Back to Journals » Journal of Inflammation Research » Volume 18
Effects of Hypoglycemic Agents on Pulmonary Diseases: A Comprehensive Narrative Review
Authors Yang Y ![]() , Wang K, Chen S
, Wang K, Chen S
Received 28 July 2025
Accepted for publication 16 December 2025
Published 24 December 2025 Volume 2025:18 Pages 18053—18078
DOI https://doi.org/10.2147/JIR.S556790
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Tara Strutt
Yu Yang,1,2 Kan Wang,3 Shuchun Chen2,4,5
1Department of Pharmacy, The Second Hospital of Hebei Medical University, Shijiazhuang, People’s Republic of China; 2Department of Endocrinology, Hebei General Hospital, Shijiazhuang, People’s Republic of China; 3Department of Endoscopy, The Fourth Hospital of Hebei Medical University, Shijiazhuang, People’s Republic of China; 4Department of Internal Medicine, Hebei Medical University, Shijiazhuang, People’s Republic of China; 5Hebei Key Laboratory of Metabolic Diseases, Hebei General Hospital, Shijiazhuang, People’s Republic of China
Correspondence: Shuchun Chen, Department of Endocrinology, Hebei General Hospital, 348 Heping West Road, Shijiazhuang, Hebei, 050051, People’s Republic of China, Tel/Fax +86 311 85988406, Email [email protected]
Abstract: Beyond glycemic control, hypoglycemic agents exhibit multifaceted effects that may influence pulmonary health in patients with diabetes mellitus. This narrative review synthesizes available evidence from preclinical and clinical studies on the impact of major hypoglycemic drug classes—including biguanides, sulfonylureas, thiazolidinediones, α-glucosidase inhibitors, DPP-4 inhibitors, SGLT-2 inhibitors, GLP-1 receptor agonists, and insulin—on pulmonary diseases. Evidence suggests that these agents exert class-specific, and often conflicting, effects: preclinical studies support their protective potential in acute lung injury, while clinical data indicate variable efficacy in asthma, COPD, and respiratory infections including COVID-19. Conversely, some agents may be associated with increased risks of lung cancer or COPD exacerbations, underscoring the need for context-specific prescribing. Mechanistic insights from animal models primarily involve modulation of inflammatory, oxidative, and immune pathways. This narrative review aims to provide a clinical framework for personalizing hypoglycemic therapy in patients with comorbid pulmonary conditions, while underscoring the need for well-designed prospective studies to resolve existing controversies.
Keywords: hypoglycemic agents, pulmonary diseases, diabetes mellitus, drug class-specific effects, mechanisms of action
Introduction
Diabetes mellitus (DM) has emerged as a global epidemic, characterized by chronic hyperglycemia that drives widespread metabolic and vascular complications. Accumulating evidence identifies the lungs —endowed with an intricate network of alveolar capillaries and high metabolic activity—as critical targets of diabetic injury, with studies consistently demonstrating reduced lung function in patients with diabetes compared to nondiabetic individuals.1–3 In addition to impairing lung function, sustained hyperglycemia.also elevates the risk of various pulmonary complications: patients with type 2 diabetes (T2DM) face a higher likelihood of developing asthma, chronic obstructive pulmonary disease (COPD), pulmonary fibrosis, and pneumonia, while preexisting pulmonary conditions (eg, COPD, asthma) are further exacerbated by diabetic metabolic disturbances.4 Pulmonary complications of diabetes not only increase hospital readmission rates and long-term mortality but also impose a heavy healthcare burden, yet they remain underrecognized in routine diabetes management.
Hyperglycemia exerts multiple adverse effects on the lungs: it promotes airway hyperresponsiveness via the Rock pathway, accelerates lung fibrosis by activating STAT3, CTGF and TGFβ, enhances cancer cell growth, chronic inflammation, cytokine release and oxidative stress through the NFκB pathway, NOX activation and ROS/RNS production, and increases susceptibility to pulmonary infections by elevating airway surface liquid glucose concentrations and impairing lung immune cell function.5 These pathways collectively disrupt pulmonary homeostasis, creating a pathological basis for hypoglycemic agents to modulate lung disease outcomes by targeting hyperglycemia-related damage.
Historically, pulmonary outcomes have been largely neglected in major diabetes trials, creating a significant gap in our understanding of how glycemic management influences respiratory health. Against this backdrop, the role of hypoglycemic agents in modulating pulmonary health has garnered growing interest. Beyond their primary glycemic control function, these drugs exhibit multifaceted actions that may influence pulmonary pathophysiology—ranging from anti-inflammatory and antioxidant actions to regulation of immune responses and cellular repair mechanisms. Currently, the armamentarium of hypoglycemic agents encompasses diverse classes: biguanides (eg, metformin), α-glucosidase inhibitors (AGIs; eg, acarbose), insulin secretagogues (eg, sulfonylureas), thiazolidinediones (TZDs; eg, rosiglitazone), dipeptidyl peptidase-4 (DPP-4) inhibitors (eg, sitagliptin), sodium-glucose cotransporter 2 (SGLT-2) inhibitors (eg, dapagliflozin), glucagon-like peptide-1 receptor agonists (GLP-1 RAs; eg, liraglutide), and insulin. Each class interacts uniquely with molecular pathways in pulmonary tissues, potentially exerting protective or deleterious effects on lung health.
The pulmonary complications of diabetes, including accelerated lung function decline and increased susceptibility to infections, fibrosis, and obstructive diseases, remain underrecognized yet carry significant prognostic implications. This oversight is compounded by the multifaceted, off-target effects of hypoglycemic agents on the lungs, which are not systematically considered in clinical guidelines. Therefore, this narrative review aims to synthesize the current preclinical and clinical evidence regarding the impact of major hypoglycemic drug classes on a spectrum of pulmonary diseases. We first establish a mechanistic framework (Figure 1), which summarizes the core pathways (inflammatory, oxidative, immune modulation) through which hypoglycemic agents regulate pulmonary pathophysiology and then critically evaluate the class-specific effects of these agents on pulmonary conditions. By clarifying these pathways, resolving conflicting findings, and highlighting knowledge gaps, this review seeks to provide a clinical framework for personalizing hypoglycemic therapy in the growing population of patients with diabetes and comorbid pulmonary conditions.
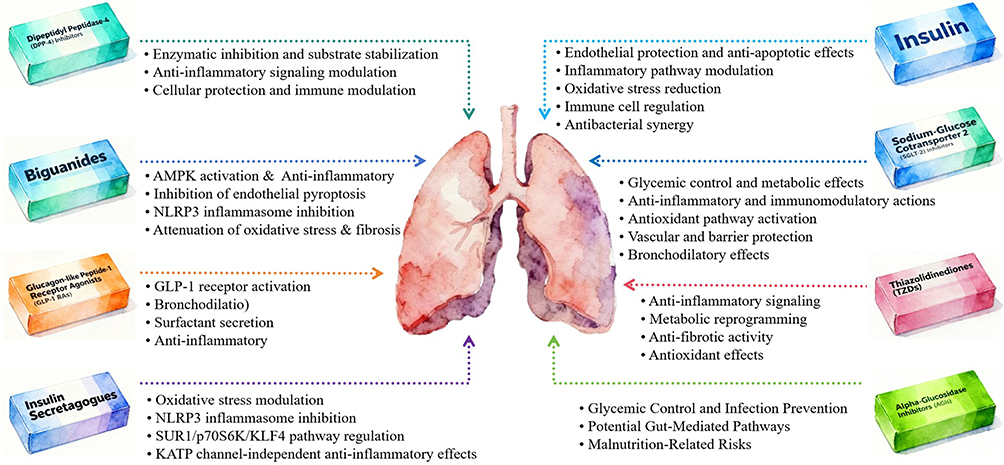 |
Figure 1 Core mechanisms of major hypoglycemic agents in regulating pulmonary pathophysiology. |
Literature Search and Selection Criteria
This narrative review synthesizes current evidence on the effects of different hypoglycemic agents in pulmonary diseases. The selection of cited literature was based on a comprehensive search of the PubMed database for relevant pre-clinical and clinical studies published up to 2025, aiming to cover key mechanistic pathways and major clinical outcomes for each drug class. Unlike a systematic review, this narrative approach does not employ strict, pre-defined inclusion/exclusion criteria or a formal risk-of-bias assessment. Instead, by evaluating their impacts on acute and chronic lung conditions—including infections (eg, COVID-19, pneumonia), fibrotic disorders, asthma, COPD, and lung cancer—this work integrates evidence from diverse study designs to clarify mechanistic pathways, resolve conflicting findings, and highlight critical knowledge gaps.
Hypoglycemic Agents
Biguanides
Biguanides, most notably metformin—a first-line oral antidiabetic agent—exhibit extensive protective effects across diverse pulmonary pathologies through multifaceted molecular mechanisms that extend beyond glycemic control. Their multifaceted actions, spanning metabolic signaling modulation, oxidative stress reduction, inflammation suppression, and mitochondrial function regulation, position metformin as a promising therapeutic adjunct in both acute and chronic lung diseases.
Mechanisms of Action
The pulmonary protective effects of metformin are rooted in its regulation of key molecular pathways and mitochondrial function. It acts as a central modulator of AMP-activated protein kinase (AMPK) and interacts with downstream signaling cascades including mammalian target of rapamycin (mTOR), sirtuin 1 (SIRT1), and nuclear factor-κB (NF-κB).6,7 Key mechanistic pathways include:
- Suppression of inflammatory responses via AMPK-dependent inhibition of mTOR, reducing the production of pro-inflammatory cytokines such as interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α).8
- Inhibition of endothelial pyroptosis through the SIRT1/NF-κB/NOD-like receptor pyrin domain-containing 3 (NLRP3) inflammasome axis, which decreases cleaved caspase-1, gasdermin D-N (GSDMD-N), and IL-1β levels in lung tissues and pulmonary endothelial cells.9
- Blockade of NLRP3 inflammasome activation independent of AMPK, by inhibiting mitochondrial ATP and DNA synthesis, thereby reducing alveolar macrophage-mediated inflammation.10
- Attenuation of oxidative stress and fibrosis via suppression of NADPH oxidase 4 (NOX4) expression, which reduces NOX4-derived reactive oxygen species (ROS) and subsequent SMAD phosphorylation in transforming growth factor-β (TGF-β)-mediated pathways.11
Effects on Pulmonary Diseases
Acute Lung Injury (ALI) and Acute Respiratory Distress Syndrome (ARDS)
Metformin exerts robust protective effects in ALI/ARDS induced by various insults. In endotoxemia-induced ALI, it alleviates pulmonary inflammation and histological damage by restoring AMPK-dependent suppression of mTOR.8 In ARDS associated with NLRP3 inflammasome activation (eg, severe COVID-19), metformin blocks NLRP3 activation and IL-1β production, mitigating alveolar inflammation.10 It also attenuates ventilator-induced lung injury in an ex vivo rabbit model by preserving alveolar-capillary permeability, reducing edema, microhemorrhage, and protein leakage in bronchoalveolar lavage fluid (BALF).12
Pulmonary Fibrosis (PF)
Metformin shows efficacy in multiple models of pulmonary fibrosis. In a murine model of radiation-induced pulmonary fibrosis, it reduces collagen deposition, TGF-β expression, and Smad2/3 phosphorylation, while mitigating alveolar structural damage and inflammatory infiltration.13 For PM2.5-induced lung injury, it suppresses systemic and pulmonary inflammation, oxidative stress, and fibrosis via an AMPKα2-independent mechanism linked to enhanced mitochondrial antioxidant enzyme activity.14 In silicosis, metformin alleviates silica-induced lung inflammation and PF at both early and late stages by reversing oxidative stress, epithelial-mesenchymal transition (EndMT), and fibroblast activation via an AMPK-dependent pathway.15
In idiopathic pulmonary fibrosis (IPF), metformin targets TGF-β-mediated pathways to inhibit myofibroblast differentiation. It activates AMPK to suppress NOX4 expression, reducing ROS production and SMAD phosphorylation—critical steps in TGF-β-induced fibrosis. In bleomycin-induced PF, metformin reduces collagen deposition and myofibroblast accumulation, with similar effects observed in fibroblasts from IPF patients.11 Additionally, it attenuates gefitinib-induced exacerbation of PF: in vitro, it inhibits TGF-β or EGFR-TKI-induced expression of fibrosis markers (Collagen1a1, α-actin) in lung fibroblasts by reducing SMAD2/3 phosphorylation; in vivo, it suppresses gefitinib-mediated worsening of bleomycin-induced PF in rats.16
Lung Cancer
Metformin exhibits complex and context-dependent effects on lung cancer, with mechanisms involving metabolic reprogramming and apoptosis induction. Lung squamous cell carcinoma (SCC) relies heavily on glycolysis. Metformin synergizes with hexokinase 2 (HK2) depletion: HK2 inhibition reduces glycolysis via AMPK/mTORC1 activation, and metformin enhances this effect by increasing oxygen respiration, thereby augmenting apoptosis in vitro and in vivo.17 In lung adenocarcinoma, metformin induces G0/G1 cell cycle arrest and mitochondria-mediated apoptosis in A549 cells by downregulating anti-apoptotic proteins (Bcl-2, Bcl-xL) and upregulating pro-apoptotic Bax.18 It also synergizes with cisplatin and etoposide in NCI-H460 cells, though cisplatin antagonizes its effects in most subtypes except adenocarcinoma.19,20
Clinical outcomes vary: in stage IV non-small cell lung cancer (NSCLC), metformin users had longer median survival (5 vs 3 months; HR 0.80);21 in diabetic NSCLC patients, metformin use was associated with prolonged overall survival (OS, 25.0 vs 11.5 months) and disease-free survival (15.6 vs 8.5 months), independent of other factors.22
Asthma
Metformin may improve outcomes in diabetic patients with concurrent asthma. Observational studies show that metformin users have a lower risk of asthma-related hospitalization (OR 0.21, 95% CI 0.07–0.63) and exacerbations (OR 0.39, 95% CI 0.19–0.79).23 A claims-based cohort study further demonstrates that metformin initiation is associated with reduced hazards of asthma exacerbations (HR 0.92, 95% CI 0.86–0.98), driven by fewer emergency department visits (HR 0.81, 95% CI 0.74–0.88) and hospitalizations (HR 0.67, 95% CI 0.50–0.91).24
Infectious Pulmonary Diseases
Metformin enhances host defense against diverse respiratory pathogens. It inhibits hyperglycemia-induced Pseudomonas aeruginosa growth in airway epithelial cultures by preserving tight junctions (claudin-1, occludin), reducing paracellular glucose flux and bacterial proliferation.25 Against Legionella pneumophila, metformin activates AMPK in macrophages, stimulates mitochondrial ROS, and enhances bactericidal activity, improving survival in murine pneumonia models.26 For tuberculosis (TB), metformin acts as a host-adjunctive therapy: it inhibits Mycobacterium tuberculosis (Mtb) intracellular growth via AMPK, promotes phagosome-lysosome fusion, and enhances the efficacy of conventional TB drugs in mice, with human cohorts showing improved Mtb control and reduced disease severity.27 In COVID-19, meta-analyses confirm that metformin is associated with lower mortality in patients with type 2 diabetes (OR 0.54, 95% CI 0.47–0.62).28,29 These meta-analyses adjusted for key covariates including age, sex, diabetes duration, and major comorbidities (eg, cardiovascular disease, chronic kidney disease), and baseline use of other antidiabetic drugs. However, residual confounding may persist from unmeasured factors such as COVID-19 disease severity at admission and high statistical heterogeneity, which limits the generalizability of the findings.
