")
Back to Journals » Clinical Ophthalmology » Volume 10
Efficacy of valproic acid for retinitis pigmentosa patients: a pilot study
Authors Iraha S, Hirami Y, Ota S, Sunagawa GA, Mandai M, Tanihara H, Takahashi M, Kurimoto Y
Received 6 April 2016
Accepted for publication 24 May 2016
Published 25 July 2016 Volume 2016:10 Pages 1375—1384
DOI https://doi.org/10.2147/OPTH.S109995
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Satoshi Iraha,1–5 Yasuhiko Hirami,1,2 Sachiko Ota,1,2 Genshiro A Sunagawa,3 Michiko Mandai,1–3 Hidenobu Tanihara,5 Masayo Takahashi,1–4 Yasuo Kurimoto1,2
1Department of Ophthalmology, Institute of Biomedical Research and Innovation Hospital, 2Department of Ophthalmology, Kobe City Medical Center General Hospital, 3Laboratory for Retinal Regeneration, RIKEN Center for Developmental Biology, Kobe, 4Application Biology and Regenerative Medicine, Graduate School of Medicine, Kyoto University, Kyoto, 5Department of Ophthalmology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan
Purpose: The purpose of this study was to examine the efficacy and safety of valproic acid (VPA) use in patients with retinitis pigmentosa (RP).
Patients and methods: This was a prospective, interventional, noncomparative case study. In total, 29 eyes from 29 patients with RP whose best-corrected visual acuities (BCVAs) in logarithm of the minimum angle of resolution (logMAR) ranged from 1.0 to 0.16 with visual fields (VFs) of ≤10° (measured using Goldmann perimeter with I4) were recruited. The patients received oral supplementation with 400 mg of VPA daily for 6 months and were followed for an additional 6 months. BCVAs, VFs (measured with the Humphrey field analyzer central 10-2 program), and subjective questionnaires were examined before, during, and after the cessation of VPA supplementation.
Results: The changes in BCVA and VF showed statistically significant differences during the internal use of VPA, compared with after cessation (P=0.001). With VPA intake, BCVA in logMAR significantly improved from baseline to 6 months (P=0.006). The mean deviation value of the VF significantly improved from baseline to 1 month (P=0.001), 3 months (P=0.004), and 6 months (P=0.004). These efficacies, however, were reversed to the baseline levels after the cessation of VPA intake. There were no significant relations between the mean blood VPA concentrations of each patient and the changes in BCVA and VF. During the internal use of VPA, 15 of 29 patients answered “easier to see”, whereas blurred vision was registered in 21 of 29 patients on cessation. No systemic drug-related adverse events were observed.
Conclusion: While in use, oral intake of VPA indicated a short-term benefit to patients with RP. It is necessary to examine the effect of a longer VPA supplementation in a controlled study design.
Keywords: retinitis pigmentosa, valproic acid, clinical trial, retina
Introduction
Retinitis pigmentosa (RP) is the term used for a group of retinal diseases that are characterized by inherited, progressive degeneration of retinal tissue, mainly rod and secondarily cone photoreceptors. The prevalence of RP is ~1:4,000 worldwide. The clinical features are night blindness and visual field (VF) loss due to loss of rod photoreceptors. Many patients fall into a classical pattern of difficulties with dark adaptation and night blindness and loss of midperipheral vision field. As the disease advances, they lose peripheral vision, eventually developing “tunnel vision” with the remaining cone photoreceptors, and finally lose central vision and visual acuities (VAs) as these cones secondarily degenerate in the macular region.1,2 Typically, it takes several years until the patients lose their central vision; therefore, it is important to protect the cone photoreceptors in the macular area for RP patients. Because RP is caused by various mutations in any of >45 responsible genes, the processes of degeneration are considered to be not uniform, and no effective treatment other than nutritional supplementation of vitamin A currently exists.
Recently, Noorwez et al3 reported that valproic acid (VPA) can increase the yield of properly folded RP mutant rhodopsins; by using their high-throughput screening method involving in silico, cell-based, and in vitro assays, the authors were able to identify pharmacological chaperones of misfolded rhodopsin. Based on the data, Clemson et al4 reported in their retrospective study that treatment with VPA in patients with RP improved VAs and VFs.
However, there were controversies over that study,5–7 and another group, Bhalla et al,8 not only claimed no improvement in VA and VF in their study but also stated that VPA may facilitate some adverse side effects. To date, various clinical studies have been performed,9,10 but no conclusion has been reached regarding the efficacy of using VPA in patients with RP.
