Back to Journals » Journal of Pain Research » Volume 19
Efficacy and Safety of Adjunctive Therapy Using Single High-Dose S-Ketamine Infusion for Fibromyalgia: A Multicenter, Prospective, Randomized, Controlled, Open-Label, Blinded-Endpoint Study Protocol
Authors Shrestha N ![]() , Liu M, Niu S, Cheng H, Sun Y, Mei S
, Liu M, Niu S, Cheng H, Sun Y, Mei S ![]() , Luo F
, Luo F ![]()
Received 26 January 2026
Accepted for publication 7 May 2026
Published 16 May 2026 Volume 2026:19 598907
DOI https://doi.org/10.2147/JPR.S598907
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor King Hei Stanley Lam
Niti Shrestha,1,* Minying Liu,2,* Shaoning Niu,3,* Hao Cheng,3,* Yongxing Sun,4,* Shenghui Mei,5,6 Fang Luo2
1Department of Anesthesia and Intensive Care, National Academy of Medical Sciences, Bir Hospital, Kathmandu, Nepal; 2Department of Pain Management, Beijing Tiantan Hospital, Capital Medical University, Beijing, People’s Republic of China; 3Department of Anesthesiology, Beijing Ditan Hospital, Capital Medical University, Beijing, People’s Republic of China; 4Department of Anesthesiology, Beijing Sanbo Brain Hospital, Capital Medical University, Beijing, People’s Republic of China; 5Department of Pharmacy, Beijing Tiantan Hospital, Capital Medical University, Beijing, People’s Republic of China; 6Department of Clinical Pharmacology, College of Pharmaceutical Sciences, Capital Medical University, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Fang Luo, Department of Pain Management, Beijing Tiantan Hospital, Capital Medical University, No. 119, Nansihuan Xilu, Fengtai District, Beijing, 100070, People’s Republic of China, Email [email protected] Shenghui Mei, Department of Pharmacy, Beijing Tiantan Hospital, Capital Medical University, No.119, Nansihuan Xilu, Fengtai District, Beijing, 100070, People’s Republic of China, Email [email protected]
Background: Fibromyalgia (FM) is a chronic pain syndrome with limited response and delayed onset to current treatments. Esketamine (ESK) shows potential as a rapid-acting analgesic in FM patients, but previous low-dose studies have had limited efficacy and are constrained by single-center designs, monotherapy approaches, and small sample sizes. The efficacy of higher-dose ESK administered as an adjunct to guideline-recommended pharmacotherapy in FM remains unclear.
Study Design: Multicenter, prospective, randomized, controlled, open-label, blinded-endpoint trial.
Methods and Analysis: A total of 92 adult FM patients will be randomized in a 1:1 ratio to either the treatment group (single intravenous infusion of ESK plus oral pregabalin and venlafaxine) or the control group (oral pregabalin and venlafaxine alone). In the treatment group, ESK will be administered as a single intravenous infusion at a dose of 1 mg/kg on the day of enrollment. Both groups will follow identical dose titration regimens for pregabalin and venlafaxine, with dose escalation individualized according to patient tolerability and clinical response. Plasma concentrations of ESK and its metabolites will be measured at the end of infusion to enable a limited characterization of systemic exposure and to support exploratory exposure-response analyses. Participants will be followed for 12 weeks. The primary outcome is the change from baseline in the mean pain intensity during the first week after treatment. The secondary outcomes include average pain intensity, worst pain intensity, and proportion of patients achieving ≥ 50% and ≥ 30% pain reduction, median pain relief time, maximal tolerated doses of pregabalin and venlafaxine, patient-reported outcomes (Revised FM Impact Questionnaire, Hospital Anxiety and Depression Scale, Short Form 36 Health survey, Multidimensional Fatigue Inventory, and Medical Outcomes Study Sleep Scale), plasma concentrations of ESK and its metabolites at the end of infusion, and adverse events throughout the study period.
Trial Registration: ClinicalTrails.gov identifier: NCT07230171. Registered on November 13, 2025 (https://www.clinicaltrials.gov/search?term=NCT07230171).
Keywords: fibromyalgia, esketamine, adjunctive therapy, blinded-endpoint
Introduction
Fibromyalgia (FM) is a chronic condition characterized by generalized musculoskeletal pain, sleep disturbance, fatigue, and frequently comorbid depression.1 Its diagnostic criteria have evolved substantially over the past three decades, with the 2018 American Pain Society of Pain Taxonomy emphasizing multisite pain count at ≥6 sites, sleep disturbance, and fatigue lasting for over 3 months, with depression as a core associated symptom.2–4 FM affects approximately 2%~4% of the population,2,5–7 yet its etiology remains unclear, impeding development of targeted etiologic therapies.2,7
Due to heterogeneity of FM, a multimodal treatment approach is essential. Non-pharmacological therapies including education, cognitive behavioral therapy, and exercise, etc,8 are the first-line therapy for the management of FM.6 Pharmacological treatment is used when non-pharmacological therapies does not work.9 The American Pain Society, as well as the European League Against Rheumatism recommended the used of antiepileptics, tricyclic antidepressants, serotonin norepinephrine reuptake inhibitors (SNRIs), and selective serotonin reuptake inhibitors.9 However, the above drugs generally require gradual dose titration and often work slightly more slowly compared with rapid-acting agents such as esketamine (ESK), leading to a decline in patient compliance. Moreover, only a small proportion of patients achieve substantial pain relief.10 Consequently, there is a pressing need for a treatment strategy for FM that provides more rapid pain relief while also increasing the proportion of patients achieving significant pain relief.
