Back to Journals » Neuropsychiatric Disease and Treatment » Volume 15
Effects of transection of cervical sympathetic trunk on cognitive function of traumatic brain injury rats
Authors Long J ![]() , He CJ
, He CJ ![]() , Dai H, Kang XG, Zou JF, Ye SL
, Dai H, Kang XG, Zou JF, Ye SL ![]() , Yu Q
, Yu Q
Received 25 December 2018
Accepted for publication 28 March 2019
Published 3 May 2019 Volume 2019:15 Pages 1121—1131
DOI https://doi.org/10.2147/NDT.S199450
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Yu-Ping Ning
Juan Long,1 Chunjing He,1 Hong Dai,2 Xinguo Kang,1 Jinfeng Zou,1 Shengli Ye,1 Qian Yu2
1Department of Pain, Guizhou Province People‘s Hospital, Guiyang, People’s Republic of China; 2Department of Neurology, Guizhou Province People‘s Hospital, Guiyang, People’s Republic of China
Objective: To observe the effects of transection of cervical sympathetic trunk (TCST) on the cognitive function of traumatic brain injury (TBI) rats and the potential mechanisms.
Methods: A total of 288 adult male SD rats were divided into 3 groups using a random number table: TBI group (n=96), TBI + TCST group (n=96) and Sham group (n=96). The water maze test was performed before TBI (T0) and at day 1 (T1), day 2 (T2), day 3 (T3), 1 week (T4), 2 weeks (T5), 6 weeks (T6) and 12 weeks (T7) after TBI. The levels of α1-adrenergic receptors (α1-ARs), α2-adrenergic receptors (α2-ARs), toll-like receptor 4 (TLR-4) and P38 in hippocampi were detected by real-time PCR. Hippocampal P38 expression was assayed by Western blot. The expressions of interleukin-6 (IL-6), tumor necrosis factor (TNF-α) and brain-derived neurotrophic factor (BDNF) were examined by immunohistochemistry. Noradrenaline (NE) expression in plasma was evaluated by ELISA. The respiratory control ratio (RCR) of brain mitochondria was detected using a Clark oxygen electrode.
Results: TCST effectively improved the cognitive function of TBI rats. TCST significantly inhibited sympathetic activity in the rats and effectively inhibited inflammatory responses. The expression of BDNF at T1-T6 in TBI+TCST group was higher than that in TBI group (P<0.05). Furthermore, P38 expression was inhibited more effectively in TBI+TCST group (P<0.05), than in TBI group (P<0.05), and the RCR of the brain was significantly higher in TBI+TCST group than in TBI group (P<0.05).
Conclusions: TCST can enhance cognitive function in TBI rats by inhibiting sympathetic activity, reducing inflammatory responses and brain edema, upregulating BDNF and improving brain mitochondrial function.
Keywords: traumatic brain injury, transection of the cervical sympathetic trunk, sympathetic nervous system, inflammatory response, mitochondrial function
Traumatic brain injury (TBI) is one of the most common diseases with high mortality in humans.1 TBI patients often show different degrees of cognitive and behavioral disorders,2,3 and clinical treatment and intervention often fail. A series of pathophysiological changes appear after TBI, such as the sympathetic nervous system response to stress, inflammatory response, brain edema, brain hypoxia-ischemia and mitochondrial dysfunction.4,5 In recent years, the autonomic nervous system has been revealed to play an important role in stress and the inflammatory responses after severe trauma.6 The sympathetic nervous system shows hyperexcitability after severe trauma and secretes numerous inflammatory cytokines, thus resulting in systemic inflammatory response syndrome (SIRS).7 Thus, effectively inhibiting stress and inflammatory responses is especially important for improving the prognosis and reducing the mortality of TBI patients. Therefore, exploring favorable treatments that can improve or even completely inhibit these secondary injuries is a significant goal for the treatment of cognitive dysfunction in TBI patients. Stellate ganglion block (SGB) can improve cellular and humoral immune functions and regulate abnormalities in the neuroendocrine system, thus relieving overactivation of the sympathetic nervous system throughout the body.8,9 SGB has been widely used for a variety of clinical diseases and has exerted favorable curative effects in recent years. Transection of cervical sympathetic trunk (TCST) of rats is regarded as a satisfactory animal model simulating long-term SGB in humans.10 Studies have suggested that unilateral SGB can improve and block the volume of blood flow to the hemibrain and thus protect brain functions.11 Studies have shown that SGB exerts a certain improvement effect on cognitive dysfunction,12,13 but the mechanism is unknown. Therefore, in this study, early TCST was conducted in rats after TBI to observe its effects on the sympathetic nervous system, inflammatory responses, brain edema and mitochondrial respiratory functions and to analyze the potential mechanism by which TCST improves cognitive functions after TBI.
Materials and methods
Experimental animals and grouping
In this experiment, 288 healthy adult naïve male Sprague–Dawley rats weighing 250–300 g were provided by the animal laboratory of Third Military Medical University (license No. for animal use: SCXK [Chongqing] 2012–0005). Animal feeding was conducted by the animal laboratory of the Third Military Medical University. The rats were housed in cages with ad libitum access to food and water and divided into the TBI group (TBI group, n=96), TBI + TCST group (TBI + TCST group, n=96) and sham operation group (Sham group, n=96) using a random number table. All procedures used in this study were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of the Third Military Medical University. All efforts were made to minimize animal suffering and the number of animals used.
Establishment of models
Sham group model: After the rat was placed under prone anesthesia, its head was shaved, the iodophor was disinfected, the median incision was made, the periosteum was removed and the left parietal bone was exposed. A dental drill was then used to create a 5 mm bone window located 1.5 mm behind the coronal suture and 2.5 mm adjacent to the midline. The intact periosteum was retained; the rat was then sutured and allowed to continue feeding.
