")
Back to Journals » International Journal of Nanomedicine » Volume 11
Effects of the protein corona on liposome–liposome and liposome–cell interactions
Authors Corbo C, Molinaro R, Taraballi F, Toledano Furman N, Sherman M, Parodi A, Salvatore F, Tasciotti E
Received 23 March 2016
Accepted for publication 4 May 2016
Published 4 July 2016 Volume 2016:11 Pages 3049—3063
DOI https://doi.org/10.2147/IJN.S109059
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Claudia Corbo,1,2 Roberto Molinaro,1 Francesca Taraballi,1 Naama E Toledano Furman,1 Michael B Sherman,3 Alessandro Parodi,1 Francesco Salvatore,2,4 Ennio Tasciotti1,5
1Center for Biomimetic Medicine, Houston Methodist Research Institute, Houston, TX, USA; 2CEINGE-Biotecnologie Avanzate s.c.a r.l., Naples, Italy; 3Department of Biochemistry and Molecular Biology, Sealy Center for Structural Biology and Molecular Biophysics, University of Texas Medical Branch, Galveston, TX, USA; 4Department of Molecular Medicine and Medical Biotechnologies, University of Naples Federico II, Naples, Italy; 5Department of Orthopedics, Houston Methodist Hospital, Houston, TX, USA
Abstract: A thorough understanding of interactions occurring at the interface between nanocarriers and biological systems is crucial to predict and interpret their biodistribution, targeting, and efficacy, and thus design more effective drug delivery systems. Upon intravenous injection, nanoparticles are coated by a protein corona (PC). This confers a new biological identity on the particles that largely determines their biological fate. Liposomes have great pharmaceutical versatility, so, as proof of concept, their PC has recently been implicated in the mechanism and efficiency of their internalization into the cell. In an attempt to better understand the interactions between nanocarriers and biological systems, we analyzed the plasma proteins adsorbed on the surface of multicomponent liposomes. Specifically, we analyzed the physical properties and ultrastructure of liposome/PC complexes and the aggregation process that occurs when liposomes are dispersed in plasma. The results of combined confocal microscopy and flow cytometry experiments demonstrated that the PC favors liposome internalization by both macrophages and tumor cells. This work provides insights into the effects of the PC on liposomes’ physical properties and, consequently, liposome–liposome and liposome–cell interactions.
Keywords: liposomes, protein corona, macrophages, cancer cells
Introduction
In the last few decades, nanoparticle (NP)-based delivery platforms have improved the therapeutic index of many drugs. In fact, by increasing their pharmaceutical bioavailability and targeting, NPs avoid systemic toxicity while retaining pharmacological activity.1 After intravenous injection, NPs adsorb a multitude of biological molecules (proteins, lipids, glycans, and metabolites) from plasma on their surface. These biomolecules continuously recycle2,3 on the NP surface, thereby forming a dynamic shell known as a “biomolecular corona.”4 Most research studying the biomolecular corona has focused on protein composition characterization:5,6 therefore, the biomolecular corona is generally called the “protein corona” (PC).7,8 This natural coating provides NPs with a new biological identity and significantly affects their fate, pharmacokinetics, and cellular interactions.
In terms of NP targeting abilities, the PC can 1) hinder the function of targeting ligands attached to the NP surface, thereby reducing their accumulation in the targeted tissue,3,9,10 and 2) modify the carrier surface, facilitating cell internalization.11 The PC can also modulate immune response.12 Several studies have characterized PC structure, function, and formation dynamics.13,14 Structurally, proteins that are weakly bound to the NP surface form the “soft corona,” while proteins strongly bound to the NP surface form the “hard corona.”15,16 Functionally, the PC is characterized by the presence of opsonins (complement proteins), immunoglobulins, and lectins,17 which match to specific receptors on leukocyte membranes. Until recently, NP opsonization was considered part of the body’s defense mechanism by inducing NP clearance from circulation and consequent accumulation in the organs of the mononuclear phagocytic system (mainly spleen and liver).18 However, in a recent study, unfolded proteins that promote macrophage uptake or their shielding by other proteins in the PC were found to reduce in vitro particle uptake by RAW264.7 macrophages.19,20 The particular context, therefore, needs to be discussed when considering cellular uptake. Although the biological identity of NPs largely determines their biological fate, their physical identity (ie, shape, size, and surface charge) strictly regulates NP–cell interactions. Indeed, large NPs (>300 nm) with a higher positive or negative surface charge (< −15 mV or > +15 mV) exhibited increased macrophage uptake, in vitro and in vivo.21,22
The scope of this work was to shed light on the interactions occurring at the interface between nanocarriers and biological systems. To this aim, we evaluated liposomes as proof-of-concept NPs, given their widespread use in the pharmaceutical field and their high versatility. While most research into the PC has studied inorganic NPs, only recently the PC of liposomes is emerging as a fundamental entity regulating the interactions of liposomes with living systems.23 The PC is believed to have a potentially key impact on targeted delivery in nanomedicine, as it could be manipulated by using different liposomal formulations. Herein, we selected multicomponent choline-based liposomes as a model system to study the PC’s effects in targeted drug delivery applications.24 We studied the correlation between the biological and physical identity of liposomes. To investigate the physical changes occurring in liposomes after PC formation, we characterized NP–PC complexes and used proteomics to analyze the PC composition. Lastly, in an attempt to reproduce the environment surrounding NPs upon systemic injection, we investigated the interactions between liposome–PC complexes and two cell lines in vitro. We studied the impact of the PC composition on liposome uptake by the main cell population responsible for clearance of foreign materials (macrophages J774 cells) and by cancer cells (breast cancer 4T1 cells).
Materials and methods
Liposome assembly
Liposomes were prepared with the thin-layer evaporation method. Briefly, dipalmitoylphosphatidylcholine, dioleoylphosphatidylcholine, and cholesterol (Avanti Polar Lipids, Alabaster, AL, USA) in 6:3:1 molar ratio were dissolved in chloroform/methanol (3:1, v/v). This choline-based lipid composition provides safe and not immunogenic liposomes, relevant for drug delivery application.25 This allowed us to study the formation of the PC focusing on the physical identity of the particles, reducing the effects of secondary factors due to the intrinsic immunogenic features of liposome components. A thin lipid film was prepared with a rotary evaporator (BUCHI Labortechnik AG, Flawil, Switzerland). The film was then hydrated with ultrapure distilled water to obtain a lipid concentration of 20 mM. Liposomes were extruded through 200-nm pore cellulose acetate membranes (Whatman, Schleicher & Schuell, Maidstone, UK) at 45°C. The unilamellar liposomes obtained were stored at 4°C until use. The same procedure was used to assemble rhodamine-labeled liposomes for cellular uptake studies. For those particles, Rhod-DOPE was also added to the lipid mixture; the entire procedure was performed in the dark.
Plasma collection
The study was conducted according to the Guide for the Care and Use of Laboratory Animals and was approved by the Houston Methodist Institutional Animal Care and Use Committee. An aseptic technique was used to withdraw blood from healthy BALB/C mice (Charles River Laboratories International, Inc., Wilmington, MA, USA) when they were sedated. Within 30 minutes after collection, using 50 mM ethylenediaminetetraacetic acid as an anticoagulant, plasma was isolated by centrifugation for 15 minutes at 1,000× g. Plasma was aliquoted and stored at −80°C until use. When required, aliquots were thawed at 4°C and then centrifuged at room temperature for 5 minutes at 15,000× g to eliminate protein aggregates. The blood volume withdrawn from the mice was calculated according to the Lee and Blaufox equation:
BV =0.06×BW+0.77 | (1) |
where BV is the blood volume (mL) and BW is the body weight (g), and the plasma volume is BV/2.26
Formation of liposome–PC complexes
Liposome–PC complexes were obtained by incubating NPs with plasma. Liposome formulation was diluted 20 times (1 mM final lipid concentration) with ultrapure water and then incubated in plasma at 1:1 liposome/plasma ratio (v/v) in order to mimic the protein concentration in vivo (50% plasma in blood).27 The incubation was done in murine plasma for 1 hour at 37°C under continuous agitation. The liposome–PC complexes were recovered after 10 minutes centrifugation at 15,000× g, and then washed three times in cold phosphate buffered saline to remove unbound proteins. We conducted a Bradford assay (Bio-Rad Laboratories, Hercules, CA, USA) on the washing solution to ensure that no additional proteins were eluted from the liposomes. The tubes were changed after each washing step to minimize contamination with plasma proteins bound to the tubes. A plasma aliquot not incubated with liposomes was subjected to the same procedure, as a control, to verify the absence of protein precipitation. All experiments were performed in triplicate.
