")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Effects of PEGylated lipid nanoparticles on the oral absorption of one BCS II drug: a mechanistic investigation
Authors Zhang X, Chen G, Zhang T, Ma Z, Wu B, Wang H
Received 27 August 2014
Accepted for publication 11 October 2014
Published 26 November 2014 Volume 2014:9(1) Pages 5503—5514
DOI https://doi.org/10.2147/IJN.S73340
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Thomas Webster
Xingwang Zhang,* Guijiang Chen,* Tianpeng Zhang, Zhiguo Ma, Baojian Wu
Division of Pharmaceutics, College of Pharmacy, Jinan University, Guangzhou, People’s Republic of China
*These authors contributed equally to this work
Abstract: Lipid nanocarriers are becoming a versatile platform for oral delivery of lipophilic drugs. In this article, we aimed to explore the gastrointestinal behaviors of lipid nanoparticles and the effect of PEGylation on oral absorption of fenofibrate (FN), a Biopharmaceutics Classification System (BCS) II model drug. FN-loaded PEGylated lipid nanoparticles (FN-PLNs) were prepared by the solvent-diffusion method and characterized by particle size distribution, morphology, Fourier transform infrared spectroscopy, and drug release. Lipolytic experiments were performed to assess the resistance of lipid nanoparticles against pancreatic lipase. Pharmacokinetics was evaluated in rats after oral administration of FN preparations. The obtained FN-PLNs were 186.7 nm in size with an entrapment efficiency of >95%. Compared to conventional lipid nanoparticles, PLNs exhibited slower drug release in the lipase-containing medium, strikingly reduced mucin binding, and suppressed lipolysis in vitro. Further, oral absorption of FN was significantly enhanced using PLNs with relative bioavailability of 123.9% and 157.0% to conventional lipid nanoparticles and a commercial formulation (Lipanthyl®), respectively. It was demonstrated that reduced mucin trapping, suppressed lipolysis, and/or improved mucosal permeability were responsible for increased oral absorption. These results facilitated a better understanding of the in vivo fate of lipid nanoparticles, and suggested the potential of PLNs as oral carriers of BCS II drugs.
Keywords: fenofibrate, lipid nanoparticles, PEGylation, oral bioavailability, absorption mechanism
Introduction
The Biopharmaceutics Classification System (BCS) defines BCS II drugs as a family of active compounds with a low aqueous solubility and high intestinal permeability.1 BCS II drugs are often plagued with inadequate absorption and highly variable bioavailability owing to low aqueous solubility. To improve the dissolution and oral bioavailability, various drug delivery systems have been explored. These systems include novel solid formulations such as nanocrystals,2,3 solid dispersions,4,5 and cyclodextrin inclusion complexes;6,7 and liquid nanocarriers such as liposomes,8 micelles,9 lipid nanoparticles,10 and micro/nanoemulsions.11,12 For solid dosage forms, three main factors, ie, solubility, dissolution rate, and intestinal permeability, govern the rate and extent of oral absorption of BCS II drugs. However, as far as liquid nanocarriers are concerned, the oral bioavailability largely depends on the types of carriers and their in vivo fates post-administration.
Lipid-based formulations could have profound influence on the absorption and transepithelial transport of lipophilic drugs.13 Generally, lipid carriers can improve the oral absorption of drug through three mechanisms: solubilize drug into the intestinal milieu; facilitate drug transport by interacting with the enterocytes; and reduce the first-pass effect by altering transport pathway (portal vein versus lymphatic duct).14 Solid lipid nanoparticles (SLNs)15 and nanostructured lipid carriers16 are the most widely used lipid vehicles. Due to involving no dissolution process, lipid nanocarriers theoretically can markedly enhance the oral absorption of BCS II drugs (eg, several or dozens of times increase in bioavailability); nevertheless, this is not always the case. For instance, although micronized fenofibrate (FN) (mean diameter size ~5 μm) showed significantly lower systemic absorption than FN-loaded SLNs (diameter size involving 90% particles <0.12 μm; P=0.05) and FN nanocrystals (DissoCubes®, diameter size involving 90% particles <0.57 μm; P=0.01), there was no significant difference in the area under the curve of plasma drug concentration vs time from 0 to 22 hours between SLNs and DissoCubes. The lipid nanocarriers (SLNs) merely increased oral bioavailability of FN by 24.34%–89.05% relative to two micronized FN suspensions (dispersed in hydroxethyl cellulose solution or sirupus, respectively).17
The particle size of drug and formulation composition are not the ultimate determinants to transepithelial absorption of BCS II drugs. The in vivo fates of nanoparticles most likely also play a key role in oral drug absorption. As is known, lipid nanoparticles will undergo a series of physiological processes after oral administration, of which lipolysis and mucin trapping are the main dispositions. Lipolysis of lipid carriers can lead to drug precipitation and formation of bulk aggregate, if unable to immediately assemble into micelles with endogenous bile salts and phospholipids.18,19 Mucin, a glycoprotein, is the major component of the mucus that covers the cells lining the surfaces of the digestive tract.20 Rapid mucus turnover leads to a large quantity of free mucin occupying the gastrointestinal (GI) lumen. Nanocarriers used for drug delivery are readily captured by free mucin and rapidly removed from the GI tract, resulting in reduced oral bioavailability.21 Upon bound with the GI mucins, especially the free ones, nanoparticles can neither readily diffuse into the absorptive epithelia nor be uptaken by epithelial cells.22,23 As a consequence, a considerable proportion of drug is orally unavailable and excreted with the feces.
Recently, PEGylated lipid nanoparticles (PLNs) used for encapsulation and delivery of therapeutic proteins have received more attention.24,25 Also, PEGylation is an effective approach to overcome the drawbacks of lipid nanoparticles as oral delivery carriers because it can reduce the capture of both mucins26 and lipolysis.27 PEGylation has shown potential in bioavailability enhancement of small-molecule and protein drugs.28,29 Although PEGylated vehicles have been extensively evaluated for the parenteral delivery of active ingredients, the efficacy of PLNs as oral carriers of BCS II drugs has not been fully established. It remains largely unclear how lipid nanoparticles (with or without polyethylene glycol [PEG] modification) are processed in the GI tract.
In this study, PEGylated and conventional lipid nanoparticles (CLNs) loading FN, a BCS II drug,30 were prepared based on different techniques and characterized by particle size, morphology, FTIR, in vitro release, lipolysis, and in vivo pharmacokinetics. It was demonstrated that PLNs had a substantial impact on drug absorption and resulted in an enhanced oral bioavailability of FN. We aimed to provide new insights into the gastrointestinal behavior of different lipid nanoparticles and elucidated the possible implications of PEGylation for oral delivery of FN.
Materials and methods
Materials
FN was obtained from Enhua Pharma Co., Ltd (Xuzhou, People’s Republic of China), and fenofibric acid was purchased from Xingyuan Chemicals (Zhengzhou, People’s Republic of China). Stiripentol (4,4-dimethyl-1-[3,4(methylenedioxy)-phenyl]-1-penten-3-ol) was from Sigma-Aldrich (St Louis, MO, USA). Lauroyl polyoxyl-6 glycerides (LPG) (Labrafil M2130 CS) and glyceryl distearate (Precirol ATO 5) were kindly provided by Gattefossé Co. (Saint Priest, Cedex, France). Lipase from porcine pancreas (>20,000 units/mg) and PEG monooleate (Mn ~860) were bought from Aladdin Reagent (Shanghai, People’s Republic of China). Tween 80 was supplied by Aladdin Reagent. High-performance liquid chromatography (HPLC)-grade methanol was provided by Merck (Darmstadt, Germany). Deionized water was prepared by a water purifier (Chengdu, People’s Republic of China). All other chemicals were of analytical grade and used as received.
