Back to Journals » Journal of Pain Research » Volume 12
Effects of the glial modulator palmitoylethanolamide on chronic pain intensity and brain function
Authors Alshelh Z, Mills EP, Kosanovic D, Di Pietro F, Macey PM ![]() , Vickers ER, Henderson LA
, Vickers ER, Henderson LA
Received 22 March 2019
Accepted for publication 9 July 2019
Published 2 August 2019 Volume 2019:12 Pages 2427—2439
DOI https://doi.org/10.2147/JPR.S209657
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Katherine Hanlon
Zeynab Alshelh,1 Emily P Mills,1 Danny Kosanovic,1 Flavia Di Pietro,1 Paul M Macey,2 E Russell Vickers,1 Luke A Henderson1
1Department of Anatomy and Histology, University of Sydney, Sydney, NSW, Australia; 2School of Nursing and Brain Research Institute, David Geffen School of Medicine, University of California at Los Angeles, Los Angeles, CA, USA
Background: Chronic neuropathic pain (NP) is a complex disease that results from damage or presumed damage to the somatosensory nervous system. Current treatment regimens are often ineffective. The major impediment in developing effective treatments is our limited understanding of the underlying mechanisms. Preclinical evidence suggests that glial changes are crucial for the development of NP and a recent study reported oscillatory activity differences within the ascending pain pathway at frequencies similar to that of cyclic gliotransmission in NP. Furthermore, there is evidence that glial modifying medications may be effective in treating NP. The aim of this Phase I open-label clinical trial is to determine whether glial modifying medication palmitoylethanolamide (PEA) will reduce NP and whether this is associated with reductions in oscillatory activity within the pain pathway.
Methods: We investigated whether 6 weeks of PEA treatment would reduce pain and infra-slow oscillatory activity within the ascending trigeminal pathway in 22 individuals (17 females) with chronic orofacial NP.
Results: PEA reduced pain in 16 (73%) of the 22 subjects, 11 subjects showed pain reduction of over 20%. Whilst both the responders and non-responders showed reductions in infra-slow oscillatory activity where orofacial nociceptor afferents terminate in the brainstem, only responders displayed reductions in the thalamus. Furthermore, functional connections between the brainstem and thalamus were altered only in responders.
Conclusion: PEA is effective at relieving NP. This reduction is coupled to a reduction in resting oscillations along the ascending pain pathway that are likely driven by rhythmic astrocytic gliotransmission.
Keywords: infra-slow oscillations, astrocytes, connectivity, thalamus, imaging
Significance statement
In this investigation, we have tested this hypothesis by conducting a Phase I clinical trial to determine the effectiveness of a glial modulator, palmitoylethanolamide (PEA) on ongoing pain intensity and oscillatory brain activity. We found that after 6 weeks of PEA, ongoing was significantly reduced in 16 of 22 subjects and that this reduction was significantly correlated to changes in infra-slow oscillatory activity within the ascending pain pathway. These data provide the first human evidence that a substance that can reduce glial activation can relieve pain without side effects, and that this is associated with a reduction in ongoing patterns of neural activity in pain pathways that are consistent with the idea that glial transmission maintains chronic neuropathic pain.
Introduction
Chronic neuropathic pain (NP) is a complex disease state that results from damage or presumed damage to the somatosensory nervous system. Current treatment regimens such as non-steroidal anti-inflammatory medication, tricyclic antidepressants and anticonvulsants are often ineffective and have significant side effects and consequently, many individuals suffer with considerable pain for years or even decades. The major impediment in developing more effective treatment regimens for NP is our limited understanding of the mechanisms responsible for the development and maintenance of NP following injury.
Critically, NP is not associated with prolonged activity increases in the classic “pain pathways” but instead is associated with altered brain rhythm. This altered rhythm is characterized by increased thalamic burst firing, thalamocortical power and infra-slow oscillations (<0.1 Hz) along the ascending pain pathway.1–3 Whilst it is not known how these altered rhythms are generated, the infra-slow oscillation increases occur at approximately the same frequency range as calcium waves in astrocytes, ie, 0.03–0.06 Hz.4 Astrocytes can modulate synaptic activity by the release of gliotransmitters and there is preclinical and postmortem human evidence of prolonged astrocyte activation in the dorsal horn/spinal trigeminal nucleus (SpV) in NP.5,6 Furthermore, preclinical studies have shown that following nerve injury, increased astrocyte activation is associated with allodynia and hyperalgesia7,8 and blocking astrocyte activation reverses this allodynia and hyperalgesia.9–11
PEA is a naturally occurring fatty acid amide that belongs to the N-acetylethanolamine family. It is thought that PEA targets alpha peroxisome proliferator-activated receptors (PPAR-α),12–16 which are expressed on neurons and astrocytes17,18 and are activated as a response to nerve damage.6,10 Interestingly, a recent case study found that in an individual without the ability to breakdown PEA, elevated serum PEA was associated with pain insensitivity, implying that endogenous PEA plays a role in pain processing.19 In addition, there have been numerous clinical trials exploring the effects of PEA in various pain states and most show pain reductions and critically, PEA has reportedly no serious side effects.20 If PEA can reduce NP without any serious side effects, this would be a major advance in the way in which we treat chronic NP and significantly reduce the economic and social burden of this debilitating condition. The aim of this investigation is to provide evidence that PEA reduces the intensity of ongoing pain and reverses the increase in infra-slow oscillatory activity within the primary afferent synapse in individuals with chronic NP.
Methods
Subjects
Twenty-two subjects with chronic orofacial NP (17 females; mean±SEM age, 50.5±3.0 years) were recruited for the study. NP subjects were diagnosed using the Liverpool criteria as having posttraumatic neuropathy.21 Subjects were excluded if they had any psychiatric conditions, any other pain conditions or did not meet standard magnetic resonance imaging (MRI) inclusion criteria.
Protocol approval and registration
Informed written consent was obtained for all procedures according to the Declaration of Helsinki, and the Human Research Ethics Committees of the University of Sydney approved the study. This trial was registered with the Australian New Zealand Clinical Trials Registry with the trial ID: ACTRN12618001637235.
Study design
All subjects completed two MRI sessions, 6 weeks apart. During the 7 days prior to the start of PEA treatment and the first MRI session, subjects kept a pain diary in which they recording the intensity of their ongoing pain, three times a day. Subjects rated the intensity of their pain using a 10 cm horizontal VAS, with 0 indicating “no pain” and 10 indicating “the most intense imaginable pain”. These 21 pain intensity scores were then averaged over the 7-day period to create a mean pain intensity score prior to PEA treatment. On the day of their first MRI scanning session, each NP subject outlined the location of their ongoing pain on a standard drawing of the head and completed a McGill Pain Questionnaire. Following their first MRI scanning session, each NP subject began PEA treatment and at 6 weeks, a second MRI session was performed. During the 7 days prior to the second MRI scan, subjects kept another pain diary recording the intensity of their ongoing pain. On the day of the second MRI scan, subjects outlined the location of their ongoing pain on a standard drawing of the head and completed a McGill Pain Questionnaire.
Interventions
NP subjects obtained the PEA from Visionary Health Compounding Chemist, Newcastle, Australia in the form of 300 mg capsules, with dosage individualized between 600 and 900 mg, three times a day. PEA was provided to the compounding chemist in a naive form and was ground with a mortar and pestle until a fluffy consistency was obtained. Analysis of the final PEA revealed that more than 75% was in an ultra-micronized state (<10 microns) with the remaining in a micronized state with particles between 10 and 50 microns in size. The PEA was then placed into gelatine capsules for oral consumption.
MRI acquisition
For each MRI scanning session, NP and control subjects lay supine on the bed of a 3 Tesla MRI scanner (Achieva; Phillips) with their head immobilized in a tight-fitting head coil. With each subject relaxed and at rest, a series of 180 gradient echo echo-planar functional MRI (fMRI) image volumes using blood oxygen level-dependent contrast were collected. Each image volume contained 35 axial slices covering the entire brain (field of view 240×240 mm; matrix size 80×78; slice thickness 4 mm; repetition time 2000 ms; echo time 30 ms; flip angle 90°). Following this, a series of 108 pseudocontinuous arterial spin labeling (pCASL) images containing 50 axial slices covering the entire brain was collected (54 label/control image pairs, field of view 240×240 mm; matrix size 100×100; slice thickness 3 mm; repetition time 5084 ms, echo time 12.7 ms, labelling time 1600 ms, slice time 36.6 ms, labeling efficiency 0.85, blood T1 relaxation time 1667ms). Finally, in each subject, a high-resolution 3D T1-weighted anatomical image set covering the entire brain was also collected (turbo field echo; field of view, 250×250 mm; matrix size 288×288; slice thickness 0.87 mm; repetition time 5600 ms; echo time 2.5 ms; flip angle 8°).
