Back to Archived Journals » Hypoxia » Volume 2
Effects of copper sulfate-oxidized or myeloperoxidase- modified LDL on lipid loading and programmed cell death in macrophages under hypoxia
Authors Vlaminck B, Calay D, Genin M, Sauvage A, Ninane N, Boudjeltia K, Raes M, Michiels C ![]()
Received 31 March 2014
Accepted for publication 16 July 2014
Published 23 September 2014 Volume 2014:2 Pages 153—169
DOI https://doi.org/10.2147/HP.S65242
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Benoit Vlaminck,1 Damien Calay,1 Marie Genin,1 Aude Sauvage,1 Noelle Ninane,1 Karim Zouaoui Boudjeltia,2 Martine Raes,1 Carine Michiels1
1Laboratory of Biochemistry and Cellular Biology (URBC), Namur Research Institute for Life Sciences (NARILIS), University of Namur, Namur, Belgium; 2Laboratory of Experimental Medicine (ULB 222 Unit), Universite Libre de Bruxelles, CHU de Charleroi, Charleroi, Belgium
Abstract: Atheromatous plaques contain heavily lipid-loaded macrophages that die, hence generating the necrotic core of these plaques. Since plaque instability and rupture is often correlated with a large necrotic core, it is important to understand the mechanisms underlying foam cell death. Furthermore, macrophages within the plaque are associated with hypoxic areas but little is known about the effect of low oxygen partial pressure on macrophage death. The aim of this work was to unravel macrophage death mechanisms induced by oxidized low-density lipoproteins (LDL) both under normoxia and hypoxia. Differentiated macrophages were incubated in the presence of native, copper sulfate-oxidized, or myeloperoxidase-modified LDL. The unfolded protein response, apoptosis, and autophagy were then investigated. The unfolded protein response and autophagy were triggered by myeloperoxidase-modified LDL and, to a larger extent, by copper sulfate-oxidized LDL. Electron microscopy observations showed that oxidized LDL induced excessive autophagy and apoptosis under normoxia, which were less marked under hypoxia. Myeloperoxidase-modified LDL were more toxic and induced a higher level of apoptosis. Hypoxia markedly decreased apoptosis and cell death, as marked by caspase activation. In conclusion, the cell death pathways induced by copper sulfate-oxidized and myeloperoxidase-modified LDL are different and are differentially modulated by hypoxia.
Keywords: Ox-LDL, myeloperoxidase, hypoxia, UPR, apoptosis, autophagy, macrophages
Introduction
Atherosclerosis is characterized by an intimal thickening of medium and large sized arteries that slowly evolves from an asymptomatic lesion, simply impairing the blood flow, to an unstable lesion that can rupture and lead to vascular accidents. It is believed that lesions develop at specific sites called diffuse intimal thickenings, enriched in proteoglycans that have high affinity for lipoproteins.1,2 These structures are also located at sites of turbulent blood flow, which, in coordination with other risk factors, tends to activate the endothelium. This activation of the endothelium initiates an inflammatory process and leads to an increase in permeability, which is believed to favor the infiltration and thus retention of lipoproteins, such as low-density lipoproteins (LDL), in the intima. There, the infiltrated LDL undergo different modifications, including hydrolysis and oxidation, that are promoted by their interactions with proteoglycans.2 By this process, they become the so-called oxidized LDL.3 Oxidized and/or enzymatically modified LDL acquire proinflammatory properties that further increase the inflammatory state of the endothelium, thereby leading to the recruitment of immune cells, such as monocytes, from the blood flow. Once infiltrated into the intima, monocytes differentiate into macrophages enhancing the inflammatory reaction. They also internalize oxidized/modified LDL through the scavenger receptors and by macropinocytosis,4,5 thereby becoming foam cells. Being stressed by the oxidized lipids, cholesterol overloading, and exposure to oxidative stress, these cells finally die, releasing debris into the plaque and resulting in the formation of a necrotic core. As the plaque evolves, macrophage death combined with defective efferocytosis worsens the accumulation of debris and apoptotic bodies, promoting a destabilizing inflammatory reaction and rendering the plaque more prone to rupture.6 In this case, macrophage death is associated with rupture and accidents, but other findings suggest the death of macrophages in the preliminary stages of plaque formation could be beneficial due to the removal of inflammatory cells.7,8 In addition, as the advanced plaques grow thicker, their core becomes hypoxic, due to the high inflammatory activity of foam cells and to the increasing distance for oxygen diffusion.9–11 Hypoxia has indeed been evidenced in plaque and is associated with areas rich in macrophages,12 influencing notably their metabolism and polarization.13,14 However, how it modulates macrophage responses to stress is not known.