Size and zeta potential experiment
A Multisizer 4 Coulter counter (Beckman Coulter Inc., Miami, FL, USA) and ZetaSizer Nano ZS (Malvern Instruments, Malvern, UK) were employed to measure the size and the zeta potential, respectively. Samples were diluted 50× with distilled water at 25°C. The results are presented as the average of five measurements.
Cryo-electron microscopy analysis
Lipid vesicles were plunge-frozen on holey film grids (R2x2 Quantifoil®; Micro Tools GmbH, Jena, Germany). A 626 cryo-specimen holder (Gatan, Inc., Pleasanton, CA) was used for imaging. Data were collected on a JEOL 2100 electron microscope (JEOL Ltd., Tokyo, Japan). Images were recorded under low electron dose conditions (5–20 electrons/Å2) using a 4,096×4,096 pixel CCD camera (UltraScan 895, GATAN, Inc., Pleasanton, CA, USA) at a nominal magnification of 20,000×.
Atomic force microscopy
Atomic force microscopy (AFM) images of the liposome and the liposome–PC complex were collected in the Scan Asyst® mode on a Multimode (Bruker Corporation, Santa Barbara, CA, USA) microscope using single-beam silicon cantilever probes (Bruker MLCT at a resonance frequency of 10 kHz, a nominal tip curvature radius of 10 nm, and a force constant of 0.04 N/m). All measurements were subjected to first-order flattening. Particle size was calculated using the Nanoscope 6.13R1 software (Digital Instruments, NY, USA). Mean values from 40 random particles in three independent experiments are reported. Quantitative AFM force mapping analysis was performed to evaluate the relative elasticity of particles in order to obtain a complete elastic property map of heterogeneous samples. We prepared samples by coating the mica surface sample holder with 0.1% (3-Aminopropyl)triethoxysilane (APTES) to stabilize the NPs (to prevent their collapse on the mica surface), and then performed the AFM analysis. Young’s modulus was calculated for six different samples corresponding to 512×512 force–separation curves obtained over a 10×10 μm area. Young’s modulus was calculated using the following equation, as reported elsewhere:28
|
|
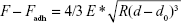
where F – Fadh is the force on the cantilever relative to the adhesion force, R is the tip end radius, d – d0 is the deformation of the sample and E* is the reduced modulus.
Protein separation by one-dimensional gel electrophoresis and in situ digestion
Proteins adsorbed on the liposomes were separated by one-dimensional sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and stained by Coomassie Brilliant Blue (Thermo Fisher Scientific, Waltham, MA, USA). Plasma proteins on the surface of liposomes were quantified by densitometric analysis with the ImageJ software Version 1.45 (imagej.en.softonic.com/). The most abundant protein bands were cut and treated as previously described.29,30 Briefly, each band was de-stained by washing in acetonitrile and 50 mM ammonium bicarbonate. Then, cysteine residues of the proteins were reduced in 10 mM dithiothreitol for 1 hour at 57°C, and alkylated with 55 mM iodoacetamide for 45 minutes at room temperature in the dark. Proteins were in situ digested using Porcine Trypsin Sequencing Grade (Promega Corporation, Fitchburg, WI, USA) overnight at 37°C at an enzyme/substrate ratio of 1:50. The next day, the supernatant was collected and larger peptides were extracted by adding acetonitrile to the bands. Enzyme activity was stopped by adding 0.5% trifluoroacetic acid. Peptides were recovered from the supernatant after centrifugation at 18,000× g for 10 minutes. The resulting peptides were extracted and identified by liquid chromatography-tandem mass spectrometry.
Mass spectrometry and identification of proteins
We used a NanoAcquity UPLC system (Waters Corporation, Milford, MA, USA) coupled inline with a Synapt HDMS (G1) mass spectrometer with an electrospray source to analyze proteins. The liquid chromatography system consisted of a 180 μm ×20 mm Symmetry C18 (5 μm particle) trapping column, and a 75 μm ×250 mm BEH130 C18 (1.7 μm particle) analytical column. The peptide mixture was resolved as previously described.31 The ProteinLynx Global Server (PLGS v2.4; Waters Corporation) was used to identify proteins. The Uniprot 2013_03 (“reviewed”) mouse proteome (16,614 entries) served as the target database. Only proteins with a minimum of two peptides, 95% confidence interval, precursor ion mass error tolerance ≤5 ppm, and fragment ion mass tolerance ≤15 ppm were considered. The following criteria were used in the database search strategy: one trypsin missed cleavage was also allowed, together with oxidation of methionines as variable modification. The only allowed fixed modification was, instead, the carbamidomethylation of cysteines. Quantitative values were normalized using 100 fmol/sample of yeast alcohol dehydrogenase as internal standard and analyzed with the Identity E algorithm.32
Cell culture
The J774 mouse macrophage cell line (ATCC® TIB-67™) and the 4T1 mouse mammary tumor cell line (ATCC® CRL2539™) were used. Cells were cultured using Dulbecco’s Modified Eagle’s Medium containing 10% (v/v) fetal bovine serum and 1% (v/v) penicillin/streptomycin solution. They were maintained in humidified incubators under 5% CO2, at 37°C and passaged at 70%–80% of confluence.
Flow cytometry
To evaluate cellular uptake of liposomes by flow cytometry, 200,000 cells/well were seeded in a twelve-well tissue culture plate. After 24 hours of cell culture expansion, cells were incubated in serum-free conditions with 0.15 mM of rhodamine-labeled bare liposomes and rhodamine-labeled liposome–PC complexes. Incubation occurred for 3 hours at 37°C. After treatment, the cells were washed with phosphate buffered saline, harvested using trypsin, and centrifuged for 10 minutes at 1,500 rpm. A solution of trypan blue (0.01%) was added to the tubes before acquisition to quench the surface fluorescence.33 The analysis was performed using a Becton Dickinson LSR II instrument equipped with Diva software. Each sample was evaluated with 10,000 events. Untreated cells were used as controls. Results are presented as averages of three independent experiments.
Confocal fluorescence microscopy
Cells were seeded onto an eight-well chamber slide (10,000 cells/well) and treated with the particles as described earlier. At the end of the experiment, cells were thoroughly washed and fixed in 4% paraformaldehyde solution. Nuclei were stained using DAPI (blue), and cell membranes were stained using WGA-AlexaFluor 488 conjugate (green). Slides were imaged using a Nikon A1 confocal imaging system and analyzed via NIS Elements AR. Images are presented as a maximum projection or as a 3D side projection of a chosen z section. For statistical analysis, images (n=3) were acquired for each sample at five different areas. For each micrograph, the area fraction for the particles signal (red) was calculated and divided by the number of nuclei counted (blue). All results are presented as mean ± SD in a graph of area fraction/cell in the different treatments.
Results and discussion
Characterization of liposomes before and after PC formation
Liposomes were incubated in plasma for 1 hour to allow liposome–PC complex formation. This incubation time was chosen because it is known that a stable PC forms over a period of 1 hour.34 Moreover, previous studies reported that 1 hour is the liposomes’ saturation point in terms of proteins adsorbed during incubation in plasma.35
Samples were analyzed before and after incubation in plasma by dynamic light scattering (DLS), zeta potential, and cryo-electron microscopy analyses to investigate their size (and size homogeneity: polydispersity index, PDI), their surface charge, and their structure, respectively. Control liposomes had a very low PDI (0.07), which is typical of homogeneous formulations, and a hydrodynamic diameter (HD) of ~140 nm (Table 1), while the liposome–PC complexes had a wide size distribution (PDI 0.21). In particular, three main populations were detected in the liposome–PC complexes sample (Table 1): 1) a population of ~60 nm in HD attributable to plasma proteins and plasma protein aggregates and/or to liposomes that had shrunk due to the elastic properties of their lipid bilayer;36,37 2) a population with an HD slightly larger (≈180 nm) than control liposomes (ie, liposomes before incubation in plasma) ascribable to PC formation; and 3) a population of particles from two to three times larger than control liposomes (HD =400 nm), which are most likely due to PC-induced liposome aggregation.
 | Table 1 DLS analysis of liposomes before and after incubation in plasma |
As given in Table 1, control liposomes had a slightly negatively charged surface (ζ-potential =−5 mV). The ζ-potential decreased to =−16 mV after the formation of the PC on the liposome surface, which is similar to the charge of plasma proteins (ζ-potential =−18 mV, data not shown).