Pain intensity changes
At the completion of the 6-week treatment period, NP subjects’ pain intensity changes were assessed. To explore the association between pain intensity reductions and changes in brain function, NP subjects were divided into those that responded to PEA treatment, ie, responders: >20% reduction in pain intensity, and non-responders: <20% reduction in pain intensity. To determine the effect size of changes in pain intensity in responders and non-responders, Cohen’s d was calculated. This is used to determine whether the effect size is small (d=0.2), medium (d=0.5) or large (d=0.8).22 Changes in pain distributions before and after treatment for each subject in the two groups separately were determined using a paired t-test (p<0.05, Bonferroni corrected).
MRI analysis; infra-slow oscillations
Using SPM12,23 all functional magnetic resonance images were motion corrected, global signal drifts removed24 and co-registered to each individual’s T1-weighted image set. Each subject’s T1-weighted anatomical image was then spatially normalized to a template in Montreal Neurological Institute (MNI) space and the parameters applied to the fMRI image sets. All images were then spatially smoothed (6 mm full-width half-maximum [FWHM] Gaussian filter). In those subjects with pain restricted to the left side of the face (n=12), resting fMRI scans were reflected across the midline so that, in all subjects, the right side was ipsilateral to ongoing pain. To perform a brainstem-specific analysis, the fMRI images were cropped to include only the brainstem and cerebellum and a mask of these regions created automatically using the SUIT toolbox.25 These brainstem/cerebellum masks were then manually adjusted to accurately encompass the brainstem and cerebellum only. Using this mask, fMRI brainstem images were spatially normalized to the Spatial Unbiased Infratentorial Template in MRI space and spatially smoothed using a 3 mm FWHM Gaussian filter.
To determine regional changes in infra-slow oscillation power, we used the SPM toolbox REST26 to calculate the sum of amplitudes of low-frequency fluctuations (ALFF) in the frequency band 0.03–0.06 Hz for the brainstem only and wholebrain fMRI image sets. ALFF values were then divided over the entire frequency range to obtain fractional ALFF (fALFF) value for each voxel. Using the wholebrain and brainstem only images, the effects of PEA treatment on fALFF were determined in all 22 NP subjects using a paired random effects procedure. In addition, the effects of PEA on fALFF values were determined in the responder and non-responder groups separately using paired random effects procedures. Following an initial threshold of p<0.001, small volume corrections were used to account for the effects of multiple comparisons (p<0.05 Bonferonni corrected). For each analysis, the six movement parameters derived from the realignment step and signal intensity derived from a 3 mm sphere located in the fourth ventricle were included as nuisance variables. For each significant cluster, fALFF values were extracted from each subject and the mean (±SEM) plotted.
We also determined the relationships between changes in fALFF over the treatment period and the associated change in pain intensity. In each voxel, the change in fALFF during PEA treatment (pre-post PEA) was calculated and voxel-by-voxel correlations between this fALFF change and the percent change in pain intensity were performed for the responder and non-responder groups separately (movement and CSF signal included as nuisance variables). Following an initial threshold of p<0.001, small volume corrections were used to account for the effects of multiple comparisons (p<0.05 Bonferonni corrected).
In addition, we also performed a functional connectivity analysis using a seed placed into the region of the SpV. This seed was based on the results of the fALFF comparison pre compared to post-PEA treatment in the responder group. Signal intensity changes from the SpV were extracted in each individual subject and correlations between this signal and every voxel in the brainstem only and wholebrain image sets determined (movement parameters and CSF signal included as nuisance variables). Connectivity values between the SpV and the ventroposterior (VP) thalamus (a cluster based on the results from the fALFF analysis) were extracted for each subject and significant differences between pre- and post-PEA treatment and between responder and non-responder groups determined using paired and two group t-tests (p<0.05, Bonferroni corrected).
MRI analysis; resting blood flow
All pCASL image sets were realigned, co-registered to each individual’s T1-weighted image set, the label and control images averaged and a mean cerebral blood flow (CBF) image created using the subtraction method using the ASL toolbox.27 Wholebrain and brainstem-only CBF maps were then normalized into MNI space and smoothed in the same manner as the fMRI images sets. For each significant cluster derived from the fALFF analysis, blood flow values were extracted for each subject and the mean values plotted for each subject before and after treatment and significance differences determined using paired t-tests (p<0.05, Bonferroni corrected).
Data availability statement
Anonymized data can be shared by request from any qualified investigator.
Results
Psychophysics
Prior to PEA treatment, NP subjects had ongoing pain of 4.5±0.4 (mean±SEM) VAS, pain duration of 4.1±1.3 years and 5 NP subjects were taking Serotonin-Noradrenaline Reuptake Inhibitors. PEA treatment resulted in no reported side effects and an overall reduction in ongoing pain intensity of 0.7±0.4 VAS or 21.2±11.4% (Table 1). Overall, PEA treatment reduced pain in 16 (73%) of the 22 NP subjects. Dividing subjects into responders (>20% pain reduction) and non-responders (<20% pain reduction) resulted in 11 subjects as responders and 11 as non-responders (Figure 1A). Responders had an average pain reduction of 2.1±0.3 VAS or 61.9±7.2% and non-responders an overall increase of 0.6±0.5 VAS or 19.6±12.8% (Figure 1B). Cohen’s d showed that as a whole group, the effect size was small (d=0.3). However, in the responders, the effect size is large (d=1.8) and in the non-responders, the effect size is small (d=0.3). Although there was a significant difference in the magnitude of pain intensity reduction between groups, there was no significant change in the area of pain in response to PEA treatment in either group (mean±SEM pixels: responders pre-PEA 1143±494, post-PEA 416±187; non-responders pre-PEA 7935±4325, post-PEA 6890±3841, both p>0.05) (Figure 1C).