Preparation of FN-loaded lipid nanoparticles
FN-PLNs were prepared using the solvent-diffusion technique. Briefly, FN, LPG and PEG monooleate (PM) were dissolved in 1.0 mL of ethanol at 45°C and then rapidly injected into 10 mL of water with a syringe. Drug and lipid materials were spontaneously assembled into nanoparticles upon the solvent diffusion into the aqueous phase. Subsequently, the residual ethanol was removed under reduced pressure by a rotatory evaporator until the nanosuspensions were condensed to an appropriate volume. Particle size and entrapment efficiency (EE) as indices were chosen to optimize the formulation of FN-PLNs using the ratio of LPG/PM as the formulation variable with an invariable FN amount (30 mg).
As a reference, FN-CLNs were prepared by the melting-emulsification method according to previously described procedures with a slight modification.31 The solid lipid (Precirol ATO 5, 360 mg) was melted at 75°C followed by the addition of FN (30 mg). Then, the completely melted mixture was dispersed in a 75°C aqueous solution (30 mL) containing 1.5% Tween-80 (w/v) for 2 minutes at a rate of 10,000 rpm with a high-speed disperser (IKA Works, Guangzhou, People’s Republic of China) to produce the coarse emulsions. Afterward, the coarse emulsions were homogenized using a microfluidizer (Nano DeBee, South Easton, MA, USA) under 20,000 psi until the particle size of the FN-CLNs were close to that of the FN-PLNs.
Characterization of FN-loaded lipid nanoparticles
Particle size of FN-loaded lipid nanoparticles was determined by dynamic light scattering using a Zetasizer Nano ZS (Malvern Instruments, Worcestershire, Malvern, UK) at 25°C. An aliquot sample (circa 0.1 mL) was diluted with deionized water to 1 mL and then subjected to laser diffraction. The data were analyzed with the build-in software for the calculation of particle size.
Morphology of FN-loaded lipid nanoparticles was observed by transmission electron microscopy (TEM) (Philips, Amsterdam, the Netherlands). The formulation for TEM detection was first diluted to approximately 25 times with water. Then, the nanoparticle suspensions were dropped on a carbon-coated copper grid and solidified to the supporter under a warming lamp. The fixed nanoparticles were photographed with Tecnai 10 TEM at an acceleration voltage of 100 kV.
EE of FN in lipid nanoparticles was determined by separating free FN from FN-loaded lipid nanoparticles. A procedure of centrifugation (5,000 g, 5 minutes) was first performed to precipitate any non-encapsulated bulk FN. Then, the supernatants (nanosuspensions) were subjected to a centrifugal force of 10,000 g for 10 minutes against a centrifugal filter device (Amicon® Ultra-0.5, MWCO 50,000; Millipore, Boston, USA). Concentrations of free FN (Mfre) in the filtrate and initial FN in nanosuspensions were quantified by HPLC. EE was defined as the ratio of entrapped FN (Ment) to total FN (Mtot) and calculated based on the equation below.
EE (%) = (1–Mfre/Mtot) ×100% | (1) |
Drug assay of in vitro/vivo samples
FN concentration of in vitro samples was determined by HPLC (Wufeng LC 100 series; Wufeng Scientific Instruments, Shanghai, People’s Republic of China). The HPLC system was composed of a dualistic pump, a degasser, an autosampler, a column heater, and a UV100 ultraviolet detector. FN was separated by a Diamonsil C18 column (5 μm, 4.6 mm ×250 mm) guarded with a precolumn at 40°C and detected at 286 nm. The injection volume was designated as 20 μL. The mobile phase consisted of 80% methanol and 20% water pumped at a flow rate of 1.0 mL/minute.
FN is a prodrug and rapidly metabolized to its major active metabolite, fenofibric acid. Original FN cannot be detected in the plasma after oral absorption. Therefore, an indirect way based on the quantification of fenofibric acid in plasma is generally adopted to evaluate the pharmacokinetics of FN.32 In this study, fenofibric acid in rat plasma was determined by the above established HPLC method with some modifications. The mobile phase with same ratio of methanol/water was adjusted to pH 3.0 using phosphoric acid. An internal standard of stiripentol, a similar compound in lipophilicity, was used to accurately quantify the plasma fenofibric acid.
FTIR
FTIR spectrum was collected to further estimate the possible interactions between FN and excipients in the lipid nanoparticles. Briefly, the samples of FN, LPG, PM, physical mixture (LPG, PM, and FN at a ratio of 9/3/1), and lyophilized FN-PLNs were ground thoroughly with KBr to obtain an infrared transparent matrix. FTIR scanning was performed on a FTIR-8400S spectrometer (Shimadzu Corporation, Kyoto, Japan). Spectra were recorded from 3,500 to 600 cm−1 with a resolution of 1.0 cm−1.
Release study in vitro
Release study of FN from lipid nanoparticles was carried out using the reverse bulk equilibrium dialysis technique. Release media with or without pancreatic lipase were comprised of a pH 6.8 buffer and 0.5% sodium dodecyl sulfate (SDS) as a solubilizer. The enzyme activity of lipase-containing medium was 100 IU/mL. Ligated dialysis bags containing 0.5 mL identical release medium, into which a glass ball was included (to ensure the bag was below the liquid surface), were put into 900 mL of release medium. After equilibrium with the bulk phase under magnetic agitation, 10 mL of FN-PLNs or FN-CLNs (3 mg/mL) were introduced and reversely dialyzed at 37°C against the dialysis bag. At predetermined intervals, the dialysis bags were withdrawn. FN concentration in dialyzates was determined, and the percentage of drug release was calculated.
Ex vivo evaluation of nanoparticles–mucin binding
In order to detect the interaction between lipid nanoparticle and mucins, ex vivo intestinal fluids were prepared using Sprague–Dawley rats. Prior to experimentation, six rats (250±10 g) were fasted for 24 hours but allowed free access to water. The rats were orally given 2 mL water by gavage before collection of intestinal fluids so as to obtain more physiological liquids. After 30 minutes, the rats were sacrificed by cervical dislocation. The intestine from duodenum to ileum of each rat was carefully isolated from the mesentery following ligation of both ends. The external wall and two ends of the isolated intestines were rinsed with physiological saline. Afterward, intestinal fluids were squeezed out from one end by hand and centrifuged at 10,000 g for 15 minutes to harvest the mucin-containing clear liquids. Particle size of ex vivo intestinal fluids was measured with a Zetasizer Nano ZS after appropriate dilution. To investigate the capture of lipid nanoparticles by mucins, FN-PLNs and FN-CLNs (100 μL) were added into 100 μL intestinal fluids and incubated at 37°C. At designated time points (0.25, 0.5, 1.0, and 2.0 hours), the particle size of the co-incubation system was determined in real time.
Lipolytic experiment
In vitro lipolysis study of FN-PLNs and FN-CLNs was performed according to a previously reported method with minor modifications under simulated fasted-state intestinal fluids.33 Briefly, 6 mL of lipid nanoparticle suspensions were added into 14 mL of digestive medium, which comprised 150 mM NaCl, 5 mM CaCl2, 5 mM cholates, 1.25 mM lecithin, 300 IU/mL pancreatic lipase, and 0.5% SDS buffered at pH 6.8 with Tris-maleate. The mixed system was incubated in a thermostated water bath at 37°C. The pH was maintained at 6.8 throughout the whole lipolysis process by manual titration of 0.2 M NaOH to neutralize the fatty acid produced by the digestion of lipids. The digestion process was considered complete when the pH change was less than 0.05 units at an interval of 15 minutes. Cumulative lipolysis percentage versus time profiles of FN-PLNs and FN-CLNs titrated by NaOH were plotted.