VPA has been widely used as an antiepileptic drug for several decades. The pharmacological basis of the antiepileptic action of VPA has been related to the regulation of the glutamate excitatory neurotransmission and/or gamma-aminobutyric acid (GABA) inhibitory neurotransmission.11 Recent studies showed that VPA is an effective inhibitor of histone deacetylases, the key enzymes for the control of histone acetylation state and hence for the epigenetic regulation of gene expression. Mainly through inhibition of histone deacetylases, VPA induces apoptosis of microglia cells12 and activates BDNF promoter.13 Moreover, VPA induces neuronal differentiation but suppresses astrocytic and oligodendrocytic differentiation of neural stem cells14 and promotes neurite outgrowth.15
In this prospective study, we examined the efficacy and safety of VPA use in Japanese patients with RP.
Patients and methods
Trial registration
This study is registered with the clinical trials registry of the Japan Medical Association Center for Clinical Trials, number JMA-IIA00053.
Ethics
In this prospective, interventional, noncomparative case study, the study protocols adhered to the tenets of the Declaration of Helsinki. Ethics committee approval was granted by the ethics committee at the Institute of Biomedical Research and Innovation. Written informed consent was obtained from each patient before any study procedures were performed. All the patients were seen at the Institute of Biomedical Research and Innovation Hospital (Kobe, Japan) from December 2010 to January 2013.
Patients
The diagnosis of RP was based on the patients’ symptoms of night blindness, ring scotoma or concentric constriction of VFs, markedly reduced or nonrecordable a- and b-wave amplitudes on electroretinogram (ERG) testing, and ophthalmoscopic findings (ie, characteristic fundus changes in attenuated retinal vessels and bone-spicule-like pigment clumping).
The inclusion criteria were the following: 20 years old or older, best-corrected visual acuities (BCVAs) of >20/200 and <20/30, and VFs of ≤10° measured using Goldmann perimeter with isopter I4.
The exclusion criteria were the following: patients with retinal diseases other than RP, including retinal degeneration secondary to inflammation or infection and retinal vascular or macular diseases; cataractous lens gradings of more than N1, C2, or P1 according to Lens Opacities Classification System III grading; previous intraocular surgery except for uncomplicated cataract extraction; women of childbearing potential who were pregnant, nursing, or planning a pregnancy; presentation of liver disease or a urea cycle disorder; patients who had drug hypersensitivity; patients who had attempted suicide or had suicidal thoughts with manic depressive illness; and patients who were using contraindication medicine.
Study protocols
Prior to treatment at the initial study visit, each patient underwent ophthalmic examinations, including BCVA measured using a Landolt chart, intraocular pressure measurements, VF measured with the Humphrey field analyzer (HFA; Zeiss-Humphrey Systems, Dublin, CA, USA) 10-2 program, slit-lamp biomicroscopy, and dilated indirect fundus ophthalmoscopy.
After baseline measurements were obtained, all the patients were instructed to receive oral supplementation of 400 mg (the lowest dosage used for anticonvulsant therapy) of VPA (Depakene-R®; Kyowa Hakko Kirin, Tokyo, Japan) daily for 6 months. The patients were followed for an additional 6 months without VPA intake.
The patients returned to our clinic for follow-up visits and were asked to report on the development of any subjective visual changes as well as any systemic adverse events. BCVAs, VFs, and ophthalmic findings were collected throughout the entire study at months 1, 3, 6 (end of supplementation), 9, and 12. Blood samples were collected to check blood counts, clinical biochemistry, and the blood concentration of VPA at months 1, 2, 3, 4, and 6 (end of supplementation).
BCVA was converted to the value of logarithm of the minimum angle of resolution (logMAR) for all analyses.
VF test results were summarized using the mean deviation (MD) value calculated by the HFA provided software. The calculation of the MD value involved averaging the differences between the measured sensitivities and the age-adjusted normal sensitivities (total deviations) at each test point.
Although not included as routine tests in our study design, microperimetry-1 (MP-1; NIDEK, Gamagori, Japan) or multifocal ERG (VERIS; Electro-Diagnostic Imaging, Inc., Redwood, CA, USA and LE-4000; Tomey, Nagoya, Japan) were also recorded in some patients at pretreatment and at 6 months or at 6 months and 12 months.
The primary end point of this study was improvement in BCVA after the 6 months of treatment with VPA, and the secondary end points were VF and the occurrence of adverse events.
Determination of sample size
The variance approximation for the estimation of sample size was obtained from previous studies that have tested a similar hypothesis.4 A sample size calculation was performed before the study, assuming a maximum dropout rate of 30%. Accordingly, we assumed that at least 30 patients were going to fail power of 80% (β-1) to detect a logMAR 0.2 difference in VA between patients before and after receiving VPA.
Statistical analysis
All parameters obtained prior to VPA treatment and at 1 month, 3 months, 6 months, 9 months, and 12 months after treatment initiation were compared using the Wilcoxon signed-rank test with Bonferroni correction. We utilized a Bonferroni adjustment for multiple comparisons, after which P-values ≤0.01 were considered as statistically significant. All statistical analyses were performed using the statistical analysis software (SPSS Inc., Chicago, IL, USA).