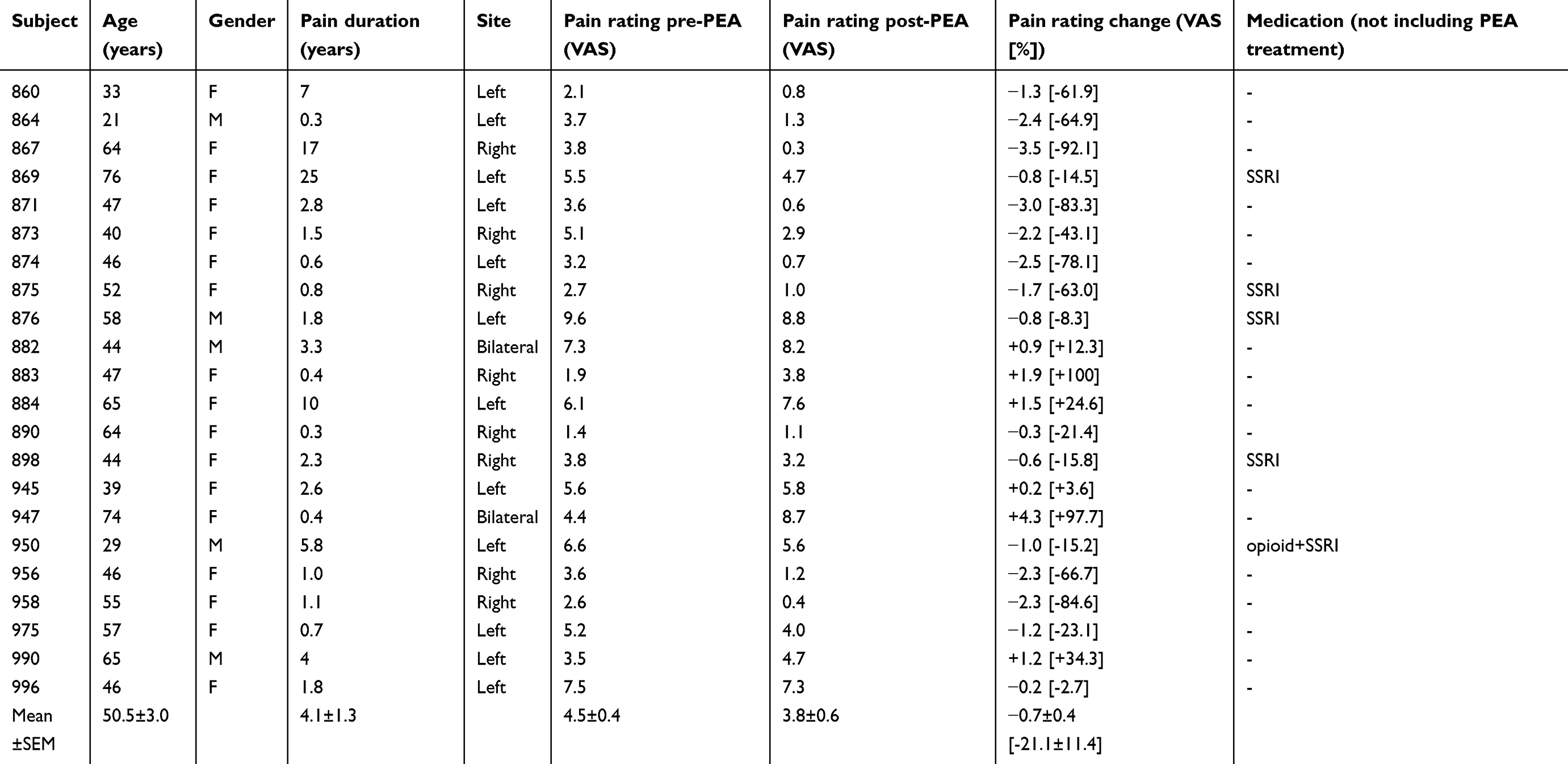 |
Table 1 Subject characteristics |
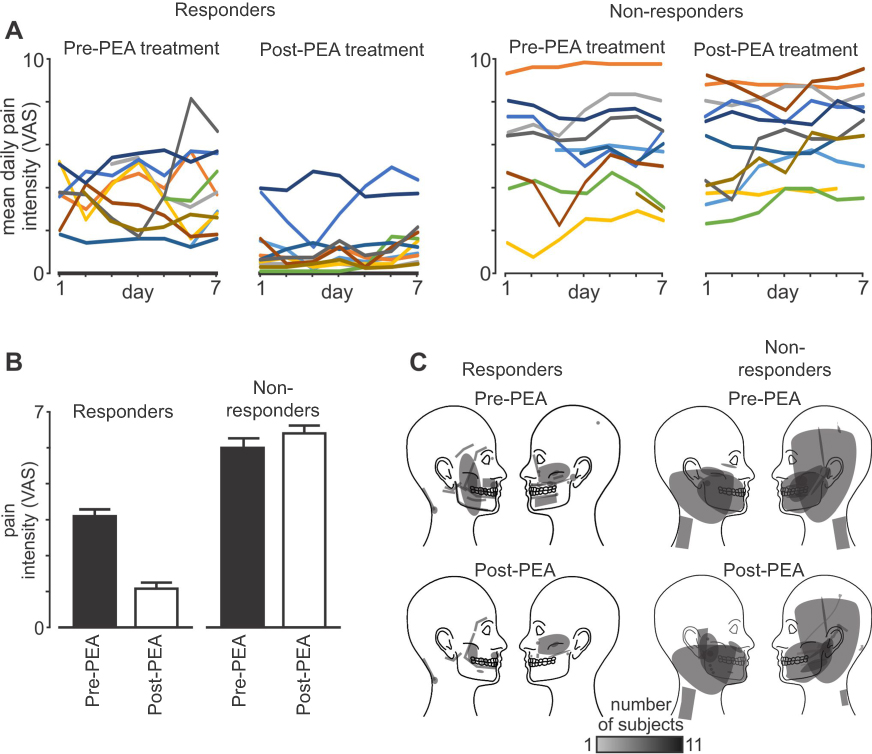 |
Figure 1 Subject psychophysics. (A) Daily pain intensity ratings in each of the 22 patients pre-palmitoylethanolamide (PEA) treatment and post-PEA treatment separated into responders (>20% pain reduction) and non-responders (<20% pain reduction). Pain intensity was rated three times daily on a 10 cm VAS; (B) mean (±SEM) pain intensity ratings pre- and post-PEA in the responder (n=11) and non-responder (n=11) groups; (C) areas of ongoing pain pre- and post-PEA in the responder and non-responder groups. |
In addition to differences in the responsiveness to PEA treatment, responders had a significantly lower pain intensity (3.4±0.3 VAS) compared with non-responders (5.6±0.7 VAS) prior to PEA treatment (p<0.05). There was, however, no significant difference in pain duration (responders 3.0±1.5 years, non-responders 5.2±2.1 years, p>0.05) or the area of perceived pain (responders 1143±494 pixels, non-responders 7935±4325 pixels, p>0.05) between groups before PEA treatment.
Infra-slow oscillatory activity and functional connectivity
PEA treatment had a significant effect on regional resting infra-slow oscillations in both the responder and non-responder groups. Remarkably, in both the responder and non-responder groups, PEA treatment significantly reduced ongoing infra-slow oscillatory activity in the region encompassing the ipsilateral (to side of ongoing pain) SpV, ie, the region where nociceptor afferents from the area of ongoing pain terminate (fALFF 0.03–0.06 Hz: responders pre-PEA 1.03±0.06 post-PEA 0.79±0.05; non-responders pre-PEA 1.14±0.07 post-PEA 0.92±0.06; Figure 2, Table 2). Whilst both responders and non-responders displayed reduced oscillatory activity in SpV, only the responders also displayed significant oscillatory activity reductions in the upstream target of SpV, ie, in the region of the VP thalamus (responders pre-PEA 0.98±0.10 post-PEA 0.84±0.08; non-responders pre-PEA 0.99±0.06 post-PEA 0.98±0.05; Figure 3). Plots of individual subject oscillatory power pre- and post-PEA shown in Figure 3 highlight the consistency of these changes in the SpV in both responder and non-responder groups and in the thalamus in responders. In addition to changes in the ascending trigeminal pathway, the non-responders displayed oscillation decreases in the midbrain including the region of the periaqueductal gray matter (PAG), the anterior and posterior cingulate cortices and increases in the precuneus. Furthermore, in the responders, PEA treatment also resulted in oscillation decreases in the dorsolateral pons, hippocampus, lateral prefrontal cortex and precuneus.