The pathophysiology of FM is multifactorial, with central sensitization and dysregulation of neurotransmitters such as serotonin and norepinephrine potentially being key mechanisms.3,5,8,11–13 This pathophysiology underlies the close association between FM and depression, with up to 40% of patients experiencing depressive symptoms at any time and lifetime prevalence rates exceeding 70%14,15 Shared pathophysiological pathways and treatment responses have led to the view that FM and depression may represent differential expressions of a common underlying process.15 The high comorbidity between FM and depression further complicates management and supports the use of agents with dual analgesic and antidepressant properties. Accordingly, drugs possessing rapid-onset antidepressant effects represent promising candidates for rapid FM therapy.
Ketamine as a noncompetitive antagonist of N-methyl-D-aspartate (NMDA) type glutamate receptor that induces dissociative effects and is used for depression therapy and pain conditions, such as FM, phantom limb pain, and chronic migraine.16,17 A recent systematic review demonstrates the effectiveness and safety of ketamine in FM patients in short term.18 However, the widespread clinical use of ketamine for chronic pain management has been limited, largely due to concerns regarding its psychotomimetic adverse effects and potential for abuse, as well as the availability of better-tolerated alternatives.19 Attention has thus shifted to its enantiomers. ESK, the S-enantiomer of ketamine, has twice as potent as the racemic mixture and a four times higher affinity for the NMDA receptor compared to the R-enantiomer.16,20,21 At subanesthetic doses, ESK has demonstrated rapid and robust antidepressant effect with acceptable short-term tolerability.16,20,22 These pharmacological properties make ESK an attractive candidate for the treatment of chronic pain conditions characterized by central sensitization, such as FM. In this context, Noppers et al demonstrated that a low-dose intravenous ESK infusion (0.5 mg/kg over 30 minutes) produced a rapid analgesic response in FM patients, with a significantly higher proportion of patients achieving more than 50% pain reduction within 45 minutes after infusion compared with midazolam.23 This finding positioned ESK as a promising strategy for rapid-onset FM treatment, particularly during the early phase of treatment when conventional pharmacotherapies have not yet reached full efficacy. In deed, combination therapy with SNRIs and pregabalin represents an effective regimen for FM, but is limited by a delayed onset of analgesic benefit, often requires weeks of dose titration.24 Therefore, a rapid onset agent, such as ESK, may bridge this initial therapeutic gap, accelerating pain relief and enhancing overall efficacy compared to standard therapy.
Notably, no prior study has investigated ESK as an adjunct to guideline-recommended pharmacotherapy for FM. Moreover, the pioneering work by Noppers et al was constrained by small sample size, monotherapy design, lack of concurrent optimization of guideline-recommended therapies, and the use of relatively low dose of ESK, which may have contributed to the limited efficacy observed, with no sustained benefit beyond one week. In contrast, a randomized trial in patients with complex regional pain syndrome employing a more intensive intravenous ESK infusion regimen (up to 0.35 mg/kg/h for 4 hours daily over multiple days) demonstrated significant and sustained reductions in pain parameters lasting up to several weeks, suggesting that greater cumulative exposure or higher-dose ESK regimens may confer more durable analgesic benefits than lower-dose infusions.25 However, evidence regarding higher-dose ESK administered as adjunctive therapy in FM remains absent. Together, these limitations underscore the need for further investigation of higher-dose ESK (eg, 1 mg/kg) administered as an adjunct to standard pharmacological treatment, which aligns with the multi-modal therapeutic principle underlying FM management.
Therefore, we designed a multicenter, prospective, randomized, controlled, open-label, blinded-endpoint trial to test whether a single high-dose (1mg/kg) ESK infusion used as adjunctive therapy to standard pharmacotherapy for FM patients, results in a greater reduction in pain intensity during the first week after treatment compared to standard pharmacotherapy alone. In addition, plasma concentrations of ESK and its metabolites will be measured to enable a limited systemic exposure and to support exploratory exposure-response analyses, rather than comprehensive pharmacokinetic modeling. If the results support our hypothesis, ESK adjunctive to standard pharmacotherapy could become a superior treatment regimen for FM patients.
Methods
Study Design and Setting
A multicenter, prospective, randomized, open-label, blinded-endpoint (PROBE) trial will be performed to investigate whether a single intravenous infusion of high-dose ESK (1mg/kg) as adjunctive therapy to standard SNRI (venlafaxine) and antiepileptic (pregabalin) results in a greater reduction in pain intensity during the first week after treatment for FM patients compared to standard treatment. The study will be conducted at Beijing Tiantan Hospital, Beijing Ditan Hospital, and Beijing Sanbo Brain Hospital. All participating pain physicians will undergo standardization training to ensure consistency in clinical assessment and protocol execution. The study protocol has been approved by the Institutional Review Board of Beijing Tiantan Hospital (KY2025-217-03) and the local ethics committees of all participating centers following the criteria required by the Declaration of Helsinki. The study has been prospectively registered at https://www.clinicaltrials.gov (NCT07230171). The flowchart is briefly shown in Figure 1, and all trial procedures are summarized in Table 1.