We used cryo-electron microscopy to evaluate the morphology of the liposome–PC complex. Single-vesicle analysis of control liposomes and liposome–PC complexes revealed that liposomes preserved their structure and shape after incubation in plasma (Figure 1). In addition, unlike control liposomes (Figure 1A), additional mass adsorbed on liposome surfaces attributable to a compact PC was observed (Figure 1B).
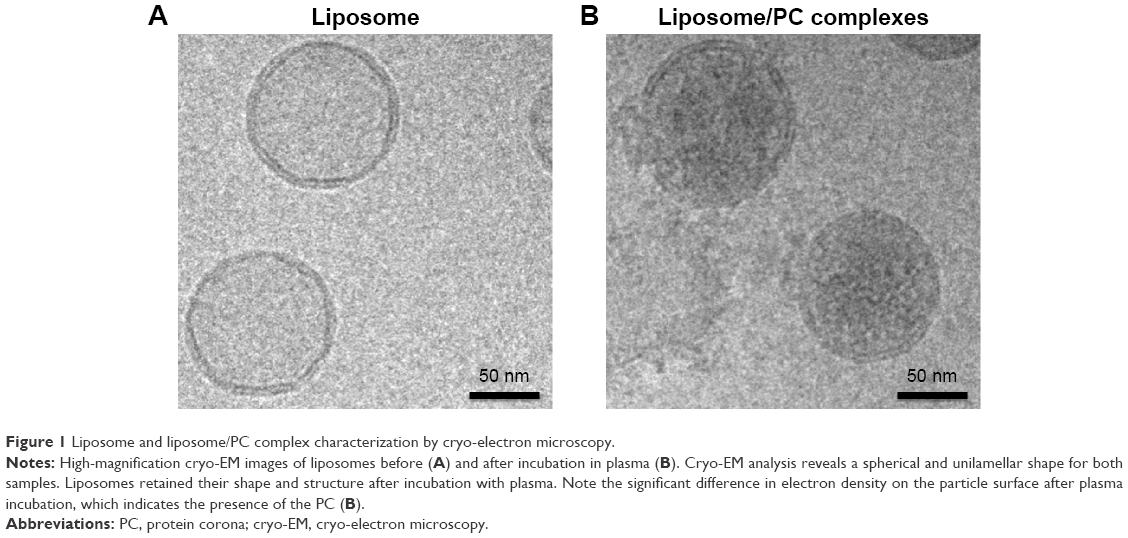 | Figure 1 Liposome and liposome/PC complex characterization by cryo-electron microscopy. |
We used AFM to identify changes in the mechanical properties of liposomes after PC formation. As shown in Figure 2A, liposome–PC complexes were larger than control liposomes (142–171 nm), which confirms our DLS results. Control liposomes resulted in smaller, homogeneous particles, while the liposome–PC complexes are relatively larger and heterogeneous. Size distribution analysis showed a large variation in particle size ranging from smaller values of 180–820 nm.
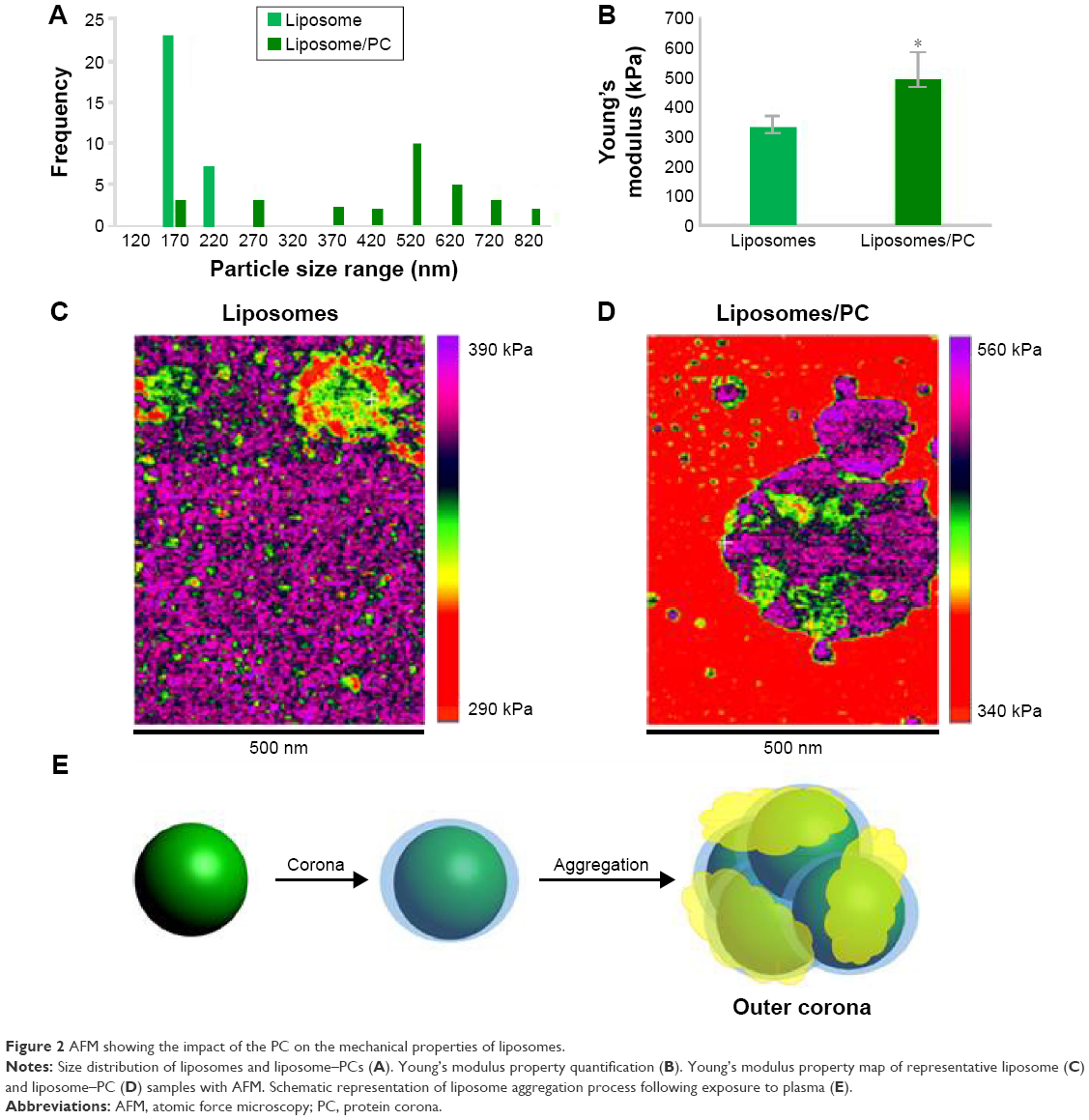 | Figure 2 AFM showing the impact of the PC on the mechanical properties of liposomes. |
Quantitative AFM analysis (Figure 2B) revealed that liposomes after incubation with plasma were significantly stiffer (P<0.005) than bare liposomes (470 kPa vs 310 kPa). The increased elastic modulus is most likely induced by the presence of particle aggregates with PC deposition. Bare liposomes contained a stiffer central area corresponding to the highest particle zone, while a more elastic region (the external ring in red) corresponded to the particle edges (Figure 2C). The liposome–PC aggregates map, instead, showed a heterogeneous elastic modulus variation over the large surface (Figure 2D).
Taken together, these data suggest that the formation of particle aggregates could be a two-step process (Figure 2E). As mentioned earlier, PC formation is a dynamic process.38 Our analysis showed a heterogeneous scenario as a snapshot of the overall mechanism. The smaller particles found in the liposome–PC complex sample represent single particle–PC complexes. AFM analysis revealed that these individual liposome/PC complexes were stiffer than bare liposomes but softer than liposome–PC aggregates (Figure S1). According to this finding, we speculate that once each liposome is covered by the first PC layer, it becomes adhesive, thereby inducing aggregation into larger clusters.39 Moreover, the analysis of the elastic map revealed another protein adsorption process on the particle cluster that we call the “outer corona” (Figure 2E). In addition, although the cluster itself is stiffer than bare liposomes, there are some areas underneath the outer corona that are less stiff (Figure S2). However, these softer areas remain stiffer than the bare liposomes (green) and are, instead, very similar in value to those of single liposome–PC complexes (Figure S1). Therefore, we hypothesize a nonhomogeneous presence of the outer corona due to this difference in topographical arrangement. We speculate that formation of the outer corona might be triggered by the size of the PC rather than by its composition (Figure S3).