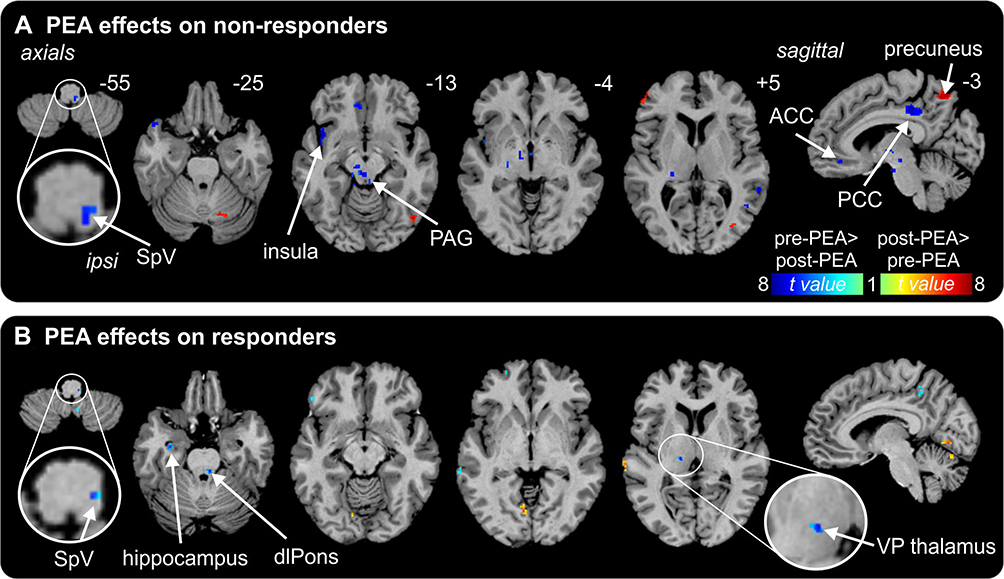 |
Figure 2 Effects of palmitoylethanolamide (PEA) on infra-slow oscillations. (A) Differences in fractional amplitude of low-frequency fluctuations (fALFF) pre-PEA compared with post-PEA treatment on non-responders. Significant increases (hot color scale) and decreases (cool color scale) in fALFF are overlaid onto a series of axial and sagittal T1-weighted anatomical slices. Slice locations in Montreal Neurological Institute space are indicated at the top right of each slice. (B) Differences in fALFF pre-PEA compared with post-PEA treatment in responders. Note that fALFF decreased in the spinal trigeminal nucleus (SpV) in both responders and non-responders and in the ventroposterior (VP) thalamus only in responders. Abbreviations: ACC, anterior cingulate cortex; dlPons, dorsolateral pons; PAG, midbrain periaqueductal gray matter; PCC, posterior cingulate cortex. |
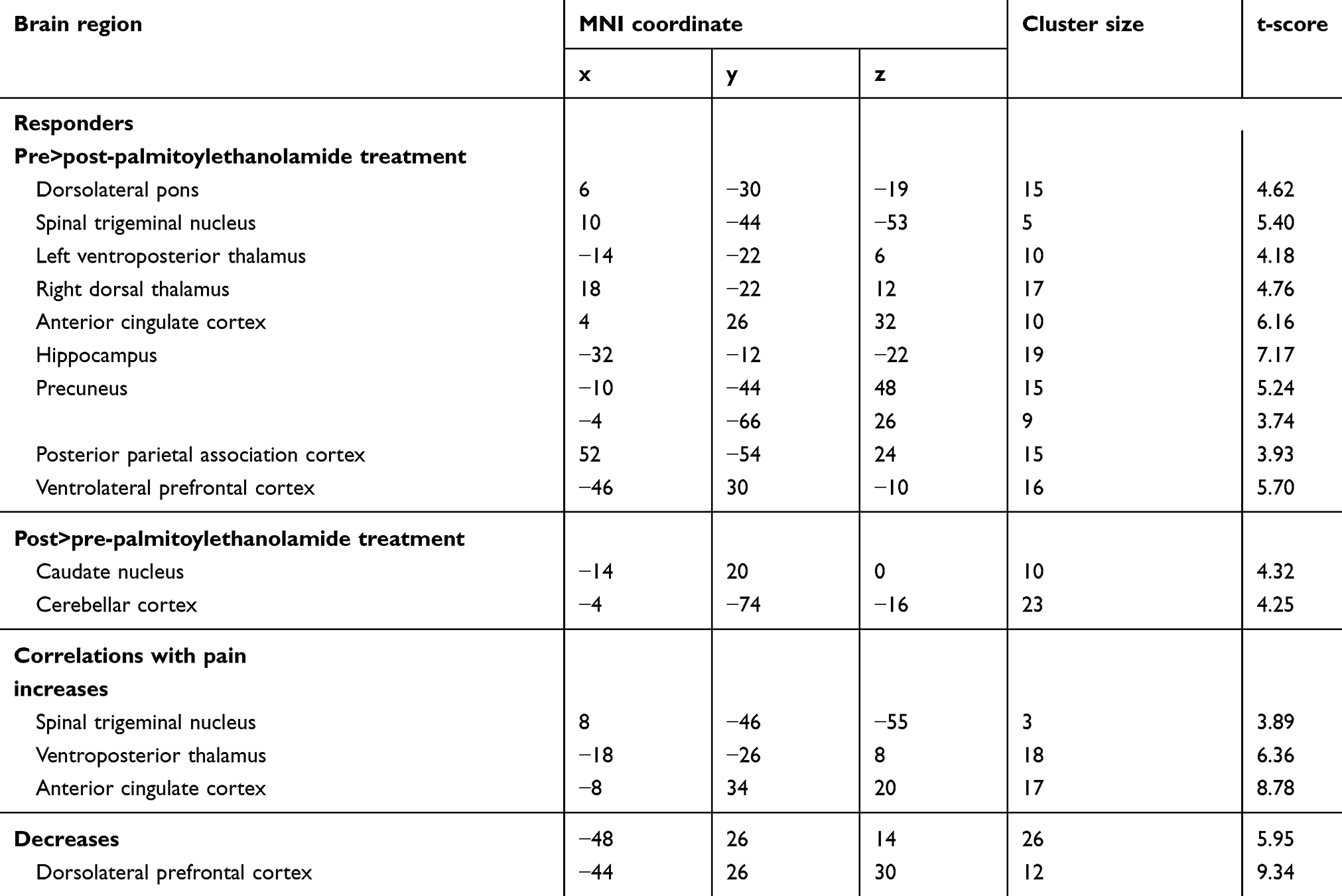 |
Table 2 Montreal Neurological Institute (MNI) coordinates, cluster size and t-score for regions of significant differences in fractional amplitude of low-frequency fluctuations between pre- and post-palmitoylethanolamide treatment |
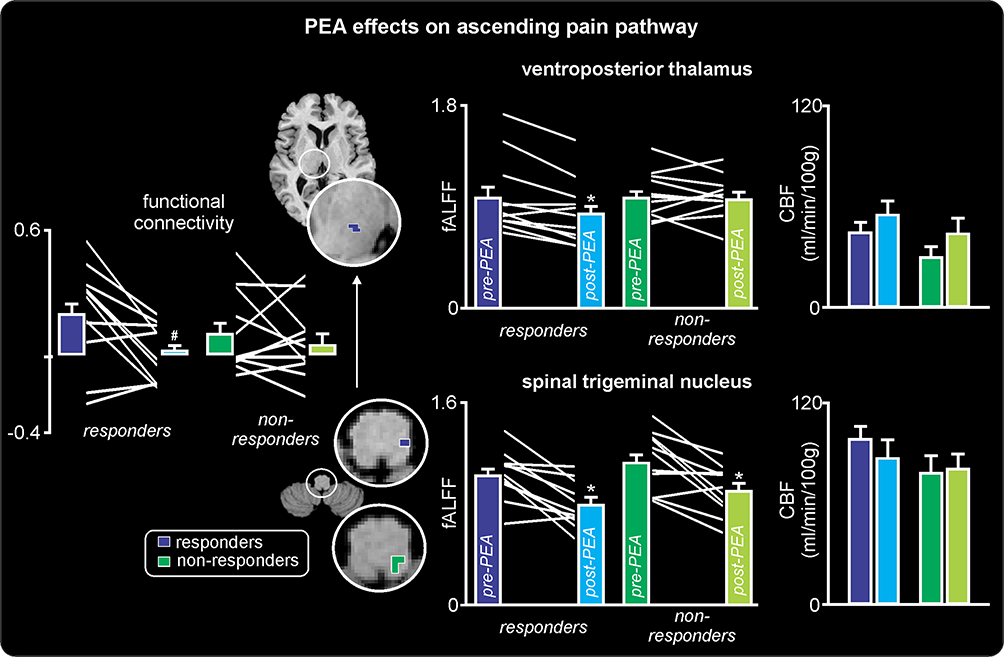 |
Figure 3 Effects of palmitoylethanolamide (PEA) on ascending pain pathway. To the right are plots of mean (±SEM) fractional amplitude of low-frequency fluctuations (fALFF) and cerebral blood flow (CBF) in responders (blue) and non-responders (green), pre- and post-PEA treatment. Individual subject values pre- and post-PEA are indicated in the fALFF plots by the white lines between the colored bars indicating the mean (±SEM). Note that both the responders and non-responders have a significant reduction in infra-slow oscillations in the spinal trigeminal nucleus, but only the responders have a decrease in the ventroposterior thalamus. In contrast, there are no changes in CBF. The regions in which fALFF decreases as a consequence of PEA treatment are color coded blue (responders) and green (non-responders) and overlaid onto axial T1-weighted anatomical slices. To the left of these overlays are plots of mean (±SEM) resting functional connectivity between the spinal trigeminal nucleus and ventroposterior thalamus. Individual subject values pre- and post-PEA are indicated in the functional connectivity plots by the white lines between the colored bars. Note that functional connectivity decreases significantly following PEA treatment in the responders only. *p<0.05 derived from voxel-by-voxel analysis; #p<0.05 derived from two-sample t-test. |
The extraction of fALFF values from the SpV and VP thalamus confirmed that PEA treatment resulted in significant reductions in infra-slow oscillations in the SpV in both responders and non-responders and reductions in the VP thalamus in the responder group only (Figure 3). Furthermore, only responders displayed a significant change in resting connectivity between the SpV and VP thalamus in response to PEA treatment. That is, prior to PEA treatment there was a significant signal covariation between the SpV and VP thalamus in responders which was subsequently completely eliminated by PEA treatment (SpV-VP thalamus connectivity: responders pre-PEA 0.18±0.07 post- PEA 0.01±0.04, p<0.05). In contrast, PEA treatment had no effect on SpV-VP thalamus connectivity in the non-responders (pre-PEA 0.09±0.06 post-PEA 0.04±0.05, p>0.05).