Bioavailability studies
All animal experiments were conducted in accordance with the Guidelines on the Care and Use of Animals for Scientific Purposes. The protocols for the animal studies were also reviewed and approved by the Experimental Animal Ethical Committee of Jinan University. Sprague–Dawley rats (250±10 g) were randomly divided into three groups (n=6). Rats were fasted for 12 hours prior to the experiments but allowed free access to water. The rats were dosed with FN-PLNs, FN-CLNs, or the reference preparation of Lipanthyl® (micronized FN, suspended with water for oral gavage) at an equivalent dose of 25 mg/kg. Blood samples (about 0.25 mL) were withdrawn from the tail vein at 0.25, 0.5, 1, 2, 4, 6, 8, and 12 hours after administration. Plasma was separated by centrifugation at 5,000 g for 5 minutes.
Fenofibric acid in rat plasma was collected by a deproteinization procedure. Typically, three-fold volume of methanol/HCl (1 M) (95/5, v/v) was added into 100 μL of plasma supplemented with 20 μL of 100 μg/mL stiripentol. The mixture was then vortexed vigorously for 5 minutes and centrifuged at 12,000 g for 10 minutes. The supernatant was transferred to centrifuge tubes followed by evaporation using a Concentrator Plus (Eppendorf, Hamburg, NY, USA). The dried residues were reconstituted in 100 μL methanol and analyzed by HPLC after centrifugation. Non-compartmental analysis was employed to process the data and extract the pharmacokinetic parameters with PKSolver 2.0 software.
Results and discussion
Preparation and characterization of FN-PLNs
In the preliminary experiment, we found that the ratio of LPG/PM had a significant impact on the formulation properties. Accordingly, an optimization was performed to obtain a desired formulation. The effects of LPG/PM ratio on particle size and EE of FN-PLNs are showed in Figure 1. Particle size together with polydispersity index of FN-PLNs decreased but EE increased as the LPG amount increased. However, these trends were reversed with the decrease of PM when the LPG amount was fixed. This indicated that there might be a synergetic effect between LPG and PM in dominating the formation of FN-PLNs and drug loading. High lipid content (ie, 360 mg) was advantageous to enhance the EE of FN, since a large proportion of lipids can load more FN molecules. Enhanced EE of lipophilic drugs utilizing high ratio lipids in the formulation has also been demonstrated in the literature.34,35
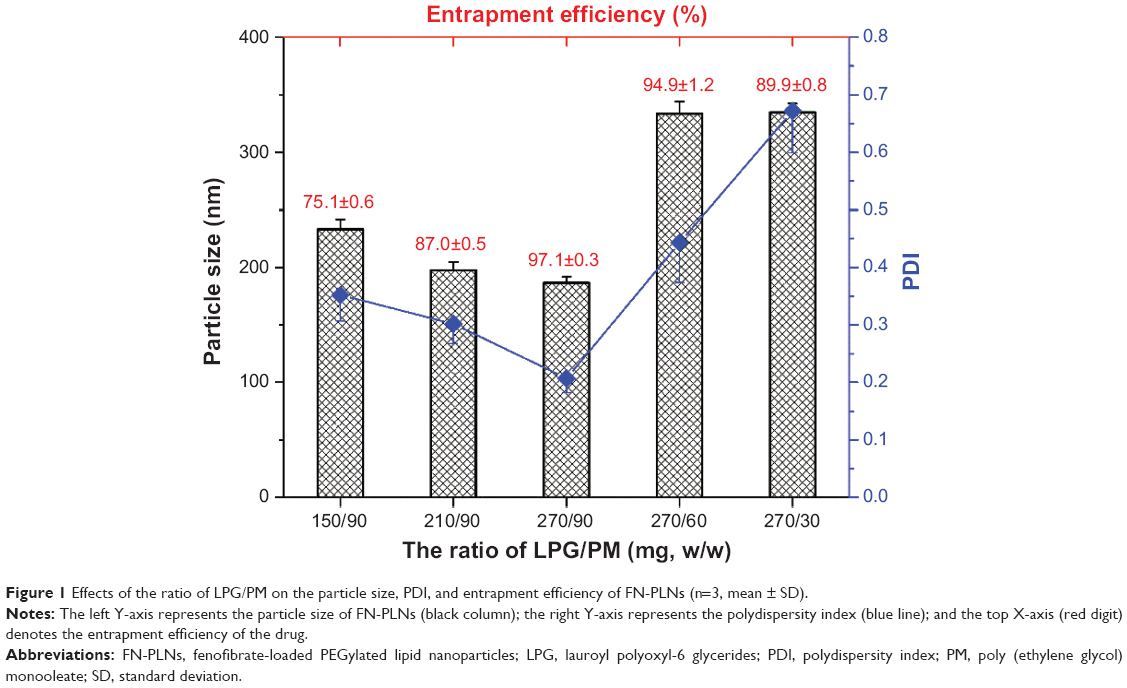 | Figure 1 Effects of the ratio of LPG/PM on the particle size, PDI, and entrapment efficiency of FN-PLNs (n=3, mean ± SD). |
The particle size of FN-PLNs prepared using the optimized formulation, consisting of 30 mg FN, 270 mg LPG, and 90 mg PM, was 186.7 nm and presented a unimodel distribution (polydispersity index =0.207) (Figure 2A). Almost all nanoparticles (99.8%) were less than 342.0 nm from the undersize curve (green line) given by build-in software, indicating no particularly large particles in the nanosuspensions. The resulting FN-PLNs were spherical as revealed by TEM (Figure 2B). The particle size was estimated to be about 200 nm from the scale bar, which was consistent with the value measured by dynamic light scattering. High EEs (more than 95%) were obtained for all investigated batches. High drug encapsulation may be accounted for by the poor solubility of FN in water (circa 5.41 μg/mL determined at 25°C) and/or the intense interactions between FN and lipid materials.36
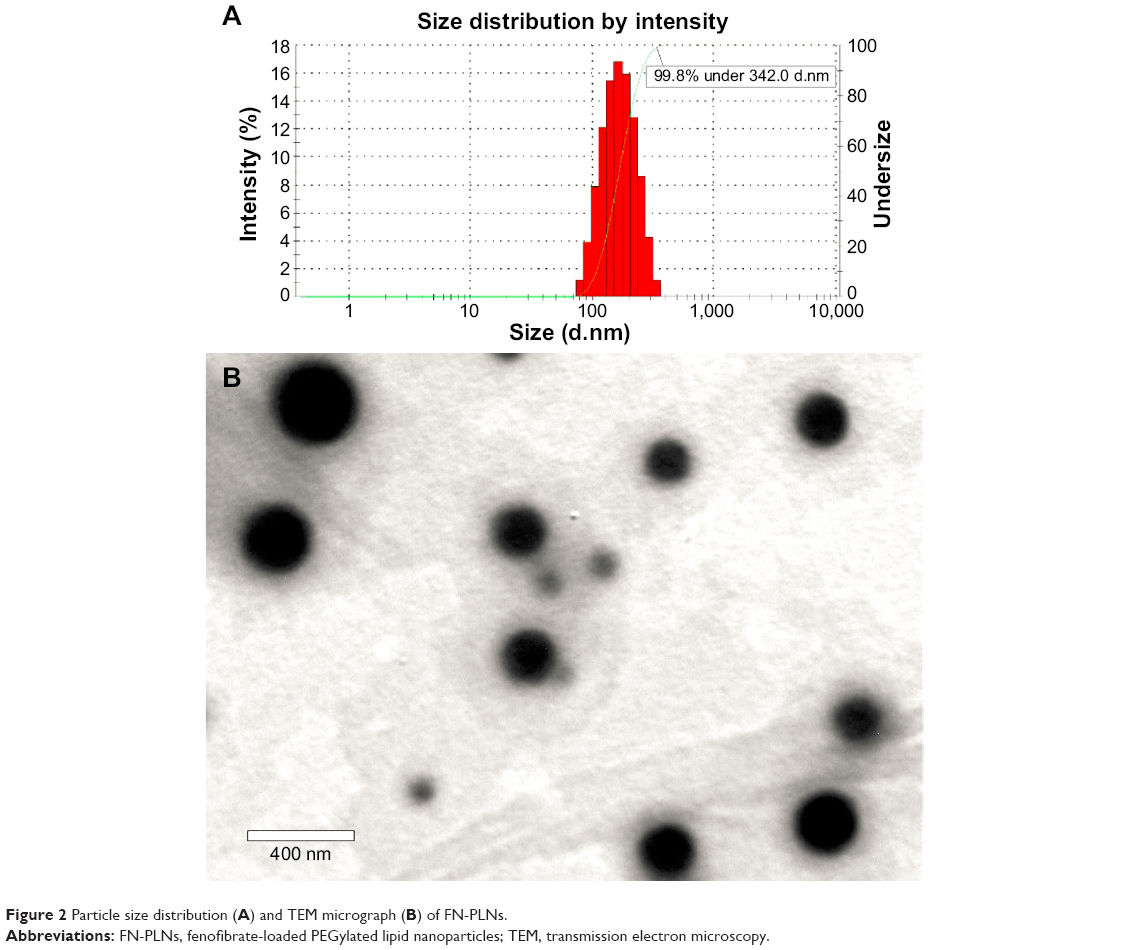 | Figure 2 Particle size distribution (A) and TEM micrograph (B) of FN-PLNs. |
FTIR
FTIR spectra of FN, LPG, PM, physical mixture, and FN-PLNs are shown in Figure 3. Significant changes in the characteristic bands of the FTIR spectra were observed, suggesting an alteration in drug micromilieu. The diagnostic absorption peaks of FN appeared at 1654.8, 1176.5, and 1014.5 cm−1, corresponding to the stretching vibrations of R-CO-R (υs), C-O-C (υs), and C-O-C (υas), respectively. The FN-associated peaks in raw drug and physical mixture were nearly identical. However, those diagnostic peaks, as marked by red lines, shifted in position or disappeared in the FTIR spectrum of FN-PLNs. Alternations of characteristic peaks suggested that there were intense molecular interactions between drug and excipients. This may be ascribable to the formation of non-covalent bonds (eg, hydrogen bond, hydrophobic interaction) between the groups containing an electronegative O atom of FN and the ether oxygen group of PLG and PM.4 These interactions might facilitate effective solubilization and incorporation of FN into the lipid materials.