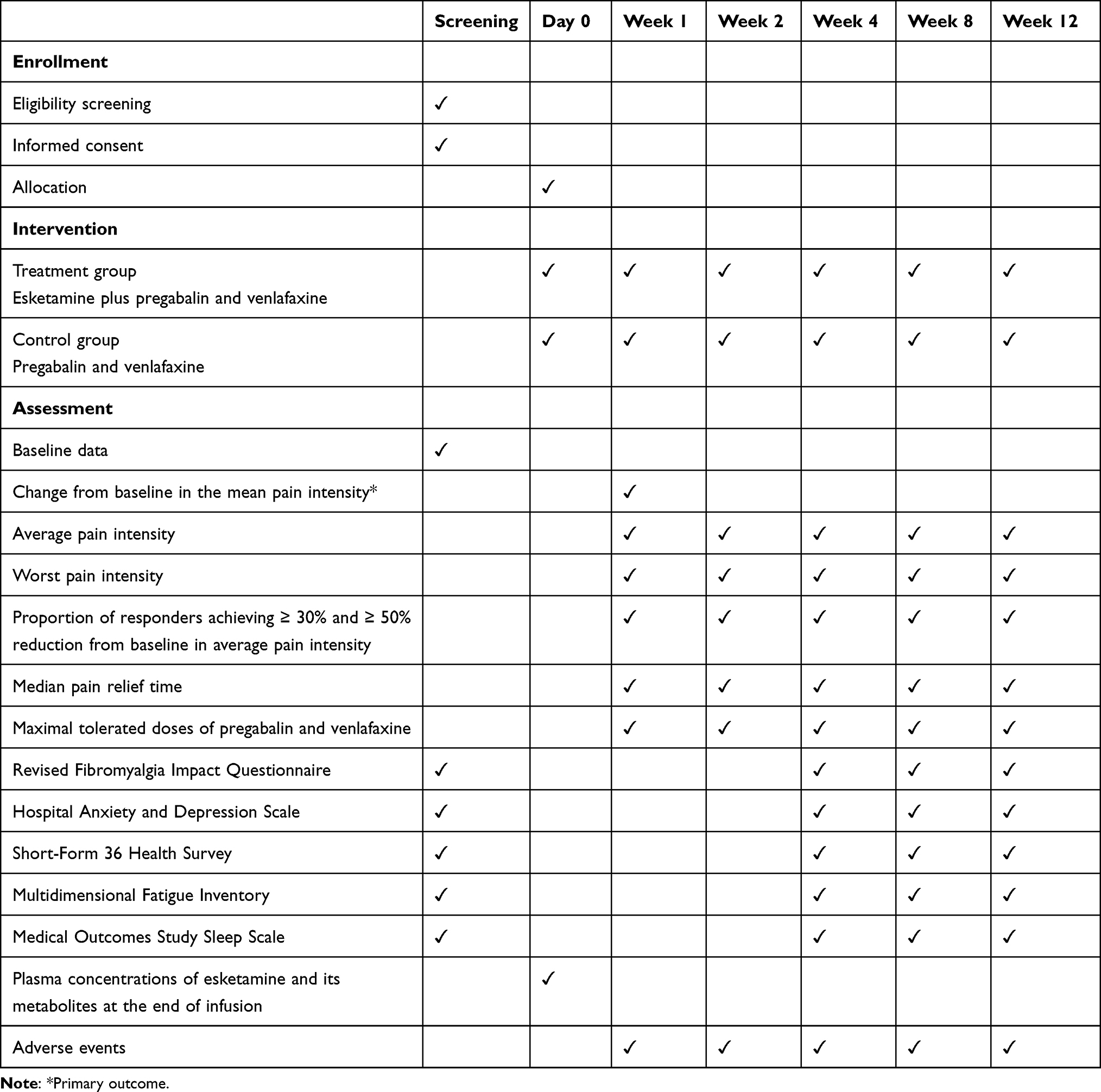 |
Table 1 The Schedule of Enrollment, Interventions, and Assessments |
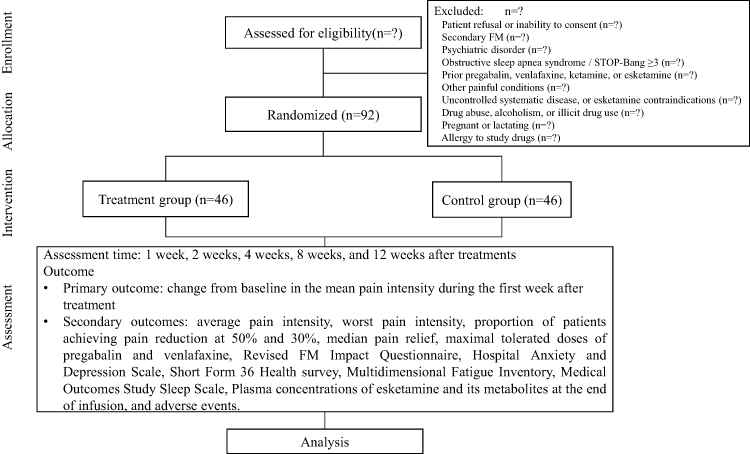 |
Figure 1 Flowchart of enrollment, allocation, intervention, and assessment. |
Recruitment and Informed Consent
Eligible patients will be screened during their initial outpatient visit by experienced pain physicians from the multicenter site, according to the predefined inclusion and exclusion criteria. Histories of prior treatment with pregabalin and/or venlafaxine for any indication, prior intravenous ketamine or ESK treatment for chronic pain, and prescription drug abuse, alcoholism, or illicit drug use will be assessed during screening through structured patient interviews and verified against available medical records where applicable. Participants meeting any of these criteria will be excluded before randomization. A screening period of up to 1 week will be allowed to ensure adequate evaluation of eligibility criteria and to provide sufficient time for participants to consider study participation. Participants who meet all inclusion criteria and do not meet any exclusion criteria, and who provide written informed consent, will be randomized after the screening period.