Results
In total, 31 patients with RP were enrolled in this study. Two patients were lost to follow-up after the 6 months visit; the data for these patients were not included in the analysis. Overall, 29 patients (12 males and 17 females) with RP completed the 12 months study period. The patients’ ages ranged from 30 years to 72 years (mean ± SD: 52.5±11.5 years). Mendelian inheritance studies disclosed 13 sporadic, eleven autosomal recessive, and five autosomal dominant patterns. The age, sex, Mendelian inheritance, BCVA, MD value of the VF, and mean blood VPA concentration are shown in Table 1.
 | Table 1 Summary of data in this study |
First, we evaluated the changes in the visual function during the VPA administration period (from baseline to 6 months) and the cessation period (from 6 months to 12 months). The median changes in the value of logMAR BCVA per month were 0.00 (interquartile range [IQR], −0.18–0.00) during administration and 0.00 (IQR, 0.00–0.20) during cessation, which showed statistically significant difference (P=0.001; Figure 1A). The median changes in the MD value of VF per month were −0.11 dB (IQR, −0.18–0.03) during administration and 0.48 dB (IQR, −0.17–0.28) during cessation, which also showed statistically significant difference (P=0.001; Figure 1B).
 | Figure 1 The changes in the visual function during the VPA administration period and the cessation period. |
Next, we evaluated the shift in VA (logMAR) and VF (dB) over time (Figure 2). The median logMAR BCVA values were 0.39 (IQR, 0.30–0.69) at baseline, 0.39 (IQR, 0.30–0.52; P=0.08) at 1 month, 0.39 (IQR, 0.30–0.61; P=0.02) at 3 months, 0.39 (IQR, 0.30–0.52; P=0.006) at 6 months, 0.39 (IQR, 0.30–0.69; P=0.14) at 9 months, and 0.39 (IQR, 0.30–0.76; P=0.62) at 12 months. Compared with baseline, the logMAR BCVA value was significantly improved at 6 months, during the period of VPA treatment (Figure 2A).
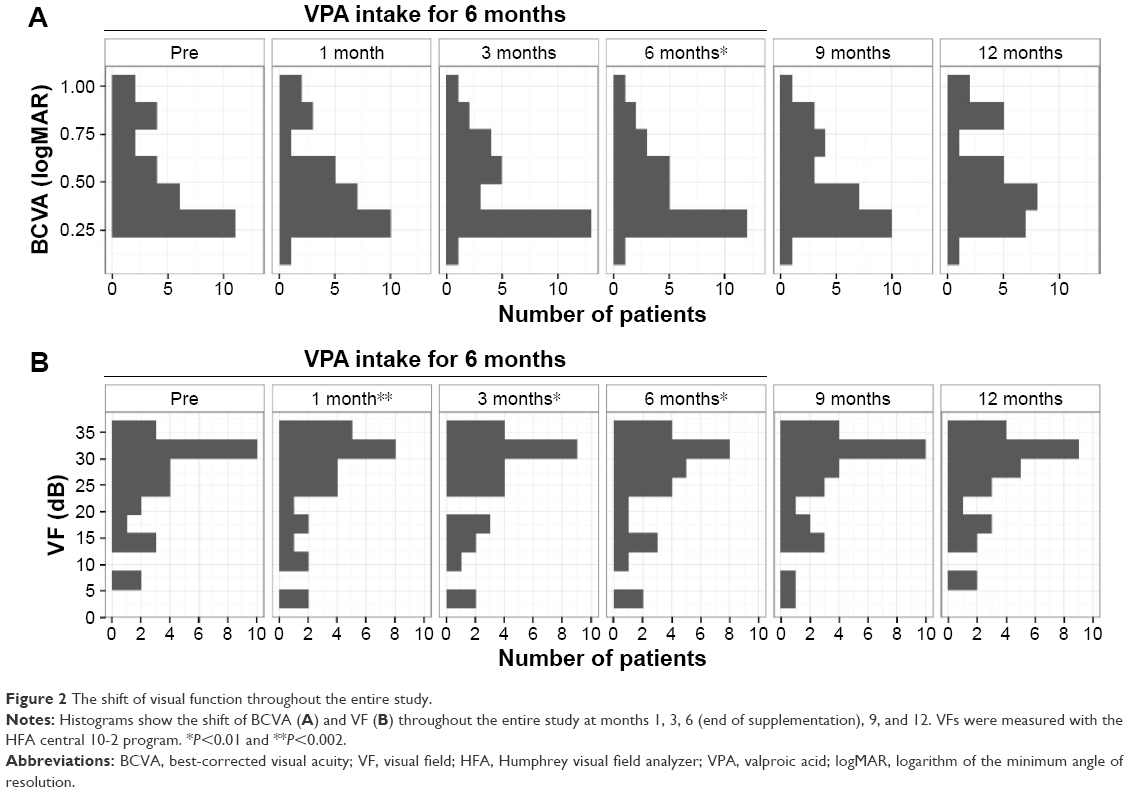 | Figure 2 The shift of visual function throughout the entire study. |
The median MD values of VF were 28.89 dB (IQR, 20.26–33.18) at baseline, 27.54 dB (IQR, 18.92–32.52; P=0.001) at 1 month, 28.14 dB (IQR, 18.29–32.60; P=0.004) at 3 months, 28.16 dB (IQR,18.59–31.96; P=0.004) at 6 months, 27.97 dB (IQR, 19.51–33.11; P=0.88) at 9 months, and 29.15 dB (IQR, 18.68–33.06; P=0.97) at 12 months. Compared with baseline, the median MD values of VF were significantly improved at 1 month, 3 months, and 6 months, corresponding to the period of VPA treatment (Figure 2B).