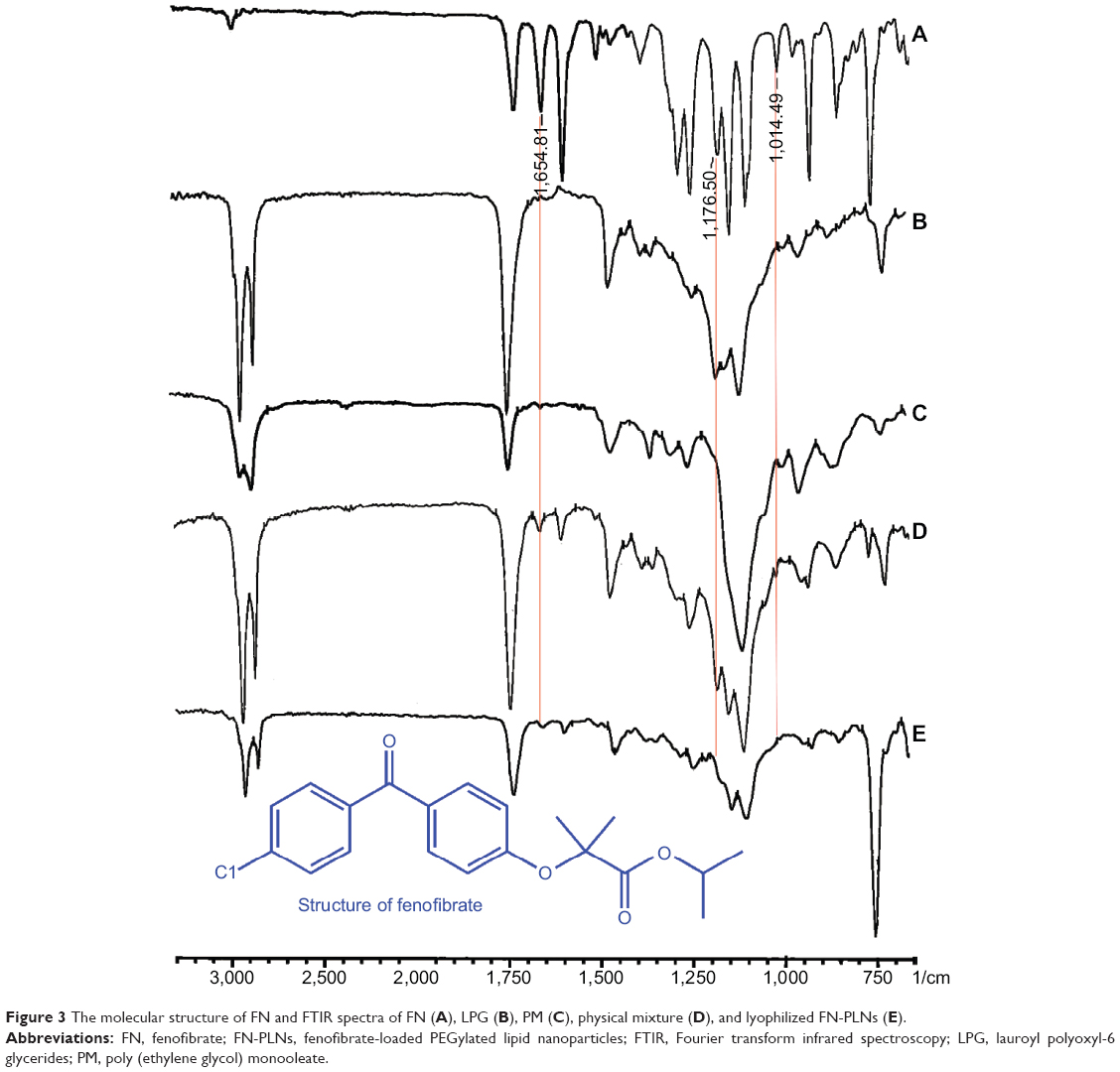 | Figure 3 The molecular structure of FN and FTIR spectra of FN (A), LPG (B), PM (C), physical mixture (D), and lyophilized FN-PLNs (E). |
Drug release
The release profiles of FN-PLNs and FN-CLNs in media with or without pancreatic lipase are shown in Figure 4. The accumulative release of FN increased as a function of time in the lipase-free medium (Figure 4A), indicating that FN could be continuously released into the bulk phase under a sink condition (containing 0.5% SDS). There was no significant difference in the release kinetics of two preparations. The f2 value (similarity factor) between two preparations was calculated to be 58, revealing that their releases were comparable. Likewise, the accumulative release of FN from the vehicles increased with the time in the lipase-containing medium. However, there was a significant difference in the release profiles between PEGylated (FN-PLNs) and non-PEGylated (FN-CLNs) formulations (Figure 4B). The calculated f2 value was merely 33. FN-CLNs exhibited a fast release as compared to FN-PLNs. The results suggested that the digestive enzyme (lipase) exerted an action on the structure of the lipid nanoparticles, thereby affecting the process of drug release.