All potential participants will receive a detailed verbal explanation of the study objectives, procedures, benefits, and potential risks, and will be given ample time to inquire about the study and consider their participation before enrollment. Informed consent will be obtained from all participants before trial participation. Participation will be entirely voluntary, and participants will retain the right to withdraw from the study at any time without any effect on their subsequent medical care.
Study Population
Inclusion Criteria
- Patients between ages over 18 years with proper cognitive function and language skills for the study;
- Patients diagnosed with FM according to the 2016 revisions to the 2010/2011 American College of Rheumatology diagnostic criteria;26
- Patients who have experienced insufficient symptom relief with non-pharmacological treatments and have not previously received recommended pharmacological treatment for FM;
- A score of ≥ 4 on the average pain intensity over 7 days on the numeric rating scale (NRS) at baseline.
Exclusion Criteria
- Patient refusal;
- Inability to sign informed consent;
- Had other secondary FM, this is, hypothyroidism, nutritional deficiency, diabetes mellitus, connective tissue disorder;
- Had psychiatric disorder, this is, schizophrenia and other psychotic disorder, bipolar disorder, or personality disorder;
- Obstructive sleep apnea syndrome or a STOP-Bang score ≥ 3;27
- History of treatment with pregabalin and/or venlafaxine for any disease;
- History of treatment with intravenous ketamine or ESK for chronic pain;
- Presence of other painful ailments such as inflammatory rheumatic disease;
- Uncontrolled diabetes, refractory hypertension, malignancies, narrow-angle glaucoma, hyperthyroidism, severe cardiovascular disease or any other contraindications to ESK;
- History of prescription drug abuse, alcoholism or illicit drug use;
- Pregnant or lactating women;
- Allergic to any of the study drugs.
Withdrawal Criteria
- Voluntary withdrawal;
- Lost to follow-up;
- Unanticipated adverse events (AEs).
Randomization and Blinding
Participants who fulfill the criteria and complete informed consent will be randomly assigned to either the treatment group (ESK plus pregabalin and venlafaxine concomitant therapy) or the control group (pregabalin and venlafaxine concomitant therapy) in a 1:1 ratio. Randomization will be performed using a computer-generated, stratified block randomization sequence (block size of 4) via a secure, centralized web-based system. To minimize variability across trial sites, randomization will be stratified by center. Allocation will be implemented in real time after eligibility confirmation and written informed consent, ensuring that investigators responsible for recruitment and treatment have no access to upcoming allocation sequences.
This study adopts an open-label design. Therefore, participants and treating physicians will be aware of treatment allocation. To minimize bias, outcome assessors will remain blinded and independent of treatment procedures, and outcome will primarily be deprived from standardized patient-reported measures and daily pain diaries. Assessors will be instructed to avoid discussing treatment allocation with participants.
Intervention
All participants will initiate combination pharmacological treatment with pregabalin and venlafaxine. Dose titration will follow a standardized titration protocol and will be individualized according to patient tolerability and clinical response, with the aim of reaching the maximal tolerated dose (MTD) or the maximum daily recommended dose. The maximum recommended daily doses will be 450 mg28 for pregabalin and venlafaxine will be 225 mg. Dose escalation will be performed gradually.29 At each contact, pain intensity and AEs will be recorded and classified as mild, moderate or severe. In case of moderate-to-severe AEs, tolerability will be assessed. If AEs are tolerable, participants will be instructed to maintain the current dose for an additional 2–3 days to allow for possible adaptation. If AEs are intolerable, the dose will be reduced to the last tolerable level, and further escalation will be attempted during subsequent follow-up contacts.
Participants assigned to the treatment group will additionally receive a single intravenous infusion of ESK on the day of enrollment. Participants will be instructed to refrain from food or drink at least 4 hours prior to ESK administration.23 Esketamine Hydrochloride Injection (Hengrui Medicine Co., Ltd, Jiangsu, China) will be prepared at a dose of 1 mg/kg, diluted in 20 mL of normal saline.30 The infusion will be administered at a rate of 30mL/h over 40 minutes under the supervision of an experienced anesthesiologist in the day-care surgery unit.30 To reduce infusion-related adverse effects, such as nausea and vomiting, ondansetron (8 mg) will be administered prophylactically. In the event of psychotomimetic symptoms (eg, hallucinations), remimazolam (0.5–2 mg) will be given as needed. Continuous monitoring, including pulse oximetry, non-invasive blood pressure, and electrocardiography, will be performed throughout the infusion. All AEs will be documented and managed according to standard clinical practice. After completion of the infusion, participants will be transferred to a recovery unit and monitored for at least 2 hours or longer if clinically indicated.23 In the absence of ESK-related adverse effects, participants will be discharged. Given the anesthetic nature of the intervention and the potential need for airway and hemodynamic support, ESK administration will be conducted exclusively in the day-care surgery unit rather than in the outpatient clinic, ensuring immediate access to appropriate equipment and trained medical personnel. In the treatment group, a venous blood sample will be collected at the end of infusion to capture early systemic exposure to ESK and to support exploratory exposure-response analyses.31
Follow-Up
Participants will undergo structured follow-up assessments combining outpatient visits and telephone consultations to monitor treatment response, safety, medication titration, and adherence over the 12-week study period.23 A screening period of up to 7 days will be conducted prior to randomization to assess eligibility and obtain informed consent. Following this period, participants will be randomized and treatment will be initiated on day 0. Scheduled post-randomization assessment time points will be conducted at day 0, week 1, week 2, week 4, week 8, and week 12. The designated research assistants will guide dose titration of pregabalin and venlafaxine, assess tolerability, and record AEs.