Neonatal Pulmonary Disorders
Metformin addresses hyperoxia-induced lung injury, a hallmark of bronchopulmonary dysplasia (BPD). In neonatal rats exposed to 100% oxygen, metformin reduces mortality, alveolar septal thickening, macrophage/neutrophil infiltration, and fibrosis (collagen III and fibrin deposition), while improving vascularization.30
Literature Conflicts, Inconsistencies, and Knowledge Gaps
Key Conflicts and Inconsistencies
- Lung cancer risk variability: Observational studies yield contradictory findings. Zhang et al reported a 29% reduced lung cancer risk (pooled RR 0.71, 95% CI 0.55–0.95) in metformin users without full adjustment for smoking status,31 while Smiechowski et al found no association (rate ratio 0.94, 95% CI 0.76–1.17) after adjusting for smoking and other confounders.32 A meta-analysis of 15 studies further confirmed no overall association (adjusted OR 0.99, 95% CI 0.87–1.12) due to high heterogeneity across study designs (cohort, case-control, RCT),33 while another meta-analysis of 15 studies reported a 15% reduced risk (OR 0.85, 95% CI 0.77–0.92)34—this protective effect was attenuated in smoking-adjusted sub-analyses (OR=0.84, 95% CI 0.61–1.06), indicating smoking status as a key confounder. Residual confounding from unmeasured factors such as smoking intensity or quit duration may explain the conflicting findings.
- Smoking status as a modifier: Sakoda et al showed an inverse association between metformin use and lung cancer only in never smokers (HR 0.57, 95% CI 0.33–0.99), but no effect in current smokers.35 However, few studies have stratified analyses by smoking intensity or quit duration, leading to unresolved uncertainty about metformin’s utility in smoking-related lung cancer risk.
- Survival outcomes in lung cancer: While a meta-analysis of 6 studies (2,350 patients) reported a modest improvement in OS with metformin (HR 0.90, 95% CI 0.84–0.96),36 a cohort study noted that metformin users who developed lung cancer had more metastatic disease and shorter survival (HR 1.47).37
Causes of Inconsistencies
Heterogeneity in study design (retrospective vs prospective), tumor subtype inclusion (squamous cell carcinoma vs adenocarcinoma), and confounding by concurrent antineoplastic therapies or smoking status explain discrepancies. For example, metformin’s synergism with HK2 depletion is specific to squamous cell carcinoma, but most epidemiological studies do not account for tumor histology.17 Smoking adjustment also plays a critical role—meta-analyses show protective effects are attenuated when controlling for smoking, highlighting unmeasured confounding.34
Knowledge Gaps
Several critical knowledge gaps persist regarding metformin’s pulmonary applications: the mechanisms underlying its divergent effects across lung cancer subtypes remain unclear, and there is a notable absence of long-term safety data in diabetic patients with idiopathic pulmonary fibrosis. Key unexplored areas include its potential drug interactions with inhaled corticosteroids for asthma, its role in post-COVID-19 pulmonary fibrosis, and its dose-dependent effects on pulmonary function in COPD patients with diabetes.
In summary, metformin demonstrates broad pulmonary protective effects, primarily mediated through AMPK activation, against acute lung injury, fibrosis, and infections, while also reducing asthma exacerbations. However, its role in lung cancer risk modulation remains uncertain due to conflicting evidence and significant confounding by smoking. It represents a favorable choice for diabetics with asthma or pulmonary fibrosis, though its use in patients with active lung cancer requires careful risk-benefit evaluation.
Insulin Secretagogues
In contrast to biguanides, insulin secretagogues—primarily sulfonylureas such as gliclazide and glibenclamide—exert pulmonary effects through distinct mechanisms. These agents stimulate insulin release from pancreatic β-cells and have been reported to show potential beneficial effects on pulmonary pathophysiology in preclinical studies, spanning anti-oxidative, anti-inflammatory, and anti-tumorigenic activities. Their mechanisms, often involving the modulation of oxidative stress, inflammatory signaling, and cellular proliferation, position them as potential therapeutics for pulmonary disorders beyond glycemic control.
Mechanisms of Action
The pulmonary effects of sulfonylureas are mediated through several key molecular pathways:
Oxidative stress modulation: Gliclazide demonstrates radioprotective effects by reducing oxidative stress markers (malondialdehyde [MDA], protein carbonyl [PC]) and increasing glutathione (GSH) levels in lung tissue.38
NLRP3 inflammasome inhibition: Glibenclamide exerts anti-inflammatory effects by blocking NLRP3 inflammasome activation, evidenced by reduced expression of NLRP3, caspase-1 p10, and pro-inflammatory cytokines (IL-1β, IL-18) in lung tissues and BALF.39,40
Sulfonylurea receptor 1 (SUR1)/p70S6K/KLF4 pathway regulation: Glibenclamide inhibits NSCLC cell growth, epithelial-mesenchymal transition, and migration by suppressing p70S6K activity and upregulating the tumor suppressor Krüppel-like factor 4 (KLF4).41
KATP channel-independent anti-inflammatory effects: Despite their KATP channel inhibitory properties, sulfonylureas demonstrate unexpected benefits in asthma models, possibly through modulation of Th2 cytokines and STAT6 signaling.42
Effects on Pulmonary Diseases
Radiation-Induced Lung Injury
Gliclazide demonstrates radioprotective effects in irradiation-induced lung injury. In a murine model of thoracic irradiation, gliclazide mitigates histopathological damage by reducing oxidative stress markers and increasing glutathione levels, thereby preserving lung tissue architecture.38
Lung Cancer and Carcinogenesis
Sulfonylureas show anti-tumorigenic effects through multiple mechanisms. Gliclazide has been identified as a candidate therapeutic for lung adenocarcinoma (LUAD), inducing cell cycle arrest and apoptosis in LUAD cells by targeting key regulators of cell division (CCNB1, CCNB2, CDK1, and AURKA).43 Glibenclamide decreases tumor incidence, mean tumor count, and pathological severity in benzo(a)pyrene (B(a)p) plus lipopolysaccharide (LPS)-induced lung carcinogenesis by downregulating NLRP3, IL-1β, and cleaved IL-1β expression.40 In NSCLC, glibenclamide inhibits cancer cell growth and migration via the SUR1/p70S6K/KLF4 pathway, with in vivo xenograft models confirming reduced tumor growth.41
Obstructive Airway Diseases
In asthma, glibenclamide unexpectedly mitigates key pathological features despite its KATP channel inhibitory properties. In an ovalbumin (OVA)-induced murine asthma model, it reduces airway hyperresponsiveness (AHR), airway inflammation, and Th2 cytokines, while downregulating VCAM-1 and phosphorylated STAT6 in lung tissues.42 For COPD in patients with type 2 diabetes, a nationwide cohort study utilizing propensity-score matching demonstrated that sulfonylurea users had significantly lower risks of all-cause mortality (adjusted HR 0.53, 95% CI 0.48–0.58), cardiovascular events (aHR 0.88, 0.81–0.96), invasive mechanical ventilation (aHR 0.57, 0.50–0.66), non-invasive positive pressure ventilation (aHR 0.74, 0.60–0.92), and bacterial pneumonia (aHR 0.78, 0.70–0.87) compared to matched non-users over a mean follow-up of 6.57 years.44 A longer cumulative duration of SU use was associated with progressively lower risks of these outcomes. However, these findings from observational data must be interpreted with caution due to potential confounding. The non-user comparator group is heterogeneous and may include both healthier patients managed by lifestyle alone and those with contraindications to oral therapy. Unmeasured factors, such as socioeconomic status, overall healthcare adherence, or prescriber bias, could influence both the choice of sulfonylurea and the pulmonary outcomes. Therefore, while suggestive, this association does not establish causality and requires validation in studies with more robust designs to control for these confounders.
Environmental and Oxidative Lung Injury
Glibenclamide exerts robust anti-inflammatory effects in oxidative and environmental insult-induced lung injury. In a murine model of ozone-induced pulmonary inflammation, glibenclamide reduces inflammatory cell infiltration and protein leakage in BALF, lowers histopathological inflammatory scores, and decreases alveolar damage through NLRP3 inflammasome inhibition.39
Infectious Diseases
In COVID-19, sulfonylureas show neutral effects on mortality in diabetic patients. A Bayesian network meta-analysis found no association between sulfonylurea use and COVID-19 mortality or severe disease risk (OR=0.97, 95% CI 0.83–1.01).29 This analysis adjusted for baseline covariates such as age, comorbidity burden, and concurrent use of other antidiabetic drugs, though potential confounding from unmeasured factors (eg, healthcare access or COVID-19 treatment intensity) cannot be fully excluded due to limited individual patient data.
Literature Conflicts, Inconsistencies, and Knowledge Gaps
Key Conflicts and Inconsistencies
- Mechanistic paradox in asthma: Despite KATP channel blockade theoretically promoting bronchoconstriction, glibenclamide demonstrates anti-asthmatic effects in preclinical models, suggesting involvement of other, non-canonical pathways that remain incompletely characterized.42
- Translation gap between preclinical and clinical evidence: While robust preclinical data support sulfonylureas’ pulmonary benefits, clinical evidence remains limited to observational studies,44 with no interventional trials confirming these effects.38–40
- Class-effect variability: Most evidence derives from glibenclamide and gliclazide, creating uncertainty about whether pulmonary effects are consistent across all sulfonylureas or agent-specific.
- Residual confounding in observational evidence: While the nationwide cohort study employed propensity-score matching to improve comparability between groups, residual confounding remains possible.44 Important clinical parameters such as COPD severity (eg, GOLD stage), smoking status, and lung function measures were not available in the administrative database and thus could not be fully adjusted for. Therefore, while this large, well-conducted study provides compelling real-world evidence, the observed associations should not be interpreted as definitive proof of causality.
Causes of Inconsistencies
The stark contrast between extensive preclinical data and limited clinical validation stems from fundamental differences in pathophysiology between animal models and human chronic diseases, as well as the predominance of retrospective study designs susceptible to confounding. The paradoxical efficacy in asthma models despite theoretical bronchoconstrictive potential suggests the involvement of alternative mechanisms (eg, NLRP3 inhibition) that may predominate in specific contexts.
Knowledge Gaps
Several key research gaps exist regarding sulfonylureas’ pulmonary effects: the anti-asthmatic mechanisms of glibenclamide independent of KATP channel blockade remain undefined, and the long-term pulmonary outcomes in diabetic patients with pre-existing lung conditions are unknown. Furthermore, their potential synergistic benefits with standard therapies, differential effects among various agents on specific diseases, and role in managing drug-induced pulmonary toxicity remain unexplored.
Adverse Effect Considerations: Hypoglycemia and Pulmonary Risk
A critical drawback of insulin secretagogues, particularly sulfonylureas, is their elevated risk of hypoglycemia. Notably, a cohort study that reported pulmonary benefits also found sulfonylurea use was associated with an increased risk of hypoglycemia compared to non-use, highlighting the critical need to balance potential benefits against this well-established risk in clinical decision-making.44 Severe hypoglycemic events may theoretically influence pulmonary risk through several mechanisms. Neuroglycopenia and altered mental status during hypoglycemia could increase the risk of aspiration pneumonia, especially in elderly or frail patients. Additionally, the systemic stress response triggered by hypoglycemia, involving catecholamine release and inflammation, might theoretically exacerbate underlying pulmonary conditions such as asthma or COPD. Although this pathway remains speculative and warrants direct investigation, it underscores the importance of meticulous glycemic management to avoid hypoglycemia when using these agents in patients with significant pulmonary comorbidity.
Taken together, these data suggest that sulfonylureas, particularly gliclazide and glibenclamide, demonstrate promising anti-inflammatory, antioxidant, and anti-tumorigenic effects in preclinical models. However, clinical translation remains limited, and their association with hypoglycemia poses a significant risk that may offset pulmonary benefits in vulnerable populations. Future studies should clarify whether these effects are class-wide or agent-specific.
