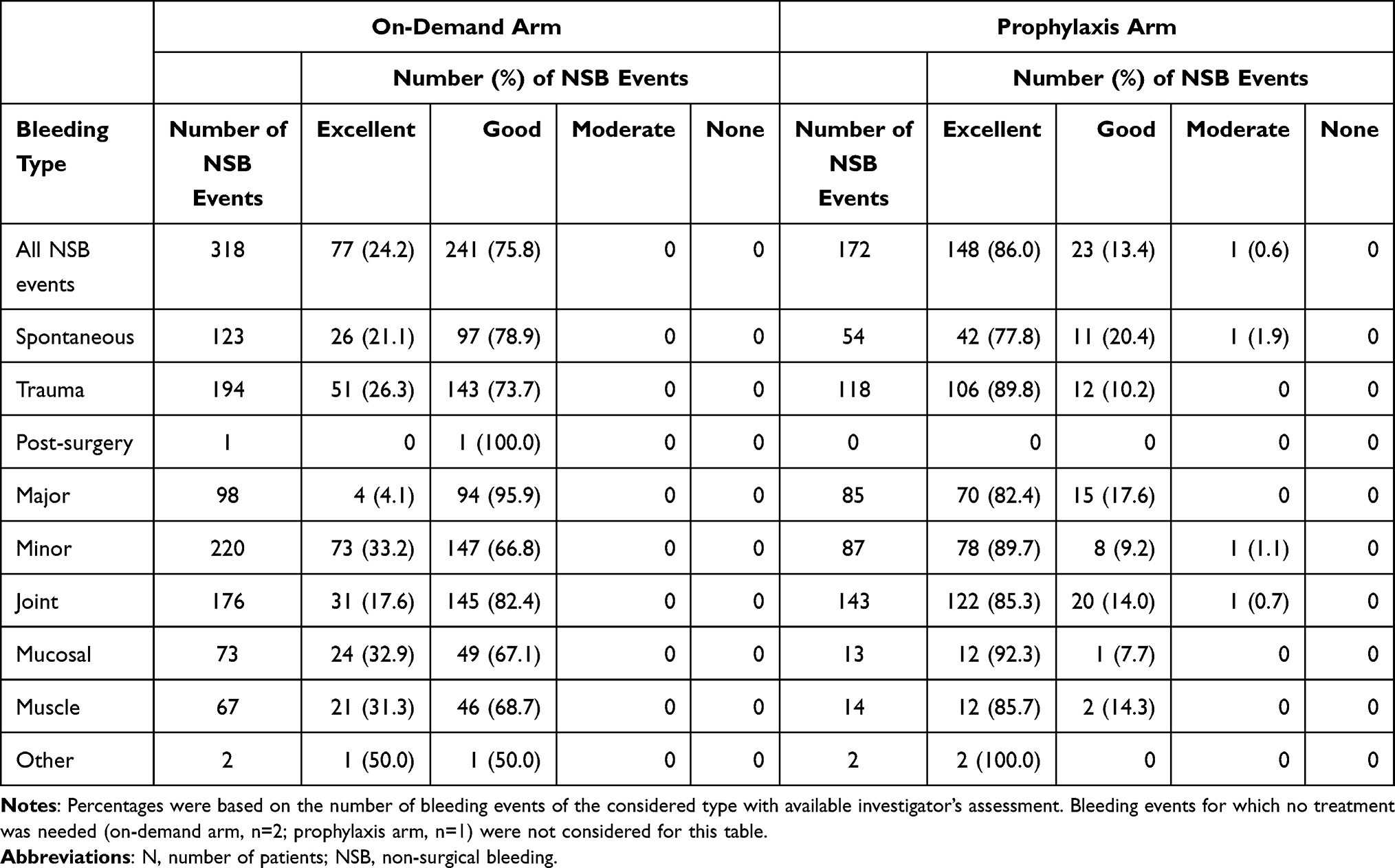 |
Table 4 Investigator’s Assessment of Hemostatic Efficacy per Bleeding Event in the On-Demand and Prophylaxis Arms |
Prophylaxis arm: The 18 patients on prophylaxis treatment received a median number of 92 infusions at a median average dose of 30.6 IU FVIII/kg per infusion (range: 21.1–58.7 IU/kg). The patients experienced 173 NSB events; fewer than 1 out of 3 events (31%) were spontaneous bleedings. More than half of these spontaneous NSB events (56%) were reported by three patients (see Table 5). The first of these patients was 9 years old, and experienced an AsBR of 24.91. Prior to study enrollment, this patient was receiving prophylaxis and had reported hemarthrosis of the right elbow, left and right ankle, left-hand finger, and both wrists. Additionally, during the study the patient was incompliant with his prophylaxis schedule. The second patient (10 years old) had an AsBR of 9.36, and prior to this study had received prophylaxis and reported hemarthrosis of both knees and both ankles. The third of these patients was 11 years old with an AsBR of 8.05; prior to his enrollment in this study, the patient had received on-demand treatment and reported hemarthrosis of the right knee, both elbows, both ankles, and the right hip. Seven of the 18 