In addition, the change in infra-slow oscillations within the ascending trigeminal pathway was not associated with changes in overall activity levels as evidenced by no change in resting blood flow in either the SpV (mL/min/100 g: responders pre-PEA 98.7±7.9 post-PEA 87.4±10.7, non-responders pre-PEA 79.9±10.5 post-PEA 81.8±9.4, p>0.05) or the VP thalamus (responders pre-PEA 43.9±6.1 post-PEA 54.8±8.5, non-responders pre-PEA 28.9±5.4 post-PEA 45.8±8.6, p<0.05).
Correlations between changes in pain intensity and changes in infra-slow oscillations revealed significant clusters in the SpV and VP thalamus overlapping those significant clusters revealed in the pre- versus post-fALFF analysis (Figure 4). In responders, the magnitude of the decrease in infra-slow oscillations was significantly positively correlated with a decrease in pain intensity during PEA treatment in both the region of the SpV (r=0.83, p<0.05) and VP thalamus (r=0.93, p<0.05). In contrast, no significant correlations occurred in non-responders in the SpV (r=0.04, p>0.05) or VP thalamus (r=0.28, p>0.05).
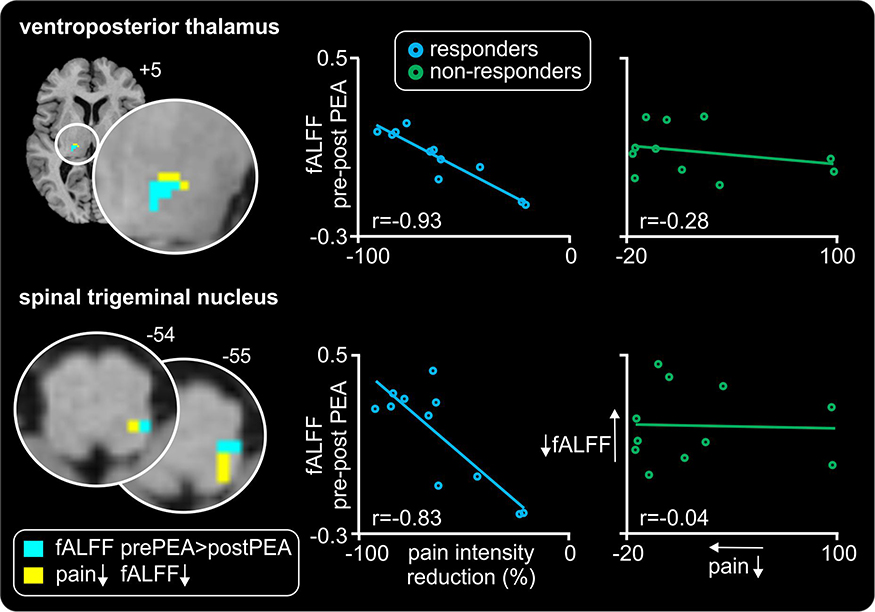 |
Figure 4 Relationships between changes in pain intensity and infra-slow oscillations as a result of palmitoylethanolamide (PEA) treatment. Significant positive linear relationships between changes in ongoing pain and fractional amplitude of low-frequency fluctuations (fALFF) in the thalamus and medulla are shown overlaid onto axial T1-weighted anatomical slices for the responder group only (yellow shading). Note no such relationship occurred in the non-responders. Slice locations in Montreal Neurological Institute space are indicated at the top right of each slice. The location of significant decreases in infra-slow oscillations (ISO) pre- versus post-PEA are also shown (light blue shading); note the overlap within the spinal trigeminal nucleus and ventroposterior thalamus. To the right are plots of change in fALFF versus percent change in pain for the SpV and VP thalamus cluster in responders and non-responders pre- versus post-PEA treatment. In responders, the greater the reduction in fALFF the greater the reduction in pain intensity. |
Discussion
This investigation describes the effects of PEA on ongoing pain and brain function in patients with chronic orofacial NP. PEA reduced pain intensity in almost 75% of patients with 50% of all patients exhibiting an overall decrease of over 20% with no recorded side effects. Additionally, PEA altered regional brain function by reducing infra-slow oscillatory activity in the ascending trigeminal pain pathway. More specifically, our results show that altered activity patterns and connectivity within the trigeminal pathway are associated with an individual’s pain relief responsiveness to PEA treatment. Whilst three previous investigations have reported NP relief as a result of PEA treatment,28–30 this is the first to show that the effectiveness of PEA is associated with altered activity patterns in the ascending pain pathway. Critically, like our study, no clinical trial using PEA for pain or any other condition has reported significant side effects.
We have previously shown that chronic orofacial NP is associated with an increase in resting infra-slow oscillations along the ascending trigeminal pain pathway, including the SpV, VP thalamus, thalamic reticular nucleus and primary somatosensory cortex.31,32 Given that there is preclinical and human postmortem evidence of chronic astrogliosis in the SpV/dorsal horn in individuals with NP and that the oscillations were at the same approximate frequencies in which astrocyte calcium and gliotransmission release have been recorded, we speculate that these oscillatory activity increases were the result of chronic astrogliosis. Astrocyte calcium can propagate to neighboring astrocytes either directly through connecting channels or indirectly through the release of gliotransmitters to produce synchronous oscillatory gliotransmitter release.33,34 This oscillatory astrocyte function can elicit long-lasting NMDA-mediated currents in surrounding neurons4 and there is recent evidence that astrocytes can elicit long-lasting increases in neural firing.35 We have proposed that following nerve injury, excess activity within primary afferent neurons results in neural death and elicits astrocyte activation at the primary afferent synapse. This results in more regular and increased oscillatory gliotransmitter release and increased infra-slow neural oscillations which are transferred along the ascending pain pathway. The recurrent nature of thalamocortical circuits results in self-sustaining thalamocortical dysrhythmia, paroxysmal events and the constant perception of pain.1,2,36 Interestingly, recent preclinical investigations have shown a gender divide regarding the main cells involved in chronic NP. In males, microglia play a large role in the initiation and maintenance of neuropathic37,38 pain and in our investigation, only one in the four males enrolled in the study responded to the treatment. Furthermore, in the previous investigation on infra-slow oscillatory activity in patients with orofacial NPn, 82% of the subjects were females. Hence, it is more likely that astrocytes might be the driving force of infra-slow oscillatory activity in the female population than it is in the male.