TBI model: This model was constructed using Feeney’s weight-dropping impact method.14 The pedal hit by the falling body attaches directly to the dura mater (area 4.6 mm×4.0 mm). Before the impact, each rat was anesthetized with an intraperitoneal injection of 10% chloral hydrate (300 mg/kg). After the rat was fixed in prone position, its head was shaved and disinfected. The median incision was made to strip the periosteum and expose the left parietal bone. A dental drill was used to drill a bone window (5 mm in diameter) located 1.5 mm behind the coronal seam and 2.5 mm adjacent to the midline to maintain the intact periosteum. The impact bar was placed on the dura mater, and 20 g weights fell from 25 cm high. According to Lu et al,15 this method was used to establish a moderate TBI in rats. The experimental TBI model was established successfully.
TCST model: After successful establishment of the TBI model, the skin from the neck was prepared, sterilized with iodine and incised at the median to isolate the left cervical sympathetic nerve. The sympathetic stem was dissociated at 3 mm superior to the sympathetic ganglia.10 After resuscitation from anesthesia, the rats showed typical signs of Horner syndrome, such as ptosis on the same side of cervical sympathetic transection, small eye fission and pupil narrowing, which indicate the successful modeling of cervical sympathetic trunk transection. Thus, the TCST model was successfully established in this experiment.
Behavioral tests
Twelve rats were randomly selected from each group and subjected to water maze training for 4 days before the models were established. Rats were placed on the platform for 10 s for adaptation prior to the experiment and then placed randomly in the tank at different quadrant locations. The rats were led to the platform, and recordings were ended after the rats reached the platform and remained there after 10 s of adaptation. Twelve rats randomly selected from each group were withdrawn from the platform before TBI (T0) or at day 1 (T1), day 2 (T2), day 3 (T3), 1 week (T4), 2 weeks (T5), 6 weeks (T6) or 12 weeks (T7) after TBI and placed in the tank. The target quadrant distance, target quadrant residence time and number of times the rat passed the platform were recorded over 2 mins. Data acquisition was completed using the water maze image supervisory system. After the behavioral indicators were tested at each time point, 6 out of 12 rats were randomly selected for immunohistochemical analysis and brain water content, PCR, ELISA, Western blot and mitochondrial RCR analyses were performed on the brain tissue, hippocampal tissue and blood samples collected from the other 6 rats.
Expressions of IL-6, TNF-α and BDNF in hippocampi by immunohistochemistry
After the behavioral tests and anesthesia, the thoracic cavities of the selected 6 rats were opened, and a syringe needle was inserted into the apex of the heart. The right auricle was cut and infused rapidly with 100 mL of physiological saline followed by 4% paraformaldehyde. The brain was removed, immersed in 4% paraformaldehyde for 72 hrs, dehydrated and embedded in paraffin wax. Then, the brain was sliced into continuous 4-μm-thick coronal sections. Each section that contained hippocampal tissue was mounted on an anti-slip glass slide that was then baked at 60°C for 2 hrs. These sections were repaired with antigen repair fluid for 20 mins, incubated and immersed in 3% H2O2 for 10 mins to block endogenous peroxidase, and washed with PBS 3 times for 3 mins. The sections were incubated with primary antibodies against interleukin-6 (IL-6) (1:400, Abcam, USA), tumor necrosis factor (TNF-α) (1:250, Abcam, USA), and brain-derived neurotrophic factor (BDNF) (1:150, Abcam, USA) at 4°C overnight and then washed with PBS 3 times for 30 mins. Polymeric reagents were added, and the sections were incubated at 37°C for 15 mins and then washed with PBS 3 times for 2 mins. Samples were stained with DAB coloring reagent and observed under a microscope. The DAB reaction was halted with tap water, and the samples were restained with hematoxylin, returned to blue with tap water, dehydrated until transparent and then mounted. Cells positively expressing IL-6, TNF-α and BDNF were identified based on the detection of yellow or brownish granules in the cytoplasm. The hippocampal tissue sections of 6 rats in each group were observed. Five nonoverlapping fields were randomly observed under a high-power microscope (10×40 times) to detect the positive expression of IL-6, TNF-α and BDNF. The integrated optical density (IOD) of IL-6 TNF-α, BDNF expression was determined by ImagePro Plus 6.0 image analysis software for statistical analysis.
Determination of brain water content
A cube of the cerebral cortex (approximately 5 mm×9 mm×9 mm×5 mm) located near the lesion was dissected, immediately weighed on the electronic balance to obtain the wet weight (WW), dried in a high-temperature drying oven at 105°C for 48 hrs, and weighed until a constant weight was obtained, ie, the dry weight (DW). The brain water content was calculated based on the Elliott formula as follows: brain water content (%)=(WW - DW)/WW ×100%.
RNA extraction and real-time PCR
Total RNA was extracted from hippocampal cells using TRIzol and reverse transcribed into cDNA. The following primers were used: P38 (sense: 5’-CGGCTTGCTCATGTCCTCAGAAC-3’, antisense: 5-‘GGAGGGCGGCTGCACATACAC-3’); toll-like receptor 4 (TLR-4) (sense: 5’-AGCCCTGTTGGATGGAAAAGC-3’, antisense: 5’-GGGTTTTAGGCGCAGAGTTTTG-3’); α1-adrenergic receptors (α1-ARs) (sense: 5’-TGGGCCATCTCCGCGCTG-3’, antisense: 5’-GCCCGGTTGGTGACGAAATC-3’); α2-adrenergic receptors (α2-ARs) (sense: 5’-TTCTGTGCCTTCGCCGGTCTTCC-3’, antisense: 5’-TCAGGGAGGGGCCGTCTTAAAG-3’). Each reaction was performed in triplicate using Maxima SYBR Green/ROX qPCR Master Mix (2×) according to the manufacturer’s protocol. The PCR conditions were as follows: predenaturation at 96°C for 6 mins; 40 cycles of denaturation at 96°C for 30 s, annealing at 57°C for 30 s, and extension at 72°C for 30 s; and a final extension step at 72°C for 10 mins. The 2 −△△CT value was calculated.