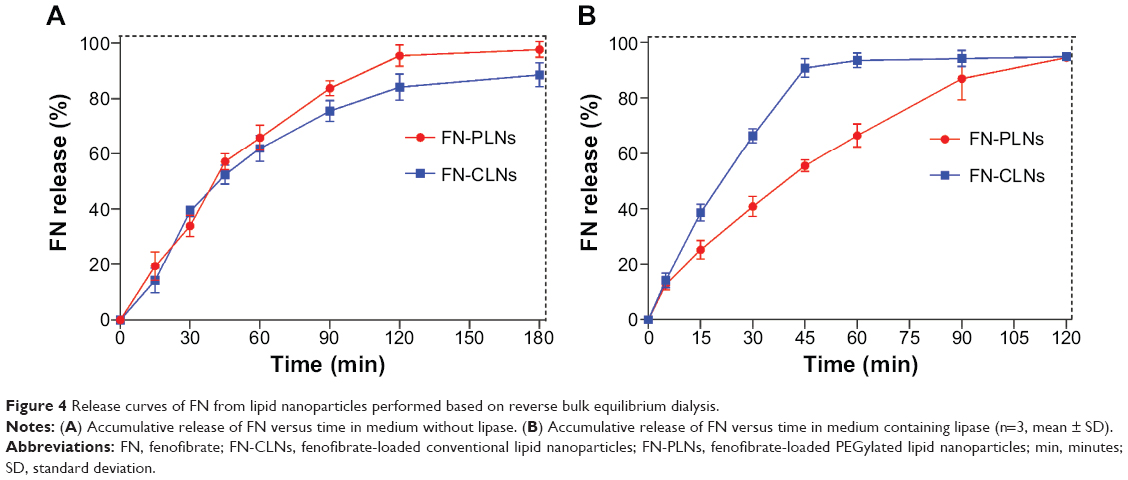 | Figure 4 Release curves of FN from lipid nanoparticles performed based on reverse bulk equilibrium dialysis. |
Degradation of lipid nanoparticle by digestive enzymes is an inevitable physiological course.37 Indeed, enzymes have less opportunity to hydrolyze target substrates unless freely accessible to the objects. PEGylation provides a feasible strategy to shield the accessibility of enzymes to nanoparticles. The hydrophilic corona of nanoparticles formed by PEG moiety reduces the degradation of lipid nanoparticles.38 Digestion of lipids will result in a more rapid drug release from lipid carriers. Therefore, FN-CLNs produced an accelerated drug release in the lipase-containing medium compared to FN-PLNs, with an accumulative release amount more than 90% within 45 minutes. In contrast, the release of FN-PLNs was relatively slow, and the accumulative release was only 55.45% at 45 minutes. The drug release from lipid nanoparticles in the lipase-containing medium correlated well the results of the lipolytic experiment below.
Interaction between lipid nanoparticles and mucin
A better understanding of interactions between nanoparticles and free mucin is highly helpful in predicting the in vivo fate and performance of nanocarriers. Figure 5 shows the particle size changes of the co-incubation system with time after addition of lipid nanoparticles into the ex vivo intestinal fluids. The particle size of the system involving FN-CLNs significantly increased as the incubation time increased. The particle size reached up to 1526.1 nm at 120 minutes, indicating considerable absorption of mucins onto the nanoparticles. Although the particle size of FN-PLNs/intestinal fluid system raised (from 339.8 nm to 409.2 nm) as well, the change was marginal compared to the system involving FN-CLNs. These facts lent strong support that PLNs can effectively mask the binding of GI free mucins.
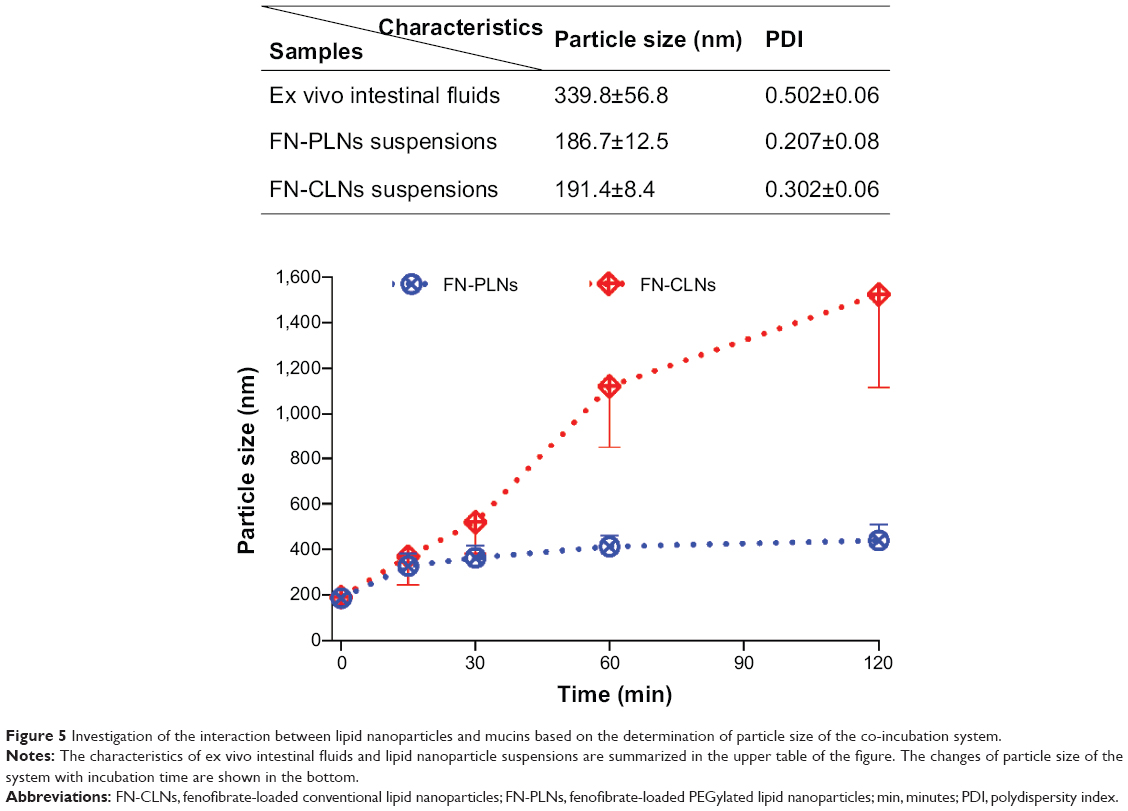 | Figure 5 Investigation of the interaction between lipid nanoparticles and mucins based on the determination of particle size of the co-incubation system. |
The polymer of PEG is very hygroscopic and easily binds to water. Each ether oxygen subunit (-CH2CH2O-) demonstrated firm association with two or three water molecules in aqueous solution.39 Mucin contains hydroxyl groups in the branched sugar chains, carboxylic acid, and sulfate groups in the terminal groups of branched chains.20 Formation of hydrogen bonds was regarded as the main adhesion mechanism for the binding of mucins to foreign particles.40 Conventional nanoparticles would be rapidly coated by soluble mucin via hydrogen bond interaction, resulting in loss of permeability and/or clearance with GI mobility before reaching the mucus layer on the epithelium. However, the intrinsic hydrogen bonds between PEG and water are required to break for mucins to bind to PEGylated nanoparticles. Therefore, PEGylation potentially circumvents the capture of nanoparticles by free mucins in the lumen, which suggests the enhancement of oral bioavailability.
Reduced lipolysis attributable to PEGylation
The in vitro lipolysis model is usually utilized to evaluate the digestive feature of lipids-based formulations in the GI tract.41 Cumulative lipolytic percentages of lipid nanoparticles versus time at pH 6.8 are shown in Figure 6. It was observed that lipolysis of FN-CLNs was relatively fast in the first 20 minutes and then turned into slow digestion. Lipolysis of FN-CLNs was nearly completed at 50 minutes with a total amount of 6.8 mL NaOH solution consumption. However, FN-PLNs exhibited a dissimilar lipolysis process in the simulated digestive medium in contrast with FN-CLNs. In the first 20 minutes, the accumulative lipolytic percentage of FN-PLNs was 48.33%, contrasting with a value of up to 78.56% for FN-CLNs. At the end point of 60 minutes, the total lipolysis of FN-PLNs was only 82.91%, which indicated that PEG-lipids possessed a moderate resistance to digestive enzyme. Although the drugs released from lipid carriers under the action of lipase can be simultaneously assembled into mixed micelles with the digestive products, phospholipids, and bile salts,37 there is still a considerable number of drugs failing to be transformed into micelles as a result of drug precipitation upon rapid lipolysis.18 A delayed and reduced hydrolysis of lipid nanoparticles using PEG-lipids will lead to favorable conditions for self-assembly of micelles as drug carriers and for transepithelial transport via intact lipid nanoparticles. Hence, PLNs had positive effects on oral drug absorption in terms of lipolysis retarding.