Consistent with this hypothesis, the present investigation found that PEA administration resulted in a reduction in both pain intensity and oscillatory activity within the ascending trigeminal pain pathway. PEA is abundant in the central nervous system and there is preclinical evidence that NP is associated with reductions in PEA levels in the ipsilateral spinal cord and that these levels increase following pain relief.39 PEA is produced by glial cells40 and has been studied primarily for its anti-inflammatory and neuroprotective effects.16,20,41 Whilst PEA can exert its effects via multiple mechanisms, it is thought that the main target of PEA is the peroxisome proliferator-activated receptor alpha (PPAR-α).12,13,15,16,42 PPAR-α is an isotope of the PPARs that can affect the transcription of a number of genes and is expressed by astrocytes and it is possible that astrocytes play a role in the anti-nociceptive effects of PEA through a PPAR-α pathway.16 Critically, it has been shown to blunt astrocyte activation in vitro and improve neuronal survival through selective PPAR-α activation.43 Indeed, PEA fails to exert its anti-nociceptive effects in PPAR-α knockout mice.12–14,42,44
We found that in all patients, oral administration of PEA resulted in significant reductions in infra-slow oscillations specifically at frequencies similar to those of astrocyte calcium waves (0.03–0.06 Hz) in the ipsilateral SpV. However, only in individuals in which PEA evoked a significant reduction in pain did a similar reduction in neural oscillations also occur in the VP thalamus. Furthermore, PEA treatment eliminated the resting neural covariation between SpV and VP thalamus only in responders. These results suggest that whilst PEA blunts the increase in resting neural oscillatory activity at the region of the primary afferent synapse in all patients, only when this reduction is then transferred to higher brain centers such as the thalamus does an analgesia result. Indeed, only in responders was the reduction in neural oscillation power significantly correlated to the reduction in pain, ie, the greater the reduction in oscillations the greater the reduction in ongoing pain. This is entirely consistent with the results from an experimental animal model of NP in which infra-slow oscillatory activity in the VP thalamus was eliminated by severing the connection between the primary afferent synapse and the thalamus.45 Furthermore, PEA decreases infra-slow oscillatory activity in the PAG which makes up part of the descending pain modulation pathway. In nerve injury models, there is increase in astrocyte activity in the PAG and other regions of the descending pain modulation pathway such as the rostroventral medulla.46,47 However, in the results of this investigation, the non-responders had decreases in infra-slow oscillatory activity in the PAG only, which may not be sufficient to modulate the descending pain pathway enough to produce pain intensity decreases.
Whilst we suggest that in individuals with chronic NP, PEA produces pain relief via its effects on astrocyte function, PEA may also exert effects via a number of other mechanisms. Within the central nervous system, PPAR-α receptors act via two main mechanisms; a rapid effect which involves activation of calcium-dependent potassium channels which has a silencing effect on neuronal firing15 and a slower effect which is comprised of neurosteroid de novo synthesis which increases allopregnanolone production.48,49 Allopregnanolone is a positive allosteric modulator of GABA action and slows the rate of recovery of GABA receptors.50,51 In addition to a potential action on neuronal GABA receptors, PEA may influence GABA receptors on astrocytes, which when activated can increase intracellular calcium levels.52 Interestingly, astrocyte calcium wave propagation is dependent upon mechanisms which include the removal of excess calcium by mitochondria to prevent IP3 receptor desensitization.53,54 It is possible that PEA increases calcium concentrations thereby reducing the propagation of astrocyte calcium waves and associated gliotransmission. Interestingly, all of the five patients in this investigation that were also taking selective serotonin reuptake inhibitors (SSRIs) reported pain relief, although four of these subjects reported reductions that were small and were categorized as non-responders. Whilst the precise mechanism of SSRIs is not known, it has been shown that they can increase the expression of GABAB receptors and alter astrocyte function directly, evoking astrocyte calcium transients in an often oscillatory manner, even when neural activity is inhibited.55,56 Given this, it is possible that SSRIs partially counteract the mechanism of PEA and thus reduce its overall analgesic effectiveness.
It is also a possibility that PEA may act on other targets in the nervous system such as cannabinoids in neurons and astrocytes,57,58 PPAR-γ59 or mast cells60 to reduce pain. Whilst these and other targets are possible, we suggest that PEA most likely targets PPAR-α in individuals with NP. As mentioned above, PEA fails to exert its anti-nociceptive effects in PPAR-α knockout mice. Furthermore, a recent study in the spared nerve injury model of NP found that whilst PEA reversed the development of thermal hyperalgesia, mechanical allodynia and cognitive impairment in wild-type mice, it had no effect in PPAR-α knockout mice.61 Similarly, in the chronic constriction model of NP, PEA treatment prevented the development of hyperalgesia and reduced the inflammatory changes in the damaged nerve, changes that were not seen following PEA treatment in PPAR-α knockout mice.62 It is possible that the effects of PEA on PPAR-α may also involve more indirect targets since it was shown that PEA increases cannabinoid receptor type 2 mRNA and protein expression via PPAR-α activation in cultured rat microglia and human macrophages.63 Whilst a role for PEA effects via microglial actions may exist, it is likely to occur in only the onset phase of NP since there is evidence that in chronic NP, microglial activation subsides following an initial increase whereas astrocyte activation persists.64,65
Interestingly, the midbrain periaqueductal gray matter and the anterior cingulate cortex have been shown to modulate cannabinoid-induced analgesia.66,67 The non-responders showed reductions in infra-slow oscillations in the PAG and the anterior cingulate cortex and no such change was evident in the responders, which may be contributing to the lack of change in pain intensity in the non-responders. Furthermore, PEA has been shown to reduce pain and negative affect through the endocannabinoid and glutamatergic system in a number of pain states.68–70 It is possible that PEA acts on multiple receptors in a number of sites to attenuate inflammation and pain, and reduce negative affect associated with nerve damage and chronic pain.60 In the non-responders, although there was no significant decrease in pain intensity, there was a significant decrease in infra-slow oscillatory activity in the anterior cingulate and the insular cortex. These regions are involved in the affective state71 and while the affective measures were not collected, it is possible that PEA modulated negative affect states in the non-responders without affecting the pain scores.
A limitation of this study is that this was an open-label clinical trial and we did not control for the potential effects of placebo. However, it is unlikely that the decrease in pain intensity in the responders was solely due to the placebo effect. In fact, a Cochrane meta-analysis showed that over a number of investigations, the effect size of a placebo response, measured as Cohen’s d, is 0.27.72 In this investigation, the effect size as a whole group is 0.3, which may indeed be due to a placebo effect. However, in the responders only, the effect size is 1.8, suggesting that the response in the responders is not a placebo effect. Nonetheless, the decreases in pain intensity correlate with a change in neural activity in the responders as a result of PEA. A future larger clinical trial investigating the effects of PEA on NP and the associated changes in brain function is needed.
Acknowledgment
The study is funded by Australia National Health and Medical Research Council (G160279 and G182968) and NGW Macintosh Memorial Fund.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Sarnthein J, Stern J, Aufenberg C, Rousson V, Jeanmonod D. Increased EEG power and slowed dominant frequency in patients with neurogenic pain. Brain. 2006;129(Pt 1):55–64. doi:10.1093/brain/awh631
2. Walton KD, Llinas RR. Frontiers in Neuroscience. Central pain as a thalamocortical dysrhythmia: a thalamic efference disconnection?. In: Kruger L, AR L, editors. Translational Pain Research: From Mouse to Man. Boca Raton (FL): CRC Press/Taylor & Francis Llc.; 2010:301-314.
3. Di Pietro F, Macey PM, Rae CD, et al. The relationship between thalamic GABA content and resting cortical rhythm in neuropathic pain. Hum Brain Mapp. 2018. accepted. doi:10.1002/hbm.23973
4. Crunelli V, Blethyn KL, Cope DW, et al. Novel neuronal and astrocytic mechanisms in thalamocortical loop dynamics. Philos Trans R Soc Lond B Biol Sci. 2002;357(1428):1675–1693. doi:10.1098/rstb.2002.1155
5. Garrison CJ, Dougherty PM, Kajander KC, Carlton SM. Staining of glial fibrillary acidic protein (GFAP) in lumbar spinal cord increases following a sciatic nerve constriction injury. Brain Res. 1991;565(1):1–7. doi:10.1016/0006-8993(91)91729-k
6. Okada-Ogawa A, Suzuki I, Sessle BJ, et al. Astroglia in medullary dorsal horn (trigeminal spinal subnucleus caudalis) are involved in trigeminal neuropathic pain mechanisms. J NeuroSci. 2009;29(36):11161–11171. doi:10.1523/JNEUROSCI.3365-09.2009
7. Colburn RW, Deleo JA, Rickman AJ, Yeager MP, Kwon P, Hickey WF. Dissociation of microglial activation and neuropathic pain behaviors following peripheral nerve injury in the rat. J Neuroimmunol. 1997;79(2):163–175.