Assessing the expression of P38 by Western blot
RIPA lysis buffer and phenylmethylsulfonyl fluoride (PMSF) were mixed at a 100:1 volume ratio to fully lyse the tissue; the resulting mixture was centrifuged at 4°C at 12,000 rpm for 15 mins, and the supernatant was then collected. The protein concentration of the sample was detected using the BCA protein assay kit. After vertical electrophoresis, the samples were transferred for 1.5 hrs, sealed for 2 hrs, incubated with the primary antibody (Abcam, 1:500) at 4°C overnight, and then incubated with the secondary antibody (1:1000) at room temperature for 1 hr. Chemiluminescence was performed with enhanced chemiluminescence (ECL) luminous liquid, and films were used for exposure.
Assessing the expression of serum noradrenaline (NE) by ELISA
Rat peripheral venous blood (1 ml) was collected, incubated at room temperature for 2 hrs and centrifuged at 2000× g for 20 mins; the supernatant was then collected to detect the level of serum NE with the ELISA kit (USA R&D) based on the kit instructions.
Determination of the brain mitochondrial respiratory control ratio (RCR)
The brain tissue in the lesioned area was dissolved in the separation medium to obtain the tissue homogenate, which was centrifuged at 12,000× g at 4°C for 10 mins 2 times; the precipitant contained the mitochondria. The mitochondria (2 mg) were added to 3 mL of reaction medium (225 mmol/L mannitol, 70 mmol/L sucrose, 1 mmol/L EDTA, 0.1% BCA, 10 mmol/L potassium phosphate, pH 7.4, temperature 25°C) and stirred until the basic concentration was stable (240 nmol/mL). Then, 20 μL of the mitochondrial suspension (approximately 2 mg) was incubated with 20 μL of substrate (disodium succinate 4 mmol/L) for 2 mins. Clark’s oxygen electrode method was then used to detect the respiratory oxygen quotient IV (R4).16 Then, 20 μL of adenosine diphosphate (ADP, 50 mmol/L) was added, and the respiratory oxygen quotient III (R3) was measured. The mitochondrial RCR was calculated using the following equation: RCR=R3/R4.
Statistical analysis
Statistical analysis was conducted with SPSS16.0. The data are all expressed as ±S. Comparisons among groups were analyzed with one-way ANOVA; multiple comparisons were analyzed with least square difference (LSD) tests. P<0.05 suggested that differences were statistically significant.
Results
Analysis of the behavioral effects of TCST on TBI rats
Behavioral indexes of rats in the three groups were measured by the water maze test. Compared with those in Sham group, rats in TBI group exhibited an increase in the target quadrant distance at T1-4 (P<0.05) (Figure 1A) and in the target quadrant time at T1-4 (P<0.05) (Figure 1B). Furthermore, the number of platform crosses at T1-6 was lower in TBI group than in Sham group (P<0.05) (Figure 1C). There were no significant differences in behavioral indicators among TBI+TCST group, Sham group or TBI group at any time point.
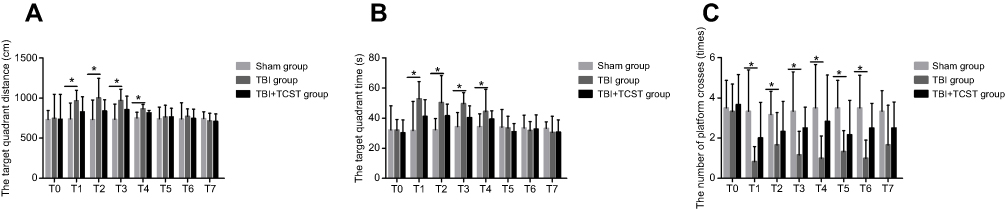 | Figure 1 The behavioral test results of rats in all groups. (A) Comparison of the distances in target quadrants at different time points in rats of all groups. (B) Comparison of the times in target quadrants at different time points in rats of all groups. (C) Comparison of the number of times the rats crossed the platform in all groups. All data are expressed as the mean±standard deviation. Six rats per group; Sham group versus TBI group *p<0.05, Sham group versus TBI+TCST group *p<0.05, TBI group versus TBI+TCST group *p<0.05. |
Effects of TCST on the sympathetic nervous system of TBI rats
Compared to that in Sham group, the plasma NE level in TBI group at T1-5 was obviously higher (P<0.05); however, there were no significant differences in the plasma NE levels between TBI+TCST group and Sham group and TBI group (Figure 2A). Hippocampal α1-ARs expression was significantly lower at T1-5 in TBI group and TBI+TCST group compared with that in Sham group (P<0.05); hippocampal α1-ARs expression was obviously lower at T1-3 in TBI group than in TBI+TCST group (P<0.05) (Figure 2B). Hippocampal α2-ARs expression was markedly lower at T1-5 in TBI group and TBI+TCST group than in Sham group (P<0.05), and hippocampal α2-ARs expression was lower at T1-3 in TBI group than in TBI+TCST group (P<0.05) (Figure 2C).