In this work, the effects of hypoxia on lipid loading and on stress responses and death mechanisms in foam cells were investigated. For the experimental model, THP-1-derived foam cells generated by incubation with oxidized LDL were used. Two models of oxidized LDL were used: the classically used copper sulfate-oxidized LDL (Ox-LDL) and the more physiological myeloperoxidase-modified LDL (Mox-LDL) because accumulating data suggest that this peroxidase plays a critical role in the pathology. Indeed, this enzyme is capable of generating the powerful oxidant hypochlorous acid, is present and active in atherosclerotic plaques,15 and is able to oxidize LDL.16 Moreover, hypochlorous acid-derived modifications are found in atheroma and atheroma-derived LDL.17–19 Copper and myeloperoxidase induce some distinct biochemical alterations of LDL.20 Actually, the oxidation of LDL with copper ions causes important modifications of both the lipid and protein moieties of the LDL.21 On the other hand, the Mox-LDL used in this work are predominantly modified only in their protein moiety, with limited modifications of the lipids.22 The malondialdehyde content, as determined by the thiobarbituric acid reactive substances assay, was clearly higher in Ox-LDL compared to native LDL, whereas it was similar in Mox-LDL. Compared to native LDL, Ox-LDL also showed an increased lipid peroxidation while it was only slightly increased in Mox-LDL.23 Mox-LDL have been reported to induce an inflammatory reaction in endothelial cells and THP-1 monocytes as well as strong oxidative stress in RAW264.7 murine macrophages.22,23
The work here shows that foam cells can be obtained with the two types of oxidized LDL – Ox-LDL or Mox-LDL – but the phenotype was much more pronounced with Mox-LDL. Hypoxia increased the lipid load in cells incubated without LDL or with Ox-LDL but not with Mox-LDL. The study of the stress pathways showed that Mox-LDL and, to a higher extent, Ox-LDL induced the unfolded protein response (UPR), which was reduced by hypoxia. Moreover, both types of LDL induced apoptosis and autophagy, with a clear protective effect of hypoxia on oxidized LDL-induced cell death.
Materials and methods
LDL isolation and oxidation
Native LDL were obtained by sequential density gradient ultracentrifugation from plasma of healthy blood donors. The concentration of LDL in phosphate-buffered saline (PBS) was adjusted to 1 mg/mL before incubation with 10 mM CuSO4 for 24 hours at 37°C. The oxidation was stopped by the addition of 25 mM butylated hydroxytoluene and incubation on ice for 1 hour. Mox-LDL were generated by mixing 8 μM of 1M HCl (final concentration: 4 mM), 45 μM of recombinant human myeloperoxidase 92.4 U/mL (final relative activity: 2.6 U/mg LDL), a volume containing 1.6 mg LDL, and 40 μM of hydrogen peroxide 50 mM (final concentration: 1 mM). The volume was adjusted to 2 mL with PBS containing 1 g/L ethylenediaminetetraacetic acid (EDTA) at pH 6.5. Recombinant human myeloperoxidase was provided by the Experimental Medicine Laboratory (ULB 222 Unit, CHU de Charleroi, Charleroi, Belgium). The oxidation reaction for the generation of Mox-LDL was immediately performed at 37°C for 5 minutes and stopped by incubation on ice to inhibit the myeloperoxidase enzymatic activity. Native LDL, Ox-LDL, and Mox-LDL were desalted against CO2-independent medium without glutamine (Lonza AG, Basel, Switzerland) by using PD-10 desalting columns (GE Healthcare Biosciences AB, Uppsala, Sweden). LDL were sterile filtered (0.2 mm), stored in the dark at 4°C, and used within 4 days.