Identification of proteins in the corona
As reported in the section Characterization of liposomes before and after PC formation, liposomes were incubated in plasma for 1 hour to allow liposome–PC complex formation. Our aim was to define proteins constituting the PC in order to understand their potential biological effect on liposomes. To this aim, the adsorbed proteins (PCs) were eluted from the liposomes, separated by 1D SDS PAGE (Figure 3A), and identified by mass spectrometry. The most abundant proteins, selected by manual visualization and densitometric analysis (Figure 3B), were excised and hydrolyzed with trypsin. The 20 most abundant proteins adsorbed on the liposome surface after exposure to murine plasma are shown in Figure 3C, while the mass spectrometry profiles of the identified proteins are listed in Table S1. Overall, most PC components were small, negatively charged proteins (>70% with molecular weight [MW] <60 kDa, Figure 4A, and >70% with isoeletric point [pI] <6.5, Figure 4B). A previous study showed that negatively charged particles preferentially attract positively charged proteins.40,41 Contrary to our expectation, negatively charged proteins preferentially adsorbed on our negatively charged liposomes. A similar trend was reported by others,42,43 and it supports the idea that predictions of PC composition exclusively based on NP charge are too simplistic. Electrostatic interactions are not the only force that drives NP–PC interactions. Many other factors (eg, protein abundance in plasma, the presence of a functional group on the NP surface, plasma type, and incubation time) affect PC composition. On the other hand, neutral and positive proteins are also present in the PC, though in small percents (Figure 4B). Considering our observation discussed above on the formation of a nonhomogenous corona, we believe that it is possible that these proteins create small positive charges or neutral patches on the particles’ surface, which facilitates aggregation by electrostatic or hydrophobic interactions.
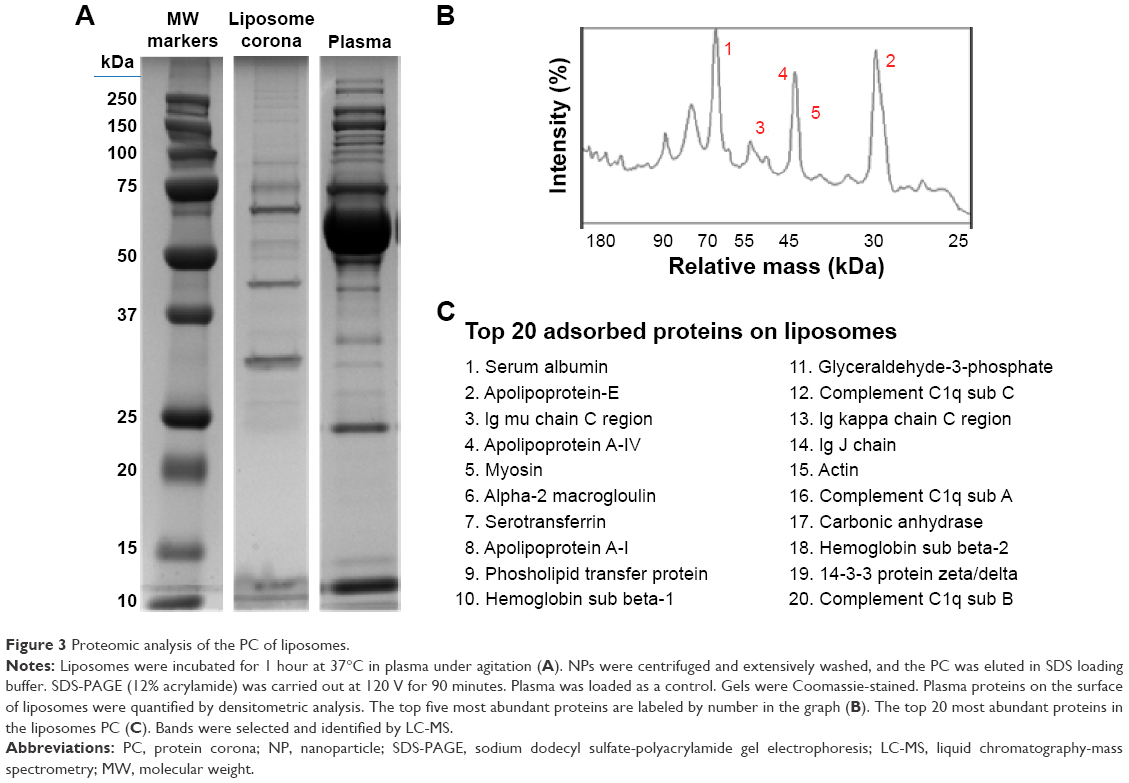 | Figure 3 Proteomic analysis of the PC of liposomes. |
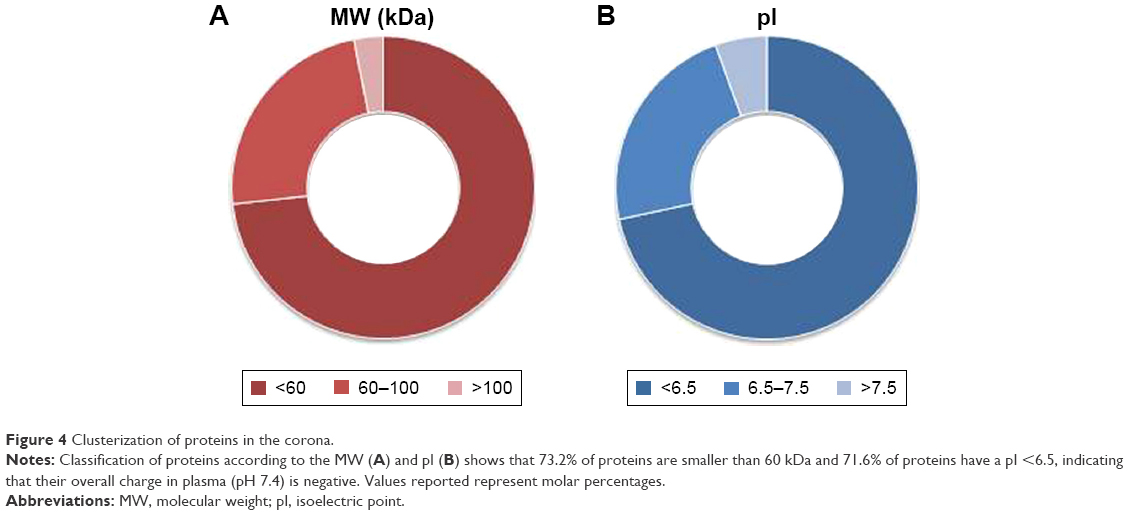 | Figure 4 Clusterization of proteins in the corona. |
In our study, albumin, apolipoproteins, and IgGs were the main PC components (Figure 3C), which confirms data obtained with other vesicle types.35 To better understand the biological impact of the PC on particle–cell interactions, we investigated liposome and liposome–PC complex uptake by macrophages and breast cancer cells. We discuss the results in the next section.
Cellular uptake of liposomes before and after PC formation
It is well recognized that the PC affects the fate of particles in the bloodstream.44 Notably, the PC can affect particle clearance by immune cells,8 thereby affecting their biodistribution properties. To obtain an overview of what could happen in the context of in vivo cancer therapy when therapeutic NPs are systemically injected, we studied their uptake by macrophages, which are responsible for particle clearance, and their internalization by cancer cells, which is essential for pharmacological efficacy. We used mouse J774 macrophages (Figure 5A) and mouse 4T1 breast cancer cells (Figure 5B) as proof-of-concept models for this in vitro study. We treated both cell lines with fluorescent particles, and then examined their interactions with NPs using confocal microscopy. The uptake of liposomes with a PC was greater than that of bare liposomes in both cell lines. Moreover, large particle aggregates formed in PC-endowed liposomes but not in bare liposomes (white arrows in Figure S4).