The mean blood concentration of VPA increased to 43.04±16.96 μg/mL at 1 month after intake. During the internal use period, the blood concentration of VPA was stable in each patient and the total mean blood concentration value of VPA within 6 months was 39.00±12.47 μg/mL. As shown in Figure 3, there were no significant relations between the mean blood VPA concentration values of each patient and the changes in BCVA (r=−0.06, P=0.73) and VF (r=−0.18, P=0.33) at 6 months.
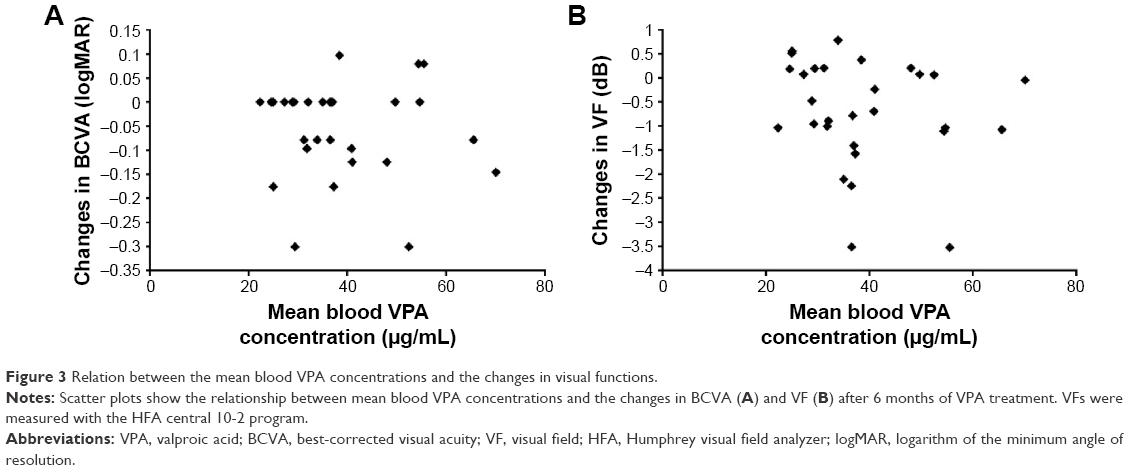 | Figure 3 Relation between the mean blood VPA concentrations and the changes in visual functions. |
The patients’ subjective visual symptoms reported during and after VPA treatment are summarized in Table 2. During intake of VPA, eleven of 29 (38%) patients felt no change in their vision, eight (28%) patients felt clearer color vision, four (14%) patients felt legibleness, and four (14%) patients felt brightness. Two patients complained of blurred vision, but it disappeared without treatment. After the cessation of VPA intake, eight (28%) patients reported having blurred vision, four (14%) patients reported seeing dimness, three (10%) patients reported having photophobia, two (7%) patients reported difficulty seeing colors, and one (3%) patient experienced fatigue.
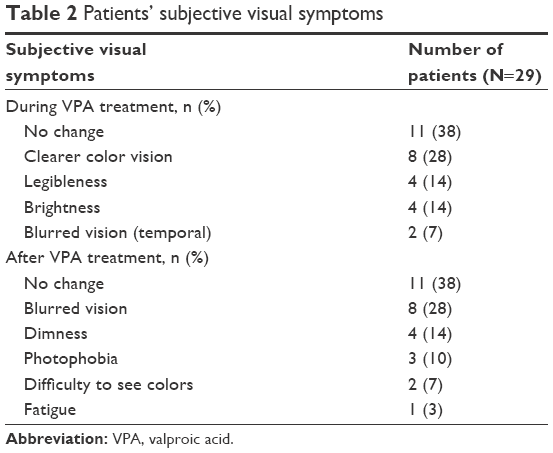 | Table 2 Patients’ subjective visual symptoms |
Although not included in the study design and statistical analysis, we performed MP-1 and multifocal ERG tests on some patients, and the preliminary results are shown in the Supplementary materials section. Some patients gained sensitivity by MP-1 at 6 months after VPA treatment (Figure S1). With multifocal ERG, the amplitudes of (P1-N1) in rings 3 and 4 (perifoveal area)16 were relatively unchanged between the two periods, whereas (P1-N1) amplitudes of ring 1 (center of the fovea) seem to increase in some patients after 6 months of treatment, whereas the changes after VPA cessation seem to be insignificant (Figure S2).