LDL were characterized as explained in Calay et al.23
Cell culture and hypoxia incubation
Human lymphoma cells THP-1 (American Type Culture Collection, Manassas, VA, USA) were cultured in Costar® 75 cm2 polystyrene flasks (Corning Incorporated, Corning, Lowell, MA, USA) with 15 mL RPMI medium complemented with 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), pyruvate, glucose, and 2-mercaptoethanol (final concentration: 4.5 g/L glucose, 10 mM HEPES, 1 mM sodium pyruvate, 0.05 mM β-mercaptoethanol) and with 10% of heat inactivated fetal calf serum (following the American Type Culture Collection’s recommendations). The cells were cultured under an atmosphere containing 5% carbon dioxide. For hypoxia experiments (1% oxygen), cells were incubated in serum-free carbon dioxide-independent medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 1 mM L-glutamine (Sigma-Aldrich Co, St Louis, MO, USA) and complemented with HEPES, pyruvate, glucose, and 2-mercaptoethanol. Normoxic control cells were incubated in the same conditions but in normal atmosphere (21% oxygen).
Cell differentiation
Depending on the experiment, monocytes were seeded at the appropriate confluence in complete medium with 150 nM of phorbol 12-myristate 13-acetate (PMA; P8139; Sigma). After 24 hours, medium with PMA was removed and fresh complete medium was added for 24 hours. After differentiation, the cells were incubated in the presence of 100 or 200 μg of LDL/mL for 72 hours in order to become foam cells.
Oil Red O (ORO) staining
Cells were seeded and differentiated in a Costar 12-well plate (Corning) at 350,000 cells/well. After incubation, the cells were washed with PBS and fixed with paraformaldehyde (Merck Millipore, Billerica, MA, USA) 4% for 2 minutes and 30 seconds. Cells were then incubated in the presence of an ORO solution (ORO 0.12% [Sigma], 2-propanol 60%, dextrin 0.4%) for 30 minutes. The cells were then washed four times with PBS and pictures were taken. Cells were then permeabilized with 96% ethanol for 30 minutes and the ethanol containing the diluted ORO was recovered and transferred to another multiwell plate. The optical density was then read at 490 nm. The permeabilized cells were then incubated in the presence of propidium iodide (P4170; Sigma) solution (PBS with propidium iodide 0.4%) for 30 minutes. Fluorescence was measured at 612 nm after excitation at 515 nm. The results are expressed as a ratio between the ORO absorbance and propidium iodide fluorescence.
Western blot analysis
Cells were seeded and differentiated in Costar 25 cm2 flasks (Corning) at 1.2 million cells/well. After incubation, cells were scrapped in 150 μL lysis buffer (40 mM Tris pH 7.5, 50 mM potassium chloride, 1 mM EDTA, 1% sodium dodecyl sulfate) containing a protease inhibitor mixture (1 tablet in 2 mL water added at a 1/25 dilution; Hoffman-La Roche Ltd, Basel, Switzerland) and phosphatase inhibitors (sodium metavanadate 25 mM, 4-nitrophenyl phosphate 250 mM, β-glycerophosphate 250 mM, sodium fluoride 125 mM at a 1/25 dilution). Western blot analysis was performed as described in Notte et al24 using a rabbit monoclonal anti-phosphorylated eIF2α antibody (119A11; Cell Signaling Technology, Inc., Danvers, MA, USA), a mouse anti-β-actin antibody (A5441; Sigma), a rabbit polyclonal anti-cleaved caspase 3 antibody (9662S; Cell Signaling), a rabbit polyclonal anti-caspase 7 antibody (9492; Cell Signaling), a rabbit polyclonal anti-caspase 4 antibody (4450S; Cell Signaling), a rabbit polyclonal anti-caspase 9 antibody (9502; Cell Signaling), a mouse monoclonal anti-caspase 8 antibody (9746; Cell Signaling), a mouse monoclonal anti-LC3 antibody (Nanotools GmbH, Munchen, Germany), or a mouse anti-p62 antibody (Abnova, Taipei, Taiwan).