patients (39%) switched to prophylaxis treatment with pdVWF/FVIII from prophylaxis with a prior treatment. One NSB event (0.6%), a minor trauma-induced oral bleed, did not require treatment and this event was not assessed for efficacy. The majority of all NSB events (159 events, 91.9%) required only one infusion for hemostatic control, the remainder required 2 infusions (13 events, 7.5%). Hemostatic efficacy was assessed by the investigator as excellent (86%) or as good (14%) (Table 4). The AsBR for patients who completed the study and the frequency of prophylaxis dosing per patient is shown in Table 5. Seven patients (47%) had no spontaneous NSB events.
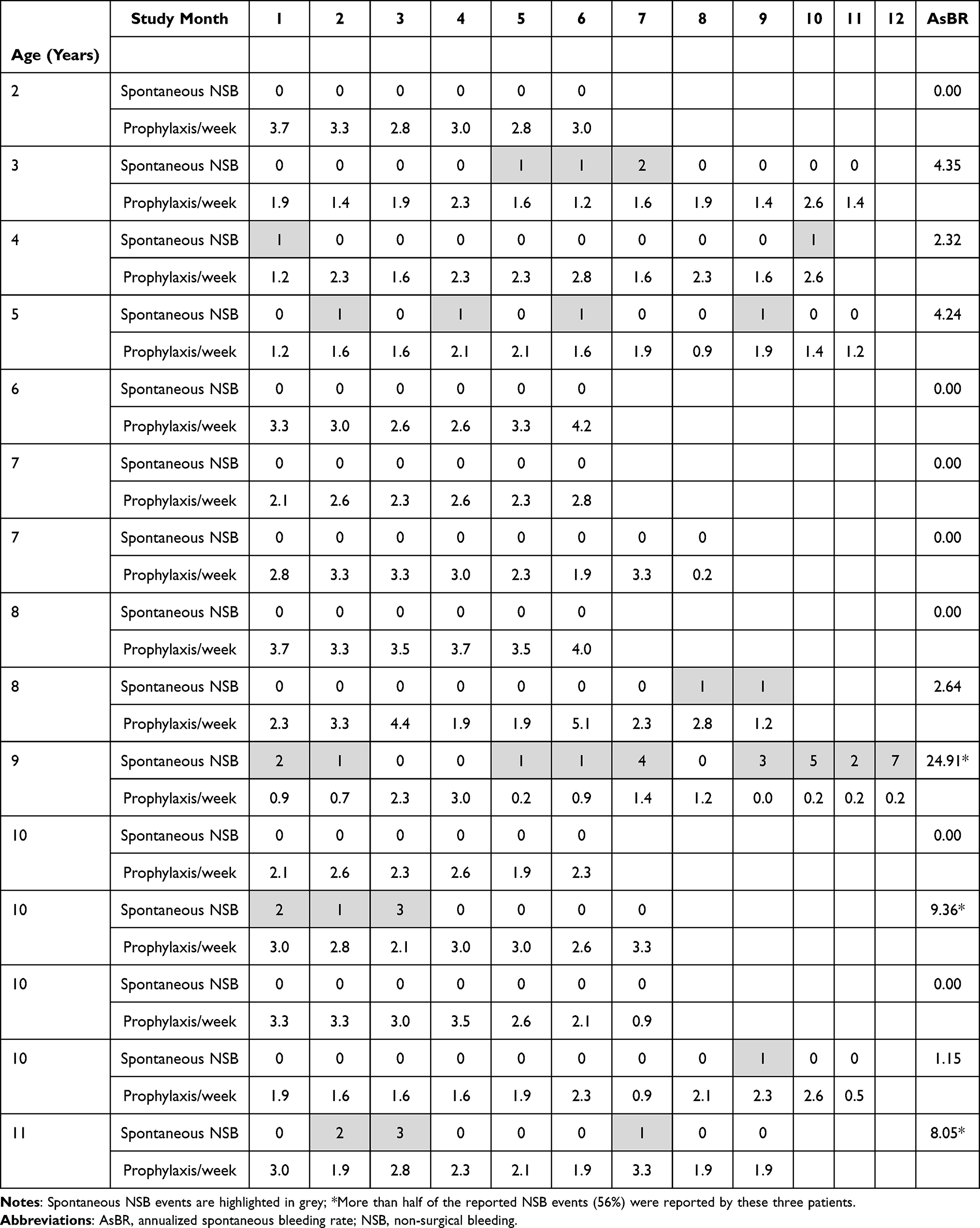 |
Table 5 Annualized Spontaneous Bleeding Rate and Prophylaxis Schedule (Number of Doses per Week per Individual Patient Who Completed the Study) |
Surgical events: During the study, five patients underwent a total of five surgeries: two major (synovectomy of the right knee and elongation of the Achilles tendon) and three minor (one dental surgery and two tooth extractions). At discharge, the investigator assessed the hemostatic efficacy for two of the minor surgical events as excellent and for the remaining major and minor surgical events as good. Blood loss during surgery was assessed as less (two surgical events) or as comparable (three surgical events) to the expected blood loss from a patient without a bleeding disorder undergoing the same procedure in all surgical events. No patient required blood product transfusions.
Safety
The median (range) number of EDs for patients in the prophylaxis arm was 91.5 (15–117), with 2 patients having <50 EDs and 16 patients having ≥50 EDs. For the on-demand arm, the median (range) was 29.0 (1–89) days, with 12 patients having <50 EDs and 5 having ≥50 EDs.
Overall, pdVWF/FVIII was well tolerated. During this study, 12 patients (66.7%) on prophylaxis treatment reported 64 treatment-emergent adverse events (TEAEs), and 11 patients (64.7%) in the on-demand treatment arm reported 33 TEAEs. The most frequently reported TEAEs overall were cough (eight patients), pyrexia (six patients), and rhinitis, FVIII inhibition, and rash (four patients each). Four patients had an adverse drug reaction (three cases of FVIII inhibition and one case of rash). Five patients experienced one SAE each (four cases of FVIII inhibition [one of which was pre-existing and so not considered related to pdVWF/FVIII] and one tibia fracture). There were no clinically relevant differences in the TEAE reporting profile between patients aged <6 years and those aged 6–<12 years.