Throughout the study period, no systemic drug-related serious adverse events were observed in the study participants; checked blood counts from collected blood samples and clinical biochemistry were within normal lesion. During the period of VPA intake, dizziness was reported in four (14%) patients. Stomatitis, alopecia, and diarrhea were each reported in one (3%) patient.
Discussion
While several reports showed that oral VPA treatment improved VAs and VFs of RP patients, the opposite results were shown in other reports. The results of existing reports in VPA treatment are summarized in Table 3.
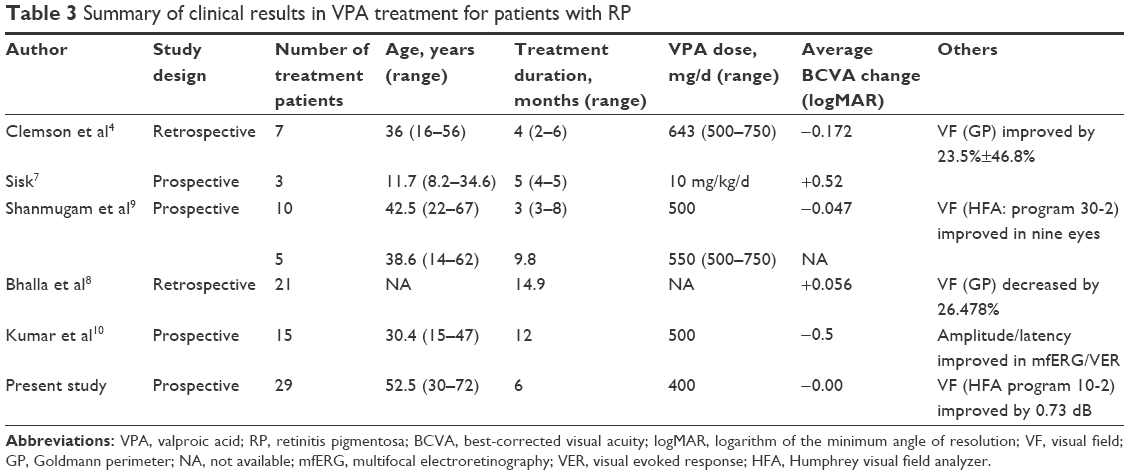 | Table 3 Summary of clinical results in VPA treatment for patients with RP |
In the present study of RP patients, the BCVA and MD values in the HFA 10-2 program significantly improved on average after 6 months of VPA intake and returned to the baseline levels after the cessation of the VPA treatment (Figure 2). Based on a large cohort study of natural courses of RP, the mean annual rates of decline of remaining BCVA and VF were estimated to be 1.0%–8.6% and 2.6%–13.5%, respectively.17 Compared to these data, our results showed some beneficial effect of VPA on RP patients at the 12-month time point even with its confined periodic effect. We cannot deny the possibility of placebo effect, but two previous studies using docosahexaenoic acid or 9-cis β-carotene had a randomized placebo treatment group without provision of multivitamin, and the placebo group in both of these studies worsened in VF by 1.4±1.32 dB with HFA between years 0 and 4 or by 0.5±4.5 cm2 with Goldmann perimetry within 90 days.18,19 Indeed, we cannot make a direct comparison between our results and these past studies, but we think that the VPA effect observed here is worthy of further investigation with a controlled study design in the future.
Nevertheless, the effect of VPA on BCVA observed in our study may not seem to be remarkable as one might expect (0.2 log units). A large cohort study showing the significant effect to slow the progression of RP by nutritional supplements such as vitamin and docosahexaenoic acid presented its efficacy by VF and ERG amplitudes but not by BCVA.20,21 Altogether, these results indicate that the change in BCVA may not be sufficiently sensitive to evaluate the efficacy of a treatment for RP patients. We also performed MP-1 and, more objectively, multifocal ERGs on a limited number of patients. Although the data collection was performed in the limited number of patients, we observed the increased P1-N1 amplitudes specifically in ring 1 central fovea in some patients after 6 months of VPA treatment. This may imply the effect of VPA on the area of remaining photoreceptors, but the overall amplitudes were very small. We need more data to objectively evaluate the effect of VPA on foveal function.