Thiazolidinediones (TZDs)
Unlike insulin secretagogues, TZDs exert their pulmonary effects primarily through peroxisome proliferator-activated receptor gamma (PPAR-γ) activation—a nuclear receptor integral to metabolic regulation and inflammatory response modulation. As PPAR-γ agonists, TZDs exhibit multifaceted effects beyond glycemic control, influencing pulmonary inflammation, remodeling, and injury through anti-inflammatory, antioxidant, and anti-fibrotic pathways.45
Mechanisms of Action
The pulmonary benefits of TZDs are predominantly mediated through PPAR-γ-dependent mechanisms:
- Anti-inflammatory signaling: TZDs suppress pro-inflammatory pathways (eg, NF-κB activation), reducing cytokines (TNF-α, IL-5), chemokines, and adhesion molecules (ICAM-1).46,47
- Metabolic reprogramming: In pulmonary arterial hypertension, pioglitazone restores fatty acid oxidation (FAO) through miRNA modulation (miR-197, miR-146b), reversing vascular remodeling.48
- Anti-fibrotic activity: PPAR-γ activation inhibits TGF-β-induced myofibroblast differentiation, fibroblast proliferation, and collagen secretion, attenuating fibrotic progression.49
- Antioxidant effects: Rosiglitazone enhances antioxidant capacity by increasing superoxide dismutase (SOD) and glutathione (GSH) levels while reducing oxidative stress markers.46
Effects on Pulmonary Diseases
Asthma and Allergic Airway Disease
Preclinical studies demonstrate promising effects of TZDs in asthma models. Inhaled ciglitazone reduces airway hyperresponsiveness (AHR), remodeling, and TGF-β synthesis,50 while rosiglitazone lowers pro-inflammatory mediators (IL-5, IgE) and enhances antioxidant defenses.46 Human asthmatic airways exhibit elevated PPAR-γ expression linked to remodeling, with corticosteroids downregulating its expression.51 However, clinical translation has been limited—a 12-week trial of pioglitazone showed no benefit for mild asthma, directly contrasting with the robust efficacy observed in murine models.52 This stark disconnect not only underscores the limitations of animal models in recapitulating human chronic inflammatory airway disease but also suggests that PPAR-γ activation may be insufficient as a monotherapy in established human asthma, or that benefits are restricted to specific, yet-to-be-defined asthma endotypes.
Neonatal Lung Development and Injury
TZDs protect against neonatal hyperoxia-induced lung injury, a model of bronchopulmonary dysplasia (BPD). Rosiglitazone prevents hyperoxia-induced neonatal rat lung injury by inhibiting lipo-to-myofibroblast transdifferentiation, preserving alveolar architecture and promoting normal lung development.53
Acute Lung Injury and Endotoxemia
In endotoxemia models, rosiglitazone reduces pulmonary edema, neutrophilia (as measured by MPO activity), oxidative stress, and pro-inflammatory factors (TNF-α, ICAM-1) via PPAR-γ-mediated NF-κB inhibition, supporting their potential utility in sepsis-related lung injury.47
Pulmonary Arterial Hypertension (PAH)
Pioglitazone demonstrates therapeutic potential in experimental PAH. It reverses vascular remodeling and right ventricular (RV) failure in SuHx rats by restoring fatty acid oxidation through miRNA modulation, with similar effects observed in human PAH samples.48
Pulmonary Fibrosis (PF)
PPAR-γ agonists exhibit consistent anti-fibrotic effects across multiple models. They inhibit fibroblast proliferation, TGF-β-induced myofibroblast differentiation, and collagen secretion, reducing bleomycin-induced fibrosis even when administered post-inflammation.49
Lung Cancer
Epidemiological evidence regarding TZDs and lung cancer risk remains inconclusive. While one cohort study linked TZD/metformin use to lower lung cancer risk,37 a subsequent meta-analysis found no significant protective effect for TZDs alone (OR 0.86, 95% CI 0.70–1.02).34
Literature Conflicts, Inconsistencies, and Knowledge Gaps
Key Conflicts and Inconsistencies
- Asthma efficacy gap: The stark contrast between consistent benefits in animal models of asthma46,50 and the lack of efficacy in human clinical trialsrepresents a major translational disconnect.52
- Lung cancer risk uncertainty: While preclinical data suggest potential anti-neoplastic effects through PPAR-γ activation, epidemiological studies show inconsistent associations with lung cancer risk.34,37
- PPAR-γ expression paradox: The finding that PPAR-γ is elevated in human asthmatic airways yet its activation does not translate to clinical benefit raises questions about receptor functionality and downstream signaling in human disease.51
Causes of Inconsistencies
The divergence between robust preclinical findings and limited clinical success stems from multiple factors: fundamental differences between animal models and human chronic inflammatory diseases; suboptimal patient stratification in clinical trials; and potential confounding by TZDs’ side effect profile (eg, fluid retention, weight gain) that may limit their utility in patients with compromised respiratory function. The context-dependent nature of PPAR-γ signaling—being influenced by disease stage, cellular microenvironment, and co-regulator availability—further complicates clinical translation.
Knowledge Gaps
Several critical research gaps persist in the application of TZDs for pulmonary diseases. The efficacy boundary between preclinical models and human asthma remains poorly defined, particularly regarding optimal dosing and patient selection. The safety and tolerability of TZDs, particularly regarding fluid retention and risk of pulmonary edema, in patients with pre-existing cardiopulmonary diseases (eg, COPD with cor pulmonale, pulmonary hypertension) remain poorly characterized and pose a major barrier to clinical trials in these populations. Furthermore, their utility in specific asthma endotypes and predictive biomarkers for treatment response are lacking, hindering targeted clinical translation.
Safety Considerations and Pulmonary Implications
TZDs promote renal sodium reabsorption and plasma volume expansion, leading to peripheral edema and increasing the risk of congestive heart failure. In patients with underlying cardiopulmonary compromise, this can precipitate or worsen pulmonary edema, presenting as increased dyspnea, which can be misattributed to progression of underlying lung diseases like COPD or asthma, thereby triggering unnecessary exacerbation management. This safety profile presents a substantial barrier to the repurposing of TZDs for chronic lung diseases. Clinicians managing diabetic patients with comorbid respiratory conditions must be vigilant about these risks, as the drug-induced pulmonary edema can mimic disease exacerbations. This necessitates careful patient selection, avoiding TZDs in patients with a history of heart failure or significant baseline edema.
Taken together, these data suggest that while thiazolidinediones exhibit robust preclinical pulmonary benefits (anti-asthma, anti-fibrosis, PAH improvement) via PPARγ activation, these effects fail to translate to human clinical outcomes. Their primary safety concern of fluid retention-induced pulmonary edema further restricts use in patients with cardiopulmonary compromise, making them an unfavorable choice for chronic lung disease repurposing.
Alpha-Glucosidase Inhibitors (AGIs)
Distinct from TZDs, AGIs modulate pulmonary health primarily through glycemic control and indirect systemic effects, demonstrating context-dependent outcomes in specific respiratory conditions. Unlike drugs with direct pulmonary molecular targets, AGIs exert their influence primarily by reducing postprandial hyperglycemia, thereby generating unique lung effects that vary significantly across different disease contexts.
Mechanisms of Action
The pulmonary effects of alpha-glucosidase inhibitors (AGIs) are primarily mediated through the following indirect mechanisms:
- Glycemic Control and Infection Prevention: By delaying carbohydrate digestion and reducing postprandial hyperglycemia, AGIs lower glucose availability in airway surfaces, thereby inhibiting bacterial growth and potentially contributing to their protective role in respiratory infections like COVID-19.54,55
- Potential Gut-Mediated Pathways: Emerging evidence suggests that AGIs may modulate gut microbiota composition and function, potentially influencing systemic immunity and inflammatory responses relevant to lung health through the gut-lung axis.
- Malnutrition-Related Risks: The proposed association with increased COPD risk may be linked to drug-induced gastrointestinal side effects (eg, malabsorption),56 which could lead to chronic malnutrition and weight loss, thereby exacerbating pulmonary vulnerability in susceptible individuals; however, this pathway remains speculative.
Effects on Pulmonary Diseases
COVID-19
AGIs demonstrate promising protective effects in patients with type 2 diabetes mellitus (T2DM) affected by COVID-19. A multi-institutional retrospective study in China, including 4922 T2DM patients with COVID-19, found that AGI use was associated with significantly lower all-cause mortality (adjusted HR 0.53, 95% CI 0.35–0.80), with similar trends observed after propensity score matching (adjusted HR 0.59, 95% CI 0.35–0.98).54 This aligns with another study showing that inpatient use of acarbose, either alone or in combination with metformin, significantly improved survival rates in T2DM patients with COVID-19: 91.2% of acarbose users survived, compared to lower survival rates in non-users.55
COPD
Contrasting with their beneficial effects in COVID-19, AGIs have been associated with an increased risk of COPD development in diabetic patients. A cohort study revealed that AGI use was associated with a nearly twofold higher risk of COPD (HR 1.964, 95% CI 1.207–2.380) compared to non-users, with a more pronounced risk observed in direct comparisons with other oral antidiabetic medications (HR 2.295, 95% CI 1.304–4.038).56 Proposed mechanisms include potential gastrointestinal side effects (eg, malabsorption) leading to malnutrition, which may exacerbate pulmonary vulnerability, though further research is needed to confirm causality.
Literature Conflicts, Inconsistencies, and Knowledge Gaps
Key Conflicts and Inconsistencies
- Disease-specific divergence: The most striking conflict involves the opposing effects of AGIs in different respiratory conditions—demonstrating protective effects in COVID-19 while being associated with increased COPD risk.54–56
- Mechanistic uncertainty: The protective mechanisms in COVID-19 remain poorly characterized, while the proposed mechanism for increased COPD risk (malabsorption-related malnutrition) lacks direct experimental validation.
- Confounding by indication: The observed associations may be influenced by prescribing patterns, as patients selected for AGI therapy may have distinct baseline characteristics that independently affect pulmonary outcomes.
Causes of Inconsistencies
The contradictory findings likely stem from fundamental differences in the pathophysiology of acute viral infection (COVID-19) versus chronic inflammatory disease (COPD), with AGIs exerting different effects in these distinct contexts. The predominance of observational evidence introduces substantial confounding, as AGI users may differ from non-users in ways that affect respiratory outcomes. Additionally, the lack of mechanistic studies specifically examining pulmonary effects of AGIs leaves critical pathways unexplored.
Knowledge Gaps
Substantial knowledge gaps persist regarding the pulmonary effects of AGIs. The molecular mechanisms underlying their protective effect in COVID-19 remain largely unknown beyond glycemic control, particularly concerning potential immunomodulatory or antiviral actions.54,55 Crucially, the nature of the association with increased COPD risk requires clarification through prospective studies that rigorously control for nutritional status, comorbidities, and prescriber bias to determine if it represents true causality or confounding.56 The effects of AGIs on other major respiratory conditions, including asthma, pulmonary fibrosis, and lung cancer, remain completely unexplored. Furthermore, potential drug-drug interactions with common pulmonary medications and the comparative impact of different AGIs (acarbose, miglitol, voglibose) on specific respiratory outcomes warrant systematic investigation to guide clinical decision-making.
Taken together, these data suggest that alpha-glucosidase inhibitors exert context-dependent pulmonary effects—reducing COVID-19 mortality via glycemic control and potential gut-lung axis modulation, yet associating with increased COPD risk (possibly linked to malabsorption). Given the lack of mechanistic clarity and conflicting disease-specific outcomes, they may be considered for diabetic patients with COVID-19 but require caution in those at high risk of COPD.
Dipeptidyl Peptidase-4 (DPP-4) Inhibitors
Dipeptidyl peptidase-4 (DPP-4) inhibitors modulate pulmonary pathophysiology through enzymatic inhibition and immunomodulatory pathways, demonstrating complex and context-dependent effects across respiratory diseases. Beyond their glycemic benefits through incretin hormone stabilization, these agents influence pulmonary health via DPP-4’s widespread expression in respiratory tissues and its role in regulating inflammatory, oxidative, and immune responses. DPP-4 is a membrane-anchored ectopeptidase with a broad spectrum of biological functions in immune regulation, cancer biology, and glucose metabolism. Growing evidence suggests that DPP-4 plays a deleterious role in respiratory diseases, and progressing our knowledge of this multi-faceted molecule may yield novel therapies for conditions such as lung cancer, asthma, and COPD.57
Mechanisms of Action
DPP-4 inhibitors exert pulmonary effects through multiple distinct yet interconnected pathways:
- Enzymatic inhibition and substrate stabilization: DPP-4 is widely distributed in human respiratory tissues, with enriched expression in distal airways, alveolar parenchyma (type I/II pneumocytes, alveolar macrophages), and vascular endothelium.58 Chronic lung diseases further upregulate DPP-4 expression, enhancing susceptibility to pathogens and exacerbating inflammation.