Telephone follow-up will be performed at weeks 1 and 2 to monitor AEs, assess initial efficacy, and provide guidance for dose titration as needed. Outpatient clinic visits will be conducted at weeks 4, 8, and 12 for comprehensive evaluation of treatment efficacy and safety. During each outpatient visit, pain intensity and multidimensional patient-reported outcome measures will be assessed, including the Revised FM Impact Questionnaire (FIQR), Hospital Anxiety and Depression Scale (HADS), Short-Form 36 Health Survey (SF-36), Multidimensional Fatigue Inventory (MFI), and Medical Outcomes Study Sleep Scale (MOS). Face-to-face consultations will also be used to review medication adherence, adjust pharmacological treatment if necessary, document AEs, and provide prescriptions for subsequent medications.
Data Collection
Baseline assessments will be performed on the day of enrollment prior to the intervention. Demographic and clinical characteristics, including age, gender, weight, height, body mass index, duration of symptoms, number of tender points, and history of over-the-counter analgesic use (eg, paracetamol, non-steroidal anti-inflammatory drugs, tramadol), will be collected. In addition, patient-reported outcome measures, including the FIQR, HADS, SF-36, MFI, and MOS, will be assessed at baseline.
At each participating center, trained research assistants will conduct patient assessments using the following instruments:
Pain intensity will be measured using the NRS, where 0 indicates no pain, and 10 represents the worst imaginable pain. All participants will be instructed to complete a daily pain diary, recording their average pain intensity and worst pain intensity over the preceding 24 hours using the NRS. In both groups, pain diary data will be collected throughout the 12-week follow-up period, with scheduled study visits at weeks 1, 2, 4, 8, and 12. These diary records will be used to determine the time to pain relief and to calculate weekly average and worst pain intensity.
The FIQR will be used to assessed health status and functional disability.32 FIQR is a 21-item self-administered questionnaire, based on symptoms reported within the preceding 7 days. FIQR total score ranges from 0 to 100, with higher values indicating worse health status.
The HADS will be used to assess anxiety (7 items) and depression (7 items).33 Scores range from 0 to 21 on each subscale, with higher scores indicating worse symptomatology.
Quality of life will be assessed via the SF-36, which provides scores for eight dimensions (physical functioning, role-physical, role-emotional, bodily pain, general health, vitality, social functioning, and mental health), and a remaining item on the perception of health change. Each score ranges from 0 to 100, with a higher score implying better health status.34
Fatigue will be evaluated using the MFI,35 a 20-item assessment tool with five domains. Higher scores indicate a higher degree of fatigue.
The MOS is a questionnaire comprising 12 items that assess various aspects of sleep using a 6-point ordinal scale (1 indicating permanence and 6 indicating absence).36
AEs will be defined as any untoward medical occurrence following study intervention, regardless of causality. All AEs will be systematically recorded at each study visit and follow-up contact using a structured approach, including both open-ended questioning and targeted symptom checklists focusing on known ESK-related AEs. All AEs will be recorded in standardized case report forms. The severity of AEs will be graded according to the Common Terminology Criteria for AEs. The causal relationship between AEs and the study intervention will be assessed using the WHO-UMC system. Safety reporting will follow CONSORT recommendations for harms.37
Data Management and Quality Control
All study data will be collected by trained investigators at each participating site using standardized case report forms specifically designed for this protocol. To ensure data accuracy, completeness, and integrity, all information will be entered into a centralized electronic database (EpiData Manager, Version 4.6.0.6; EpiData Association, Denmark) through an independent double-data entry process. Discrepancies between the two entries will be identified automatically and resolved by verification against the original source documents.
An independent Data and Safety Monitoring Committee will be established to provide objective oversight of trial conduct and participant safety. The committee will convene at six-month intervals to review cumulative safety data, assess data quality and protocol adherence, and evaluate the overall progress of the study. Based on these reviews, the committee will issue formal recommendations to the Steering Committee regarding continuation, modification, or early termination of the trial.
Outcomes
Primary Outcome
The primary outcome will be the change from baseline in the mean pain intensity during the first week after treatment, calculated as the average of daily 24-hour pain intensity scores recorded in patient diaries over 7 consecutive days using NRS.
Secondary outcomes
- Average pain intensity at weeks 1, 2, 4, 8, and 12;
- Worst pain intensity at weeks 1, 2, 4, 8, and 12;
- Proportion of patients achieving pain reduction at 50% and 30% at weeks 1, 2, 4, 8, and 12;
- Median pain relief time, defined as the time of 50% decrease in NRS score from baseline.
- The MTDs of pregabalin and venlafaxine;
- FIQR, HADS, SF-36, MFI, and MOS scale at weeks 4, 8, and 12;
- Plasma concentrations of ESK and its metabolites at the end of infusion;
- AEs throughout the trial.
Patient and Public Involvement
Patients and the public were not involved in the conceptualization, design, or conduct of this study. Recruitment will be managed via physician referral and screened by clinical investigators. Findings will be shared through academic publications and professional presentations. Participants may request a lay summary of the study results after trial completion.