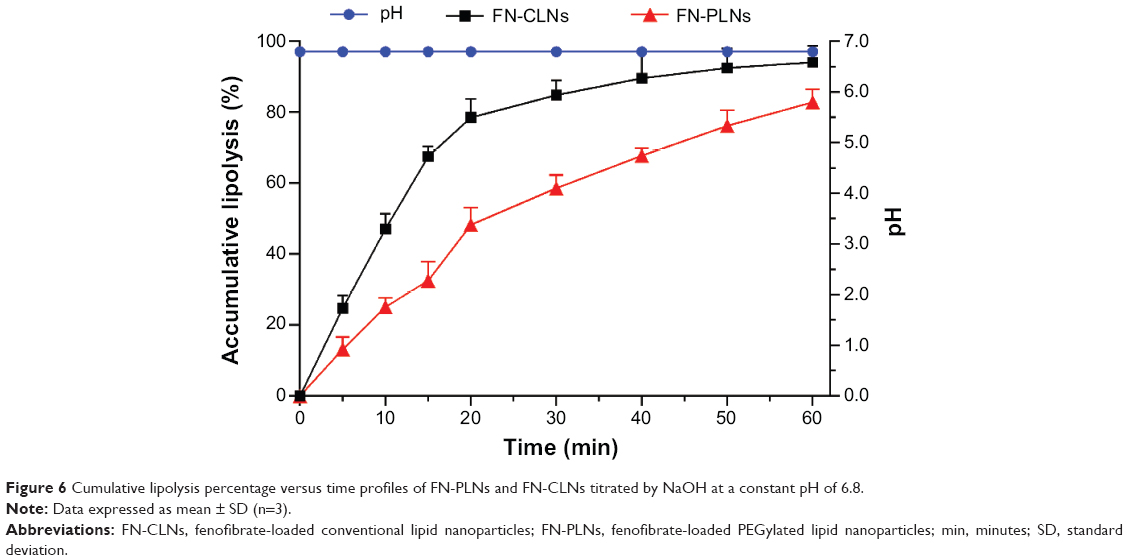 | Figure 6 Cumulative lipolysis percentage versus time profiles of FN-PLNs and FN-CLNs titrated by NaOH at a constant pH of 6.8. |
Enhanced bioavailability of FN-PLNs
Drug plasma concentration versus time plots after oral administration of FN-CLNs, FN-PLNs, and Lipanthyl are shown in Figure 7. Main pharmacokinetic parameters calculated based on a non-compartmental model are shown in Table 1. The pharmacokinetic curves for all groups were characterized by an obvious absorption phase followed by a slow elimination phase. The peak times (Tmax) of three preparations were approximately 1.75–2.25 hours, which were not significantly different among each other. Similarly, there were no significant differences in the half-lives (T1/2) for the three preparations. However, distinct peak concentrations (Cmax) were yielded after administration of FN-CLNs, FN-PLNs, and the commercial formulation. The Cmax of FN-PLNs reached 8.76 μg/mL, which was 1.52- and 1.29-fold as much as FN-CLNs and Lipanthyl, respectively. Compared to micronized FN and FN-CLNs, FN-PLNs demonstrated increased blood drug levels at almost each time point. The relative bioavailability of FN-PLNs and FN-CLNs were 156.95% and 126.67%, respectively. Also, there was a significant difference in bioavailability between FN-PLNs and FN-CLNs according to one-way analysis of variance at the P-value of 0.05. These results turned out that CLNs can enhance the oral bioavailability of FN, however, PLNs can further reinforce the performance of such drug carriers. The in vitro in vivo correlation (IVIVC) was evaluated using the data of release in lipase-containing medium and plasma FN concentration of the absorption phase (0.25, 0.5, 1.0, and 2.0 hours). Figure 8 gives the linear regression equations of drug release versus plasma FN level. FN-PLNs showed a good IVIVC with a coefficient of 0.9955, whereas the IVIVC of FN-CLNs was relatively lower. This indicated that the integrity of lipid nanoparticles in the GI tract was associated with the subsequent absorption.
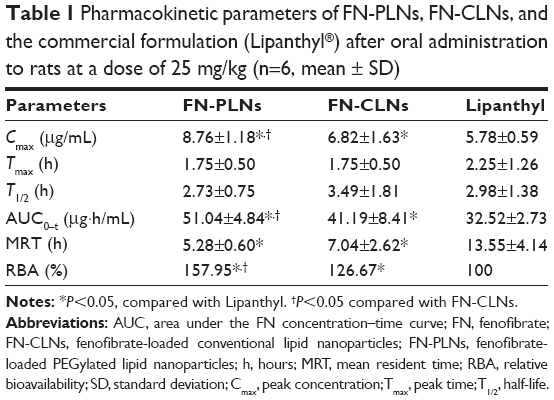 | Table 1 Pharmacokinetic parameters of FN-PLNs, FN-CLNs, and the commercial formulation (Lipanthyl®) after oral administration to rats at a dose of 25 mg/kg (n=6, mean ± SD) |
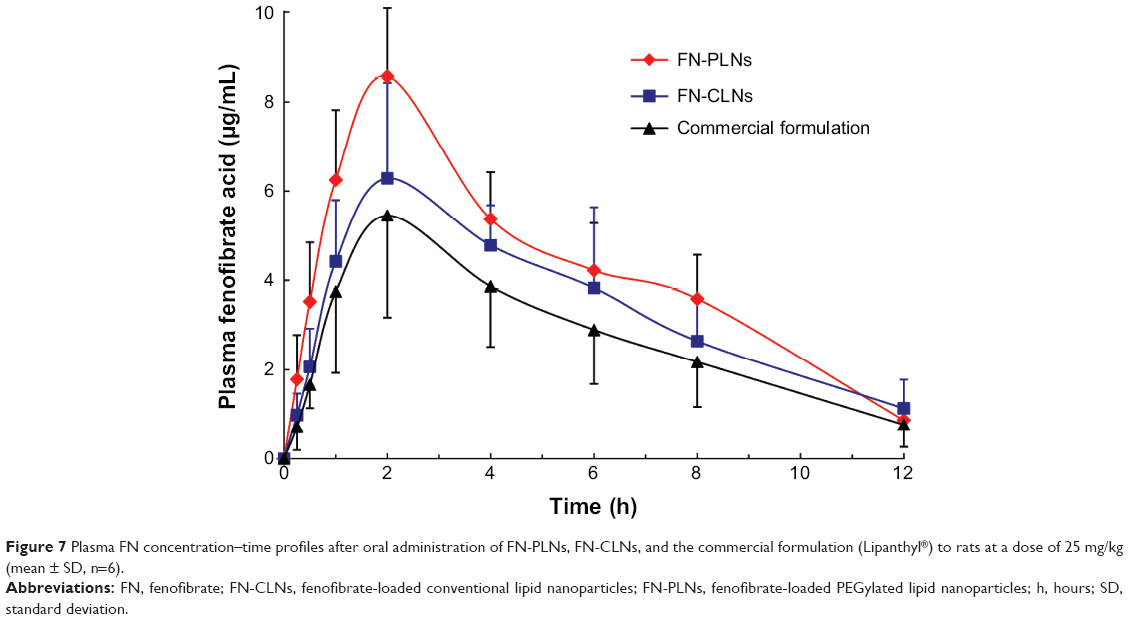 | Figure 7 Plasma FN concentration–time profiles after oral administration of FN-PLNs, FN-CLNs, and the commercial formulation (Lipanthyl®) to rats at a dose of 25 mg/kg (mean ± SD, n=6). |
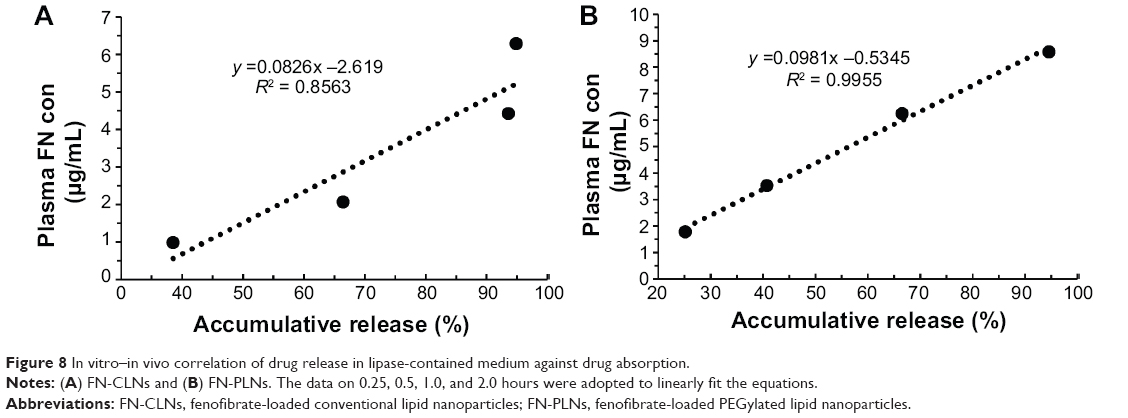 | Figure 8 In vitro–in vivo correlation of drug release in lipase-contained medium against drug absorption. |
Generally, it is believed that lymphatic transport is predominantly responsible for the post-enterocytes absorption of lipophilic drugs when lipid-based formulations are employed.13 However, the behavior of pre-enterocytes also plays a critical role in overall drug absorption. Drugs are not able to be absorbed until they traverse the mucosal layer and approach the enterocytes. The materials for the fabrication of FN-PLNs are lower than 2,000 in molecular weight and can be assimilated slightly by the small intestine. Although we cannot distinguish the effect of lipid components on the absorption of FN, enhanced oral bioavailability was substantially achieved through PLNs. The result provided evidence that PEGylated nanoparticles can exert a definite effect on the absorption of delivered drugs, such as FN.