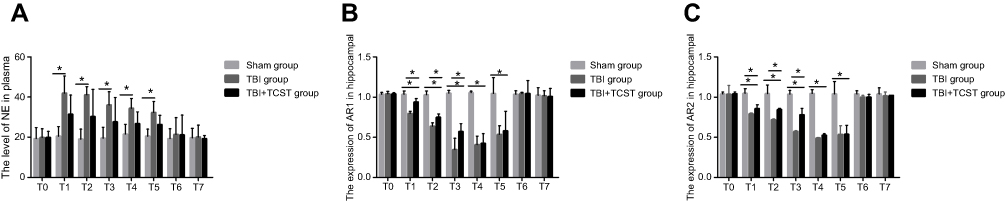 | Figure 2 The effects of TCST on the sympathetic nervous system of TBI rats. (A) Comparison of plasma NE levels in rats of each group at each time point. (B) Comparison of hippocampal AR1 expression in rats of each group at each time point (2B). (C) Comparison of hippocampal AR2 expression in rats of each group at each time point. All data are expressed as the mean±standard deviation. Six rats per group; Sham group versus TBI group *p<0.05, Sham group versus TBI + TCST group *p<0.05, TBI group versus TBI+TCST group *p<0.05. |
Analysis of the effects of TCST on the inflammatory responses of hippocampi in TBI rats
Compared with that in Sham group and TBI+TCST group, the expression of hippocampal IL-6 was markedly increased in TBI group at T1-6 (P<0.05). The expression of IL-6 was higher in TBI+TCST group than in Sham group at T1-4 (P<0.05) (Figures 3 and 5A). The expression of hippocampal TNF-α was significantly higher in TBI group at T1-6 than in Sham group and TBI+TCST group, and the expression of TNF-α was higher in TBI+TCST group than in Sham group at T1-4 (P<0.05) (Figures 4 and 5B).
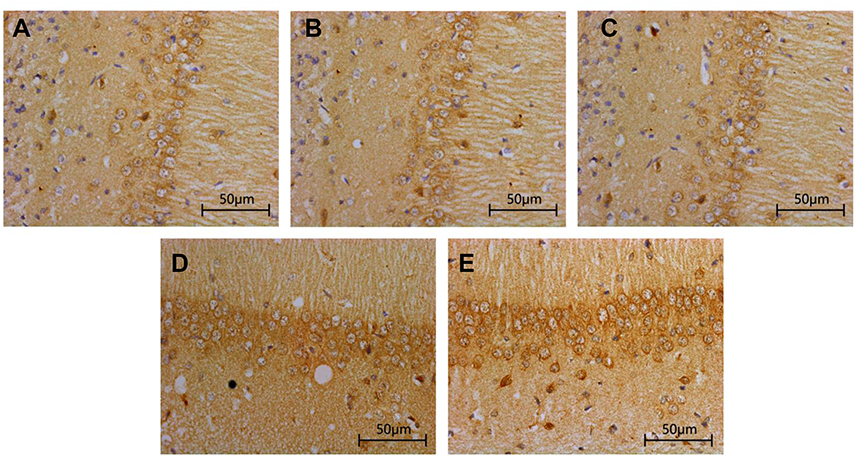 | Figure 3 Effects of TCST on the inflammatory responses of hippocampi in TBI rats. Immunohistochemical staining of IL-6 in the hippocampi of TBI rats after TCST (400×s). A-E: (A–C) The visual fields at time T0 in Sham group, TBI group and TBI+TCST group under an optical microscope. Minor hippocampal IL-6 expression was observed at T0 in the three groups. (D) The visual fields at all time points except for T5-7 in TBI+TCST group under an optical microscope. The expression of IL-6 was increased in TBI+TCST group compared to that in the other groups. (E) The visual fields at all time points except for T7 in TBI group under an optical microscope. The expression of IL-6 was increased more significantly in TBI group than in the other two groups. Six rats per group. |
 | Figure 4 The effects of TCST on the inflammatory responses of hippocampi in TBI rats. Immunohistochemical staining of TNF-α in the hippocampi of TBI rats after TCST (400×). A–E: (A–C) The visual fields at time T0 in Sham group, TBI group and TBI+TCST group under an optical microscope. Minor hippocampal TNF-α expression was observed at T0 in the three groups. (D) The visual fields at all time points except for T5-7 in TBI+TCST group under an optical microscope. The expression of TNF-α was increased in TBI+TCST group compared to that in the other groups. (E) The visual fields at all time points except for T7 in TBI group under an optical microscope. The expression of TNF-α was increased more significantly in TBI group than in the other two groups. Six rats per group. |
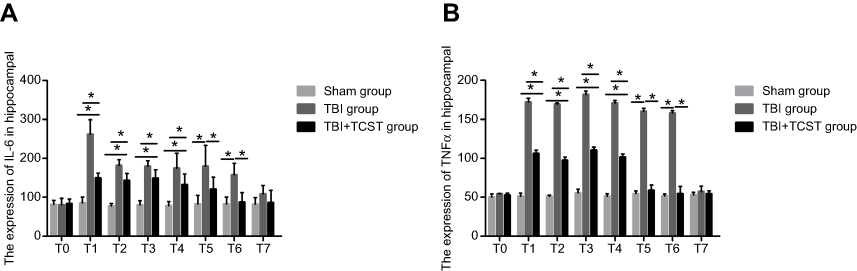 | Figure 5 The effects of TCST on the inflammatory responses of hippocampi in TBI rats. Comparison of hippocampal IL-6 (A) and TNF-α (B) expression in rats of each group at each time point. All data are expressed as the mean±standard deviation. Six rats per group; Sham group versus TBI group *p<0.05, Sham group versus TBI+TCST group *p<0.05, TBI group versus TBI+TCST group *p<0.05. |
Brain water content
The brain water content in TBI group at T1-4 was obviously higher than that in Sham group and TBI+TCST group (P<0.05). No significant differences in the brain water contents of Sham group and TBI+TCST group were observed at any time point (Table 1).