8. Aldskogius H, Kozlova EN. Central neuron–glial and glial–glial interactions following axon injury. Prog Neurobiol. 1998;55(1):1–26.
9. Miyoshi K, Obata K, Kondo T, Okamura H, Noguchi K. Interleukin-18-mediated microglia/astrocyte interaction in the spinal cord enhances neuropathic pain processing after nerve injury. J NeuroSci. 2008;28(48):12775–12787. doi:10.1523/JNEUROSCI.3512-08.2008
10. Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114(1–2):149–159. doi:10.1016/j.pain.2004.12.022
11. Zhuang ZY, Wen YR, Zhang DR, et al. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci. 2006;26(13):3551–3560. doi:10.1523/JNEUROSCI.5290-05.2006
12. D’Agostino G, La Rana G, Russo R, et al. Central administration of palmitoylethanolamide reduces hyperalgesia in mice via inhibition of NF-kappaB nuclear signalling in dorsal root ganglia. Eur J Pharmacol. 2009;613(1–3):54–59. doi:10.1016/j.ejphar.2009.04.022
13. D’Agostino G, La Rana G, Russo R, et al. Acute intracerebroventricular administration of palmitoylethanolamide, an endogenous peroxisome proliferator-activated receptor-alpha agonist, modulates carrageenan-induced paw edema in mice. J Pharmacol Exp Ther. 2007;322(3):1137–1143. doi:10.1124/jpet.107.123265
14. Lo Verme J, Fu J, Astarita G, et al. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol. 2005;67(1):15–19. doi:10.1124/mol.104.006353
15. LoVerme J, Russo R, La Rana G, et al. Rapid broad-spectrum analgesia through activation of peroxisome proliferator-activated receptor-alpha. J Pharmacol Exp Ther. 2006;319(3):1051–1061. doi:10.1124/jpet.106.111385
16. Raso GM, Esposito E, Vitiello S, et al. Palmitoylethanolamide stimulation induces allopregnanolone synthesis in c6 cells and primary astrocytes: involvement of peroxisome-proliferator activated receptor-α. J Neuroendocrinol. 2011;23(7):591–600. doi:10.1111/j.1365-2826.2011.02152.x
17. Moreno S, Farioli-Vecchioli S, Cerù MP. Immunolocalization of peroxisome proliferator-activated receptors and retinoid x receptors in the adult rat CNS. Neuroscience. 2004;123(1):131–145.
18. Bento-Abreu A, Tabernero A, Medina JM. Peroxisome proliferator-activated receptor-alpha is required for the neurotrophic effect of oleic acid in neurons. J Neurochem. 2007;103(3):871–881. doi:10.1111/j.1471-4159.2007.04807.x
19. Habib AM, Okorokov AL, Hill MN, et al. Microdeletion in a FAAH pseudogene identified in a patient with high anandamide concentrations and pain insensitivity. Br J Anaesth. 2019;123(2):e249-e253.
20. Hesselink JMK, Hekker TAM. Therapeutic utility of palmitoylethanolamide in the treatment of neuropathic pain associated with various pathological conditions: a case series. J Pain Res. 2012;5:437–442. doi:10.2147/JPR.S32143
21. Nurmikko TJ, Eldridge P. Trigeminal neuralgia—pathophysiology, diagnosis and current treatment. Br J Anaesth. 2001;87(1):117–132. doi:10.1093/bja/87.1.117
22. Cohen J. Statistical Power Analysis for the Behavioral Sciences. Hillsdale (NJ): L. Erlbaum Associates; 1988.
23. Friston KJ, Holmes AP, Worsley KJ, Poline JP, Frith CD, Frackowiak RS. Statistical parametric maps in functional imaging: a general linear approach. Hum Brain Mapp. 1994;2(4):189–210. doi:10.1002/hbm.460020402
24. Macey PM, Macey KE, Kumar R, Harper RM. A method for removal of global effects from fMRI time series. Neuroimage. 2004;22(1):360–366. doi:10.1016/j.neuroimage.2003.12.042
25. Diedrichsen J. A spatially unbiased atlas template of the human cerebellum. Neuroimage. 2006;33(1):127–138. doi:10.1016/j.neuroimage.2006.05.056
26. Song XW, Dong ZY, Long XY, et al. REST: a toolkit for resting-state functional magnetic resonance imaging data processing. PLoS One. 2011;6(9):e25031. doi:10.1371/journal.pone.0025031
27. Wang Z, Aguirre GK, Rao H, et al. Empirical optimization of ASL data analysis using an ASL data processing toolbox: aSLtbx. Magn Reson Imaging. 2008;26(2):261–269. doi:10.1016/j.mri.2007.07.003
28. Hesselink JMK. New targets in pain, non-neuronal cells, and the role of palmitoylethanolamide. Open Pain J. 2012;5:12–23. doi:10.2174/1876386301205010012
29. P D. Associazione tra pregabalin e palmitoiletanolamide per il trattamento del dolore neuropatico. Pathos. 2010;7(4):9–14.
30. Gabrielsson L, Mattsson S, Fowler CJ. Palmitoylethanolamide for the treatment of pain: pharmacokinetics, safety and efficacy. Br J Clin Pharmacol. 2016;82(4):932–942. doi:10.1111/bcp.13020
31. Alshelh Z, Di Pietro F, Youssef AM, et al. Chronic neuropathic pain: it’s about the rhythm. J NeuroSci. 2016;36(3):1008–1018. doi:10.1523/JNEUROSCI.2768-15.2016
32. Alshelh Z, Di Pietro F, Mills EP, et al. Altered regional brain T2 relaxation times in individuals with chronic orofacial neuropathic pain. NeuroImage. 2018;19:167-173.
33. Cornell-Bell AH, Finkbeiner SM, Cooper MS, Smith SJ. Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science. 1990;247(4941):470–473. doi:10.1126/science.1967852
34. Scemes E, Giaume C. Astrocyte calcium waves: what they are and what they do. Glia. 2006;54(7):716–725. doi:10.1002/glia.20374
35. Deemyad T, Lüthi J, Spruston N. Astrocytes integrate and drive neural activity. Nature Communications. 2018;9.
36. Hughes SW, Lorincz ML, Parri HR, Crunelli V. Infraslow (<0.1 Hz) oscillations in thalamic relay nuclei basic mechanisms and significance to health and disease states. Prog Brain Res. 2011;193:145–162. doi:10.1016/B978-0-444-53839-0.00010-7
37. Sorge RE, Mapplebeck JC, Rosen S, et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci. 2015;18(8):1081–1083. doi:10.1038/nn.4053
38. Coraggio V, Guida F, Boccella S, et al. Neuroimmune-Driven Neuropathic Pain Establishment: A Focus on Gender Differences. Int J Mol Sci. 2018;19(1):281. doi:10.3390/ijms19010281
39. Guasti L, Richardson D, Jhaveri M, et al. Minocycline treatment inhibits microglial activation and alters spinal levels of endocannabinoids in a rat model of neuropathic pain. Mol Pain. 2009;5:35.