One patient (<1 year old) had a pre-existing FVIII inhibitor detected in the pre-treatment PK blood sample (30.4 BU/mL), which the investigator considered related to prior FVIII (treatment given for a total of 20 EDs), and was withdrawn from the study. Two patients from the prophylaxis arm developed a low titer, transient inhibitor. The first patient (6 years old) developed a low titer FVIII inhibitor of 1.9 BU/mL in the third month of treatment following 18 EDs with pdVWF/FVIII. At the time of inhibitor development, the patient had a total of 38 EDs to FVIII (20 EDs with a prior FVIII product and 18 EDs with pdVWF/FVIII), and experienced an additional 7 EDs with pdVWF/FVIII before the inhibitor was confirmed and the patient withdrew from prophylaxis treatment. The inhibitor was confirmed at month 4 (1.7 BU/mL), but had decreased to normal values (<0.6 BU/mL) at months 6 and 10. DNA analysis identified a mutation in exon 25 of the FVIII gene. The low number of EDs represented a risk factor for inhibitor development; no other risk factors were identified. The second patient (10 years old) developed a low titer FVIII inhibitor of 2.1 BU/mL in month 1, following a total of 30 EDs to FVIII (22 EDs with a prior FVIII and 8 EDs with pdVWF/FVIII). The patient experienced a further 7 EDs with pdVWF/FVIII prior to confirmation of the inhibitor and withdrawal from prophylaxis treatment. The patient’s low titer inhibitor persisted to month 8, with titers of 1.9–2.8 BU/mL. Risk factors for inhibitor development in this patient included an intron 22 inversion in the FVIII gene, a mutation associated with a higher risk of inhibitor development, and the low number of EDs to FVIII products; no other risk factors were identified. Three bleeding events occurred during the time period between inhibitor testing and withdrawal from prophylaxis treatment in these two patients. The hemostatic efficacy of pdVWF/FVIII for these bleeds was also excellent despite the presence of the inhibitor. Both inhibitors were transient and the titer decreased to normal levels again (<0.6 BU/mL). One patient (4 years old) from the on-demand arm developed a high titer FVIII inhibitor (461 BU/mL) during the treatment phase after 8 EDs. The high titer inhibitor was confirmed on two subsequent occasions (2048 BU/mL, 1556 BU/mL), and the patient was withdrawn from the study. The patient was found to have an intron 22 inversion in the FVIII gene, with no other identified risk factors for inhibitor development. Prior to the study, the patient had been treated with a prior FVIII product for a total of 48 EDs, but had not developed inhibitors. The Independent Data Monitoring Committee concluded that the development of inhibitors did not constitute a safety concern for the product, as the development of an inhibitor is an adverse event associated with FVIII replacement and this incidence was in concordance with information from the scientific literature. No other safety findings of concern were observed. None of the patients experienced a thromboembolic event or anaphylactic reaction, and there were no events of suspected transmission of infectious agents.
Discussion
This PK, efficacy, and safety study was designed to investigate the use of pdVWF/FVIII for the prevention and treatment of bleeding events in FVIII pre-treated pediatric patients (0 to 12 years of age) with severe hemophilia A. pdVWF/FVIII is a plasma-derived, high-concentration, low-volume, high-purity concentrate which contains VWF:FVIII in a ratio ~2.4:1.20 It was anticipated that in children pdVWF/FVIII would have a similar PK, efficacy and safety profile to that in the adult population.