Effect of PEGylation on oral absorption
For oral particulates, particularly lipid nanoparticles, destruction of structure by digestive enzymes and nonspecific adhesion by secreted mucins, which potentially cause drug recrystallization and inactive transepithelial transport, are the major limiting factors for drug absorption.35,42 PLNs can maintain intact nanoarchitecture of lipid nanoparticles over a relatively long time and reduce the adhesion of free mucins as transporting across the GI tract.43 The reasons to justify are the reduced accessibility of enzymes and the shielding effect of highly hydrated PEG corona against mucins. Moreover, PEGylated nanoparticles have the potential to break through the mucosal layer, above which an unstirred water layer is associated. It is of high importance to penetrate into the submucosa as the transcellular route is one of the essential patterns of BCS II drug transport. The underlying mechanisms for enhanced oral bioavailability of FN using PLNs was schematically illustrated in Figure 9.
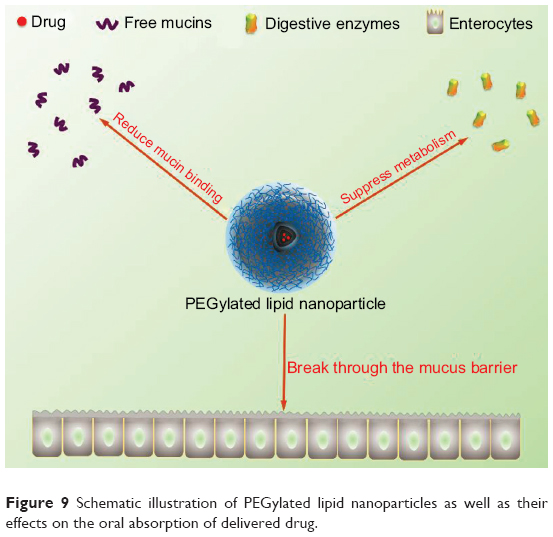 | Figure 9 Schematic illustration of PEGylated lipid nanoparticles as well as their effects on the oral absorption of delivered drug. |
Enhanced oral bioavailability or intestinal permeability using PEGylated nanoparticles has been documented in the literature. For example, PEGylated poly(anhydride) nanoparticles were developed as carriers for oral delivery of paclitaxel.44 Permeability and bioavailability of paclitaxel were significantly improved when delivered with PEGylated nanoparticles. Equally, PEGylated SLNs significantly increased the oral absorption of doxorubicin relative to SLNs.45 PEGylation increased the permeation ability of SLNs in a mucus-secreting Caco-2/HT29 coculture monolayer, whereas conventional SLNs were strongly trapped by highly viscoelastic mucus. However, these studies were focused on the mucus permeability of nanoparticles rather than the pre-enterocytes fate. This study first elucidated the GI behavior of lipid nanoparticles using an ex vivo model. Taken together, our data provide new mechanistic insights on how PLNs affect the absorption of BCS II drug.
Conclusion
PEGylated nanoparticles present attractive properties as vehicles of lipophilic drugs for the enhancement of oral bioavailability. In the present work, FN-PLNs and FN-CLNs were prepared by a solvent-diffusion technique and melting-emulsification method, respectively. Compared to FN-CLNs, FN-PLNs exhibited a slow drug release in the lipase-containing medium and possessed a suppressed lipolysis in the simulated digestive medium. Although FN-CLNs produced an increased bioavailability relative to the commercial formulation, the improvement of bioavailability by FN-PLNs was more evident in contrast with FN-CLNs. We believe these observations underline the importance of the GI fate of lipid nanoparticles in oral delivery of lipophilic drugs. Our study would provide some useful information for further enhancement of oral bioavailability of BCS II drugs such as FN, using a rational strategy.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (81373496).
Disclosure
The authors report no conflicts of interest in this work.