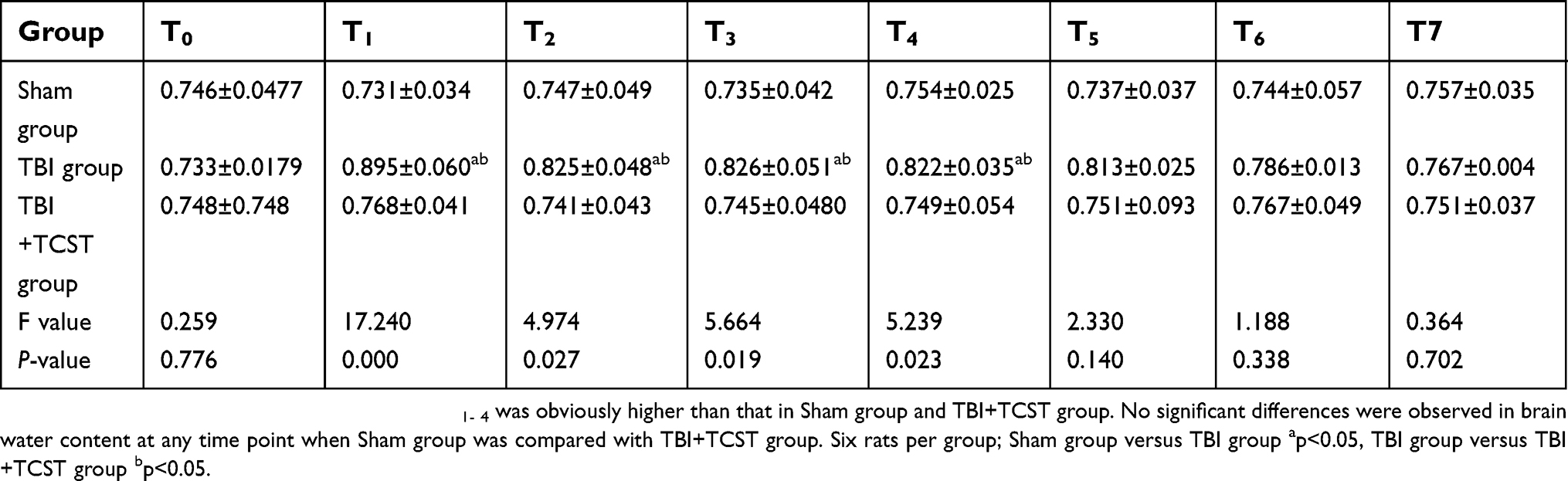 | Table 1 Comparison of brain water contents in rats from each group at each time point (g, ± SD) |
Changes in the expression of hippocampal BDNF in rats from each group
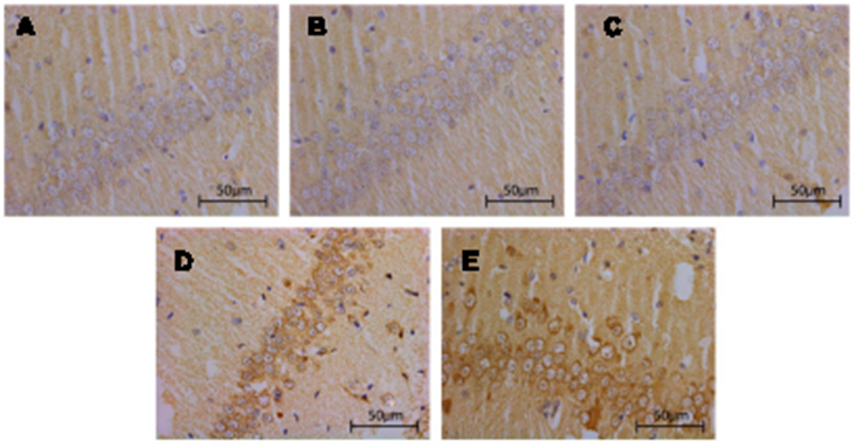
Compared with that in Sham group and TBI+TCST group, the expression of hippocampal BDNF was significantly lower in TBI group at T1-7 (P<0.05). The expression of BDNF in TBI+TCST group at T1-6 was lower than that in Sham group (P<0.05) (Figures 6 and 7).
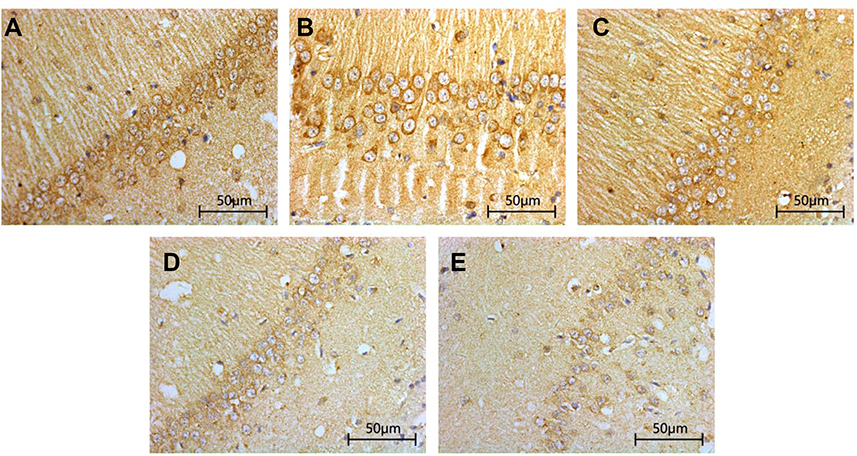 | Figure 6 Changes in the hippocampal expression of BDNF in rats of each group. Immunohistochemical staining of BDNF in the hippocampi of TBI rats after TCST (×400). A–E: (A–C) The visual fields at time T0 in Sham group, TBI group and TBI+TCST group under an optical microscope. The expression of BNDF was increased at T0 in the three groups. (D) The visual fields at all time points except for T7 in TBI+TCST group under an optical microscope. The expression of BNDF was increased in TBI+TCST group compared to that in the other two groups. (E) The visual fields at all time points in TBI group under an optical microscope. The expression of BNDF was increased more significantly in TBI group than in the other two groups. Six rats per group. |
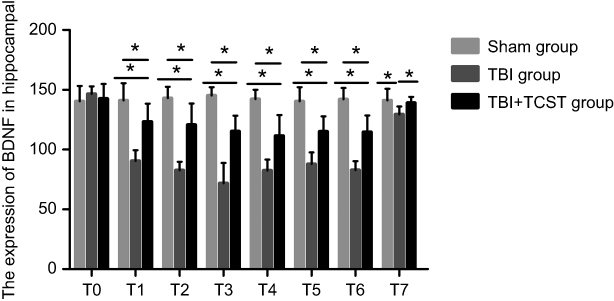 | Figure 7 Changes in the hippocampal expression of BDNF in rats of each group. Comparison of hippocampal BNDF expression in rats of each group at each time point. All data are expressed as the mean±standard deviation. Six rats per group; Sham group versus TBI group *p<0.05, Sham group versus TBI+TCST group *p<0.05, TBI group versus TBI+TCST group *p<0.05. |
Assessing the expression of TLR-4 and P38 mRNA by RT-PCR and the expression of P38 by WB
The RT-PCR results showed that compared with that in Sham group and TBI+TCST group, the expression of hippocampal TLR-4 was obviously higher in TBI group at T1-6 (P<0.05). TLR-4 expression was also higher in TBI+TCST group at T1-6 than in Sham group (P<0.05) (Figure 8A). The expression of hippocampal P38 mRNA was obviously higher in TBI group at T1-6 than in Sham group and TBI+TCST group (P<0.05), and the expression of P38 mRNA was higher in TBI+TCST group than in Sham group at T1-4 (P<0.05) (Figure 8B). Western blot showed that the hippocampal expression of the P38 protein was markedly higher in TBI group than in Sham group and TBI+TCST group at T1-6 (P<0.05). The protein expression of P38 was higher in TBI+TCST group than in Sham group at T1-4 (P<0.05) (Figure 9).