- Anti-inflammatory signaling modulation: Trelagliptin suppresses TLR4/NF-κB signaling,59 Saxagliptin balances the Nrf-2/HO-1 antioxidantand NF-κB pro-inflammatory pathways,60 and Omarigliptin activates the AMPK pathway and upregulates SOCS1 to reduce mucus overproduction.61
- Cellular protection and immune modulation: Linagliptin activates the Epac1/AKT pathway to preserve the pulmonary microvascular barrier,62 Vildagliptin enhances immune surveillance by boosting macrophage-mediated NK cell activity and TRAIL cytotoxicity,63 and multiple inhibitors collectively attenuate pathological processes like endothelial-to-mesenchymal transition and extracellular matrix deposition.64,65
Effects on Pulmonary Diseases
Asthma and COPD
DPP-4 contributes to airway inflammation and remodeling in asthma, stimulating fibroblast and smooth muscle cell proliferation.66 While sitagliptin alleviates inflammation and remodeling in murine models,67 clinical trials show no significant advantage over other glucose-lowering agents.68 Similarly, in COPD, DPP-4 inhibitors reduce neutrophilic inflammation in preclinical models but demonstrate limited clinical efficacy in preventing exacerbations.69,70
Acute and Chronic Lung Injury
DPP-4 inhibitors demonstrate consistent benefits in experimental lung injury models. Multiple agents protect against LPS-induced ALI through anti-inflammatory and antioxidant mechanisms.59–62 In pulmonary fibrosis, vildagliptin attenuates extracellular matrix deposition and EndMT in both LPS- and bleomycin-induced models.64,65 Sitagliptin additionally alleviates pulmonary arterial remodeling in experimental hypertension.71
Lung Cancer and Infectious Diseases
Vildagliptin exhibits anti-tumor activity by enhancing macrophage-mediated NK cell function.63 In COVID-19, evidence remains conflicting—one meta-analysis associated DPP-4 inhibitors with increased mortality,28 while another found that DPP-4 inhibitors were associated with reduced mortality,29 reflecting substantial methodological heterogeneity. The marked heterogeneity in these findings likely stems from significant differences in study designs, patient populations (eg, varying baseline disease severity, comorbidities), and concurrent treatments. Unmeasured confounding remains a critical limitation, as patients prescribed DPP-4 inhibitors may systematically differ from those on other glucose-lowering regimens in ways that independently influence COVID-19 prognosis.
Rare Adverse Pulmonary Events
While generally well-tolerated, DPP-4 inhibitors have been very rarely associated with idiosyncratic pulmonary reactions. A notable but isolated case report describes a hemodialysis patient with diabetic nephropathy who developed sarcoid-like lung granulomas following vildagliptin administration. The granulomas resolved completely within 4 months of drug discontinuation, suggesting a causal link.72 It is crucial to emphasize that such events are exceedingly rare, and their clinical prevalence should not be overstated.
Literature Conflicts, Inconsistencies, and Knowledge Gaps
Key Conflicts and Inconsistencies
The literature on DPP-4 inhibitors reveals significant conflicts primarily centered on their efficacy in chronic respiratory diseases and their role in COVID-19 outcomes. The most prominent inconsistency involves the stark contrast between robust preclinical benefits in asthma and COPD models and the limited clinical efficacy observed in human trials.67–70 Similarly, meta-analyses present conflicting evidence regarding COVID-19 mortality, with one reporting increased risk and another suggesting potential benefits.28,29 Additional discrepancies include drug-specific effect variations, where vildagliptin demonstrates unique anti-fibrotic and anti-cancer properties not consistently observed with other DPP-4 inhibitors.63–65
These conflicts stem from methodological heterogeneity between controlled animal studies and heterogeneous clinical populations, inadequate patient stratification by DPP-4 expression levels, and pharmacokinetic differences affecting tissue distribution among various inhibitors.
Causes of Inconsistencies
The observed inconsistencies stem from multiple interrelated factors, including fundamental disparities between preclinical models and human diseases, inadequate patient stratification by DPP-4 expression levels, significant pharmacokinetic variations among individual DPP-4 inhibitors leading to differential tissue distribution, and substantial residual confounding in observational studies, where factors influencing both drug prescription and disease outcome can create spurious associations.
Knowledge Gaps
Substantial knowledge gaps persist across multiple domains. The effects of DPP-4 inhibitors in cystic fibrosis patients remain unexplored despite known DPP-4 upregulation in this condition.58 Long-term outcomes in idiopathic pulmonary fibrosis and potential interactions with biologic therapies or corticosteroids are unknown. The efficacy across different asthma endotypes and in addressing post-COVID-19 pulmonary sequelae requires systematic evaluation. Additionally, risk factors for rare adverse events such as granulomatous reactions remain uncharacterized.72 These gaps highlight the need for stratified clinical trials based on DPP-4 expression patterns, comparative effectiveness studies among different DPP-4 inhibitors, and dedicated investigations in understudied pulmonary conditions where DPP-4 dysregulation may play a pathogenic role.
Taken together, these data suggest that DPP-4 inhibitors modulate pulmonary inflammation and fibrosis in preclinical settings, but their clinical impact on chronic respiratory diseases is modest and inconsistent. Conflicting COVID-19 outcomes and rare adverse events further complicate their pulmonary risk-benefit profile, underscoring the need for biomarker-guided patient selection.
Sodium-Glucose Cotransporter 2 (SGLT-2) Inhibitors
SGLT-2 inhibitors, including empagliflozin, dapagliflozin, and canagliflozin, represent a novel class of oral hypoglycemic agents that reduce plasma glucose by inhibiting renal glucose reabsorption. Beyond their metabolic benefits, emerging evidence demonstrates these agents exert multifaceted protective effects on pulmonary health through both direct actions on pulmonary tissues and indirect systemic mechanisms. The expression of SGLT transporters in lungs and pulmonary vasculature provides an anatomical basis for their direct pulmonary effects.73,74
Potential Pulmonary Risks and Safety Considerations
Despite the robust profile of pulmonary benefits, a comprehensive assessment must consider theoretical risks related to the established systemic effects of SGLT-2 inhibitors. Volume depletion, resulting from their natriuretic and diuretic actions, could theoretically impair respiratory function in vulnerable populations, such as patients with severe chronic obstructive pulmonary disease (COPD) or pre-existing respiratory failure, by potentially altering mucus clearance or pulmonary hemodynamics. Additionally, while euglycemic diabetic ketoacidosis (DKA) is a recognized metabolic complication, its direct impact on pulmonary physiology (eg, through acid-base disturbances) remains a theoretical concern. Crucially, large-scale meta-analyses of randomized controlled trials (RCTs), which provide the highest quality of safety evidence, have not identified a significant increase in respiratory adverse events, respiratory failure, or other pulmonary-specific harms attributable to these mechanisms.75–77 This suggests that while these theoretical risks warrant appropriate clinical vigilance, particularly in high-risk individuals, they do not translate into a significant signal against the overall backdrop of pulmonary benefits observed in broad patient populations with type 2 diabetes.
Mechanisms of Action
SGLT-2 inhibitors protect against pulmonary diseases through several interconnected pathways:
- Glycemic control and metabolic effects: By reducing blood and bronchoalveolar lavage fluid glucose concentrations, these agents create a less favorable environment for bacterial growth in the lungs, as demonstrated by dapagliflozin’s reduction of Pseudomonas aeruginosa burden in diabetic mice.78
- Anti-inflammatory and immunomodulatory actions: Empagliflozin and canagliflozin suppress immune cell infiltration and pro-inflammatory cytokine mRNA expression in allergic asthma models.79 Empagliflozin additionally normalizes T helper 1 (Th1) and Th17 cell differentiation in obesity-related airway dysfunction.80
- Antioxidant pathway activation: Dapagliflozin significantly reduces oxidative stress markers (MDA) while increasing total antioxidant capacity in experimental asthma.81
- Vascular and barrier protection: Empagliflozin protects against pulmonary ischemia/reperfusion injury via activation of extracellular signal-regulated kinases 1 and 2 (ERK1/2) signaling pathway.82
- Bronchodilatory effects: Dapagliflozin mitigates bronchospasm through activation of the NO/sGC/cGMP pathway and downregulation of S100A4.81
Effects on Pulmonary Diseases
Collectively, the protective effects of SGLT-2 inhibitors on pulmonary health are supported by a hierarchy of evidence. The most robust support comes from meta-analyses of large, randomized controlled trials (RCTs), which consistently show reduced risks of several respiratory diseases. These findings are complemented by supportive data from observational studies and compelling mechanistic insights from preclinical models.
Infectious Respiratory Diseases
SGLT-2 inhibitors demonstrate consistent benefits against pulmonary infections. A large real-world cohort study from Hong Kong showed that, compared with DPP-4 inhibitors, SGLT-2 inhibitor use was associated with a lower incidence of pneumonia (adjusted HR 0.63) and sepsis (adjusted HR 0.52), as well as reduced infection-related mortality.83 This is strongly supported by a meta-analysis of 26 randomized controlled trials (RCTs), which found that SGLT-2 inhibitors significantly reduced the risk of pneumonia (pooled RR 0.87) and septic shock (pooled RR 0.65) compared to placebo.84 More robustly, meta-analyses of large randomized controlled trials (RCTs) confirm these agents reduce the risk of infectious respiratory diseases including bronchitis and pneumonia.75 Their protective role extends to viral infections, with multiple meta-analyses showing lower mortality among patients with T2DM and COVID-19.28,29
Obstructive Airway Diseases
In asthma, SGLT-2 inhibitors exert comprehensive protective effects across multiple experimental models.79,81 Clinically, meta-analyses of large RCTs provide consistent, high-quality evidence that SGLT-2 inhibitors reduce the risk of asthma incidence and exacerbations.68,75,76 For COPD and obstructive airway diseases (OAD), observational data suggest that SGLT-2 inhibitor use is associated with a lower risk of incident OAD and reduced rate of OAD exacerbations compared to DPP-4 inhibitor use.85 A large population-based cohort study specifically conducted in patients with T2DM and comorbid COPD confirmed that SGLT-2 inhibitors are associated with a 38% decreased risk of severe exacerbations compared to DPP-4 inhibitors.70 These findings are reinforced by meta-analyses of large trials, which report that SGLT-2 inhibitors reduce the risk of COPD and related exacerbations.75–77
Obesity-Related Respiratory Complications
SGLT-2 inhibitors effectively address obesity-induced respiratory pathophysiology. Empagliflozin reduces airway hyperresponsiveness and lung fibrosis in high-fat diet-induced obese mice, while lowering pro-inflammatory mediators (IL-17, TGF-β1, IL-1β) and normalizing Th1/Th17 cell differentiation.80 These findings support their potential in managing obesity-related asthma and metabolic airway dysfunction.
Other Respiratory Conditions
Beyond common respiratory diseases, meta-analyses of RCTs associate SGLT-2 inhibitors are associated with reduced risks of sleep apnea syndrome, acute pulmonary edema, and respiratory failure.75–77,86,87 Empagliflozin specifically protects against pulmonary ischemia/reperfusion injury by improving respiratory function, attenuating lung edema and structural damage, and inhibiting apoptosis.82
Literature Conflicts, Inconsistencies, and Knowledge Gaps
Key Conflicts and Inconsistencies
Despite generally consistent beneficial effects, some uncertainties remain. The relative contribution of direct pulmonary actions versus systemic metabolic benefits remains debated, particularly whether observed clinical benefits primarily result from glycosuria-induced glycemic improvement or direct anti-inflammatory effects on lung tissues. Additionally, while most studies report class-effects, potential differences among individual SGLT-2 inhibitors require further comparative investigation.
Causes of Inconsistencies
The observed inconsistencies primarily stem from the complex interplay between direct pulmonary effects and systemic metabolic improvements, coupled with methodological variations across studies. The uncertain expression patterns and functional significance of SGLT transporters in different lung cell types create fundamental mechanistic ambiguity.73,74 Clinical heterogeneity in study populations, including variations in diabetes duration, glycemic control, and concurrent respiratory conditions, significantly influences outcomes.75,76,83 Additionally, residual confounding in observational studies, where factors influencing both drug prescription and disease outcome can create spurious associations, contributes to the current uncertainties. The predominant focus on class effects rather than drug-specific comparisons, along with variations in follow-up duration across studies, further contributes to the current uncertainties.
Knowledge Gaps
Several important questions warrant further research. The expression patterns and functional roles of SGLT transporters in different human lung cell types need comprehensive characterization. The efficacy of SGLT-2 inhibitors in non-diabetic pulmonary diseases remains largely unexplored despite compelling mechanistic rationale. Long-term effects on lung function decline and pulmonary vascular health require prospective evaluation. Potential interactions between SGLT-2 inhibitors and pulmonary medications commonly used in chronic respiratory diseases represent another unexplored area. Finally, biomarker development to identify patient subgroups most likely to benefit from SGLT-2 inhibitor therapy could enhance personalized treatment approaches.
In conclusion, SGLT-2 inhibitors currently present the most consistent and broad-spectrum pulmonary benefits among hypoglycemic agents. Their efficacy in reducing pneumonia, asthma exacerbations, and COPD-related outcomes is supported by high-quality evidence from RCT meta-analyses. While the interplay between direct pulmonary actions and systemic metabolic benefits warrants further investigation, their favorable risk-benefit profile makes them a prime consideration for diabetic patients with comorbid respiratory conditions.
Glucagon-Like Peptide-1 Receptor Agonists (GLP-1 RAs)
Glucagon-like peptide-1 receptor agonists (GLP-1 RAs) have emerged as important therapeutic agents not only for type 2 diabetes management but also for their potential benefits in pulmonary diseases. These agents exert multifaceted effects on pulmonary physiology and disease pathophysiology through multiple mechanisms, positioning them as promising therapeutic options for various respiratory conditions, particularly in patients with concurrent metabolic dysfunction.