The PK evaluation demonstrated comparable PK profiles for the children aged <6 years and aged 6 to 12 years. Differences were noted between the PK results from our pediatric study and those in the study of adult/adolescent hemophilia A patients:20 IR of FVIII was lower in pediatric patients (0.016 [IU/mL]/[IU/kg]) than in adults/adolescents (0.021 [IU/mL]/[IU/kg]), t1/2 was shorter (9.78 vs 13.4 h) and CL was higher (5.44 vs 3.92 mL/h/kg). It has been confirmed that weight-normalized CL of FVIII decreases during growth and continues to decline slightly during adulthood. Elimination t1/2, which is inversely related to CL, thus follows an opposite trend.22,23 The differences in t1/2 between our pediatric study and the adult/adolescent study may have an effect on the dose of FVIII required to maintain a desired trough level during prophylaxis and indicates that the dose of FVIII per kilogram body weight required for adequate prophylaxis probably changes throughout a subject’s life. The variance in t1/2, however, suggests that if the FVIII dose for prophylaxis is to be based on t1/2, then t1/2 should be measured in each patient, rather than be based on the patient’s age. Interestingly, a recent study in pediatric patients with hemophilia A reported that genetic determinants of VWF clearance and FVIII binding can modify FVIII PK, further supporting an individualized approach to dosing.24
This study, as well as the adult/adolescent study, showed that, despite slight age-related differences in the observed PK parameters, efficacy results were very similar across all age groups. The hemostatic efficacy of pdVWF/FVIII was assessed by the investigator as excellent or good for all NSB and surgical events without any relevant differences between patients aged <6 or 6 to 12 years. These hemostatic efficacy results are also in line with those seen in adult/adolescent previously treated patients (PTPs) with hemophilia A treated with the same pdVWF/FVIII in whom the investigator assessed the efficacy as either excellent or good in at least 96% of the bleeding events.20 Comparable hemostatic efficacy (excellent or good in 96.7% of bleeding events) was also reported by investigators in a study of adult/adolescent PTPs with severe hemophilia A who were treated with a lower concentration plasma-derived VWF/FVIII (1:1) concentrate (Wilate®, Octapharma, Switzerland).25
Prophylaxis is considered the optimal treatment strategy for pediatric patients with severe hemophilia to prevent bleeding.26,27 A prophylactic regimen must account for each patient’s unique bleeding pattern, PK profile, adherence to treatment and level of physical activity. In this study, 18 patients experienced 173 NSB events when they were on a prophylaxis regimen; 97 NSB events (56%) were reported by three patients who had a high AsBR. Of these, one patient was non-compliant with his prophylaxis regimen and the other two patients already had hemarthrosis at enrollment and experienced their bleeds during the first 3 months of the study. Seven patients switched to prophylaxis with pdVWF/FVIII from a prior product. These data clearly demonstrate the benefit of secondary prophylaxis, although the demanding medical regimen can lead to imperfect compliance.
Safety results in this study were reflective of the pediatric population, with cough and pyrexia being the most frequently reported TEAEs. Two patients in the prophylaxis arm developed a transient low titer FVIII inhibitor and one patient in the on-demand arm developed a high titer inhibitor during the study; additionally, another patient in the on-demand arm was diagnosed with a pre-existing inhibitor. Both inhibitor patients in the on-demand arm withdrew from the study, no other TEAEs led to withdrawal from the study and no deaths occurred during the study. With the exception of the development of inhibitors in three patients, safety results were comparable with those seen in adult/adolescent PTPs with hemophilia A;20 comparable safety results were also reported in adult/adolescent PTPs with severe hemophilia A who were treated with a lower concentration plasma-derived VWF:FVIII (1:1).25
Despite knowledge of several well-established risk factors for inhibitor development, why some patients develop an inhibitor and others do not remains unclear. In previously untreated patients with severe hemophilia A, the first 50 EDs are when patients are at highest risk of inhibitor development.28 The three patients who developed inhibitors during our study only had limited exposure to FVIII products prior to the study (20, 22, and 48 EDs, respectively). Mutations such as null mutations, large deletions, nonsense mutations, and intron 22 inversions are also associated with a high prevalence of inhibitors.29 This was evidenced in our study as the two patients who developed a low titer inhibitor during prophylaxis treatment with pdVWF/FVIII had a mutation in exon 25 and an intron 22 inversion of the FVIII gene, respectively, while the patient who developed a high titer inhibitor during on-demand treatment with pdVWF/FVIII had an intron 22 inversion of the FVIII gene. Although genetics play a role, the discordance in inhibitor development between monozygotic twins observed in an international study of brothers with hemophilia A demonstrates that other treatment-related variables may also play a role in inhibitor development.30 Age at first infusion (<6 months vs >12 months) and prophylaxis versus no prophylaxis have also been identified as potential risk factors, with a higher rate of inhibitor development in those who receive their first FVIII infusion before 6 months of age31 and a reduced risk of inhibitor development in those who receive regular prophylaxis.32 However, later analyses have offered conflicting results, with some studies showing no effects on inhibitor development linked to these factors, and so it is not clear whether or not first infusion at an early age or regular prophylaxis offer a protective effect.33 In our study, the patient with a pre-existing FVIII inhibitor, who developed FVIII inhibitors after a FVIII product taken between screening and start of treatment with pdVWF/FVIII, was at high risk because he had started his first FVIII on-demand treatment at a very young age (<8 months) and all four patients had less than 50 EDs prior to study entry. Although the published studies designed to associate treatment conditions with inhibitor development are indicative, the association between FVIII administration and inhibitor formation is hard to predict until the genetic factors that underlie inhibitor development are better understood and can be used to properly stratify patients.