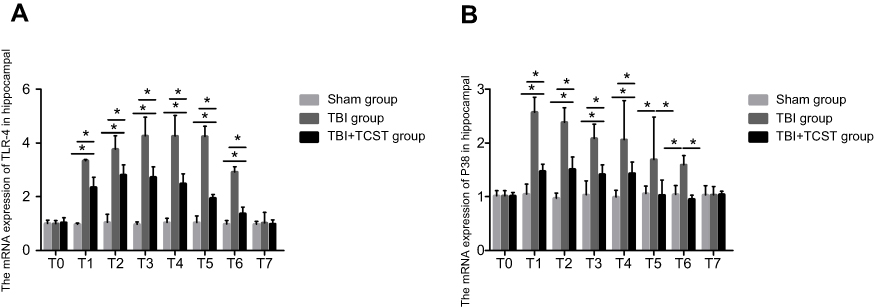 | Figure 8 Analysis of TLR-4 and P38 mRNA expression by RT-PCR. Comparison of TLR-4 mRNA levels in rats of each group at each time point (A). Comparison of P38 mRNA levels in rats of each group at each time point (B). All data are expressed as the mean±standard deviation. Six rats per group; Sham group versus TBI group *p<0.05, Sham group versus TBI+TCST group *p<0.05, TBI group versus TBI+TCST group *p<0.05. |
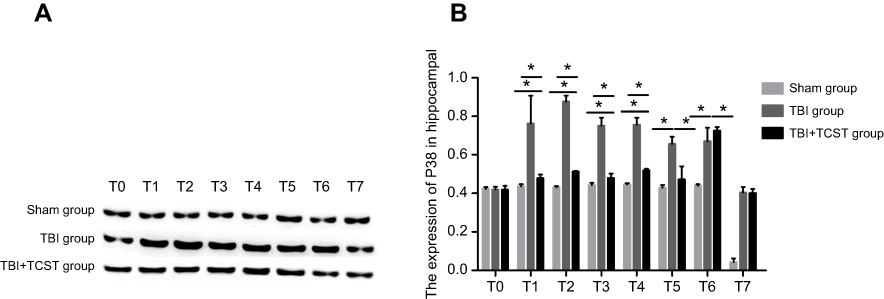 | Figure 9 Analysis of P38 mRNA expression by WB. P38 expression was notably increased in the hippocampi of TBI group at T1-6 and significantly higher than that in TBI+TCST group; P38 expression in TBI+TCST group was still substantially higher than that in Sham group. P38 expression was not statistically significant different among the three groups at T7 (A). Comparison of the P38 protein expression among rats of each group at each time point (B). All data are expressed as the mean±standard deviation. Six rats per group; Sham group versus TBI group *p<0.05, Sham group versus TBI+TCST group *p<0.05, TBI group versus TBI+TCST group *p<0.05. |
Changes in mitochondrial respiratory function
Compared with those in Sham group and TBI+TCST group, the mitochondrial respiratory function was obviously reduced in TBI group at T1-6 (P<0.05). The mitochondrial respiratory function was also lower in TBI+TCST group than in Sham group at T1-5 (P<0.05) (Figure 10), as shown by the measurement of respiratory function with Clark’s oxygen electrode.
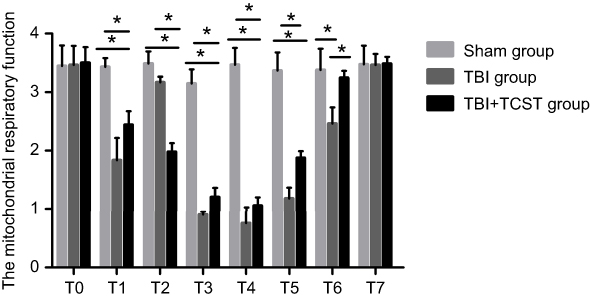 | Figure 10 Comparison of the mitochondrial respiratory functions of rats of each group at each time point. All data are expressed as the mean±standard deviation. Six rats per group; Sham group versus TBI group *p<0.05, Sham group versus TBI+TCST group *p<0.05, TBI group versusTBI+TCST group *p<0.05. |
Discussion
A cervical sympathetic block can improve local tissue hypoxia-ischemia and dysbolism by dilating blood vessels in its control area, resulting in better protection from cerebral ischemia-reperfusion injury and a reduction in the tone of the sympathetic nerve center in the hypothalamus caused by stress.10,15 TBI patients undergo TCST, which significantly increases plasma calcitonin gene-related peptide (CGRP) expression while obviously reducing NF-kBp65 expression.9 In our studies, early after TBI, TCST improved the cognitive function of rats after TBI.