Statistical Plan
Sample size calculation was performed using the PASS software (V 15.0; NCSS, LLC, USA). The primary efficacy outcome for this study is the change from baseline in the mean pain intensity during the first week after treatment. Given the lack of existing data comparing ESK plus standard therapy versus standard therapy alone in FM, estimates were conservatively derived from prior ketamine trials.38 In the randomized trial by Noppers et al, weekly FM Impact Questionnaire pain scores were assessed during follow-up. However, detailed numerical data for the week-1 change from baseline were not systematically reported.23 Therefore, we conservatively assumed a mean reduction of 3.0 points in the ESK combination group and 1.5 points in the control group during the first week after treatment, with an assumed standard deviation of 2.0. Using a two-sample t-test with unequal variances, a two-sided significance level of 0.05, 90% power, and an allocation ratio of 1:1, 39 patients per group were required. To account for an anticipated dropout rate of 15% during treatment and follow-up, the final sample size was increased to 46 patients per group, resulting in a total sample size of 92 patients.
The final conclusion will be determined in line with the intention-to-treat principle in accordance with the CONSORT guideline. All analyses will be performed using both the modified intention-to-treat and per-protocol principles. All randomized patients will be included in the efficacy analyses if they have received at least one dose of the study medication and have at least one post-baseline assessment of any efficacy parameter. For the safety population analysis, all randomized patients who receive at least one dose of the study medication will be included.
Categorical variables will be presented as numbers and percentages. Analyses will be performed using the Chi-square test or Fisher’s exact test. Continuous variables will be presented as a mean and standard deviation or median and interquartile range, as appropriate. Normality will be assessed using the Shapiro–Wilk test. Between-group comparisons will be performed using Student’s t test for normally distributed variables and the Mann–Whitney U-test for non-normally distributed variables.
The primary outcome, defined as the change from baseline in the mean pain intensity during the first week after treatment, will be analyzed using an analysis of covariance model, with treatment group as a fixed effect and baseline pain intensity as a covariate. The adjusted between-group difference in least squares means and corresponding 95% confidence intervals will be reported. Pre-defined subgroups analyses will be conducted according to age, sex, course of FM, baseline pain intensity. Treatment-by-subgroup interactions will be evaluated using interaction terms in the regression models. P < 0.05 will be considered statistically significant for treatment–covariate interactions. These analyses are exploratory in nature, as the study is not powered to detect differences within subgroups. Baseline characteristics will be summarized descriptively. No routine adjustment for baseline imbalances will be performed. If substantial and clinically relevant imbalances are observed, adjusted analyses will be conducted as sensitivity analyses. Moreover, missing data will be handled using appropriate statistical methods, such as multiple imputation under the assumption that data are missing at random.
In addition, patients achieving a ≥ 50% reduction in pain intensity from baseline were defined as responders. To investigate the relationship between ESK exposure and analgesic efficacy, plasma concentrations at the end of infusion were treated as surrogate indicators of systemic exposure. Concentrations will be compared between responders and non-responders using Student’s t tests or Mann–Whitney U-tests, as appropriate. In addition, exploratory analyses were performed to examine the association between plasma ESK concentrations and changes in pain intensity using Pearson or Spearman correlation coefficients, depending on the distribution of the data. All tests will be performed with SPSS (V 27.0; IBM Corporation; Armonk, NY, USA). A two-sided P value < 0.05 will be considered statistically significant.
Discussion
The multicenter, randomized, controlled trial evaluates the efficacy and safety of adjunctive single high-dose ESK infusion in patients with FM using a rigorous design with a relatively larger sample size. Given the limitations of oral pharmacotherapies, notably their delayed onset and modest rates of substantial pain relief, this study aims to determine whether the addition of ESK can provide more rapid and effective symptom control compared to conventional medication escalation alone.
To reconcile methodological rigor with feasibility, an open-label design is adopted while implementing blinded-endpoint assessment to minimize observer bias in outcome evaluation. The primary outcome is the change from baseline in the mean pain intensity during the first week after treatment. This endpoint is adapted from a randomized, double-blind, multicenter, placebo-controlled phase III trial of pregabalin in patients with FM,39 and is consistent with outcome measures used in other randomized trials of neuropathic pain, including studies of mirogabalin and crisugabalin.40,41 Given the rapid onset of analgesic effects observed with ESK in previous studies,23 the change in mean pain intensity during the first week after treatment is considered an appropriate endpoint to capture early treatment response.
Despite its prospective design, this study has several constraints. The open-label protocol risks introducing performance or ascertainment bias, though the blinded-endpoint mitigates some interpretive bias. Additionally, the 12-week treatment window precludes conclusions about long-term outcomes. Extended follow-up studies with larger populations will be essential to confirm these preliminary findings. Finally, plasma ESK concentrations will be measured only at the end of infusion to capture early systemic exposure.31 This design aligns with prior study.42 However, due to the limited sampling point, the exposure-response analyses will be considered exploratory. Confirmatory conclusions will require future investigations with more intensive pharmacokinetic sampling. Future research should specifically investigate the optimal therapeutic regimen, including dosage, treatment duration, and session intervals.
Study Status
The trial has been initiated and is currently in the early recruitment phase. No outcome data have been analyzed at the time of manuscript submission. The protocol has been revised in response to peer review to improve methodological clarity and rigor.
Data Sharing Statement
This is the protocol only; no participant data are included.
Patient Consent for Publication
Obtained.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agreed to be accountable for all aspects of the work.
Funding
This work was supported by the National Key Research and Development Program of China (No. 2022YFC3602200, No. 2022YFC3602201, No. 2022YFC3602202, No. 2022YFC3602203, No. 2022YFC3602205), and the National Health Commission Capacity Building and Continuing Education Centre Cancer Pain Management (No. PMT1001-1).