Limitations to our study include the low patient numbers, the disparity in age between the patients in the prophylaxis versus on-demand arms and the uneven distribution of patients with more or less than 50 EDs within these treatment arms. There are also few studies of VWF/FVIII concentrates in patients with hemophilia A, particularly in pediatrics. Further studies to confirm our observed efficacy, safety and PK parameters of VWF/FVIII in pediatric patients with severe hemophilia A would be beneficial.
Conclusions
In summary, pdVWF/FVIII was observed to be efficacious as both on-demand and prophylaxis therapy in pediatric patients with hemophilia A, with the hemostatic efficacy assessed as either excellent or good in all cases. Two patients developed a transient low titer FVIII inhibitor. One high-risk patient developed a high titer inhibitor associated with pdVWF/FVIII treatment which is within the expected incidence range of minimally pre-treated pediatric patients. No other safety findings of concern were observed. The efficacy and safety profile of pdVWF/FVIII was similar to that of the adult/adolescent population. These results provide evidence for use of pdVWF/FVIII to treat and prevent bleeding events in pediatric patients with hemophilia A, supporting the favorable benefit-risk profile of this concentrate.
Abbreviations
AE, adverse event; AsBR, annualized spontaneous bleeding rate; AUC, area under the concentration curve; CL, total clearance of the drug from the body; Cmax, maximum plasma concentration; FVIII, factor VIII; FVIII:C, factor VIII: co-agulant activity; IR, incremental recovery; MRT, mean residence time; NSB, non-surgical bleeding; PK, pharmacokinetics; SAE, serious adverse event; SD, standard deviation; t1/2, half-life; TEAE, treatment-emergent adverse event; tmax, time to maximum concentration; Vss, volume of distribution at steady state; VWF, von Willebrand factor.
Data Sharing Statement
CSL will only consider requests to share Individual Patient Data (IPD) that are received from systematic review groups or bona-fide researchers. CSL will not process or act on IPD requests until 12 months after article publication on a public website. An IPD request will not be considered by CSL unless the proposed research question seeks to answer a significant and unknown medical science or patient care question. Applicable country-specific privacy and other laws and regulations will be considered and may prevent sharing of IPD.
Requests for use of the IPD will be reviewed by an internal CSL review committee. If the request is approved, and the researcher agrees to the applicable terms and conditions in a data sharing agreement, IPD that has been appropriately anonymized will be made available. Supporting documents including study protocol and Statistical Analysis Plan will also be provided.
For information on the process and requirements for submitting a voluntary data sharing request for IPD, please contact CSL at [email protected].
Acknowledgments
The authors would like to thank Olga Aleinikova and Liudmyla Vashchenko for their input in this clinical study. Editorial support was provided by Meridian HealthComms, and was funded by CSL Behring, Germany.
Funding
This study was supported by a grant from CSL Behring, Australia.
Disclosure
Claudia Djambas Khayat has received investigator fees from CSL Behring. Tobias Rogosch and Wilfried Seifert are employees of CSL Behring. Genadi Iosava, Irina Romashevskaya, Oleksandra Stasyshyn, Maria Teresa Pompa and Marta Julia Lopez have no conflicts of interest.