40. Walter L, Franklin A, Witting A, Moller T, Stella N. Astrocytes in culture produce anandamide and other acylethanolamides. J Biol Chem. 2002;277(23):20869–20876. doi:10.1074/jbc.M110813200
41. Keppel Hesselink M. J. New targets in pain, non-neuronal cells, and the role of palmitoylethanolamide. Open Pain J. 2012;5:1. doi:10.2174/1876386301205010012
42. LoVerme J, La Rana G, Russo R, Calignano A, Piomelli D. The search for the palmitoylethanolamide receptor. Life Sci. 2005;77(14):1685–1698. doi:10.1016/j.lfs.2005.05.012
43. Scuderi C, Valenza M, Stecca C, Esposito G, Carratu MR, Steardo L. Palmitoylethanolamide exerts neuroprotective effects in mixed neuroglial cultures and organotypic hippocampal slices via peroxisome proliferator-activated receptor-alpha. J Neuroinflammation. 2012;9:49. doi:10.1186/1742-2094-9-49
44. Haller VL, Cichewicz DL, Welch SP. Non-cannabinoid CB1, non-cannabinoid CB2 antinociceptive effects of several novel compounds in the PPQ stretch test in mice. Eur J Pharmacol. 2006;546(1–3):60–68. doi:10.1016/j.ejphar.2006.07.024
45. Iwata M, LeBlanc BW, Kadasi LM, Zerah ML, Cosgrove RG, Saab CY. High-frequency stimulation in the ventral posterolateral thalamus reverses electrophysiologic changes and hyperalgesia in a rat model of peripheral neuropathic pain. Pain. 2011;152(11):2505–2513. doi:10.1016/j.pain.2011.07.011
46. Dubový P, Klusáková I, Hradilová-Svíženská I, Joukal M, Boadas-Vaello P. Activation of astrocytes and microglial cells and CCL2/CCR2 upregulation in the dorsolateral and ventrolateral nuclei of periaqueductal gray and rostral ventromedial medulla following different types of sciatic nerve injury. Front Cell Neurosci. 2018;12:40. doi:10.3389/fncel.2018.00040
47. Wei F, Guo W, Zou S, Ren K, Dubner R. Supraspinal glial–neuronal interactions contribute to descending pain facilitation. J NeuroSci. 2008;28(42):10482–10495. doi:10.1523/JNEUROSCI.3593-08.2008
48. Sasso O, Russo R, Vitiello S, et al. Implication of allopregnanolone in the antinociceptive effect of N-palmitoylethanolamide in acute or persistent pain. Pain. 2012;153(1):33–41. doi:10.1016/j.pain.2011.08.010
49. Sasso O, La Rana G, Vitiello S, et al. Palmitoylethanolamide modulates pentobarbital-evoked hypnotic effect in mice: involvement of allopregnanolone biosynthesis. European Neuropsychopharmacol. 2010;20(3):195–206. doi:10.1016/j.euroneuro.2009.09.003
50. Neigh GN, Merrill L. Chapter 3 - sex differences in neuroanatomy and neurophysiology: implications for brain function, behavior, and neurological disease. In: Sex Differences in Physiology. Boston: Academic Press; 2016:17–44.
51. Tsutsui K, Haraguchi S. Subchapter 96C - Allopregnanolone. In: Handbook of Hormones. San Diego: Academic Press; 2016:
52. Meier Silke D, Kafitz Karl W, Rose Christine R. Developmental profile and mechanisms of GABA‐induced calcium signaling in hippocampal astrocytes. Glia. 2008;56(10):1127–1137. doi:10.1002/glia.20684
53. Simpson PB, Mehotra S, Langley D, Sheppard CA, Russell JT. Specialized distributions of mitochondria and endoplasmic reticulum proteins define Ca2+ wave amplification sites in cultured astrocytes. J Neurosci Res. 1998;52(6):672–683. doi:10.1002/(SICI)1097-4547(19980615)52:6<672::AID-JNR6>3.0.CO;2-5
54. Boitier E, Rea R, Duchen MR. Mitochondria exert a negative feedback on the propagation of intracellular Ca2+ waves in rat cortical astrocytes. J Cell Biol. 1999;145(4):795–808. doi:10.1083/jcb.145.4.795
55. Schipke CG, Heuser I, Peters O. Antidepressants act on glial cells: sSRIs and serotonin elicit astrocyte calcium signaling in the mouse prefrontal cortex. J Psychiatr Res. 2011;45(2):242–248. doi:10.1016/j.jpsychires.2010.06.005
56. Sands SA, McCarson KE, Enna SJ. Relationship between the antinociceptive response to desipramine and changes in GABAB receptor function and subunit expression in the dorsal horn of the rat spinal cord. Biochem Pharmacol. 2004;67(4):743–749. doi:10.1016/j.bcp.2003.10.008
57. Re G, Barbero R, Miolo A, Di Marzo V. Palmitoylethanolamide, endocannabinoids and related cannabimimetic compounds in protection against tissue inflammation and pain: potential use in companion animals. Vet J. 2007;173(1):21–30. doi:10.1016/j.tvjl.2005.10.003
58. Navarrete M, Díez A, Araque A. Astrocytes in endocannabinoid signalling. Philos Trans Royal Soc B. 2014;369(1654):20130599. doi:10.1098/rstb.2013.0599
59. Paterniti I, Impellizzeri D, Crupi R, et al. Molecular evidence for the involvement of PPAR-delta and PPAR-gamma in anti-inflammatory and neuroprotective activities of palmitoylethanolamide after spinal cord trauma. J Neuroinflammation. 2013;10:20. doi:10.1186/1742-2094-10-151
60. Costa B, Comelli F, Bettoni I, Colleoni M, Giagnoni G. The endogenous fatty acid amide, palmitoylethanolamide, has anti-allodynic and anti-hyperalgesic effects in a murine model of neuropathic pain: involvement of CB1, TRPV1 and PPARγ receptors and neurotrophic factors. PAIN. 2008;139(3):541–550. doi:10.1016/j.pain.2008.06.003
61. Boccella S, Cristiano C, Romano R, et al. Ultra-micronized palmitoylethanolamide rescues the cognitive decline-associated loss of neural plasticity in the neuropathic mouse entorhinal cortex-dentate gyrus pathway. Neurobiol Dis. 2019;121:106–119. doi:10.1016/j.nbd.2018.09.023
62. Di Cesare Mannelli L, D’Agostino G, Pacini A, et al. Palmitoylethanolamide is a disease-modifying agent in peripheral neuropathy: pain relief and neuroprotection share a PPAR-alpha-mediated mechanism. Mediators Inflamm. 2013;2013(328797). doi:10.1155/2013/328797
63. Guida F, Luongo L, Boccella S, et al. Palmitoylethanolamide induces microglia changes associated with increased migration and phagocytic activity: involvement of the CB2 receptor. Sci Rep. 2017;7(1):375. doi:10.1038/s41598-017-00342-1
64. Shi Y, Gelman BB, Lisinicchia JG, Tang SJ. Chronic-pain-associated astrocytic reaction in the spinal cord dorsal horn of human immunodeficiency virus-infected patients. J Neurosci. 2012;32(32):10833–10840. doi:10.1523/JNEUROSCI.5628-11.2012
65. Tanga FY, Raghavendra V, DeLeo JA. Quantitative real-time RT-PCR assessment of spinal microglial and astrocytic activation markers in a rat model of neuropathic pain. Neurochem Int. 2004;45(2–3):397–407. doi:10.1016/j.neuint.2003.06.002
66. Calejesan AA, Kim SJ, Zhuo M. Descending facilitatory modulation of a behavioral nociceptive response by stimulation in the adult rat anterior cingulate cortex. Eur J Pain. 2000;4(1):83–96. doi:10.1053/eujp.1999.0158
67. Yamashita H, Zeredo JL, Kaida K, Kimoto M, Asahina I, Toda K. Stress-induced modulation of nociceptive responses in the rat anterior cingulate cortex. J Integr Neurosci. 2013;12(02):235–246. doi:10.1142/S0219635213500143
68. Guida F, Boccella S, Belardo C, et al. Altered gut microbiota and endocannabinoid system tone in vitamin D deficiency-mediated chronic pain. Brain Behav Immun. 2019. doi:10.1016/j.bbi.2019.04.006
69. Guida F, Luongo L, Marmo F, et al. Palmitoylethanolamide reduces pain-related behaviors and restores glutamatergic synapses homeostasis in the medial prefrontal cortex of neuropathic mice. Mol Brain. 2015;8(1). doi:10.1186/s13041-015-0139-5
70. Luongo L, Guida F, Boccella S, et al. Palmitoylethanolamide reduces formalin-induced neuropathic-like behaviour through spinal glial/microglial phenotypical changes in mice. CNS Neurol Disord Drug Targets. 2013;12(1):45–54. doi:10.2174/1871527311312010009
71. Baliki MN, Chialvo DR, Geha PY, et al. Chronic pain and the emotional brain: specific brain activity associated with spontaneous fluctuations of intensity of chronic back pain. J NeuroSci. 2006;26(47):12165–12173. doi:10.1523/JNEUROSCI.3576-06.2006
72. Price DD, Hirsh A, Robinson ME, et al. 5.64 - psychological modulation of pain. In: Masland RH, Albright TD, Albright TD, editors. The Senses: A Comprehensive Reference. New York (NY): Academic Press; 2008:975–1002.
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.