Mechanisms of Action: Distinguishing Direct and Indirect Pathways
The pulmonary benefits of GLP-1 RAs are mediated through a combination of direct receptor-mediated actions on lung tissues and indirect systemic effects, with the relative contribution of each pathway being a key area of investigation. Direct effects are substantiated by the expression of GLP-1 receptors on relevant pulmonary cells, including pulmonary arterial smooth muscle cells,88 type II pneumocytes,89 and large nucleated cells in lung tissue.90 Functionally, this direct pathway is evidenced by studies demonstrating GLP-1 RA-induced bronchodilation in human isolated airways via the cAMP/PKA pathway, an effect observed independently of systemic metabolic changes.91 Furthermore, GLP-1 exerts multiple direct pulmonary effects, including stimulating airway macromolecule secretion, relaxing pulmonary arteries,88 promoting surfactant secretion in type II pneumocytes,89 and exerting potent anti-inflammatory effects in murine models by inactivating NF-κB via a PKA-dependent mechanism.92 Crucially, clinical evidence from the LIRALUNG trial, which utilized a short treatment duration specifically designed to minimize weight loss, demonstrated that liraglutide still improved lung function and alveolar-capillary integrity, strongly supporting the existence of direct receptor-mediated effects in humans.93
In parallel, indirect effects secondary to systemic metabolic improvements represent another significant pathway. Substantial weight loss induced by GLP-1 RAs can mechanically enhance lung function by reducing chest wall fat and abdominal pressure, thereby improving lung compliance and volumes, as seen in longer-term studies.94 Concurrently, improved glycemic control may mitigate systemic and pulmonary oxidative stress and inflammation.
Therefore, the current evidence supports a dual mode of action. Direct effects on bronchial tone, surfactant homeostasis, and pulmonary inflammation are well-established in preclinical and mechanistic clinical studies. These are complemented by substantial indirect benefits arising from systemic metabolic improvements, with the overall clinical outcome likely reflecting the summation of both pathways.
Effects on Pulmonary Diseases
Obstructive Pulmonary Diseases
GLP-1 RAs demonstrate significant therapeutic potential in obstructive lung diseases. In a murine model of experimental obstructive lung disease, GLP-1 reduces mortality and improves lung function by mitigating the severity of acute exacerbations.95 Clinical evidence supports these findings, with real-world studies indicating that GLP-1 RA users experience fewer exacerbations of chronic lower respiratory diseases compared to users of DPP-4 inhibitors.96 Long-term observational studies further suggest that GLP-1 RAs may offer a novel therapeutic perspective for airway disorders.97 Specifically, in obese patients with COPD, 40 weeks of liraglutide treatment improves select pulmonary function parameters,94 while liraglutide increases forced vital capacity in patients with T2DM—an effect associated with reduced circulating surfactant protein D, indicating beneficial modulation of alveolar-capillary barrier function.93
Asthma and Bronchial Hyperresponsiveness
GLP-1 RAs show promise in asthma management through multiple mechanisms. The GLP-1 receptor has been identified as a novel target for treating bronchial hyperresponsiveness, with activation of the cAMP-dependent protein kinase A (PKA) pathway in human airways mediating this effect.91 This positions GLP-1 RAs as a potential therapeutic class for asthma, particularly in patients with T2DM and comorbid bronchial hyperresponsiveness. In a murine model of OVA-induced asthma, GLP-1 potently ameliorates airway inflammation and excessive mucus secretion through a PKA-dependent mechanism that inactivates NF-κB.92 However, it is worth noting that GLP-1 RAs did not significantly affect the incidence of asthma in clinical studies, which adds complexity to their role in this disease.68
Other Respiratory Conditions
Emerging evidence suggests potential benefits of GLP-1 RAs in additional respiratory contexts. The improvement in forced vital capacity observed with liraglutide treatment indicates potential utility in restrictive lung disease patterns.93 Additionally, the reduction in circulating surfactant protein D levels suggests beneficial effects on alveolar-capillary membrane integrity, which may have implications for interstitial lung diseases.
Literature Conflicts, Inconsistencies, and Knowledge Gaps
Key Conflicts and Inconsistencies
A central conflict for GLP-1 RAs involves the disconnect between their potent anti-inflammatory and bronchodilatory effects in preclinical modelsand their neutral effect on asthma incidence in large cardiovascular outcome trials.68,91,92,95 This suggests that their primary benefit in respiratory disease may be limited to specific phenotypes, such as obesity-associated asthma or COPD, where metabolic dysfunction is a key driver. Additionally, the relative contribution of direct pulmonary actions versus systemic metabolic benefits to observed clinical improvements remains unclear.
Causes of Inconsistencies
The observed discrepancies primarily stem from translational challenges between animal models and human diseases, methodological limitations in clinical trial design, and the difficulty in distinguishing direct pulmonary effects from systemic metabolic benefits. Preclinical studies utilize acute inflammatory models in controlled settings, while clinical populations comprise heterogeneous patients with chronic, multifactorial respiratory conditions.68,95 The predominant focus on metabolic endpoints in large trials, with pulmonary outcomes typically assessed secondarily, limits detection of respiratory-specific effects.68 Furthermore, substantial metabolic improvements and weight loss induced by GLP-1 RAs confound the attribution of observed pulmonary benefits to direct receptor-mediated mechanisms versus indirect systemic effects.92,93 Inadequate patient stratification by GLP-1 receptor expression or specific disease endotypes also contributes to these inconsistencies.
Knowledge Gaps
Key knowledge gaps persist, foremost being the need to quantify the contribution of direct pulmonary actions versus indirect systemic effects. Future research must prioritize clinical trials in non-diabetic populations and mechanistic studies to dissect this interplay. The efficacy in non-obese patients, long-term safety in severe respiratory disease, and potential interactions with inhaled medications also require investigation. Finally, establishing the optimal therapeutic regimen for specific lung conditions through dedicated trials is essential.
Taken together, these data suggest that GLP-1 RAs improve pulmonary function in obese COPD patients and alleviate airway hyperresponsiveness in metabolic asthma via direct bronchodilatory and indirect weight-loss-mediated effects. Their neutral impact on asthma incidence in large trials underscores benefit specificity to metabolic/obesity-related phenotypes, supporting prioritization for diabetic patients with obesity-associated respiratory comorbidities.
Insulin
Insulin represents a fundamental therapy for both type 1 diabetes and advanced type 2 diabetes, with complex and dual effects on pulmonary health. While demonstrating protective roles in acute pulmonary conditions, insulin therapy has also been associated with potential risks in chronic respiratory diseases and lung cancer, reflecting its multifaceted interactions with pulmonary pathophysiology.
Mechanisms of Action
Insulin influences pulmonary physiology through several key mechanisms:
- Endothelial protection and anti-apoptotic effects: Intensive insulin therapy attenuates lung injury by improving pulmonary microvascular endothelial cell dysfunction and reducing cell apoptosis.98
- Inflammatory pathway modulation: In a septic rat model, hyperglycemia was associated with increased lung damage and elevated levels of high mobility group box 1 (HMGB1) in serum. Insulin therapy significantly reduced this lung damage and decreased serum HMGB1 levels, potentially via inhibition of NF-κB activation.99
- Oxidative stress reduction: Insulin effectively prevents elevation of plasma nitrate/nitrite and methyl guanidine in LPS-induced acute lung injury.100
- Immune cell regulation: In a trauma model, insulin reduced leukocyte sequestration in the lungs and end-organ edema, indicating an endothelial protective effect. Notably, this benefit was achieved without impairing neutrophil function—in fact, neutrophil respiratory burst activity was enhanced in insulin-treated animals.101
- Antibacterial synergy: Insulin enhances the antibacterial effects of linezolid against Staphylococcus aureus through modulation of the TLR2/MAPKs/NLRP3 pathway.102
Effects on Pulmonary Diseases
Acute Lung Injury and Critical Illness
Insulin demonstrates consistent protective effects across various models of acute lung injury. In burn-induced lung injury, intensive insulin therapy attenuates ALI by preserving pulmonary microvascular endothelial integrity.98 In sepsis models, insulin therapy significantly alleviates lung damage by reducing HMGB1 levels through NF-κB inhibition.99 For LPS-induced ALI, insulin prevents elevation of cytotoxic metabolites and mitigates lung damage.100 In trauma models, insulin reduces leukocyte sequestration and exerts endothelial protective effects.101 Perioperative administration during lung resection does not impair respiratory function, supporting its safety for surgical patients.103
Pulmonary Infections
Insulin demonstrates therapeutic utility in pulmonary infections, particularly in diabetic patients. The combination of insulin and linezolid exerts significant antibacterial effects against Staphylococcus aureus while providing anti-inflammatory activity via the TLR2/MAPKs/NLRP3 pathway.102 This combination strategy offers improved glucose control alongside enhanced antimicrobial efficacy.
In the context of COVID-19, observational studies have identified an association between insulin use and increased mortality among T2DM patients with COVID-19 suggesting potential risks in severe viral respiratory infections that warrant careful consideration.28
Chronic Lung Diseases
Contrasting with its protective acute effects, insulin therapy shows concerning associations with chronic respiratory conditions. In patients with COPD, insulin therapy is associated with higher incidence of acute exacerbations, pneumonia, and ventilator use, though notably without significant increase in mortality.104 T2DM patients using insulin face an increased risk of asthma, highlighting the complex relationship between insulin therapy and chronic airway diseases.105
Lung Cancer
Accumulating evidence links insulin therapy to increased lung cancer risk. Human insulin use has been significantly associated with elevated lung cancer risk in T2DM patients,106 with mechanistic studies confirming that insulin promotes the development of non-small cell lung cancer by activating the phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) signaling pathway.107 These findings underscore the importance of careful risk-benefit assessment when considering long-term insulin therapy in diabetic patients.
However, this association is subject to important methodological limitations. Significant confounding by indication is a primary concern, as patients requiring insulin often have more severe diabetes and comorbidities that independently increase cancer risk. The potential roles of insulin dose and treatment duration also remain unclear. While mechanistically plausible, the direct contribution of insulin to human lung carcinogenesis remains uncertain. Therefore, current evidence suggests a correlation rather than confirms causality, underscoring the need for careful risk-benefit evaluation in clinical practice.
Literature Conflicts, Inconsistencies, and Knowledge Gaps
Key Conflicts and Inconsistencies
The most significant conflict involves insulin’s dual nature—demonstrating protective effects in acute lung injury models while being associated with adverse outcomes in chronic conditions.98–101,104–106 This paradox highlights the context-dependent nature of insulin’s pulmonary effects. The association with increased COVID-19 mortality contrasts with its established benefits in other infectious settings, suggesting infection-specific considerations.28,102 Additionally, while mechanistic studies confirm insulin’s ability to promote NSCLC development through PI3K/Akt activation, the clinical significance of this finding in diverse patient populations requires further clarification.107
Causes of Inconsistencies
Several factors contribute to these conflicting observations. First, methodological heterogeneity across studies presents a major challenge—controlled animal models of acute injury differ substantially from the complex, multifactorial nature of human chronic diseases.98–101,104–106 Second, confounding by indication significantly affects observational studies, as patients requiring insulin therapy typically have longer diabetes duration, more severe metabolic dysregulation, and greater comorbidity burden, all of which independently influence pulmonary outcomes. Third, temporal and dose-dependent effects may explain the divergence between acute benefits and chronic risks, with short-term insulin exposure providing protective signaling versus long-term exposure potentially promoting pathological processes. Fourth, disease-specific pathophysiology determines insulin’s effects, as demonstrated by its differential impact in various infectious contexts.28,102 Finally, technical variations in study design, including differences in insulin formulations (human insulin vs analogs), glycemic targets, and concomitant medications, further complicate cross-study comparisons.
Knowledge Gaps
Critical unanswered questions include the precise mechanisms underlying insulin’s differential effects in acute versus chronic pulmonary conditions. The optimal insulin dosing and glycemic targets for patients with specific pulmonary comorbidities remain undefined. Long-term pulmonary safety data, particularly regarding cancer risk with modern insulin analogs, requires further investigation. Additionally, potential interactions between insulin and commonly used pulmonary medications represent an unexplored area. The development of predictive biomarkers to identify patients at highest risk for adverse pulmonary outcomes during insulin therapy could significantly enhance personalized treatment approaches. Furthermore, the impact of different insulin administration regimens (basal-bolus vs other strategies) on respiratory outcomes warrants systematic evaluation.
Taken together, these data suggest that insulin exerts context-dependent pulmonary effects—protecting against acute lung injury and bacterial infections via endothelial protection and anti-inflammation, yet associating with increased risks of chronic lung disease exacerbations and lung cancer (confounded by diabetes severity). Its use requires individualized risk-benefit assessment: favorable for acute critical illness but cautious monitoring in patients with chronic respiratory conditions or long-term cancer risk.
Summary of Evidence
To provide a clear and comparative overview of the evidence discussed in detail above, Table 1 summarizes the key pulmonary effects, mechanisms, and levels of evidence for the eight major classes of hypoglycemic agents.