Disclosure
The authors declare that they have no competing interests in this work.
References
1. Yunus M, Masi AT, Calabro JJ, Miller KA, Feigenbaum SL. Primary fibromyalgia (fibrositis): clinical study of 50 patients with matched normal controls. Semin Arthritis Rheum. 1981;11(1):151–11. doi:10.1016/0049-0172(81)90096-2
2. Bair MJ, Krebs EE. Fibromyalgia. Ann Intern Med. 2020;172(5):ITC33–ITC48. doi:10.7326/AITC202003030
3. Sarzi-Puttini P, Giorgi V, Marotto D, Atzeni F. Fibromyalgia: an update on clinical characteristics, aetiopathogenesis and treatment. Nat Rev Rheumatol. 2020;16(11):645–660. doi:10.1038/s41584-020-00506-w
4. Häuser W, Fitzcharles M-A. Facts and myths pertaining to fibromyalgia. Dialogues Clin Neurosci. 2018;20(1):53–62. doi:10.31887/DCNS.2018.20.1/whauser
5. Häuser W, Ablin J, Fitzcharles M-A, et al. Fibromyalgia. Nat Rev Dis Primers. 2015;1(1):15022. doi:10.1038/nrdp.2015.22
6. Macfarlane GJ, Kronisch C, Dean LE, et al. EULAR revised recommendations for the management of fibromyalgia. Ann Rheum Dis. 2017;76(2):318–328. doi:10.1136/annrheumdis-2016-209724
7. Skrabek RQ, Galimova L, Ethans K, Perry D. Nabilone for the treatment of pain in fibromyalgia. J Pain. 2008;9(2):164–173. doi:10.1016/j.jpain.2007.09.002
8. Clauw DJ. Fibromyalgia: a clinical review. JAMA. 2014;311(15):1547–1555. doi:10.1001/jama.2014.3266
9. Chinn S, Caldwell W, Gritsenko K. Fibromyalgia pathogenesis and treatment options update. Curr Pain Headache Rep. 2016;20(4):25. doi:10.1007/s11916-016-0556-x
10. Häuser W, Ablin J, Perrot S, Fitzcharles M-A. Management of fibromyalgia: practical guides from recent evidence-based guidelines. Pol Arch Intern Med. 2017;127(1):47–56. doi:10.20452/pamw.3877
11. Marques AP, Santo AD, Berssaneti AA, Matsutani LA, Yuan SLK. Prevalence of fibromyalgia: literature review update. Rev Bras Reumatol Engl Ed. 2017;57(4):356–363. doi:10.1016/j.rbr.2016.10.004
12. Bradley LA. Pathophysiology of fibromyalgia. Am J Med. 2009;122(12 Suppl):S22–S30. doi:10.1016/j.amjmed.2009.09.008
13. Hawkins RA. Fibromyalgia: a clinical update. J Am Osteopath Assoc. 2013;113(9):680–689. doi:10.7556/jaoa.2013.034
14. Buskila D, Cohen H. Comorbidity of fibromyalgia and psychiatric disorders. Curr Pain Headache Rep. 2007;11(5):333–338. doi:10.1007/s11916-007-0214-4
15. Gracely RH, Ceko M, Bushnell MC. Fibromyalgia and depression. Pain Res Treat. 2012;2012:486590. doi:10.1155/2012/486590
16. Correia-Melo FS, Leal GC, Vieira F, et al. Efficacy and safety of adjunctive therapy using esketamine or racemic ketamine for adult treatment-resistant depression: a randomized, double-blind, non-inferiority study. J Affect Disord. 2020;264:527–534. doi:10.1016/j.jad.2019.11.086
17. Orhurhu V, Orhurhu MS, Bhatia A, Cohen SP. Ketamine infusions for chronic pain: a systematic review and meta-analysis of randomized controlled trials. Anesth Analg. 2019;129(1):241–254. doi:10.1213/ANE.0000000000004185
18. Graven-Nielsen T, Kendall SA, Henriksson KG, et al. Ketamine reduces muscle pain, temporal summation, and referred pain in fibromyalgia patients. Pain. 2000;85(3):483–491. doi:10.1016/S0304-3959(99)00308-5
19. Niesters M, Martini C, Dahan A. Ketamine for chronic pain: risks and benefits. Br J Clin Pharmacol. 2014;77(2):357–367. doi:10.1111/bcp.12094
20. Bahji A, Vazquez GH, Zarate CA. Comparative efficacy of racemic ketamine and esketamine for depression: a systematic review and meta-analysis. J Affect Disord. 2021;278:542–555. doi:10.1016/j.jad.2020.09.071
21. Daly EJ, Singh JB, Fedgchin M, et al. Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry. 2018;75(2):139–148. doi:10.1001/jamapsychiatry.2017.3739
22. Wajs E, Aluisio L, Holder R, et al. Esketamine nasal spray plus oral antidepressant in patients with treatment-resistant depression: assessment of long-term safety in a Phase 3, open-label study (SUSTAIN-2). J Clin Psychiatry. 2020;81(3):19m12891. doi:10.4088/JCP.19m12891
23. Noppers I, Niesters M, Swartjes M, et al. Absence of long-term analgesic effect from a short-term S-ketamine infusion on fibromyalgia pain: a randomized, prospective, double blind, active placebo-controlled trial. Eur J Pain. 2011;15(9):942–949. doi:10.1016/j.ejpain.2011.03.008
24. Giorgi V, Bazzichi L, Batticciotto A, et al. Fibromyalgia: one year in review 2023. Clin Exp Rheumatol. 2023;41(6):1205–1213. doi:10.55563/clinexprheumatol/257e99