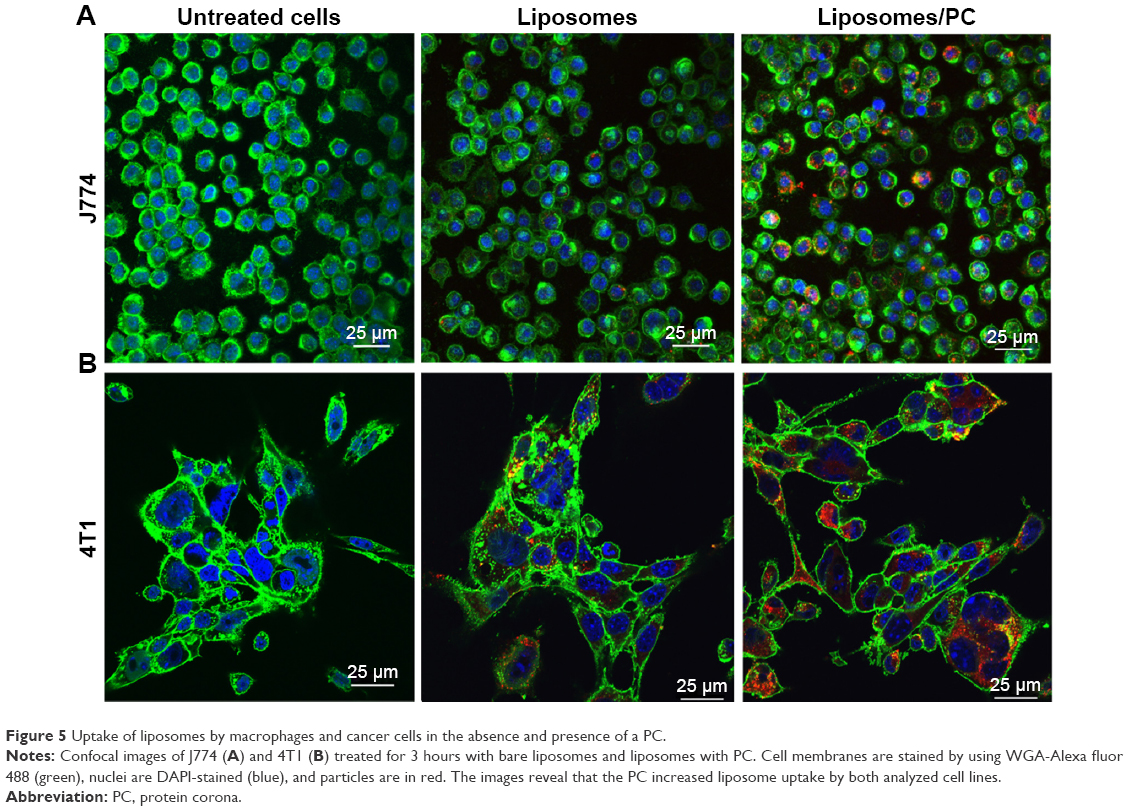 | Figure 5 Uptake of liposomes by macrophages and cancer cells in the absence and presence of a PC. |
To determine the fraction of particles internalized by J774 and 4T1 cells, we carried out a flow cytometry analysis, in which cells were quenched with trypan blue to obscure the fluorescence associated with the cell surface. As shown in Figure 6, the quantitative analysis confirmed the increased uptake of liposomes with a PC. These results concur with studies of NP uptake by immune45 and tumor cells.46
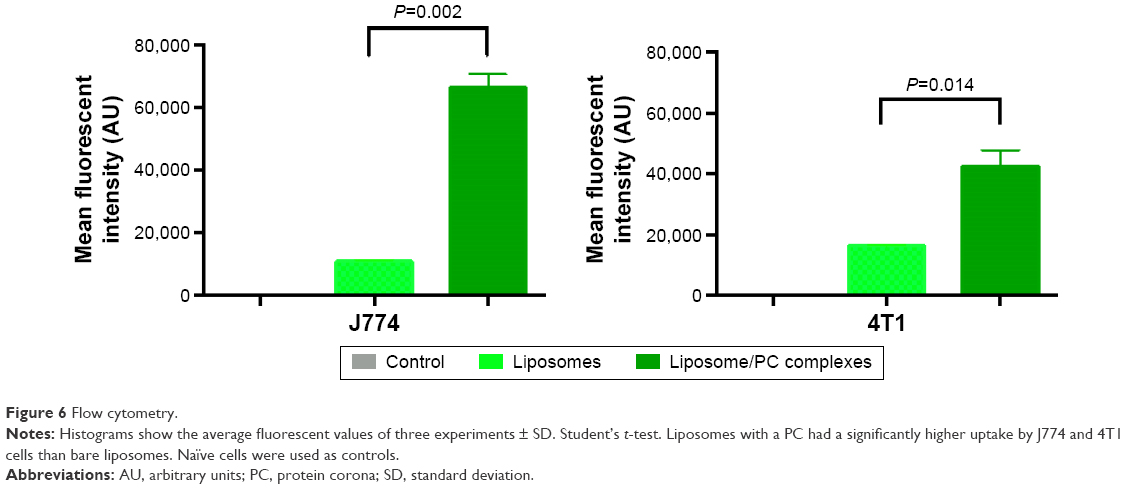 | Figure 6 Flow cytometry. |
Data on the impact of the PC on the biological fate of NPs are controversial. Since PC formation is a highly dynamic process, it is difficult to envisage a unique explanation of the interfacial events that occur between the PC and biological components. For several decades, cell opsonization was considered the body’s first defense mechanism against foreign entities,47 but recent studies have shown that the PC can shield particles from the action of adsorbed serum components (IgG and complement factors) that normally favor particle clearance.19,20 Here we report increased liposome uptake by both J774 and 4T1 cells after plasma incubation. We hypothesize that complement factors and apolipoproteins present in the PC are mainly responsible for this effect. Complement proteins, together with immunoglobulins and coagulation factors, mediate particle uptake through interactions with complement Fc and Toll-like receptors on the macrophage surface.47 On the other hand, apolipoproteins belong to the family of lipoproteins, which are key players in the cholesterol metabolism process, through which they stimulate cancer cell growth.48 Cholesterol availability is crucial for the proliferation of rapidly dividing cancer cells. Breast cancer cells, like other cancer cells, express B-class scavenger receptors on their surface,49 which are intermediate in lipid transfer between lipoproteins and cells.50 We believe that the functional motifs of these proteins are exposed on the PC, in the correct orientation to perform their function and to be recognized by their specific cell receptors, allowing PC-mediated cell internalization. These findings explain the impact of the PC, ie, the liposome’s new biological identity, on cellular uptake. On the other hand, our DLS, cryo-EM, and AFM experiments conducted to characterize liposomes provide insights into the impact of the PC also on the liposome’s new physical identity. We found that PC formation induces liposome aggregation, which implies the presence of clusters of NPs (Table 1, Figure 2) that coexist with single liposome–PC complexes, consistent with what has been demonstrated elsewhere.51,52 This means that cells interact not only with single particles surrounded by a PC through a receptor-mediated pathway but also with particles in the form of larger aggregates. The latter are likely to be internalized via phagocytosis, particularly in the case of macrophages, which would explain why we observed a major uptake by cells (Figure 7).