Immunofluorescence staining
Cells were seeded and differentiated in a Costar 24-well plate (Corning) at 100,000 cells/well on 13 mm cover glass. After the incubation, cells were fixed for 30 minutes with 4% paraformaldehyde at room temperature. The cells were then rinsed three times with PBS and permeabilized with PBS bovine serum albumin 1% (GE Healthcare, Wauwatosa, WI, USA) containing Tween 20 0.5% (Sigma) for 40 minutes. Cells were then incubated for 1 hour at room temperature with primary anti-adipophilin antibody (dilution 1/10, mouse monoclonal, AP125; PROGEN Biotechnik GmbH, Heidelberg, Germany). After having been rinsed three times with PBS bovine serum albumin 1%, cells were then incubated for 1 hour at room temperature in the dark with anti-mouse secondary antibody (coupled to Alexa 568 dilution 1/500) and the lipid probe BODIPY® 493/503 at 20 μg/mL (D3922; Thermo Fisher). Cells were then rinsed three times with PBS and mounted with Mowiol® (Sigma).
XBP1 splicing assay
Cells were seeded and differentiated in Costar 75 cm2 culture flasks (Corning) at 3.6 million cells/well. After 72 hours of incubation, total RNA was extracted using the TRIzol® reagent (Thermo Fisher). RNA was then retrotranscribed using Roche’s retrotranscription kit and used for the assay as explained in DuRose et al.25 Quantification of the integrated optical density was performed with the ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Transmission electron microscope (TEM) analysis
Cells were seeded and differentiated in a Costar 12-well plate (Corning) at 600,000 cells/well. Cells were then fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate (pH 7.4) for 2 hours and postfixed in 1% osmium tetroxide in 0.1 M sodium cacodylate for 1 hour at 4°C. Cells were then dehydrated in ethanol (30%, 50%, 70%, 85%, and 100%) and embedded in LX112 resin. Blocks were cut on a Nova ultramicrotome (Leica Microsystems, Wetzlar, Germany) to obtain an ultrathin section of 60 nm. Sections were then stained with uranyl acetate and lead citrate and were finally observed on a Tecnai™ 10 TEM (FEI, Hillsboro, OR, USA).
3-(4,5-dimethylthiazol-2-yl)- 2,5-diphenyltetrazolium bromide (MTT) mitochondrial activity assay
Cells were seeded and differentiated in a Costar 24-well plate (Corning) at 100,000 cells/well. After incubation, 500 μL of PBS 0.1 M containing MTT (2.5 mg/mL; M2128; Sigma) was added in each well and the cells were incubated for 2 hours in the dark at 37°C in the presence of 5% carbon dioxide. The medium was then removed and cells were lysed in a solution containing 20% sodium dodecyl sulfate and 33% N, N-dimethylformamide (pH 4.7). The optical density was then read at 570 nm.
Validation on peripheral blood monocytic cells (PBMCs)
PBMCs were taken from a single donor for the different experiments and purified by centrifugation on a Ficoll gradient. Blood sampling was approved by the CHU de Charleroi’s ethics committee (Comite d’Ethique ISPPC: OM008). The studies conformed to the principles outlined in the Declaration of Helsinki. Written consent was obtained from the donors. In a 50 mL tube (227261; Greiner Bio-One, Frickenhausen, Germany), 15 mL of freshly removed blood was mixed with 15 mL of Hank’s Buffered Salt Solution (HBSS; BE10-547f; Lonza). Ficoll (10 mL; Histopaque®-1077; Sigma) was then added to the mix at the bottom of the tube, and the tube containing both mixed blood with HBSS and Ficoll was centrifuged at 400× g for 30 minutes at room temperature. After centrifugation, an interfacial layer containing the monocytes was taken up and mixed in 10 mL of HBSS in 15 mL tubes (188261; Greiner). The tube was then centrifuged at 1,500 rpm for 15 minutes. After centrifugation, the HBSS was removed and the cells were resuspended in fresh HBSS. Cells were then submitted to a second centrifugation identical to the first. After centrifugation, the HBSS was removed and the cells were resuspended with RPMI 1640 supplemented with nonessential amino acids, pyruvate, and 