In this study, there were no significant relationship between the mean blood VPA concentrations of each patient and the changes in BCVA and VF. First of all, VPA dosage used in this study was relatively low in order to minimize adverse effect of VPA, and the plasma VPA levels in our patients were below the therapeutic range (45–100 mg/L). With this low concentration, albumin binding sites on VPA were unsaturated, and therefore, VPA binds to albumin at variable degrees, making its pharmacological behavior difficult to estimate.22–24 If we use the therapeutic range of VPA, it might be able to show clearer effect of VPA on BCVA or VF.
We also observed a significant improvement in MD values of HFA 10-2 programs with VPA treatment. In the original report by Clemson et al, their studies were based on an in vitro experiment that VPA acts as a molecular chaperone of rhodopsin proteins that increases the yield of properly folded mutant rhodopsins. Therefore, these authors suggested a potential effect of VPA on autosomal dominant RP, targeting improperly folded mutant rhodopsins, in rod photoreceptors. Our current study included RP patients with seemingly various causal genes with 13 sporadic, eleven autosomal recessive, and five autosomal dominant hereditary patterns. Our HFA examination with central 10-2 program may also represent cone photoreceptor functions. Additionally, close hearing of patient’s subjective symptoms also suggested some improvement in cone photoreceptor function: 16 of 29 (55%) patients felt it was easier to see during the period of VPA intake, whereas after cessation, eye discomfort was registered in 18 of 29 (62%) patients and half of the patients made some description related to color vision. Stasheff et al25,26 found that after degeneration started, ganglion cells exhibited hyperactivity, firing spontaneously at rates many times greater than normal in rd1 and rd10 mice, strains with closely related RP. Because the pharmacological basis of the antiepileptic action of VPA has been related to reduction in neuronal excitability by the increase in GABAergic activity, it is possible that VPA reduced hyperactivity of ganglion cells. Kimura et al27 also reported that VPA reduced retinal ganglion cell death in a mouse model of normal tension glaucoma. In this report, they indicated that VPA exerts neuroprotective effects through suppression of oxidative stress and stimulation of cell survival signaling. Therefore, patients may have felt that it was easier to see with reduced visual “noise” from spontaneous firing of ganglion cells or with neuroprotective effects. This phenomenon may also explain why BCVA and VF were improved in our study with various types of genetic patterns. However, this hypothesis was based on subjective symptoms of VPA-treated RP patients, and we need further objective evaluation of cone-related functions, including color vision or contrast sensitivity, in VPA-treated patients.
VPA has been widely used as an antiepileptic drug for several decades, and the use of VPA monotherapy in the treatment of epilepsy was not associated with VF defects.28,29 However, abnormal color visions in epileptic adolescents treated with VPA were reported.30,31 Sisk7 reported three cases with complications of VPA treatment, two of which had severe decrease in BCVA; the two patients were 8 years and 15 years of age and received 10 mg/kg/d of VPA for 4–5 months. Bhalla et al8 reported that 12 (39%) of 31 VPA-treated patients reported systemic side effects, of whom nine (29%) discontinued VPA intake due to side effects. In this study, we did not observe a severe decline in visual functions or systemic adverse events to discontinue VPA treatment. One possible explanation is that our patients were all at the age of 30 years or older and the dosage of VPA was relatively low (400 mg/d). It is still necessary to carefully observe the patients’ systemic conditions during VPA treatment and to exercise caution when using VPA for young RP patients.
Conclusion
In this prospective study, we found the following: 1) while in use, oral intake of VPA was suggestive of a short-term benefit to patients with RP and 2) regardless of the genotype, there were no systemic drug-related adverse events. It is necessary to examine the effect of a longer VPA supplementation in a controlled study design.
Acknowledgments
The context of this article was partially presented at ARVO 2014 Annual Meeting on Monday, May 5, 2014, and the abstract has been published in Investigative Ophthalmology & Visual Science (April 2014, Vol 55, 1390).
Disclosure
The authors report no conflicts of interest in this work.