References
Ku MS. Use of the biopharmaceutical classification system in early drug development. AAPS J. 2008;10(1):208–212. | ||
Xu Y, Liu X, Lian R, et al. Enhanced dissolution and oral bioavailability of aripiprazole nanosuspensions prepared by nanoprecipitation/homogenization based on acid-base neutralization. Int J Pharm. 2012;438(1–2):287–295. | ||
Kayaert P, Van den Mooter G. An investigation of the adsorption of hydroxypropylmethyl cellulose 2910 5 mPa s and polyvinylpyrrolidone K90 around Naproxen nanocrystals. J Pharm Sci. 2012;101(10):3916–3923. | ||
Zhang X, Sun N, Wu B, Lu Y, Guan T, Wu W. Physical characterization of lansoprazole/PVP solid dispersion prepared by fluid-bed coating technique. Powder Technol. 2008;182(3):480–485. | ||
Sun N, Zhang X, Lu Y, Wu W. In vitro evaluation and pharmacokinetics in dogs of solid dispersion pellets containing Silybum marianum extract prepared by fluid-bed coating. Planta Med. 2008;74(2):126–132. | ||
Zhang X, Wu D, Lai J, Lu Y, Yin Z, Wu W. Piroxicam/2-hydroxypropyl-beta-cyclodextrin inclusion complex prepared by a new fluid-bed coating technique. J Pharm Sci. 2009;98(2):665–675. | ||
Rong WT, Lu YP, Tao Q, et al. Hydroxypropyl-sulfobutyl-beta-cyclodextrin improves the oral bioavailability of edaravone by modulating drug efflux pump of enterocytes. J Pharm Sci. 2014;103(2):730–742. | ||
Yi C, Fu M, Cao X, et al. Enhanced oral bioavailability and tissue distribution of a new potential anticancer agent, Flammulina velutipes sterols, through liposomal encapsulation. J Agric Food Chem. 2013;61(25):5961–5971. | ||
Zhang X, Wang H, Zhang T, Zhou X, Wu B. Exploring the potential of self-assembled mixed micelles in enhancing the stability and oral bioavailability of an acid-labile drug. Eur J Pharm Sci. 2014;62:301–308. | ||
Zhang X, Zhang T, Zhou X, et al. Enhancement of oral bioavailability of tripterine through lipid nanospheres: preparation, characterization, and absorption evaluation. J Pharm Sci. 2014;103(6):1711–1719. | ||
Ryu JK, Yoo SD. Preparation and evaluation of bicyclol microemulsions for enhanced oral bioavailability. Drug Dev Ind Pharm. 2012;38(11):1313–1318. | ||
Gorain B, Choudhury H, Kundu A, et al. Nanoemulsion strategy for olmesartan medoxomil improves oral absorption and extended antihypertensive activity in hypertensive rats. Colloids Surf B Biointerfaces. 2014;115:286–294. | ||
Porter CJ, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6(3):231–248. | ||
Trevaskis NL, Charman WN, Porter CJ. Lipid-based delivery systems and intestinal lymphatic drug transport: a mechanistic update. Adv Drug Deliv Rev. 2008;60(6):702–716. | ||
Harde H, Das M, Jain S. Solid lipid nanoparticles: an oral bioavailability enhancer vehicle. Expert Opin Drug Deliv. 2011;8(11):1407–1424. | ||
Gaba B, Fazil M, Ali A, Baboota S, Sahni JK, Ali J. Nanostructured lipid (NLCs) carriers as a bioavailability enhancement tool for oral administration. Drug Deliv. Epub 2014 Mar 27. | ||
Hanafy A, Spahn-Langguth H, Vergnault G, et al. Pharmacokinetic evaluation of oral fenofibrate nanosuspensions and SLN in comparison to conventional suspensions of micronized drug. Adv Drug Deliv Rev. 2007;59(6):419–426. | ||
Porter CJ, Kaukonen AM, Boyd BJ, Edwards GA, Charman WN. Susceptibility to lipase-mediated digestion reduces the oral bioavailability of danazol after administration as a medium-chain lipid-based microemulsion formulation. Pharm Res. 2004;21(8):1405–1412. | ||
Porter CJ, Kaukonen AM, Taillardat-Bertschinger A, et al. Use of in vitro lipid digestion data to explain the in vivo performance of triglyceride-based oral lipid formulations of poorly water-soluble drugs: studies with halofantrine. J Pharm Sci. 2004;93(5):1110–1121. | ||
Perez-Vilar J, Hill RL. The structure and assembly of secreted mucins. J Biol Chem. 1999;274(45):31751–31754. | ||
Hwang SJ, Park H, Park K. Gastric retentive drug-delivery systems. Crit Rev Ther Drug Carrier Syst. 1998;15(3):243–284. | ||
Bansil R, Turner BS. Mucin structure, aggregation, physiological functions and biomedical applications. Curr Opin Colloid Interface Sci. 2006;11(2–3):164–170. | ||
Crater JS, Carrier RL. Barrier properties of gastrointestinal mucus to nanoparticle transport. Macromol Biosci. 2010;10(12):1473–1483. | ||
Angelova A, Angelov B, Drechsler M, Garamus VM, Lesieur S. Protein entrapment in PEGylated lipid nanoparticles. Int J Pharm. 2013;454(2):625–632. | ||
Angelova A, Angelov B, Drechsler M, Lesieur S. Neurotrophin delivery using nanotechnology. Drug Discov Today. 2013;18(23–24):1263–1271. | ||
Wang YY, Lai SK, Suk JS, Pace A, Cone R, Hanes J. Addressing the PEG mucoadhesivity paradox to engineer nanoparticles that “slip” through the human mucus barrier. Angew Chem Int Ed Engl. 2008;47(50):9726–9729. | ||
García-Fuentes M, Torres D, Alonso MJ. Design of lipid nanoparticles for the oral delivery of hydrophilic macromolecules. Colloids Surf B Biointerfaces. 2003;27(2–3):159–168. | ||
Webster L, Blonsky ER, Matz P, et al. Efficacy, safety and pharmacokinetics of oral nktr-118 in patients with opioid-induced constipation: results of a randomized, double-blind, placebo-controlled phase 2 study. Am J Gastroenterol. 2009;104:S174–S174. | ||
Hazra P, Adhikary L, Dave N, et al. Development of a process to manufacture PEGylated orally bioavailable insulin. Biotechnol Prog. 2010;26(6):1695–1704. | ||
Reddy MS, Haq SMF, Apte SS. solubility enhancemet of fenofibate, A BCS II drug, by self emulsifing drug delivery systems. Int Res J Pharm. 2011;2(11):173–177. | ||
Aji Alex MR, Chacko AJ, Jose S, Souto EB. Lopinavir loaded solid lipid nanoparticles (SLN) for intestinal lymphatic targeting. Eur J Pharm Sci. 2011;42(1–2):11–18. | ||
Chen Y, Lu Y, Chen J, et al. Enhanced bioavailability of the poorly water-soluble drug fenofibrate by using liposomes containing a bile salt. Int J Pharm. 2009;376(1–2):153–160. | ||
Porter CJ, Charman WN. In vitro assessment of oral lipid based formulations. Adv Drug Deliv Rev. 2001;50 Suppl 1:S127–S147. | ||
Shangguan M, Lu Y, Qi J, et al. Binary lipids-based nanostructured lipid carriers for improved oral bioavailability of silymarin. J Biomater Appl. 2014;28(6):887–896. | ||
Paliwal R, Rai S, Vaidya B, et al. Effect of lipid core material on characteristics of solid lipid nanoparticles designed for oral lymphatic delivery. Nanomedicine. 2009;5(2):184–191. | ||
Borkar N, Xia D, Holm R, et al. Investigating the correlation between in vivo absorption and in vitro release of fenofibrate from lipid matrix particles in biorelevant medium. Eur J Pharm Sci. 2014;51:204–210. | ||
Qi J, Lu Y, Wu W. Absorption, disposition and pharmacokinetics of solid lipid nanoparticles. Curr Drug Metab. 2012;13(4):418–428. | ||
Feeney OM, Williams HD, Pouton CW, Porter CJ. ‘Stealth’ lipid-based formulations: Poly(ethylene glycol)-mediated digestion inhibition improves oral bioavailability of a model poorly water soluble drug. J Control Release. 2014;192:219–227. | ||
Harris JM, Chess RB. Effect of pegylation on pharmaceuticals. Nat Rev Drug Discov. 2003;2(3):214–221. | ||
Svensson O, Thuresson K, Arnebrant T. Interactions between drug delivery particles and mucin in solution and at interfaces. Langmuir. 2008;24(6):2573–2579. | ||
Larsen AT, Sassene P, Müllertz A. In vitro lipolysis models as a tool for the characterization of oral lipid and surfactant based drug delivery systems. Int J Pharm. 2011;417(1–2):245–255. | ||
Lai SK, Wang YY, Hanes J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Adv Drug Del Rev. 2009;61(2):158–171. | ||
Plapied L, Duhem N, des Rieux A, Préat V. Fate of polymeric nanocarriers for oral drug delivery. Curr Opin Colloid Interface Sci. 2011;16(3):228–237. | ||
Zabaleta V, Ponchel G, Salman H, Agüeros M, Vauthier C, Irache JM. Oral administration of paclitaxel with pegylated poly(anhydride) nanoparticles: Permeability and pharmacokinetic study. Eur J Pharm Biopharm. 2012;81(3):514–523. | ||
Yuan H, Chen CY, Chai GH, Du YZ, Hu FQ. Improved transport and absorption through gastrointestinal tract by PEGylated solid lipid nanoparticles. Mol Pharm. 2013;10(5):1865–1873. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.