References
1. Mannucci PM, Tuddenham EG. The hemophilias – from royal genes to gene therapy. N Engl J Med. 2001;344(23):1773–1779. doi:10.1056/NEJM200106073442307
2. Lieuw K. Many factor VIII products available in the treatment of hemophilia A: an embarrassment of riches? J Blood Med. 2017;8:67–73. doi:10.2147/JBM.S103796
3. White GC
4. Berntorp E, Astermark J, Bjorkman S, et al. Consensus perspectives on prophylactic therapy for haemophilia: summary statement. Haemophilia. 2003;9(Suppl 1):1–4. doi:10.1046/j.1365-2516.9.s1.17.x
5. Ljung RC. Prophylactic infusion regimens in the management of hemophilia. Thromb Haemost. 1999;82(2):525–530. doi:10.1055/s-0037-1615875
6. Iorio A, Marchesini E, Marcucci M, et al. Clotting factor concentrates given to prevent bleeding and bleeding-related complications in people with hemophilia A or B. Cochrane Database Syst Rev. 2011;(9):CD003429. doi:10.1002/14651858.CD003429.pub4
7. Mannucci PM. The role of natural VWF/FVIII complex concentrates in contemporary haemophilia care: a guideline for the next decade. Haemophilia. 2012;18(Suppl 2):2–7. doi:10.1111/j.1365-2516.2012.02794.x
8. Calvez T, Chambost H, Claeyssens-Donadel S, et al. Recombinant factor VIII products and inhibitor development in previously untreated boys with severe hemophilia A. Blood. 2014;124(23):3398–3408. doi:10.1182/blood-2014-07-586347
9. Srivastava A, Santagostino E, Dougall A, et al. WFH guidelines for the management of hemophilia. Haemophilia. 2020;26(Suppl 6):1–158. doi:10.1111/hae.14046
10. Ettingshausen CE, Kreuz W. Recombinant vs. plasma-derived products, especially those with intact VWF, regarding inhibitor development. Haemophilia. 2006;12(Suppl 6):102–106. doi:10.1111/j.1365-2516.2006.01373.x
11. Garagiola I, Palla R, Peyvandi F. Risk factors for inhibitor development in severe hemophilia A. Thromb Res. 2018;168:20–27. doi:10.1016/j.thromres.2018.05.027
12. Gringeri A. Factor VIII safety: plasma-derived versus recombinant products. Blood Transfus. 2011;9(4):366–370. doi:10.2450/2011.0092-10
13. Gouw SC, van der Bom JG, Ljung R, et al. Factor VIII products and inhibitor development in severe hemophilia A. N Engl J Med. 2013;368(3):231–239. doi:10.1056/NEJMoa1208024
14. Chalmers EA, Brown SA, Keeling D, et al. Early factor VIII exposure and subsequent inhibitor development in children with severe haemophilia A. Haemophilia. 2007;13(2):149–155. doi:10.1111/j.1365-2516.2006.01418.x
15. Goudemand J, Rothschild C, Demiguel V, et al. Influence of the type of factor VIII concentrate on the incidence of factor VIII inhibitors in previously untreated patients with severe hemophilia A. Blood. 2006;107(1):46–51. doi:10.1182/blood-2005-04-1371
16. Gringeri A, Monzini M, Tagariello G, et al. Occurrence of inhibitors in previously untreated or minimally treated patients with haemophilia A after exposure to a plasma-derived solvent-detergent factor VIII concentrate. Haemophilia. 2006;12(2):128–132. doi:10.1111/j.1365-2516.2006.01201.x
17. Mancuso ME, Mannucci PM, Rocino A, et al. Source and purity of factor VIII products as risk factors for inhibitor development in patients with hemophilia A. J Thromb Haemost. 2012;10(5):781–790. doi:10.1111/j.1538-7836.2012.04691.x
18. Wight J, Paisley S. The epidemiology of inhibitors in haemophilia A: a systematic review. Haemophilia. 2003;9(4):418–435. doi:10.1046/j.1365-2516.2003.00780.x
19. Ammollo CT, Semeraro F, Vitulli A, et al. FVIII/VWF complex displays a greater pro-haemostatic activity than FVIII preparations devoid of VWF: study in plasma and cell-based models. Haemophilia. 2020;26(4):e151–e160. doi:10.1111/hae.14008
20. Skotnicki A, Lissitchkov TJ, Mamonov V, et al. Efficacy, safety and pharmacokinetic profiles of a plasma-derived VWF/FVIII concentrate (VONCENTO®) in subjects with haemophilia A (SWIFT-HA study). Thromb Res. 2016;137:119–125. doi:10.1016/j.thromres.2015.10.014
21. Committee for Medicinal Products for Human Use (CHMP). Guideline on the clinical investigation of recombinant and human plasma-derived factor VIII products; 2019. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-clinical-investigation-recombinant-human-plasma-derived-factor-viii-products-revision-2_en.pdf.