References
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006; 368(9549):1795–1809. | ||
Gregory-Evans K, Pennesi ME, Weleber RG. Retinitis pigmentosa and allied disorders. In: Ryan SJ, editor. RETINA. 5th ed. Philadelphia, PA: SAUNDERS; 2013:761–835. | ||
Noorwez SM, Ostrov DA, McDowell JH, Krebs MP, Kaushal S. A high-throughput screening method for small-molecule pharmacologic chaperones of misfolded rhodopsin. Invest Ophthalmol Vis Sci. 2008;49(7):3224–3230. | ||
Clemson CM, Tzekov R, Krebs M, Checchi JM, Bigelow C, Kaushal S. Therapeutic potential of valproic acid for retinitis pigmentosa. Br J Ophthalmol. 2011;95(1):89–93. | ||
van Schooneveld MJ, van den Born LI, van Genderen M, Bollemeijer JG. The conclusions of Clemson et al concerning valproic acid are premature. Br J Ophthalmol. 2011;95(1):153. | ||
Sandberg MA, Rosner B, Weigel-DiFranco C, Berson EL. Lack of scientific rationale for use of valproic acid for retinitis pigmentosa. Br J Ophthalmol. 2011;95(5):744. | ||
Sisk RA. Valproic acid treatment may be harmful in non-dominant forms of retinitis pigmentosa. Br J Ophthalmol. 2012;96(8):1154–1155. | ||
Bhalla S, Joshi D, Bhullar S, Kasuga D, Park Y, Kay CN. Long-term follow-up for efficacy and safety of treatment of retinitis pigmentosa with valproic acid. Br J Ophthalmol. 2013;97(7):895–899. | ||
Shanmugam PM, Minija CK, Ramanjulu R, Tekwani P, Saxena M. Effect of short-term oral valproic acid on vision and visual field in retinitis pigmentosa. Ophthalmol Ther. 2012;1(1):6. | ||
Kumar A, Midha N, Gogia V, Gupta S, Sehra S, Chohan A. Efficacy of oral valproic acid in patients with retinitis pigmentosa. J Ocul Pharmacol Ther. 2014;30(7):580–586. | ||
Monti B, Polazzi E, Contestabile A. Biochemical, molecular and epigenetic mechanisms of valproic acid neuroprotection. Curr Mol Pharmacol. 2009;2(1):95–109. | ||
Dragunow M, Greenwood JM, Cameron RE, et al. Valproic acid induces caspase 3-mediated apoptosis in microglial cells. Neuroscience. 2006;140(4):1149–1156. | ||
Yasuda S, Liang MH, Marinova Z, Yahyavi A, Chuang DM. The mood stabilizers lithium and valproate selectively activate the promoter IV of brain-derived neurotrophic factor in neurons. Mol Psychiatry. 2009;14(1):51–59. | ||
Hsieh J, Nakashima K, Kuwabara T, Mejia E, Gage FH. Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proc Natl Acad Sci U S A. 2004;101(47):16659–16664. | ||
Yuan PX, Huang LD, Jiang YM, Gutkind JS, Manji HK, Chen G. The mood stabilizer valproic acid activates mitogen-activated protein kinases and promotes neurite growth. J Biol Chem. 2001;276(34): 31674–31683. | ||
Piao CH, Kondo M, Tanikawa A, Terasaki H, Miyake Y. Multifocal electroretinogram in occult macular dystrophy. Invest Ophthalmol Vis Sci. 2000;41(2):513–517. | ||
Berson EL, Rosner B, Weigel-DiFranco C, Dryja TP, Sandberg MA. Disease progression in patients with dominant retinitis pigmentosa and rhodopsin mutations. Invest Ophthalmol Vis Sci. 2002;43(9): 3027–3036. | ||