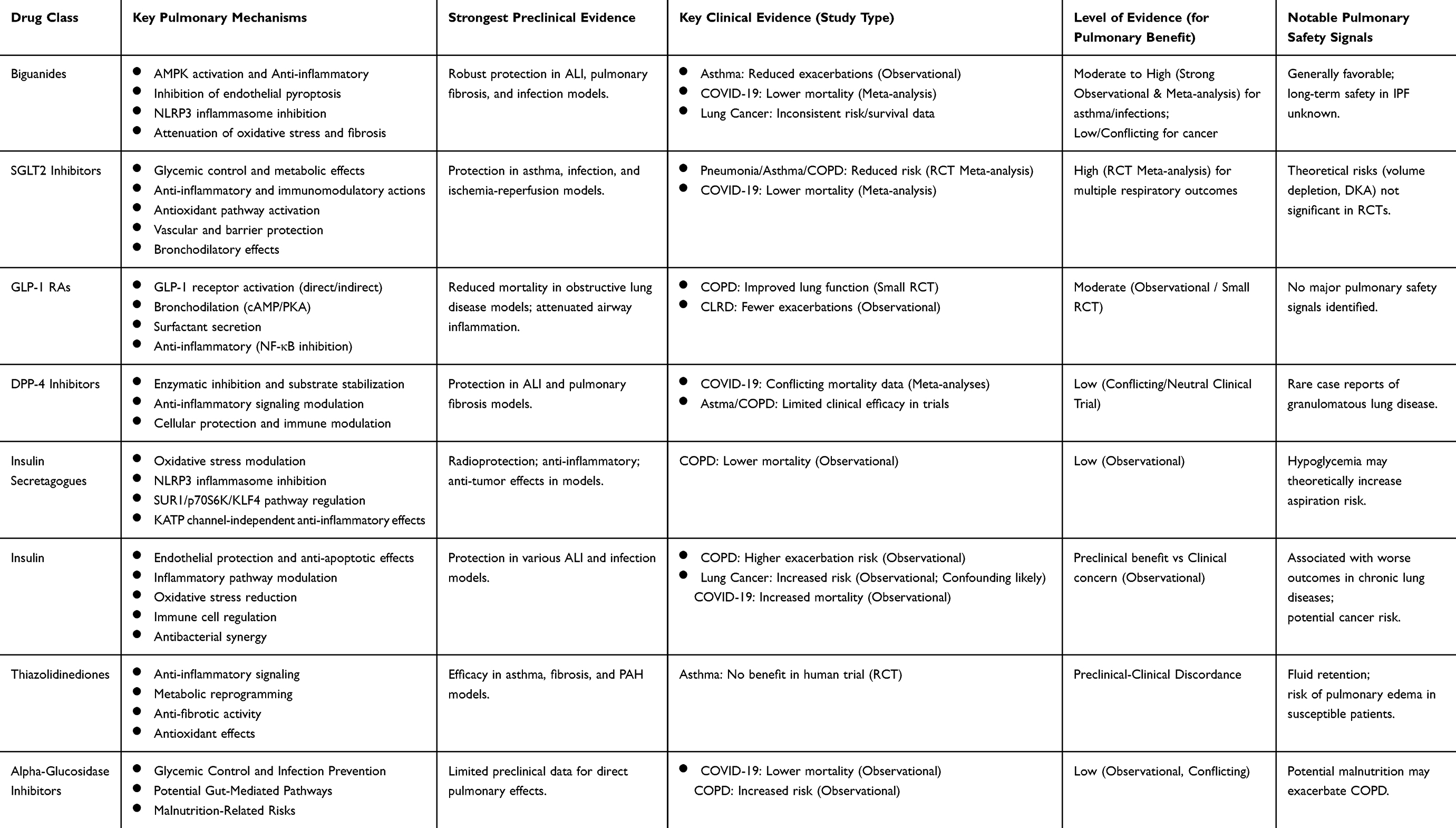 |
Table 1 Summary of Pulmonary Effects and Evidence for Major Hypoglycemic Agent Classes |
Discussion and Future Perspectives
This review broadly summarizes evidence on the pulmonary effects of eight major classes of hypoglycemic agents—Biguanides, Insulin Secretagogues, Thiazolidinediones (TZDs), α-Glucosidase Inhibitors (AGIs), Dipeptidyl Peptidase-4 (DPP-4) inhibitors, Sodium-Glucose Cotransporter 2 (SGLT-2) inhibitors, Glucagon-Like Peptide-1 Receptor Agonists (GLP-1 RAs), and Insulin—revealing a complex landscape of multifaceted, context-dependent effects that underscore the bidirectional crosstalk between diabetes and pulmonary health. Across all agents, inflammatory modulation, oxidative stress mitigation, and metabolic microenvironment regulation emerge as core mechanisms, often independent of direct glycemic control, while class-specific differences in molecular targets and pathway engagement drive divergent clinical outcomes. Novel agents—SGLT-2 inhibitors, GLP-1 RAs, and DPP-4 inhibitors—exhibit the most robust and targeted pulmonary effects: SGLT-2 inhibitors stand out with versatile actions, including reducing bronchoalveolar lavage fluid (BALF) glucose to suppress pathogen proliferation, activating vascular protective (ERK1/2) and antioxidant (Nrf-2/HO-1) pathways, and mitigating inflammation via NF-κB inhibition, which explains their consistent benefits across infectious, inflammatory, and ischemic pulmonary conditions. GLP-1 RAs focus on immune modulation, particularly inhibiting Th1/Th17 polarization and attenuating airway remodeling, while DPP-4 inhibitors target lung tissue-expressed DPP-4 to protect epithelial and endothelial barriers and suppress NLRP3 inflammasome activation. In contrast, traditional agents exert more indirect or context-dependent effects: Metformin (Biguanides) centers on AMPK activation, reducing asthma-related hospitalizations but showing inconsistent lung cancer risk across studies; Insulin Secretagogues (eg, sulfonylureas) modulate oxidative stress yet suffer from class heterogeneity, with gliclazide demonstrating anti-tumor effects not observed with glibenclamide; TZDs and AGIs lack strong clinical evidence, with TZDs showing preclinical anti-asthmatic potential that fails to translate to human benefit and AGIs protecting against COVID-19 mortality while potentially increasing COPD risk; Insulin itself exhibits dual effects, offering acute endothelial protection in acute lung injury (ALI) but posing chronic PI3K/Akt-mediated lung cancer risk, with outcomes dependent on dose and disease context.
Efficacy across pulmonary diseases further stratifies these agents: SGLT-2 inhibitors demonstrate the strongest evidence of benefit, consistently reducing the risk of pneumonia, COVID-19 mortality, asthma exacerbations, and COPD acute episodes, alongside additional protection against pulmonary ischemia/reperfusion (I/R) injury and sleep apnea. GLP-1 RAs and DPP-4 inhibitors fall into the moderate evidence category, with GLP-1 RAs improving lung function in obese COPD patients and alleviating asthma airway hyperresponsiveness, while DPP-4 inhibitors suppress LPS-induced ALI and lung cancer growth but lack consistent validation for chronic respiratory conditions. Traditional agents show weak or conflicting evidence: Metformin reduces asthma-related hospitalizations but no overall lung cancer benefit, Insulin Secretagogues reduce COPD mortality with limited data on other outcomes, and AGIs and TZDs exhibit inconsistent or isolated effects requiring further validation. Three overarching themes emerge from this synthesis: the bidirectional crosstalk of the metabolic-respiratory axis, where chronic hyperglycemia and obesity drive pulmonary dysfunction via advanced glycation end-product (AGE) accumulation, oxidative stress, and inflammatory dysregulation—pathways targeted by hypoglycemic agents to restore homeostasis; the critical role of target-phenotype matching, as drug efficacy depends on pulmonary tissue expression of targets (eg, SGLT-2 in pulmonary vasculature, GLP-1 receptors in airway smooth muscle) and alignment with disease pathophysiology (eg, SGLT-2 inhibitors for infections, GLP-1 RAs for obstructive diseases); and the favorable benefit-risk balance of novel agents over traditional drugs, attributed to their direct pulmonary targets that minimize off-target risks such as insulin’s cancer potential or AGIs’ COPD association.
Despite substantial progress, key limitations persist in the current evidence base. Studies suffer from significant heterogeneity in design (retrospective vs prospective), and critical variability in outcome definitions—for example, “COPD exacerbation” ranges from symptom worsening requiring oral corticosteroids to hospitalization, while “pneumonia” is defined by radiological findings alone in some studies versus combined clinical (fever, cough) and microbiological confirmation in others. This inconsistency complicates cross-study comparisons. Additionally, publication bias is a notable concern, particularly in preclinical research, where positive findings (eg, drug-induced reduction in experimental lung injury) are more likely to be published than neutral or negative results, potentially overestimating therapeutic potential. Most research aggregates agents by class, masking drug-specific effects that may be clinically relevant, while key subgroups—including pediatric patients, ethnic minorities, and frail elderly individuals with multiple comorbidities—are underrepresented or excluded from trials, limiting generalizability to these populations. A critical gap is the lack of head-to-head randomized controlled trials (RCTs) between drug classes specifically powered for pulmonary endpoints; most evidence compares single classes to placebo or “other hypoglycemics,” preventing definitive conclusions about superior efficacy for specific respiratory conditions. Furthermore, the interplay between systemic metabolic effects (eg, weight loss) and direct pulmonary actions (eg, endothelial protection) remains poorly understood, leaving critical mechanistic gaps.
These limitations directly inform clinical decision-making when selecting hypoglycemic agents for patients with diabetes and pulmonary comorbidities. First, the heterogeneity of endpoints necessitates that clinicians interpret study findings in the context of their local clinical practice—for example, a drug shown to reduce “mild COPD exacerbations” (symptom-driven) may not be prioritized for patients at high risk of hospitalization. Second, awareness of publication bias in preclinical research should guide clinicians to prioritize high-quality clinical evidence (eg, large RCTs, real-world cohorts) over mechanistic studies, especially when managing severe pulmonary conditions like COVID-19-associated ARDS. Third, the lack of head-to-head RCTs underscores the need for individualized therapy: factors such as baseline respiratory disease subtype (eg, infectious vs obstructive), comorbidities (eg, heart failure, chronic kidney disease), and patient tolerability (eg, SGLT-2 inhibitors in volume-sensitive COPD patients) should take precedence over class-level assumptions. Fourth, the paucity of subgroup data requires cautious use in pediatric, ethnic minority, and frail elderly patients—close monitoring for adverse events (eg, hypoglycemia in older adults on insulin, dehydration in SGLT-2 inhibitor users with advanced COPD) and shared decision-making with patients are critical to balancing potential benefits and uncertainty. Collectively, these considerations emphasize that while hypoglycemic agents offer meaningful pulmonary benefits, their use must be grounded in context-specific evidence and patient-centered factors.
To address these limitations and optimize clinical practice, future research should prioritize several key directions: (1) Head-to-head comparative trials are needed to directly compare SGLT-2 inhibitors and GLP-1 RAs in specific pulmonary conditions, such as COVID-19-associated ARDS and severe asthma, to define optimal first-line therapy for diabetic patients with pulmonary comorbidities. (2) Expanded population studies should validate the multifaceted effects of novel agents by including non-diabetic populations with pulmonary diseases (eg, idiopathic pulmonary fibrosis [IPF], COPD), thereby clarifying benefits independent of glycemic control. (3) Biomarker-driven stratification research must identify predictive markers (eg, pulmonary SGLT-2 expression, PPAR-γ levels) to select patients most likely to benefit from specific agents, resolving conflicting evidence and enabling personalized care. (4) Mechanistic deep-diving should utilize single-cell sequencing and spatial transcriptomics to map cell-type-specific drug targets in lung tissues. A priority within this effort is to dissect the role of pulmonary macrophages, clarifying how diabetes perturbs their M1/M2 polarization and metabolic states, and how hypoglycemic agents can restore homeostatic immune function to resolve inflammation and promote repair. (5) Research in special populations—including the elderly, pregnant, or renally impaired patients—is essential to evaluate safety and efficacy in vulnerable groups and to assess long-term risks (eg, insulin’s lung cancer potential) and benefits (eg, SGLT-2 inhibitors’ effect on IPF progression). (6) Drug-drug interaction studies should investigate combinations of hypoglycemic agents with pulmonary therapies (eg, inhaled corticosteroids, anti-fibrotics) to optimize combination regimens for comorbid patients.
Conclusion
This narrative review provides a comprehensive framework for understanding the multifaceted role of hypoglycemic agents in pulmonary health, synthesizing evidence that points to promising, and often more targeted, benefits (SGLT-2 inhibitors, GLP-1 RAs, DPP-4 inhibitors) across infectious, inflammatory, obstructive, and fibrotic pulmonary diseases. These agents’ effects are rooted in shared mechanisms of inflammation modulation, oxidative stress mitigation, and metabolic regulation, alongside class-specific molecular targets that drive their divergent efficacy profiles. Traditional agents, in contrast, exhibit more indirect or context-dependent outcomes, with greater variability in benefit-risk balance. The key clinical implication of this synthesis is the need for personalized therapy, tailoring hypoglycemic drug selection to a patient’s specific pulmonary comorbidity, disease subtype, and biological profile—for example, prioritizing SGLT-2 inhibitors for COVID-19 or COPD and GLP-1 RAs for obesity-related asthma. Resolving existing conflicts and knowledge gaps through targeted comparative trials, biomarker research, mechanistic dissection, and long-term safety studies will further refine this personalized approach.
Ultimately, this review underscores the critical interplay between hypoglycemic therapy and pulmonary health, offering valuable guidance for managing diabetic patients with comorbid respiratory conditions and setting the stage for future research to advance patient-centered care in this growing clinical population. Based on the current evidence, no class of hypoglycemic agents should be universally avoided solely for pulmonary reasons; instead, clinicians should individualize therapy based on the patient’s specific respiratory profile, comorbidity burden, and overall treatment goals, while proactively monitoring pulmonary outcomes.