22. Bjorkman S, Blanchette VS, Fischer K, et al. Comparative pharmacokinetics of plasma- and albumin-free recombinant factor VIII in children and adults: the influence of blood sampling schedule on observed age-related differences and implications for dose tailoring. J Thromb Haemost. 2010;8(4):730–736. doi:10.1111/j.1538-7836.2010.03757.x
23. Bjorkman S, Oh M, Spotts G, et al. Population pharmacokinetics of recombinant factor VIII: the relationships of pharmacokinetics to age and body weight. Blood. 2012;119(2):612–618. doi:10.1182/blood-2011-07-360594
24. Swystun LL, Ogiwara K, Rawley O, et al. Genetic determinants of VWF clearance and FVIII binding modify FVIII pharmacokinetics in pediatric hemophilia A patients. Blood. 2019;134(11):880–891. doi:10.1182/blood.2019000190
25. Klukowska A, Windyga J, Batorova A. Clinical efficacy of a novel VWF-containing FVIII concentrate, Wilate®, in the prophylaxis and treatment of bleeding episodes in previously treated haemophilia A patients. Thromb Res. 2011;127(3):247–253. doi:10.1016/j.thromres.2010.11.030
26. Valentino LA. Considerations in individualizing prophylaxis in patients with haemophilia A. Haemophilia. 2014;20(5):607–615. doi:10.1111/hae.12438
27. Petrini P, Valentino LA, Gringeri A, et al. Individualizing prophylaxis in hemophilia: a review. Expert Rev Hematol. 2015;8(2):237–246. doi:10.1586/17474086.2015.1002465
28. Giangrande PLF, Hermans C, O’Mahony B, et al. European principles of inhibitor management in patients with haemophilia. Orphanet J Rare Dis. 2018;13(1):66. doi:10.1186/s13023-018-0800-z
29. Oldenburg J, Schroder J, Brackmann HH, et al. Environmental and genetic factors influencing inhibitor development. Semin Hematol. 2004;41(1 Suppl 1):82–88. doi:10.1053/j.seminhematol.2003.11.016
30. Astermark J, Berntorp E, White GC, et al. The Malmo International Brother Study (MIBS): further support for genetic predisposition to inhibitor development in hemophilia patients. Haemophilia. 2001;7(3):267–272. doi:10.1046/j.1365-2516.2001.00510.x
31. van der Bom JG, Mauser-Bunschoten EP, Fischer K, et al. Age at first treatment and immune tolerance to factor VIII in severe hemophilia. Thromb Haemost. 2003;89(3):475–479. doi:10.1055/s-0037-1613376
32. Gouw SC, van der Bom JG, Marijke van den Berg H. Treatment-related risk factors of inhibitor development in previously untreated patients with hemophilia A: the CANAL Cohort Study. Blood. 2007;109(11):4648–4654. doi:10.1182/blood-2006-11-056291
33. Peyvandi F, Garagiola I. Product type and other environmental risk factors for inhibitor development in severe hemophilia A. Res Pract Thromb Haemost. 2018;2(2):220–227. doi:10.1002/rth2.12094
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.