2-β-mercaptoethanol (final concentration: nonessential amino acids 1%; 1 mM sodium pyruvate; 50 μM 2-β-mercaptoethanol) in silicone tubes (BD Vacutainer® 368500; BD, Franklin Lakes, NJ, USA). During this last phase, a portion of the cellular suspension was taken to perform cell counting using Cell-Dyn 3200 (Abbott Laboratories, Abbott Park, IL, USA). Cells were then transferred at the optimal density and at the support required for the relevant experiments in RPMI 1640, supplemented as described above. The cells were left for 2 hours 30 minutes in their culture plate, then the medium was removed and the cells were incubated in the presence of RPMI 1640, supplemented as described above, with a 10% autologous heat inactivated serum containing recombinant M-CSF (216-MCC/CF; R&D Systems, Inc., Minneapolis, MN, USA) at a final concentration of 20 ng/mL. After 36 hours, the medium was removed and the new complete medium (as described above and with 10% serum) with M-CSF at a concentration of 20 ng/mL was added. The medium was removed 36 hours later, the cells were washed with PBS, and the complete medium with M-CSF at a concentration of 20 ng/mL was added. Four days later, the medium was removed and the cells were washed with PBS, ready to be used for experiments. During the experiments, cells were incubated without M-CSF in a carbon dioxide-independent medium supplemented with glutamine, nonessential amino acids, pyruvate, and 2-β-mercaptoethanol (final concentration: glutamine 1 mM, nonessential amino acids 1%, 1 mM sodium pyruvate, 50 μM 2-β-mercaptoethanol). The experiments were then performed the same way as the THP-1-derived macrophage experiments except that ORO was seeded at 500,000 cells/well instead of 350,000 cells/well.
Statistical analyses
SigmaPlot® software (Systat Software, Inc., San Jose, CA, USA) was used for the analysis. Data are presented as mean ± one standard deviation and were evaluated by a two-way analysis of variance, using the Holm–Sidak test as the post hoc test.
Results
Effects of hypoxia on lipid load
Foam cells are characterized by a large number of lipid droplets. In order to evidence these lipid droplets and to assess the overall lipid load, ORO staining as well as fluorescent labeling with the BODIPY 493/503 probe were performed. The abundance and subcellular localization of the PAT family protein adipophilin was also assessed. This protein is now recognized as a marker of lipid accumulation and is found around the lipid droplets in foam cells where its abundance is proportional to the lipid load.26,27
The cells were incubated in the presence of 50 and 100 μg/mL LDL for 24, 48, and 72 hours. The amount of neutral lipids was assessed by ORO staining. Adipophilin and lipid droplets were visualized by double immunofluorescence labeling. Cells were incubated in the presence of Ox-LDL, Mox-LDL, and native LDL, which were used as a negative control. After 24 and 48 hours, in the presence of 50 or 100 μg of LDL/mL, lipid loading was not markedly increased, neither was the abundance of adipophilin (data not shown). However, Figure 1 shows that cells incubated for 72 hours, especially in the presence of 100 μg/mL LDL, displayed an increased lipid load (Figure 1A) as well as an important number of lipid droplets surrounded by a thick layer of adipophilin (Figure 1C). Surprisingly, an important lipid load characteristic of foam cells was observed in cells incubated in the presence of native LDL. The lipid load probably results from internalization of the LDL progressively oxidized/modified by macrophages during the 72-hour incubation. This is why, from now, these LDL will not be considered as native anymore but as cell-modified LDL (CM-LDL). Cells incubated in the presence of oxidized/modified LDL also displayed numerous lipid droplets, but the lining of adipophilin was particularly obvious with the Mox-LDL compared with the Ox-LDL. These results were validated using PBMC-derived macrophages and similar results were obtained (Figure 1B).