Data Sharing Statement
Data supporting the findings of this study are available from the corresponding authors upon reasonable request.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was funded by the Hebei Provincial Key R&D Program Project (22377713D).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Klein OL, Meltzer D, Carnethon M, Krishnan JA. Type II diabetes mellitus is associated with decreased measures of lung function in a clinical setting. Respir Med. 2011;105(7):1095–1098. doi:10.1016/j.rmed.2011.03.010
2. van den Borst B, Gosker HR, Zeegers MP, Schols AMWJ. Pulmonary function in diabetes: a meta analysis. Chest. 2010;138(2):393–406. doi:10.1378/chest.09-2622
3. Davis WA, Knuiman M, Kendall P, Grange V, Davis TME. Fremantle Diabetes Study. Glycemic exposure is associated with reduced pulmonary function in type 2 diabetes: the Fremantle Diabetes Study. Diabetes Care. 2004;27(3):752–757. doi:10.2337/diacare.27.3.752
4. Ehrlich SF, Quesenberry CP, Van Den Eeden SK, et al. Patients diagnosed with diabetes are at increased risk for asthma, chronic obstructive pulmonary disease, pulmonary fibrosis, and pneumonia but not lung cancer. Diabetes Care. 2010;33(1):55–60. doi:10.2337/dc09-0880
5. Kolahian S, Leiss V, Nürnberg B. Diabetic lung disease: fact or fiction? Rev Endocr Metab Disord. 2019;20(3):303–319. doi:10.1007/s11154-019-09516-w
6. Apostolova N, Iannantuoni F, Gruevska A, et al. Mechanisms of action of metformin in type 2 diabetes: effects on mitochondria and leukocyte-endothelium interactions. Redox Biol. 2020;34:101517. doi:10.1016/j.redox.2020.101517
7. Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60(9):1577–1585. doi:10.1007/s00125-017-4342-z
8. Wu K, Tian R, Huang J, et al. Metformin alleviated endotoxemia-induced acute lung injury via restoring AMPK-dependent suppression of mTOR. Chem Biol Interact. 2018;291:1–6. doi:10.1016/j.cbi.2018.05.018
9. Zhang Y, Zhang H, Li S, et al. Metformin alleviates LPS-induced acute lung injury by regulating the SIRT1/NF-κB/NLRP3 pathway and inhibiting endothelial cell pyroptosis. Front Pharmacol. 2022;13:801337. doi:10.3389/fphar.2022.801337
10. Xian H, Liu Y, Nilsson AR, et al. Metformin inhibition of mitochondrial ATP and DNA synthesis abrogates NLRP3 inflammasome activation and pulmonary inflammation. Immunity. 2021;54(7):1463–1477. doi:10.1016/j.immuni.2021.05.004
11. Sato N, Takasaka N, Yoshida M, et al. Metformin attenuates lung fibrosis development via NOX4 suppression. Respir Res. 2016;17(1):107. doi:10.1186/s12931-016-0420-x
12. Tsaknis G, Siempos II, Kopterides P, et al. Metformin attenuates ventilator-induced lung injury. Crit Care. 2012;16(4):R134. doi:10.1186/cc11439
13. Wang J, Wang Y, Han J, et al. Metformin attenuates radiation-induced pulmonary fibrosis in a murine model. Radiat Res. 2017;188(1):105–113. doi:10.1667/RR14708.1
14. Gao J, Yuan J, Wang Q, et al. Metformin protects against PM2.5-induced lung injury and cardiac dysfunction independent of AMP-activated protein kinase α2. Redox Biol. 2020;28:101345. doi:10.1016/j.redox.2019.101345
15. Cheng D, Xu Q, Wang Y, et al. Metformin attenuates silica-induced pulmonary fibrosis via AMPK signaling. J Transl Med. 2021;19(1):349. doi:10.1186/s12967-021-03036-5
16. Li L, Huang W, Li K, et al. Metformin attenuates gefitinib-induced exacerbation of pulmonary fibrosis by inhibition of TGF-β signaling pathway. Oncotarget. 2015;6(41):43605–43619. doi:10.18632/oncotarget.6186
17. Guo W, Kuang Y, Wu J, et al. Hexokinase 2 depletion confers sensitization to metformin and inhibits glycolysis in lung squamous cell carcinoma. Front Oncol. 2020;10:52. doi:10.3389/fonc.2020.00052
18. Wang J, Gao Q, Wang D, et al. Metformin inhibits growth of lung adenocarcinoma cells by inducing apoptosis via the mitochondria-mediated pathway. Oncol Lett. 2015;10(3):1343–1349. doi:10.3892/ol.2015.3450
19. Teixeira SF, Guimarães I Dos S, Madeira KP, et al. Metformin synergistically enhances antiproliferative effects of cisplatin and etoposide in NCI-H460 human lung cancer cells. J Bras Pneumol. 2013;39(6):644–649. doi:10.1590/S1806-37132013000600002
20. Ashinuma H, Takiguchi Y, Kitazono S, et al. Antiproliferative action of metformin in human lung cancer cell lines. Oncol Rep. 2012;28(1):8–14. doi:10.3892/or.2012.1763
21. Lin JJ, Gallagher EJ, Sigel K, et al. Survival of patients with stage IV lung cancer with diabetes treated with metformin. Am J Respir Crit Care Med. 2015;191(4):448–454. doi:10.1164/rccm.201407-1395OC
22. Xu T, Li D, He Y, et al. Prognostic value of metformin for non-small cell lung cancer patients with diabetes. World J Surg Oncol. 2018;16(1):60. doi:10.1186/s12957-018-1362-1
23. Li CY, Erickson SR, Wu CH. Metformin use and asthma outcomes among patients with concurrent asthma and diabetes. Respirology. 2016;21(7):1210–1218. doi:10.1111/resp.12818
24. Wu TD, Keet CA, Fawzy A, et al. Association of metformin initiation and risk of asthma exacerbation. A claims-based cohort study. Ann Am Thorac Soc. 2019;16(12):1527–1533. doi:10.1513/AnnalsATS.201812-897OC
25. Patkee WR, Carr G, Baker EH, et al. Metformin prevents the effects of Pseudomonas aeruginosa on airway epithelial tight junctions and restricts hyperglycaemia-induced bacterial growth. J Cell Mol Med. 2016;20(4):758–764. doi:10.1111/jcmm.12784
26. Kajiwara C, Kusaka Y, Kimura S, et al. Metformin mediates protection against legionella pneumonia through activation of AMPK and mitochondrial reactive oxygen species. J Immunol. 2018;200(2):623–631. doi:10.4049/jimmunol.1700474
27. Singhal A, Jie L, Kumar P, et al. Metformin as adjunct antituberculosis therapy. Sci Transl Med. 2014;6(263):263ra159. doi:10.1126/scitranslmed.3009885
28. Nguyen NN, Ho DS, Nguyen HS, et al. Preadmission use of antidiabetic medications and mortality among patients with COVID-19 having type 2 diabetes: a meta-analysis. Metabolism. 2022;131:155196. doi:10.1016/j.metabol.2022.155196
29. Chen Y, Lv X, Lin S, et al. The association between antidiabetic agents and clinical outcomes of COVID-19 patients with diabetes: a Bayesian network meta-analysis. Front Endocrinol. 2022;13:895458. doi:10.3389/fendo.2022.895458
30. Chen X, Walther FJ, Sengers RMA, et al. Metformin attenuates hyperoxia-induced lung injury in neonatal rats by reducing the inflammatory response. Am J Physiol Lung Cell Mol Physiol. 2015;309(3):L262–L270. doi:10.1152/ajplung.00389.2014
31. Zhang Z, Bi Y, Li S, et al. Reduced risk of lung cancer with metformin therapy in diabetic patients: a systematic review and meta-analysis. Am J Epidemiol. 2014;180(1):11–14. doi:10.1093/aje/kwu124
32. Smiechowski BB, Azoulay L, Yin H, et al. The use of metformin and the incidence of lung cancer in patients with type 2 diabetes. Diabetes Care. 2013;36(1):124–129. doi:10.2337/dc12-0740
33. Nie SP, Chen H, Zhuang MQ, et al. Anti-diabetic medications do not influence risk of lung cancer in patients with diabetes mellitus: a systematic review and meta-analysis. Asian Pac J Cancer Prev. 2014;15(16):6863–6869. doi:10.7314/apjcp.2014.15.16.6863
34. Wu Y, Liu HB, Shi XF, et al. Conventional hypoglycaemic agents and the risk of lung cancer in patients with diabetes: a meta-analysis. PLoS One. 2014;9(6):e99577. doi:10.1371/journal.pone.0099577
35. Sakoda LC, Ferrara A, Achacoso NS, et al. Metformin use and lung cancer risk in patients with diabetes. Cancer Prev Res. 2015;8(2):174–179. doi:10.1158/1940-6207.CAPR-14-0291
36. Tian RH, Zhang YG, Wu Z, et al. Effects of metformin on survival outcomes of lung cancer patients with type 2 diabetes mellitus: a meta-analysis. Clin Transl Oncol. 2016;18(6):641–649. doi:10.1007/s12094-015-1412-x
37. Mazzone PJ, Rai H, Beukemann M, et al. The effect of metformin and thiazolidinedione use on lung cancer in diabetics. BMC Cancer. 2012;12:410. doi:10.1186/1471-2407-12-410
38. Amiri FT, Arzani S, Farzipour S, et al. Radioprotective effects of gliclazide against irradiation-induced cardiotoxicity and lung injury through inhibiting oxidative stress. Med Oncol. 2022;39(12):199. doi:10.1007/s12032-022-01803-y
39. Shao H, Huang L, Duan S, et al. Glyburide attenuates ozone-induced pulmonary inflammation and injury by blocking the NLRP3 inflammasome. Environ Toxicol. 2020;35(8):831–839. doi:10.1002/tox.22919
40. Li M, Liu H, Shao H, et al. Glyburide attenuates B(a)p and LPS-induced inflammation-related lung tumorigenesis in mice. Environ Toxicol. 2021;36(8):1713–1722. doi:10.1002/tox.23293
41. Xu K, Sun G, Li M, et al. Glibenclamide targets sulfonylurea receptor 1 to inhibit p70S6K activity and upregulate KLF4 expression to suppress non-small cell lung carcinoma. Mol Cancer Ther. 2019;18(11):2085–2096. doi:10.1158/1535-7163.MCT-18-1181
42. Cui W, Zhang S, Cai Z, et al. The antidiabetic agent glibenclamide protects airway hyperresponsiveness and inflammation in mice. Inflammation. 2015;38(2):835–845. doi:10.1007/s10753-014-9993-z
43. Cheng Y, Hou K, Wang Y, et al. Identification of prognostic signature and gliclazide as candidate drugs in lung adenocarcinoma. Front Oncol. 2021;11:665276. doi:10.3389/fonc.2021.665276
44. Yen FS, Wei JCC, Yu TS, et al. Sulfonylurea use in patients with type 2 diabetes and COPD: a nationwide population-based cohort study. Int J Environ Res Public Health. 2022;19(22):15013. doi:10.3390/ijerph192215013
45. Belvisi MG, Hele DJ, Birrell MA. Peroxisome proliferator-activated receptor gamma agonists as therapy for chronic airway inflammation. Eur J Pharmacol. 2006;533(1–3):101–109. doi:10.1016/j.ejphar.2005.12.048
46. El-Naa MM, El-Refaei MF, Nasif WA, et al. In-vivo antioxidant and anti-inflammatory activity of rosiglitazone, a peroxisome proliferator-activated receptor-gamma (PPAR-γ) agonists in animal model of bronchial asthma. J Pharm Pharmacol. 2015;67(10):1421–1430. doi:10.1111/jphp.12445
47. Liu D, Zeng BX, Zhang SH, et al. Rosiglitazone, an agonist of peroxisome proliferator-activated receptor gamma, reduces pulmonary inflammatory response in a rat model of endotoxemia. Inflamm Res. 2005;54:464–470. doi:10.1007/s00011-005-1379-0
48. Legchenko E, Chouvarine P, Borchert P, et al. PPARγ agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci Transl Med. 2018;10(438):eaao0303. doi:10.1126/scitranslmed.aao0303
49. Milam JE, Keshamouni VG, Phan SH, et al. PPAR-gamma agonists inhibit profibrotic phenotypes in human lung fibroblasts and bleomycin-induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;294(5):L891–L901. doi:10.1152/ajplung.00333.2007
50. Honda K, Marquillies P, Capron M, et al. Peroxisome proliferator-activated receptor gamma is expressed in airways and inhibits features of airway remodeling in a mouse asthma model. J Allergy Clin Immunol. 2004;113(5):882–888. doi:10.1016/j.jaci.2004.02.036
51. Benayoun L, Letuve S, Druilhe A, et al. Regulation of peroxisome proliferator-activated receptor gamma expression in human asthmatic airways: relationship with proliferation, apoptosis, and airway remodeling. Am J Respir Crit Care Med. 2001;164(8 Pt 1):1487–1494. doi:10.1164/ajrccm.164.8.2101070
52. Anderson JR, Mortimer K, Pang L, et al. Evaluation of the PPAR-γ agonist pioglitazone in mild asthma: a double-blind randomized controlled trial. PLoS One. 2016;11(8):e0160257. doi:10.1371/journal.pone.0160257
53. Rehan VK, Wang Y, Patel S, et al. Rosiglitazone, a peroxisome proliferator-activated receptor-gamma agonist, prevents hyperoxia-induced neonatal rat lung injury in vivo. Pediatr Pulmonol. 2006;41:558–569. doi:10.1002/ppul.20407
54. Xu K, He W, Yu B, et al. Effects of different treatments for type 2 diabetes mellitus on mortality of coronavirus disease from 2019 to 2021 in China: a multi-institutional retrospective study. Mol Biomed. 2024;5(1):18. doi:10.1186/s43556-024-00183-1