Hoffman DR, Locke KG, Wheaton DH, Fish GE, Spencer R, Birch DG. A randomized, placebo-controlled clinical trial of docosahexaenoic acid supplementation for Xlinked retinitis pigmentosa. Am J Ophthalmol. 2004;137(4):704–718. | ||
Rotenstreich Y, Belkin M, Sadetzki S, et al. Treatment with 9-cis β-carotene-rich powder in patients with retinitis pigmentosa: a randomized crossover trial. JAMA Ophthalmol. 2013;131(8):985–992. | ||
Berson EL, Rosner B, Sandberg MA, et al. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993;111(6):761–772. | ||
Berson EL, Rosner B, Sandberg MA, et al. Further evaluation of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment: subgroup analyses. Arch Ophthalmol. 2004;122(9):1306–1314. | ||
Gidal BE, Pitterle ME, Spencer NW, Maly MM. Relationship between valproic acid dosage, plasma concentration and clearance in adult monotherapy patients with epilepsy. J Clin Pharm Ther. 1995; 20(4):215–219. | ||
Sriboonruang T, Panomvana D, Chamchitchun S, Silpakit O. The impact of dosage of sustained-release formulation on valproate clearance and plasma concentration in psychiatric patients: analysis based on routine therapeutic drug monitoring data. J Clin Psychopharmacol. 2011;31(1):115–119. | ||
Cloyd JC, Dutta S, Cao G, et al; Depacon Study Group. Valproate unbound fraction and distribution volume following rapid infusions in patients with epilepsy. Epilepsy Res. 2003;53(1–2):19–27. | ||
Stasheff SF, Shankar M, Andrews MP. Developmental time course distinguishes changes in spontaneous and light-evoked retinal ganglion cell activity in rd1 and rd10 mice. J Neurophysiol. 2011;105(6): 3002–3009. | ||
Stasheff SF. Emergence of sustained spontaneous hyperactivity and temporary preservation of OFF responses in ganglion cells of the retinal degeneration (rd1) mouse. J Neurophysiol. 2008;99(3):1408–1421. | ||
Kimura A, Guo X, Noro T, et al. Valproic acid prevents retinal degeneration in a murine model of normal tension glaucoma. Neurosci Lett. 2015;588:108–113. | ||
Sorri I, Rissanen E, Mantyjarvi M, Kalviainen R. Visual function in epilepsy patients treated with initial valproate monotherapy. Seizure. 2005;14(6):367–370. | ||
Ozkul Y, Gurler B, Uckardes A, Bozlar S. Visual functions in epilepsy patients on valproate monotherapy. J Clin Neurosci. 2002;9(3):247–250. | ||
Verrotti A, Lobefalo L, Priolo T, et al. Color vision in epileptic adolescents treated with valproate and carbamazepine. Seizure. 2004;13(6):411–417. | ||
Verrotti A, Lobefalo L, Tocco AM, et al. Color vision and macular recovery time in epileptic adolescents treated with valproate and carbamazepine. Eur J Neurol. 2006;13(7):736–741. |
Supplementary materials
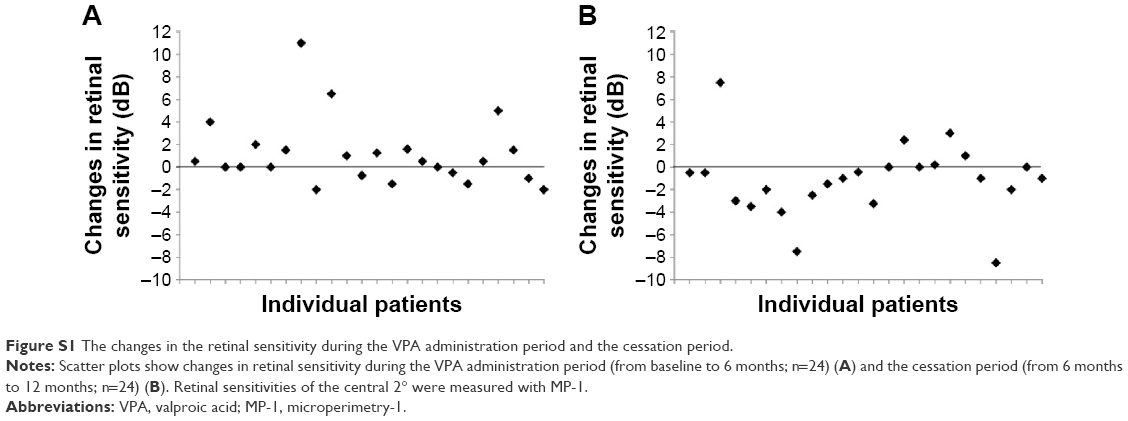 | Figure S1 The changes in the retinal sensitivity during the VPA administration period and the cessation period. |
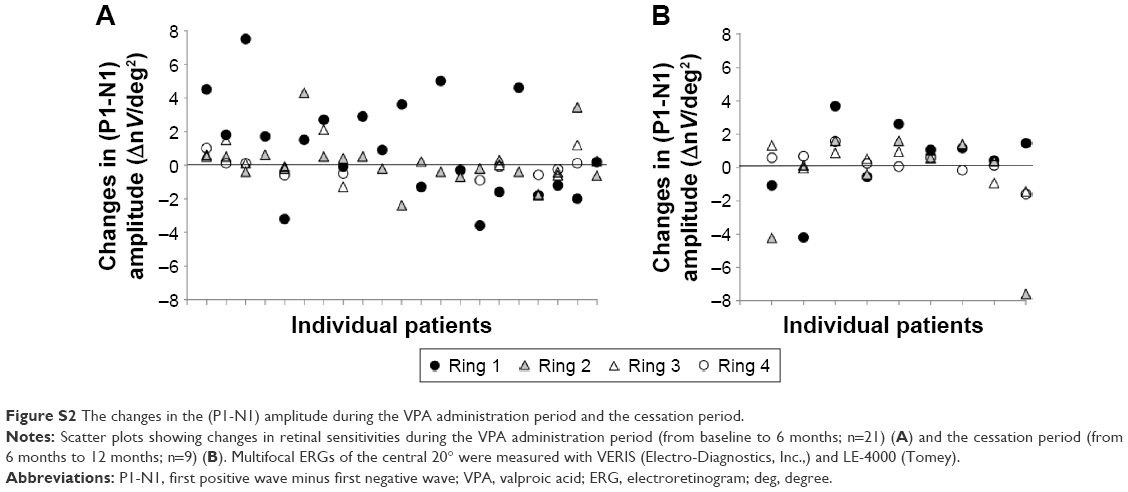 | Figure S2 The changes in the (P1-N1) amplitude during the VPA administration period and the cessation period. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.