When the body is in a stressed state, the central and peripheral sympathetic nervous systems are activated to release NE, which can cause the release of inflammatory cytokines by binding to adrenergic receptors on nerve cells.10,17 Excessive concentrations of NE are also associated with the level of brain injury and have been suggested to affect the prognosis of patients.3 Similarly, TLR-4 after TBI can mediate inflammatory responses by increasing the release of damage-related molecules from injured brain tissues.18,19 The chemotactic factors of these inflammatory mediators also increase the number of inflammatory cells and immune cell infiltration. The excessive release of inflammatory factors leads to inflammatory cascade responses and further damages the brain tissue.20–22
Our studies revealed that hippocampal NE expression was significantly increased in TBI group at T1-5 after TBI, and the hippocampal expression of TLR-4 was obviously increased in TBI group and TBI+TCST group at T1-6, suggesting that TBI could cause sympathetic tonus. Increased hippocampal NE expression is indicative of the occurrence of inflammatory responses; however, we found that TCST treatment can inhibit the sympathetic nervous system tension after TBI and reduce plasma NE levels, thereby reducing the release of IL-6 and TNF-α in the hippocampus and effectively reducing the post-TBI inflammatory response and preventing further brain damage. Excessive inflammatory response after TBI can aggravate brain damage, edema and cell death, thereby aggravating secondary brain injury.21–23 Our study showed that the inflammatory response was well controlled after TCST and that brain edema was further improved. Noradrenergic receptor activation in the prefrontal cortex of rats increases the level of arousal and can improve cognitive function.24 Hilakivi et al found that AR1 antagonists can prolong the sleep time of cats,25 while AR1 agonists shorten their sleep time and maintain the awakening state. In our study, the expression of AR1 and AR2 in the rat hippocampus was reduced to some extent immediately after TBI and upregulated after TCST treatment and 3 days after injury. These findings indicate that TCST intervention after TBI can effectively control the inflammatory response and improve cerebral edema in the acute phase and even the subacute phase after craniocerebral injury, which can improve the prognosis of TBI.
Studies have shown that TBI-induced mitochondrial damage can be significantly inhibited in TBI rats treated with the P38 inhibitor SB203580.26 In a cerebral ischemic injury study, P38 was shown to be closely related to mitochondrial dysfunction; therefore, inhibition of P38 will provide brain neuron protection.27 TCST was able to inhibit the expression of P38 at T1-6 after injury. Although the expression of P38 was higher in TBI+TCST group than in Sham group at T1-4, the RCR was obviously lower in TBI group than in TBI+TCST group at T1-6, suggesting that TCST can improve the respiratory function of mitochondria by inhibiting the sympathetic tone, inflammatory responses and the expression of P38, as demonstrated in our studies.
Produced by neurons and glial cells, BDNF, a neurotrophic factor, can regulate the development and maturation of the nervous system, maintain proteins involved in nerve functions, promote the rebirth of nervous processes and the formation of new synapses around ischemic foci and in normal brain tissue, and block neuronal apoptosis by a variety of mechanisms after brain injury, thus showing obvious protection of hypoxic-ischemic neurons during brain injury.28 In our experiment, although the expression of BDNF in TBI+TCST group was lower than that in Sham group at T1-6, it was obviously higher than that in TBI group at T1-7, suggesting that TCST can improve TBI by upregulating the expression of BDNF, and the duration of this brain protection can last up to 12 weeks after TBI.
The experimental behavioral results demonstrated that the target quadrant distance of rats in TBI+TCST group was shorter than that in TBI group at T1-4. Furthermore, the target quadrant time was also shorter at T1-4 in TBI+TCST group than in TBI group, and the number of times the rats passed the platform was greater at T1-6 in TBI+TCST group than in TBI group. However, no obvious differences were observed in the above 3 indexes between TBI+TCST group and Sham group. These results suggest that TCST can improve cognitive dysfunction to a certain extent after TBI.
In summary, TCST can improve the cognitive dysfunction of rats after TBI. This effect may be exerted via inhibition of the sympathetic nerve tone, thus inhibiting inflammatory responses, reducing brain edema and improving brain mitochondrial respiratory function, and upregulating the expression of BDNF. The effects of TCST on the cognitive function of rats from 1 day to 12 weeks after TBI were observed and analyzed in this study. The long-term cognitive function and specific mechanisms, however, need to be studied further.
Acknowledgment
This study is financially supported by the Doctoral Scientific Fund Project of Guizhou Provincial Peoples’ Hospital (GZSYBS[2015]06) and Youth Scientific Fund Project of Guizhou Provincial Peoples’ Hospital (GZSYQN[2016]04).
Disclosure
The authors have no conflicts of interest to declare in this work.
References
1. Hiebert JB, Shen Q, Thimmesch AR, Pierce JD. Traumatic brain injury and mitochondrial dysfunction. Am J Med Sci. 2015;350(2):132–138. doi:10.1097/MAJ.0000000000000506
2. Xiao H, Yang Y, Xi JH, Chen ZQ. Structural and functional connectivity in traumatic brain injury. Neural Regen Res. 2015;10(12):2062–2071. doi:10.4103/1673-5374.172328
3. Sun HY, Li Q, Chen XP, Tao LY. Mismatch negativity, social cognition, and functional outcomes in patients after traumatic brain injury. Neural Regen Res. 2015;10(4):618–623. doi:10.4103/1673-5374.155437
4. Algattas H, Huang JH. Traumatic brain injury pathophysiology and treatments: early, intermediate, and late phases post-injury. Int J Mol Sci. 2013;15(1):309–341. doi:10.3390/ijms15010309
5. Di Battista AP, Rhind SG, Hutchison MG, et al. Inflammatory cytokine and chemokine profiles are associated with patient outcome and the hyperadrenergic state following acute brain injury. J Neuroinflammation. 2016;13:40. doi:10.1186/s12974-016-0500-3
6. Yang C, Yan J, Wang HY, et al. Effects of bilateral adrenalectomy on the innate immune responses following trauma in rats. Injury. 2011;42(9):905–912.