55. Li J, Wei Q, McCowen KC, et al. Inpatient use of metformin and acarbose is associated with reduced mortality of COVID-19 patients with type 2 diabetes mellitus. Endocrinol Diabetes Metab. 2022;5(1):e00301. doi:10.1002/edm2.301
56. Wu SW, Ho YC, Luo CW, et al. Oral treatment for diabetes using α-glucosidase inhibitors was a risk factor for chronic obstructive pulmonary disease: a cohort study. Int J Med Sci. 2021;18(3):778–784. doi:10.7150/ijms.55361
57. Zou H, Zhu N, Li S. The emerging role of dipeptidyl-peptidase-4 as a therapeutic target in lung disease. Expert Opin Ther Targets. 2020;24(2):147–153. doi:10.1080/14728222.2020.1721468
58. Meyerholz DK, Lambertz AM, McCray PB. Dipeptidyl peptidase 4 distribution in the human respiratory tract: implications for the Middle East respiratory syndrome. Am J Pathol. 2016;186(1):78–86. doi:10.1016/j.ajpath.2015.09.014
59. Zhou J, Peng Z, Wang J. Trelagliptin alleviates lipopolysaccharide (LPS)-induced inflammation and oxidative stress in acute lung injury mice. Inflammation. 2021;44(4):1507–1517. doi:10.1007/s10753-021-01435-w
60. Guo K, Jin F. Dipeptidyl peptidase-4 (DPP-4) inhibitor saxagliptin alleviates lipopolysaccharide-induced acute lung injury via regulating the Nrf-2/HO-1 and NF-κB pathways. J Invest Surg. 2021;34(7):695–702. doi:10.1080/08941939.2019.1680777
61. Ma L, Chang E, Ruan X, et al. The protective effects of omarigliptin against lipopolysaccharide (LPS)-induced inflammatory response and expression of mucin 5AC (MUC5AC) in human bronchial epithelial cells. Mol Immunol. 2022;141:108–115. doi:10.1016/j.molimm.2021.11.013
62. Zhang N, Tang S, Zhang J, et al. The dipeptidyl peptidase-4 inhibitor linagliptin ameliorates LPS-induced acute lung injury by maintenance of pulmonary microvascular barrier via activating the Epac1/AKT pathway. Biomed Pharmacother. 2022;155:113704. doi:10.1016/j.biopha.2022.113704
63. Jang JH, Janker F, Meester ID, et al. The CD26/DPP4-inhibitor vildagliptin suppresses lung cancer growth via macrophage-mediated NK cell activity. Carcinogenesis. 2019;40(2):324–334. doi:10.1093/carcin/bgz009
64. Liu Y, Qi Y. Vildagliptin, a CD26/DPP4 inhibitor, ameliorates bleomycin-induced pulmonary fibrosis via regulating the extracellular matrix. Int Immunopharmacol. 2020;87:106774. doi:10.1016/j.intimp.2020.106774
65. Suzuki T, Tada Y, Gladson S, et al. Vildagliptin ameliorates pulmonary fibrosis in lipopolysaccharide-induced lung injury by inhibiting endothelial-to-mesenchymal transition. Respir Res. 2017;18(1):177. doi:10.1186/s12931-017-0660-4
66. Shiobara T, Chibana K, Watanabe T, et al. Dipeptidyl peptidase-4 is highly expressed in bronchial epithelial cells of untreated asthma and it increases cell proliferation along with fibronectin production in airway constitutive cells. Respir Res. 2016;17:28. doi:10.1186/s12931-016-0342-7
67. Nader MA. Inhibition of airway inflammation and remodeling by sitagliptin in murine chronic asthma. Int Immunopharmacol. 2015;29(2):761–769. doi:10.1016/j.intimp.2015.08.043
68. Wang A, Tang H, Zhang N, et al. Association between novel glucose-lowering drugs and risk of asthma: a network meta-analysis of cardiorenal outcome trials. Diabet Res Clin Pract. 2022;183:109080. doi:10.1016/j.diabres.2021.109080
69. Kotnala S, Kim Y, Rajput C, et al. Contribution of dipeptidyl peptidase 4 to non-typeable Haemophilus influenzae-induced lung inflammation in COPD. Clin Sci. 2021;135(17):2067–2083. doi:10.1042/CS20210099
70. Pradhan R, Lu S, Yin H, et al. Novel antihyperglycaemic drugs and prevention of chronic obstructive pulmonary disease exacerbations among patients with type 2 diabetes: population based cohort study. BMJ. 2022;379:e071380. doi:10.1136/bmj-2022-071380
71. Xu J, Wang J, He M, et al. Dipeptidyl peptidase IV (DPP-4) inhibition alleviates pulmonary arterial remodeling in experimental pulmonary hypertension. Lab Invest. 2018;98(10):1333–1346. doi:10.1038/s41374-018-0080-1
72. Sada K, Wada J, Morinaga H, et al. Sarcoid-like lung granulomas in a hemodialysis patient treated with a dipeptidyl peptidase-4 inhibitor. Clin Kidney J. 2014;7(2):182–185. doi:10.1093/ckj/sft172
73. Chao EC, Henry RR. SGLT2 inhibition--a novel strategy for diabetes treatment. Nat Rev Drug Discov. 2010;9(7):551–559. doi:10.1038/nrd3180
74. Powell DR, DaCosta CM, Gay J, et al. Improved glycemic control in mice lacking Sglt1 and Sglt2. Am J Physiol Endocrinol Metab. 2013;304(2):E117–E130. doi:10.1152/ajpendo.00439.2012
75. Wang Y, Zhou X. The relationship between use of SGLT2is and incidence of respiratory and infectious diseases and site-specific fractures: a meta-analysis based on 32 large RCTs. Eur J Clin Pharmacol. 2024;80(4):563–573. doi:10.1007/s00228-024-03631-7
76. Yin DG, Qiu M, Duan XY. Association between SGLT2is and cardiovascular and respiratory diseases: a meta-analysis of large trials. Front Pharmacol. 2021;12:724405. doi:10.3389/fphar.2021.724405
77. Qiu M, Zhao LM, Zhan ZL. Comprehensive analysis of adverse events associated with SGLT2is: a meta-analysis involving nine large randomized trials. Front Endocrinol. 2021;12:743807. doi:10.3389/fendo.2021.743807
78. Åstrand A, Wingren C, Benjamin A, et al. Dapagliflozin-lowered blood glucose reduces respiratory Pseudomonas aeruginosa infection in diabetic mice. Br J Pharmacol. 2017;174(9):836–847. doi:10.1111/bph.13741
79. Lee YE, Im DS. SGLT2 inhibitors empagliflozin and canagliflozin ameliorate allergic asthma responses in mice. Int J Mol Sci. 2024;25(14):7567. doi:10.3390/ijms25147567
80. Park HJ, Han H, Oh EY, et al. Empagliflozin and dulaglutide are effective against obesity-induced airway hyperresponsiveness and fibrosis in a murine model. Sci Rep. 2019;9(1):15601. doi:10.1038/s41598-019-51648-1
81. Tabaa MME, Fattah AMK, Shaalan M, et al. Dapagliflozin mitigates ovalbumin-prompted airway inflammatory-oxidative successions and associated bronchospasm in a rat model of allergic asthma. Expert Opin Ther Targets. 2022;26(5):487–506. doi:10.1080/14728222.2022.2077723
82. Huang D, Ju F, Du L, et al. Empagliflozin protects against pulmonary ischemia/reperfusion injury via an extracellular signal-regulated kinases 1 and 2-dependent mechanism. J Pharmacol Exp Ther. 2022;380(3):230–241. doi:10.1124/jpet.121.000956
83. Wu MZ, Chandramouli C, Wong PF, et al. Risk of sepsis and pneumonia in patients initiated on SGLT2 inhibitors and DPP-4 inhibitors. Diabetes Metab. 2022;48(6):101367. doi:10.1016/j.diabet.2022.101367
84. Li HL, Tse YK, Chandramouli C, et al. Sodium-glucose cotransporter 2 inhibitors and the risk of pneumonia and septic shock. J Clin Endocrinol Metab. 2022;107(12):3442–3451. doi:10.1210/clinem/dgac558
85. Au PCM, Tan KCB, Lam DCL, et al. Association of sodium-glucose cotransporter 2 inhibitor vs dipeptidyl peptidase-4 inhibitor use with risk of incident obstructive airway disease and exacerbation events among patients with type 2 diabetes in Hong Kong. JAMA Network Open. 2023;6(1):e2251177. doi:10.1001/jamanetworkopen.2022.51177
86. Qiu M, Ding LL, Zhan ZL, et al. Use of SGLT2 inhibitors and occurrence of noninfectious respiratory disorders: a meta-analysis of large randomized trials of SGLT2 inhibitors. Endocrine. 2021;73(1):31–36. doi:10.1007/s12020-021-02644-x
87. Zou HT, Yang GH, Cai YJ, et al. Are high- or low-dose SGLT2 inhibitors associated with cardiovascular and respiratory adverse events? A meta-analysis. J Cardiovasc Pharmacol. 2022;79:655–662. doi:10.1097/FJC.0000000000001222
88. Richter G, Feddersen O, Wagner U, et al. GLP-1 stimulates secretion of macromolecules from airways and relaxes pulmonary artery. Am J Physiol. 1993;265(4 Pt 1):L374–L381. doi:10.1152/ajplung.1993.265.4.L374
89. Vara E, Arias-Díaz J, Garcia C, et al. Glucagon-like peptide-1(7-36) amide stimulates surfactant secretion in human type II pneumocytes. Am J Respir Crit Care Med. 2001;163(4):840–846. doi:10.1164/ajrccm.163.4.9912132
90. Bullock BP, Heller RS, Habener JF. Tissue distribution of messenger ribonucleic acid encoding the rat glucagon-like peptide-1 receptor. Endocrinology. 1996;137(7):2968–2978. doi:10.1210/endo.137.7.8770921
91. Rogliani P, Calzetta L, Capuani B, et al. Glucagon-like peptide 1 receptor: a novel pharmacological target for treating human bronchial hyperresponsiveness. Am J Respir Cell Mol Biol. 2016;55(6):804–814. doi:10.1165/rcmb.2015-0311OC
92. Zhu T, Wu XL, Zhang W, et al. Glucagon like peptide-1 (GLP-1) modulates OVA-induced airway inflammation and mucus secretion involving a protein kinase A (PKA)-dependent nuclear factor-κB (NF-κB) signaling pathway in mice. Int J Mol Sci. 2015;16(9):20195–20211. doi:10.3390/ijms160920195
93. López-Cano C, Ciudin A, Sánchez E, et al. Liraglutide improves forced vital capacity in individuals with type 2 diabetes: data from the randomized crossover LIRALUNG study. Diabetes. 2022;71(2):315–320. doi:10.2337/db21-0688
94. Dogan ADA, Hilberg O, Hess S, et al. Respiratory effects of treatment with a glucagon-like peptide-1 receptor agonist in patients suffering from obesity and chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis. 2022;17:405–414. doi:10.2147/COPD.S350133
95. Viby NE, Isidor MS, Buggeskov KB, et al. Glucagon-like peptide-1 (GLP-1) reduces mortality and improves lung function in a model of experimental obstructive lung disease in female mice. Endocrinology. 2013;154(12):4503–4511. doi:10.1210/en.2013-1666
96. Albogami Y, Cusi K, Daniels MJ, et al. Glucagon-like peptide 1 receptor agonists and chronic lower respiratory disease exacerbations among patients with type 2 diabetes. Diabetes Care. 2021;44(6):1344–1352. doi:10.2337/dc20-1794
97. Rogliani P, Matera MG, Calzetta L, et al. Long-term observational study on the impact of GLP-1R agonists on lung function in diabetic patients. Respir Med. 2019;154:86–92. doi:10.1016/j.rmed.2019.06.015
98. Zhang WF, Zhu XX, Hu DH, et al. Intensive insulin treatment attenuates burn-initiated acute lung injury in rats: role of the protective endothelium. J Burn Care Res. 2011;32(3):e51–58. doi:10.1097/BCR.0b013e318217f8ae
99. Hagiwara S, Iwasaka H, Hasegawa A, et al. Effects of hyperglycemia and insulin therapy on high mobility group box 1 in endotoxin-induced acute lung injury in a rat model. Crit Care Med. 2008;36:2407–2413. doi:10.1097/CCM.0b013e318180b3ba
100. Chen HI, Yeh DY, Liou HL, et al. Insulin attenuates endotoxin-induced acute lung injury in conscious rats. Crit Care Med. 2006;34:758–764. doi:10.1097/01.CCM.0000201902.37115.22
101. Donnelly M, Condron C, Murray P, et al. Modulation of the glycemic response using insulin attenuates the pulmonary response in animal trauma model. J Trauma. 2007;63:351–357. doi:10.1097/01.ta.0000251599.80602.d1
102. Ni L, Li Y, Zhang H, et al. The combination of insulin and linezolid ameliorates Staphylococcus aureus pneumonia in individuals with diabetes via the TLR2/MAPKs/NLRP3 pathway. Int J Biol Macromol. 2023;242(Pt 1):124750. doi:10.1016/j.ijbiomac.2023.124750
103. Ručka Z, Koutná I, Tesařová L, et al. Intravenous insulin therapy during lung resection does not affect lung function or surfactant proteins. BMC Pulm Med. 2014;14:155. doi:10.1186/1471-2466-14-155
104. Yen FS, Chang SH, Wei JCC, et al. Respiratory outcomes of insulin use in patients with COPD: a nationwide population-based cohort study. Pharmaceuticals. 2023;16(5):643. doi:10.3390/ph16050643
105. Rayner LH, Mcgovern A, Sherlock J, et al. The impact of therapy on the risk of asthma in type 2 diabetes. Clin Respir J. 2019;13(5):299–305. doi:10.1111/crj.13011
106. Tseng CH. Human insulin therapy is associated with an increased risk of lung cancer: a population-based retrospective cohort study. Front Endocrinol. 2019;10:443. doi:10.3389/fendo.2019.00443
107. Jiang J, Ren HY, Geng GJ, et al. Oncogenic activity of insulin in the development of non-small cell lung carcinoma. Oncol Lett. 2018;15(1):447–452. doi:10.3892/ol.2017.7347
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.