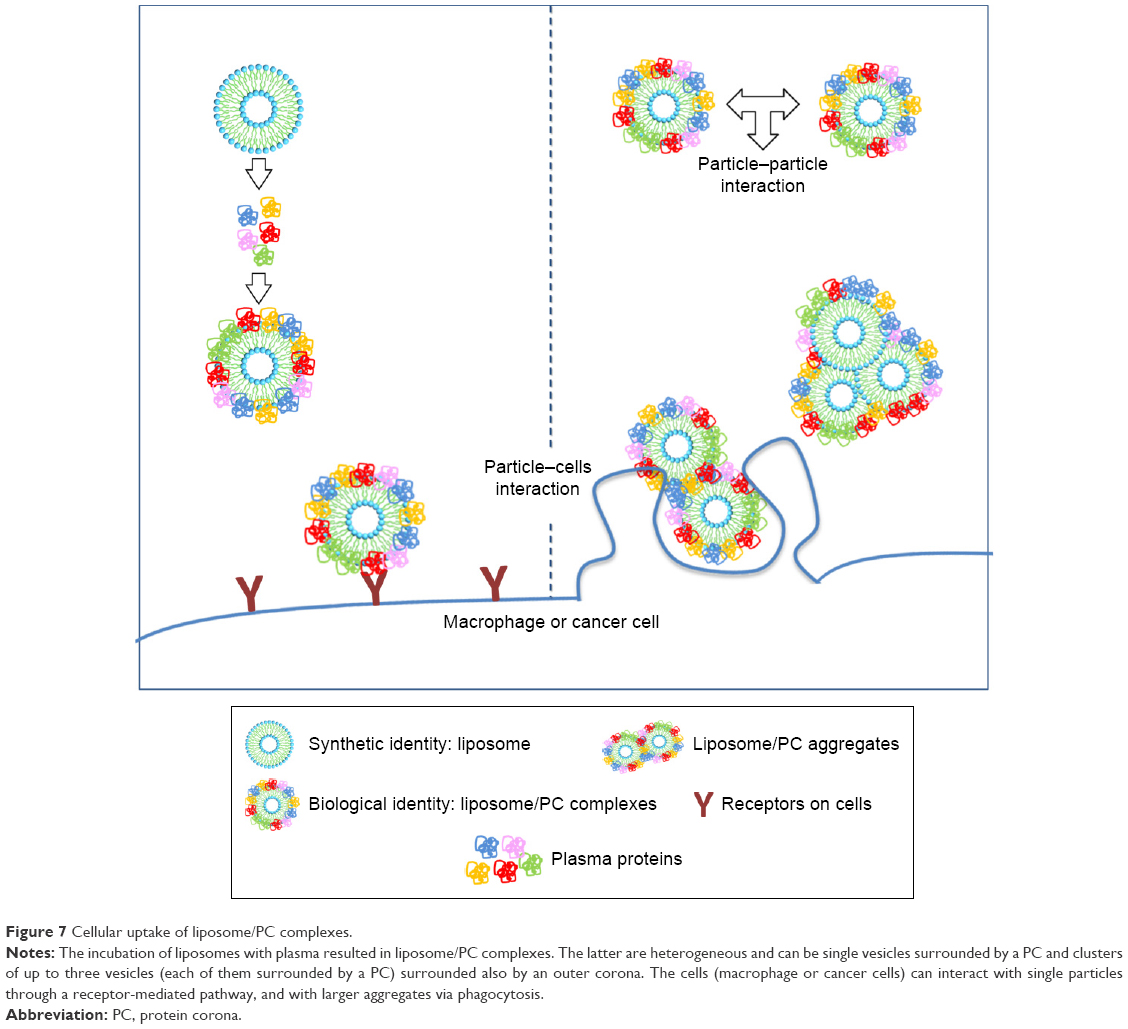 | Figure 7 Cellular uptake of liposome/PC complexes. |
Conclusion
In the last decade, the biological components of synthetic NPs have attracted the attention of the scientific community because of the fundamental role they play in the interaction between particles and cells. Several assertions have been made to describe the PC’s composition and correlate it to NP uptake by immune and cancer cells. Thus far, the results have been controversial due to the variability of the experimental settings (particle chemical composition, plasma source, plasma concentration, particle incubation time, cell lines, etc).8 In this study, we reported the structure, composition, and biological effect of PCs adsorbed on multicomponent liposomes. We found that the formation of a PC on liposomes makes their surface adhesive, thus inducing the formation of particle clusters that coexist with single vesicles. We also showed that internalization of liposome–PC complexes by both macrophages and cancer cells was greater than that of control liposomes. The presence of a PC strongly affects the NP’s biological response. This can be due to 1) the presence of molecules (ie, immunoglobulins and apolipoproteins) in the PC that recognize specific receptors on the surface of immune and cancer cells, and/or 2) the formation of NP cluster that induce phagocytosis by the cells, or 3) a combination of these effects.
We shed light on the impact of the PC first on particle–particle and then on particle–cell interactions. Our observations suggest that to predict the fate of a NP in vivo, one must not only know the PC composition and eventually the orientation of proteins, but should also carefully consider the physical changes (size, surface, and charge) that the NP undergoes when surrounded by a PC and the consequent impact on particle–particle interactions. This study shows that the efficacy of liposomal formulations must be reinterpreted, as it is strongly affected by PC formation in plasma. Classical characterization of nanodelivery systems should include studies on PC formation and be combined with experiments that simulate the tendency of NPs to aggregate after exposure to plasma proteins. We believe that such studies will hopefully help us interpret and predict in vivo data and to design more effective therapeutic carriers.
Acknowledgments
The authors would like to thank Kemi Cui and HMRI Advanced Cellular and Tissue Microscope Core Facility for traditional confocal scanning services, David Haviland and the HMRI Flow Cytometry Core Facility for flow cytometry setup and acquisition, and DA Engler, RK Matsunami, and the HMRI Proteomics Programmatic Core Laboratory for mass spectrometry analyses. The authors acknowledge the Sealy Center for Structural Biology and Molecular Biophysics at the University of Texas Medical Branch at Galveston for providing research resources. We thank Jean Ann Gilder (Scientific Communication srl. Naples, Italy) and Megan Livingston for editing the text. We thank Associazione Bianca Garavaglia, Via C Cattaneo 8, 21052 Busto Arsizio, Varese, Italy. This work was supported by grants RF-2010-2318372 and RF-2010-2305526 from Italian Ministry of Health, and by grants 1R21CA173579-01A1 from NIH/NCI, 5U54CA143837 PSOC Pilot project from NIH/NCI, W81XWH-12-10414 BCRP Innovator Expansion from Department of Defense, William Randolph Hearst Foundation, and The Regenerative Medicine Program Cullen Trust for Health Care to ET and by POR Campania FSE 2007–2013 Project DIAINTECH, Italy (to FS).
Disclosure
The authors report no conflicts of interest in this work.
References
Singh R, Lillard JW. Nanoparticle-based targeted drug delivery. Exp Mol Pathol. 2009;86(3):215–223. | ||
Lundqvist M. Nanoparticles: tracking protein corona over time. Nat Nanotechnol. 2013;8(10):701–702. | ||
Tenzer S, Docter D, Kuharev J, et al. Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat Nanotechnol. 2013;8(10):772–781. | ||
Lynch I, Dawson KA. Protein-nanoparticle interactions. Nano Today. 2008;3(1):40–47. | ||
Mirsadeghi S, Dinarvand R, Ghahremani MH, et al. Protein corona composition of gold nanoparticles/nanorods affects amyloid beta fibrillation process. Nanoscale. 2015;7(11):5004–5013. | ||
O’Connell D, Bombelli FB, Pitek A, Monopoli M, Cahill D, Dawson KA. Characterization of the bionano interface and mapping extrinsic interactions of the corona of nanomaterials. Nanoscale. 2015;7(37):15268–15276. | ||