7. Steinman L. Elaborate interactions between the immune and nervous systems. Nat Immunol. 2004;5(6):575–581. doi:10.1038/ni1078
8. Shin JH, Kim DW, Yang JY, Lee WI. Efficacy of stellate ganglion block in cholinergic urticaria with acquired generalized hypohidrosis. Korean J Pain. 2012;25(4):278–280. doi:10.3344/kjp.2012.25.4.278
9. Yang X, Shi Z, Li X, Li J. Impacts of stellate ganglion block on plasma NF-κB and inflammatory factors of TBI patients. Int J Clin Exp Med. 2015;8(9):15630–15638.
10. Hu N, Wu Y, Chen BZ, Han JF, Zhou MT. Protective effect of stellate ganglion block on delayed cerebral vasospasm in an experimental rat model of subarachnoid hemorrhage. Brain Res. 2014;1585:63–71. doi:10.1016/j.brainres.2014.08.012
11. Kim EM, Yoon KB, Lee JH, Yoon DM, Kim DH. The effect of oxygen administration on regional cerebral oxygen saturation after stellate ganglion block on the non-blocked side. Pain Physician. 2013;16(2):117–124.
12. Chun-Jing H, Shan O, Guo-Dong L, Hao-Xiong N, Yi-Ran L, Ya-Ping F. Effect of cervical sympathetic block on cerebral vasospasm after subarachnoid hemorrhage in rabbits. Acta cirurgica brasileira. 2013;28(2):89–93.
13. Liu GD, He CJ. Stellate ganglion block promotes recovery of Bell‘s palsy in patients with diabetes mellitus. Acta Otolaryngol. 2014;134(6):652–655. doi:10.3109/00016489.2014.880794
14. Feeney DM, Boyeson MG, Linn RT, Murray HM, Dail WG. Responses to cortical injury: i methodology and local effects of contusions in the rat. Brain Res. 1981;211(1):67–77.
15. Lu X, Bao X, Li J, et al. High-frequency repetitive transcranial magnetic stimulation for treating moderate traumatic brain injury in rats: a pilot study. Exp Ther Med. 2017;13(5):2247–2254. doi:10.3892/etm.2017.4283
16. Sarioglu Y, Utkan T, Akgun M, Duzcan E, Utkan NZ. Effects of deferoxamine and sympathectomy on endothelin-1-induced contraction and acetylcholine-induced relaxation following subarachnoid hemorrhage in carotid artery. Gen Pharmacol. 1997;28(1):145–151.
17. Starkie RL, Rolland J, Angus DJ, Anderson MJ, Febbraio MA. Circulating monocytes are not the source of elevations in plasma IL-6 and TNF-alpha levels after prolonged running. Am J Physiol Cell Physiol. 2001;280(4):C769–C74. doi:10.1152/ajpcell.2001.280.4.C769
18. Famakin BM, Mou Y, Ruetzler CA, Bembry J, Maric D, Hallenbeck JM. Disruption of downstream MyD88 or TRIF Toll-like receptor signaling does not protect against cerebral ischemia. Brain Res. 2011;1388:148–156. doi:10.1016/j.brainres.2011.02.074
19. Kong Y, Le Y. Toll-like receptors in inflammation of the central nervous system. Int Immunopharmacol. 2011;11(10):1407–1414. doi:10.1016/j.intimp.2011.04.025
20. Desborough JP. The stress response to trauma and surgery. Br J Anaesth. 2000;85(1):109–117.
21. Catania A, Lonati C, Sordi A, Gatti S. Detrimental consequences of brain injury on peripheral cells. Brain Behav Immun. 2009;23(7):877–884. doi:10.1016/j.bbi.2009.04.006
22. Loane DJ, Byrnes KR. Role of microglia in neurotrauma. Neurotherapeutics. 2010;7(4):366–377. doi:10.1016/j.nurt.2010.07.002
23. Impellizzeri D, Campolo M, Bruschetta G, et al. Traumatic brain injury leads to development of Parkinson‘s disease related pathology in mice. Front Neurosci. 2016;10:458. doi:10.3389/fnins.2016.00458
24. Lapiz MD, Morilak DA. Noradrenergic modulation of cognitive function in rat medial prefrontal cortex as measured by attentional set shifting capability. Neuroscience. 2006;137(3):1039–1049. doi:10.1016/j.neuroscience.2005.09.031
25. Hilakivi I, Leppavuori A. Effects of methoxamine, and alpha-1 adrenoceptor agonist, and prazosin, an alpha-1 antagonist, on the stages of the sleep-waking cycle in the cat. Acta Physiol Scand. 1984;120(3):363–372. doi:10.1111/j.1748-1716.1984.tb07396.x
26. Yang H, Gu ZT, Li L, et al. SIRT1 plays a neuroprotective role in traumatic brain injury in rats via inhibiting the p38 MAPK pathway. Acta Pharmacol Sin. 2017;38(2):168–181. doi:10.1038/aps.2016.130
27. Zhang XM, Zhang L, Wang G, et al. Suppression of mitochondrial fission in experimental cerebral ischemia: the potential neuroprotective target of p38 MAPK inhibition. Neurochem Int. 2015;90:1–8. doi:10.1016/j.neuint.2015.06.010
28. Sanders RD, Sun P, Patel S, Li M, Maze M, Ma D. Dexmedetomidine provides cortical neuroprotection: impact on anaesthetic-induced neuroapoptosis in the rat developing brain. Acta Anaesthesiol Scand. 2010;54(6):710–716. doi:10.1111/j.1399-6576.2009.02177.x
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.