25. Schwartzman RJ, Alexander GM, Grothusen JR, Paylor T, Reichenberger E, Perreault M. Outpatient intravenous ketamine for the treatment of complex regional pain syndrome: a double-blind placebo controlled study. Pain. 2009;147(1–3):107–115. doi:10.1016/j.pain.2009.08.015
26. Wolfe F, Clauw DJ, Fitzcharles M-A, et al. 2016 Revisions to the 2010/2011 fibromyalgia diagnostic criteria. Semin Arthritis Rheum. 2016;46(3):319–329. doi:10.1016/j.semarthrit.2016.08.012
27. Chung F, Abdullah HR, Liao P. STOP-bang questionnaire: a practical approach to screen for obstructive sleep apnea. Chest. 2016;149(3):631–638. doi:10.1378/chest.15-0903
28. Mease PJ, Farmer MV, Palmer RH, Gendreau RM, Trugman JM, Wang Y. Milnacipran combined with pregabalin in fibromyalgia: a randomized, open-label study evaluating the safety and efficacy of adding milnacipran in patients with incomplete response to pregabalin. Ther Adv Musculoskelet Dis. 2013;5(3):113–126. doi:10.1177/1759720X13483894
29. Gilron I, Chaparro LE, Tu D, et al. Combination of pregabalin with duloxetine for fibromyalgia: a randomized controlled trial. Pain. 2016;157(7):1532–1540. doi:10.1097/j.pain.0000000000000558
30. Wang S, Deng C-M, Zeng Y, et al. Efficacy of a single low dose of esketamine after childbirth for mothers with symptoms of prenatal depression: randomised clinical trial. BMJ. 2024;385:e078218 doi:10.1136/bmj-2023-078218.
31. Peltoniemi MA, Hagelberg NM, Olkkola KT, Saari TI. Ketamine: a review of clinical pharmacokinetics and pharmacodynamics in anesthesia and pain therapy. Clin Pharmacokinet. 2016;55(9):1059–1077 doi:10.1007/s40262-016-0383-6.
32. Bennett RM, Friend R, Jones KD, Ward R, Han BK, Ross RL. The revised fibromyalgia impact questionnaire (FIQR): validation and psychometric properties. Arthritis Res Ther. 2009;11(4):R120. doi:10.1186/ar2783
33. Bocéréan C, Dupret E. A validation study of the hospital anxiety and depression scale (Hads) in a large sample of French employees. BMC Psychiatry. 2014;14(1):354. doi:10.1186/s12888-014-0354-0
34. Jenkinson C, Coulter A, Wright L. Short form 36 (SF36) health survey questionnaire: normative data for adults of working age. BMJ. 1993;306(6890):1437–1440. doi:10.1136/bmj.306.6890.1437
35. Munguía-Izquierdo D, Segura-Jiménez V, Camiletti-Moirón D, et al. Multidimensional fatigue inventory: Spanish adaptation and psychometric properties for fibromyalgia patients. The Al-Andalus study. Clin Exp Rheumatol. 2012;30(6 Suppl 74):94–102.
36. Hays RD, Martin SA, Sesti AM, Spritzer KL. Psychometric properties of the medical outcomes study sleep measure. Sleep Med. 2005;6(1):41–44. doi:10.1016/j.sleep.2004.07.006
37. Ioannidis JPA, Evans SJW, Gøtzsche PC, et al. Better reporting of harms in randomized trials: an extension of the CONSORT statement. Ann Intern Med. 2004;141(10):781–788. doi:10.7326/0003-4819-141-10-200411160-00009
38. de Carvalho JF, de Sena EP. Ketamine in fibromyalgia: a systematic review. Adv Rheumatol. 2024;64(1):54. doi:10.1186/s42358-024-00393-9
39. Ohta H, Oka H, Usui C, Ohkura M, Suzuki M, Nishioka K. A randomized, double-blind, multicenter, placebo-controlled Phase III trial to evaluate the efficacy and safety of pregabalin in Japanese patients with fibromyalgia. Arthritis Res Ther. 2012;14(5):R217. doi:10.1186/ar4056
40. Baba M, Matsui N, Kuroha M, Wasaki Y, Ohwada S. Mirogabalin for the treatment of diabetic peripheral neuropathic pain: a randomized, double-blind, placebo-controlled phase III study in Asian patients. J Diabetes Investig. 2019;10(5):1299–1306. doi:10.1111/jdi.13013
41. Zhang D, Lei T, Qin L, et al. Efficacy and safety of crisugabalin (HSK16149) in adults with postherpetic neuralgia: a phase 3 randomized clinical trial. JAMA Dermatol. 2024;160(11):1182–1191. doi:10.1001/jamadermatol.2024.3410
42. Kobayashi Y, Nagashima W, Tokura T, et al. Duloxetine plasma concentrations and its effectiveness in the treatment of nonorganic chronic pain in the orofacial region. Clin Neuropharmacol. 2017;40(4):163–168. doi:10.1097/WNF.0000000000000225
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
The Efficacy and Safety of Crisugabalin (HSK16149) in Fibromyalgia Patients: Protocol for a Prospective Randomized Open Blinded-Endpoint (PROBE) Study
Liu M, Li R, Zhou H, Xie Z, He Y, Ren F, Feng X, Fan B, Li S, Luo F, Zhang D
Journal of Pain Research 2026, 19:580694
Published Date: 3 March 2026