Zanganeh S, Spitler R, Erfanzadeh M, Alkilany AM, Mahmoudi M. Protein corona: opportunities and challenges. Int J Biochem Cell Biol. 2016;75:143–147. | ||
Corbo C, Molinaro R, Parodi A, Toledano Furman NE, Salvatore F, Tasciotti E. The impact of nanoparticle protein corona on cytotoxicity, immunotoxicity and target drug delivery. Nanomedicine. 2016;11(1):81–100. | ||
Salvati A, Pitek AS, Monopoli MP, et al. Transferrin-functionalized nanoparticles lose their targeting capabilities when a biomolecule corona adsorbs on the surface. Nat Nanotechnol. 2013;8(2):137–143. | ||
Mirshafiee V, Mahmoudi M, Lou K, Cheng J, Kraft ML. Protein corona significantly reduces active targeting yield. Chem Commun (Camb). 2013;49(25):2557–2559. | ||
Qiu Y, Liu Y, Wang L, et al. Surface chemistry and aspect ratio mediated cellular uptake of Au nanorods. Biomaterials. 2010;31(30):7606–7619. | ||
Ge C, Du J, Zhao L, et al. Binding of blood proteins to carbon nanotubes reduces cytotoxicity. Proc Natl Acad Sci U S A. 2011;108(41):16968–16973. | ||
Ge C, Tian J, Zhao Y, Chen C, Zhou R, Chai Z. Towards understanding of nanoparticle–protein corona. Arch Toxicol. 2015;89(4):519–539. | ||
Sahneh FD, Scoglio CM, Monteiro-Riviere NA, Riviere JE. Predicting the impact of biocorona formation kinetics on interspecies extrapolations of nanoparticle biodistribution modeling. Nanomedicine (Lond). 2015;10(1):25–33. | ||
Milani S, Baldelli Bombelli F, Pitek AS, Dawson KA, Rädler J. Reversible versus irreversible binding of transferrin to polystyrene nanoparticles: soft and hard corona. ACS Nano. 2012;6(3):2532–2541. | ||
Liu W, Rose J, Plantevin S, Auffan M, Bottero J-Y, Vidaud C. Protein corona formation for nanomaterials and proteins of a similar size: hard or soft corona? Nanoscale. 2013;5(4):1658–1668. | ||
Karmali PP, Simberg D. Interactions of nanoparticles with plasma proteins: implication on clearance and toxicity of drug delivery systems. Expert Opin Drug Deliv. 2011;8(3):343–357. | ||
Moghimi SM, Szebeni J. Stealth liposomes and long circulating nanoparticles: critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog Lipid Res. 2003;42(6):463–478. | ||
Mirshafiee V, Kim R, Park S, Mahmoudi M, Kraft ML. Impact of protein pre-coating on the protein corona composition and nanoparticle cellular uptake. Biomaterials. 2016;75:295–304. | ||
Caracciolo G, Palchetti S, Colapicchioni V, et al. Stealth effect of biomolecular corona on nanoparticle uptake by immune cells. Langmuir. 2015;31(39):10764–10773. | ||
He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31(13):3657–3666. | ||
Kulkarni SA, Feng S-S. Effects of particle size and surface modification on cellular uptake and biodistribution of polymeric nanoparticles for drug delivery. Pharm Res. 2013;30(10):2512–2522. | ||
Caracciolo G. The protein corona effect for targeted drug delivery. Bioinspired Biomimetic Nanobiomater. 2013;2(1):54–57. | ||
Mahon E, Salvati A, Bombelli FB, Lynch I, Dawson KA. Designing the nanoparticle–biomolecule interface for “targeting and therapeutic delivery”. J Control Release. 2012;161(2):164–174. | ||
Van Rooijen N, van Nieuwmegen R. Liposomes in immunology: multilamellar phosphatidylcholine liposomes as a simple, biodegradable and harmless adjuvant without any immunogenic activity of its own. Immunol Commun. 1980;9(3):243–256. | ||
Lee H, Blaufox M. Blood volume in the rat. J Nucl Med. 1985;26(1):72–76. | ||
Monopoli MP, Walczyk D, Campbell A, et al. Physical−chemical aspects of protein corona: relevance to in vitro and in vivo biological impacts of nanoparticles. J Am Chem Soc. 2011;133(8):2525–2534. | ||
Maugis D. Contact, Adhesion and Rupture of Elastic Solids. Vol. 130. Berlin: Springer Science & Business Media; 2013. | ||
Corbo C, Orrù S, Gemei M, et al. Protein cross-talk in CD133+ colon cancer cells indicates activation of the Wnt pathway and upregulation of SRp20 that is potentially involved in tumorigenicity. Proteomics. 2012;12(12):2045–2059. | ||
Caterino M, Corbo C, Imperlini E, et al. Differential proteomic analysis in human cells subjected to ribosomal stress. Proteomics. 2013;13(7):1220–1227. | ||
Corbo C, Parodi A, Evangelopoulos M, et al. Proteomic profiling of a biomimetic drug delivery platform. Curr Drug Targets. 2015;16(13):1540–1547. | ||
Li G-Z, Vissers JP, Silva JC, Golick D, Gorenstein MV, Geromanos SJ. Database searching and accounting of multiplexed precursor and product ion spectra from the data independent analysis of simple and complex peptide mixtures. Proteomics. 2009;9(6):1696–1719. | ||
Nuutila J, Lilius EM. Flow cytometric quantitative determination of ingestion by phagocytes needs the distinguishing of overlapping populations of binding and ingesting cells. Cytometry A. 2005;65(2):93–102. | ||
Walczyk D, Bombelli FB, Monopoli MP, Lynch I, Dawson KA. What the cell “sees” in bionanoscience. J Am Chem Soc. 2010;132(16):5761–5768. | ||
Barrán-Berdón AL, Pozzi D, Caracciolo G, et al. Time evolution of nanoparticle–protein corona in human plasma: relevance for targeted drug delivery. Langmuir. 2013;29(21):6485–6494. | ||
Wolfram J, Suri K, Yang Y, et al. Shrinkage of pegylated and non-pegylated liposomes in serum. Colloids Surf B Biointerfaces. 2014;114:294–300. | ||
Hadjidemetriou M, Al-Ahmady Z, Mazza M, Collins RF, Dawson K, Kostarelos K. In vivo biomolecule corona around blood-circulating, clinically used and antibody-targeted lipid bilayer nanoscale vesicles. ACS Nano. 2015;9(8):8142–8156. | ||
Miclaus T, Bochenkov VE, Ogaki R, Howard KA, Sutherland DS. Spatial mapping and quantification of soft and hard protein coronas at silver nanocubes. Nano Lett. 2014;14(4):2086–2093. | ||
Gräfe C, Weidner A, vd Lühe M, et al. Intentional formation of a protein corona on nanoparticles-serum concentration affects protein corona mass, surface charge, and nanoparticle-cell interaction. Int J Biochem Cell Biol. 2016;75:196–202. | ||
Nel AE, Mädler L, Velegol D, et al. Understanding biophysicochemical interactions at the nano–bio interface. Nat Mater. 2009;8(7):543–557. | ||
Mahmoudi M, Lynch I, Ejtehadi MR, Monopoli MP, Bombelli FB, Laurent S. Protein−nanoparticle interactions: opportunities and challenges. Chem Rev. 2011;111(9):5610–5637. | ||
Walkey CD, Chan WC. Understanding and controlling the interaction of nanomaterials with proteins in a physiological environment. Chem Soc Rev. 2012;41(7):2780–2799. | ||
Tenzer S, Docter D, Rosfa S, et al. Nanoparticle size is a critical physicochemical determinant of the human blood plasma corona: a comprehensive quantitative proteomic analysis. ACS Nano. 2011;5(9):7155–7167. | ||
Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson KA. Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proc Natl Acad Sci U S A. 2008;105(38):14265–14270. | ||
Yan Y, Gause KT, Kamphuis MM, et al. Differential roles of the protein corona in the cellular uptake of nanoporous polymer particles by monocyte and macrophage cell lines. ACS Nano. 2013;7(12):10960–10970. | ||
Mulik RS, Monkkonen J, Juvonen RO, Mahadik KR, Paradkar AR. Transferrin mediated solid lipid nanoparticles containing curcumin: enhanced in vitro anticancer activity by induction of apoptosis. Int J Pharm. 2010;398(1–2):190–203. | ||
Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11(9):785–797. | ||
Johnson WJ, Mahlberg FH, Rothblat GH, Phillips MC. Cholesterol transport between cells and high-density lipoproteins. Biochim Biophys Acta. 1991;1085(3):273–298. | ||
Cao WM, Murao K, Imachi H, et al. A mutant high-density lipoprotein receptor inhibits proliferation of human breast cancer cells. Cancer Res. 2004;64(4):1515–1521. | ||
Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271(5248):518–520. | ||
Palchetti S, Colapicchioni V, Digiacomo L, et al. The protein corona of circulating PEGylated liposomes. Biochim Biophys Acta. 2016;1858(2):189–196. | ||
Pozzi D, Caracciolo G, Digiacomo L, et al. The biomolecular corona of nanoparticles in circulating biological media. Nanoscale. 2015;7(33):13958–13966. |
Supplementary materials
 | Figure S1 AFM elastic map of a single liposome/PC complex. |
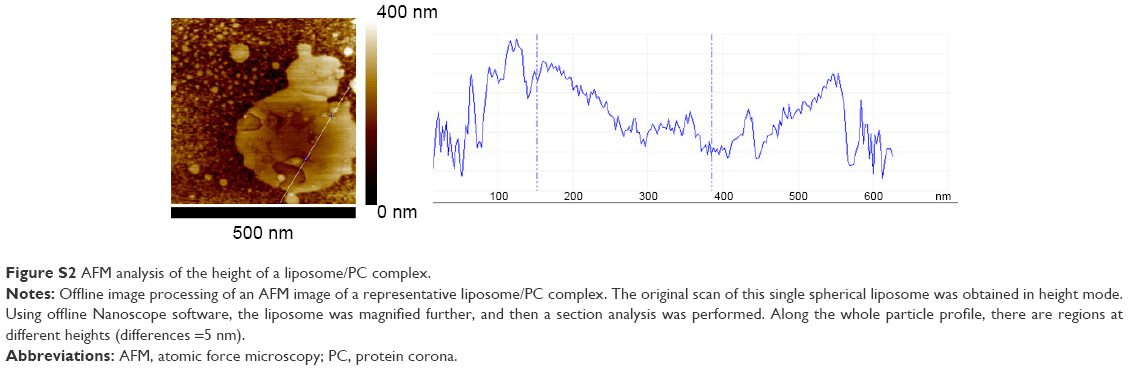 | Figure S2 AFM analysis of the height of a liposome/PC complex. |
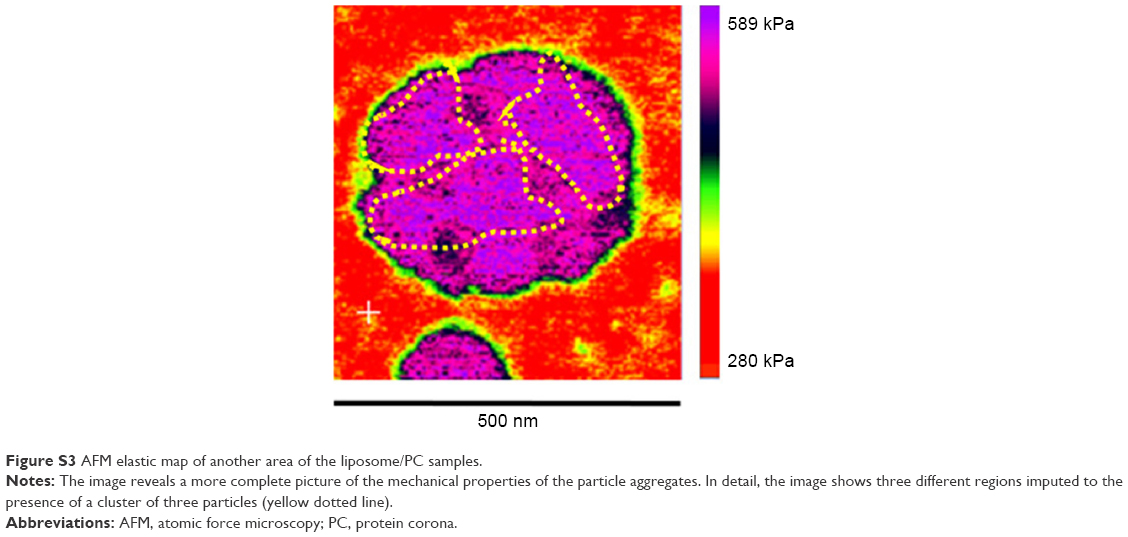 | Figure S3 AFM elastic map of another area of the liposome/PC samples. |
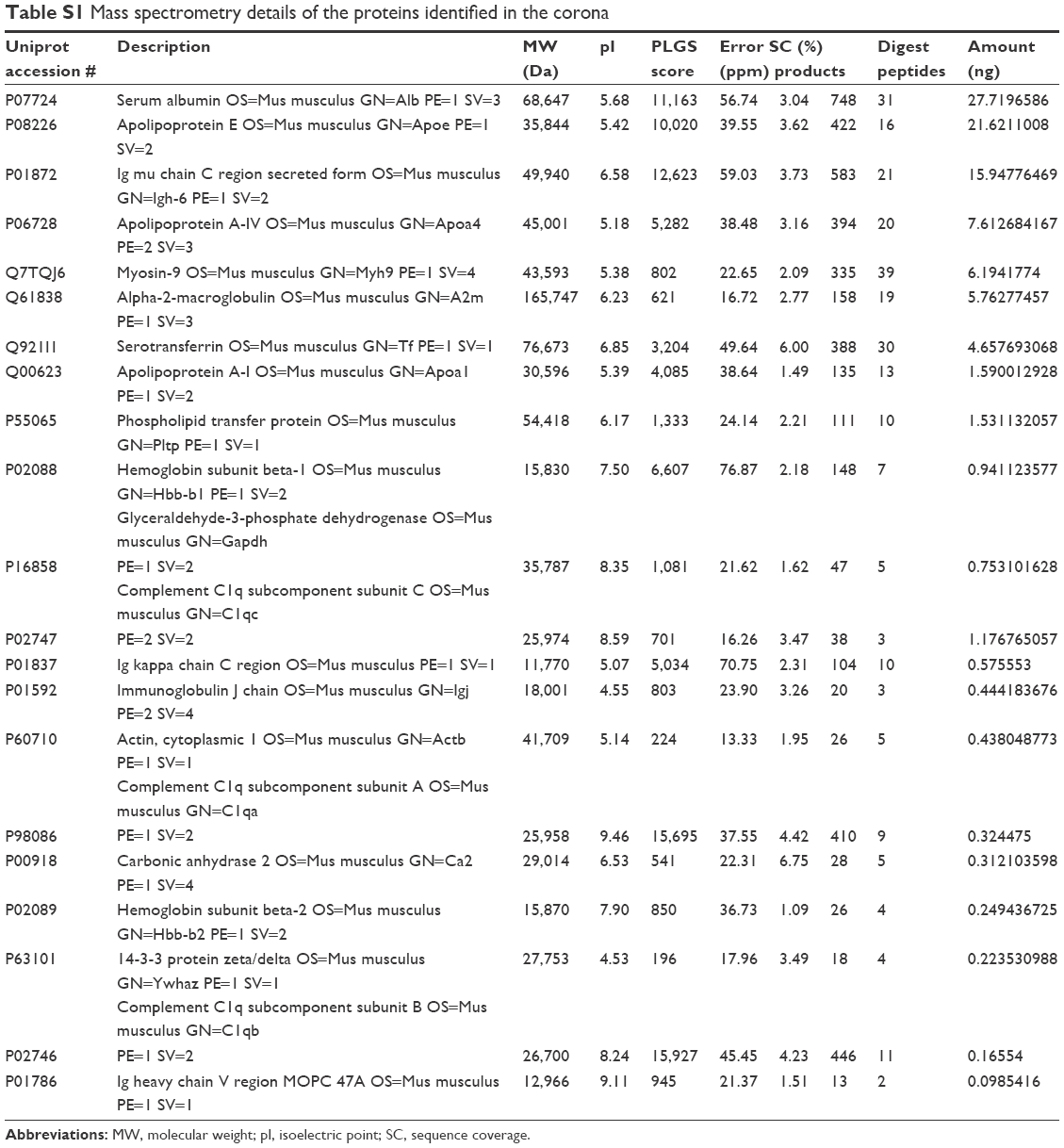 | Table S1 Mass spectrometry details of the proteins identified in the corona |
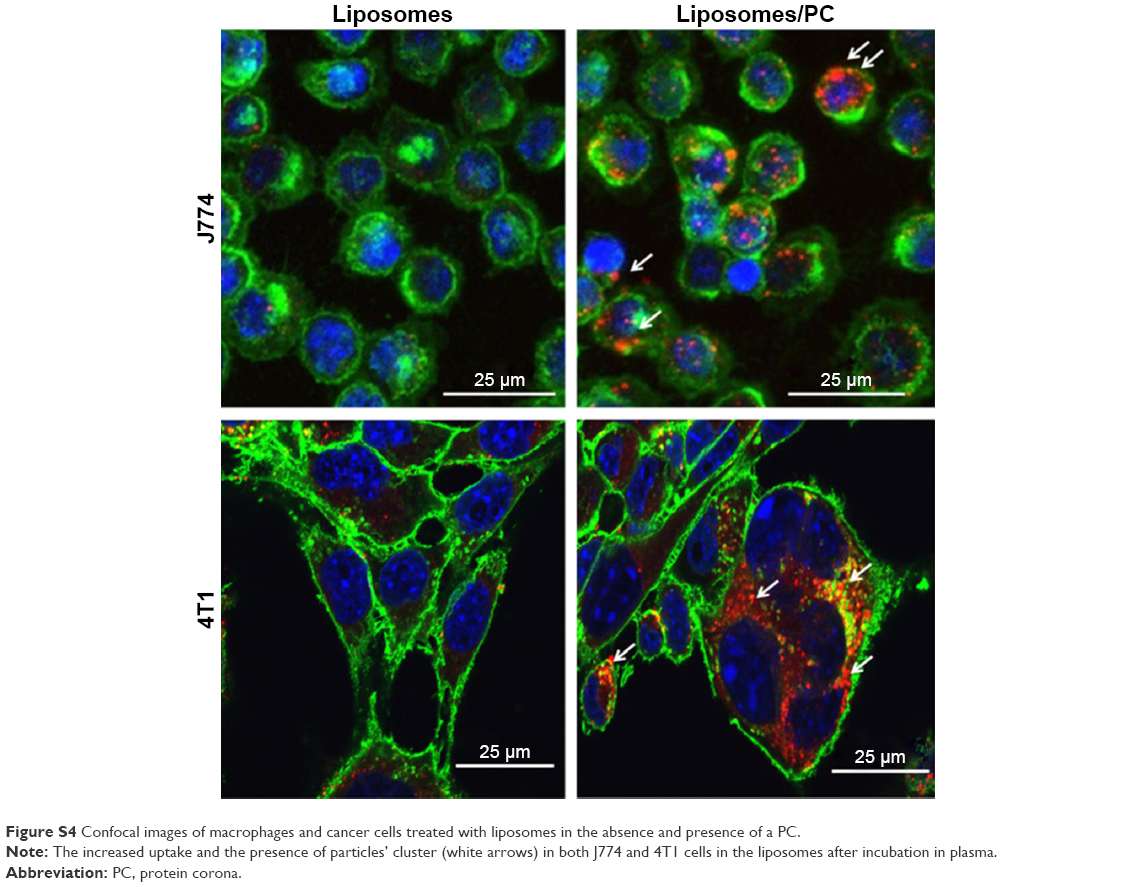 | Figure S4 Confocal images of macrophages and cancer cells treated with liposomes in the absence and presence of a PC. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.