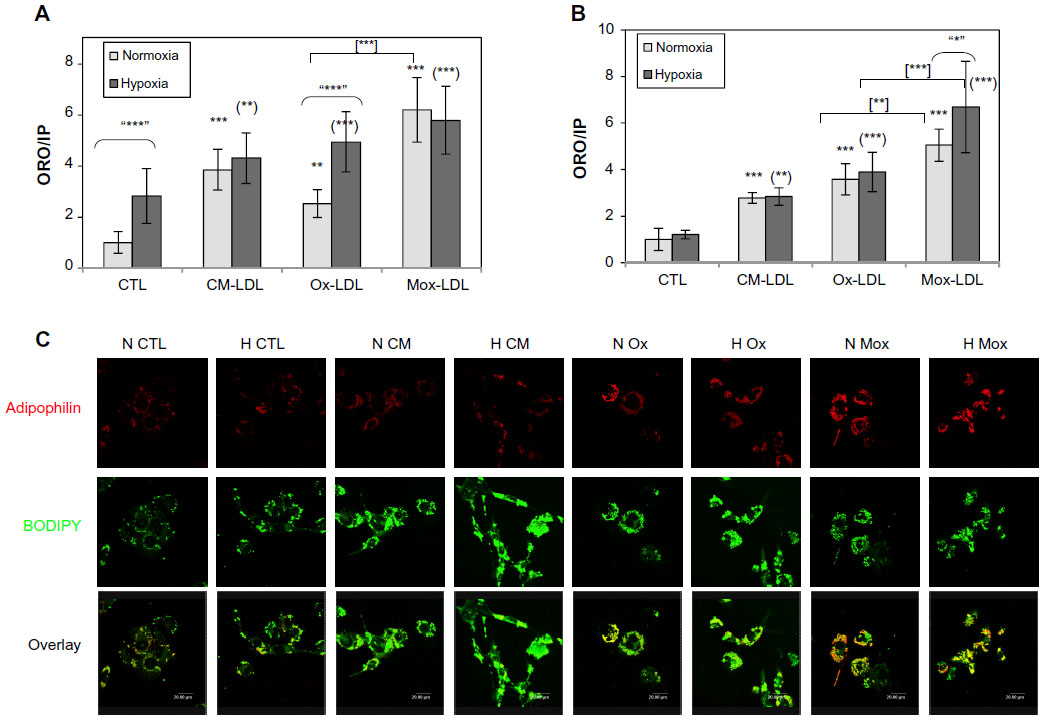 | Figure 1 Effect of hypoxia on lipid loading and on adipophilin abundance and localization. |
The possible effects of hypoxia on lipid load were evaluated in order to demonstrate that both THP-1-derived and PBMC-derived macrophages were responsive to hypoxia in the experimental conditions. The messenger RNA expression of two HIF-target genes (VEGF and BNIP3) was quantified. The results show that both types of macrophages did respond to hypoxia (data not shown).
Macrophages were incubated under hypoxia or normoxia in the presence of 100 μg/mL of the different types of LDL for 72 hours. The lipid load as well as the amount and subcellular localization of adipophilin were then assessed. As described by Bostrom et al28 and by Crucet et al,29 hypoxia alone increased lipid load. The cells incubated in the presence of CM-LDL displayed a marked lipid load, similar under hypoxia and normoxia. Cells incubated in the presence of Ox-LDL accumulated more lipids under hypoxia. Finally, the cells incubated in the presence of Mox-LDL displayed the highest lipid load, which was not influenced by hypoxia (Figure 1A). Immunofluorescence staining (Figure 1C) showed the presence of several lipid droplets lined by a low amount of adipophilin in the control cells, and numerous droplets surrounded by a larger amount of adipophilin in the cells incubated in the presence of each type of LDL under normoxia, especially in the presence of Mox-LDL. Hypoxia seemed to increase the amount of lipids in control cells and in the cells incubated in the presence of Ox-LDL compared to normoxia. There was no clear difference between hypoxia and normoxia for the cells incubated in the presence of CM-LDL or Mox-LDL. The effect of hypoxia was also tested on PBMC-derived macrophages. In contrast to what was observed with the THP-1-derived macrophages, no further increase in the lipid load in the cells incubated under hypoxia was evidenced compared with cells incubated under normoxia, whatever the type of LDL used (Figure 1B).
Stress and death pathways
Toxicity of the different types of LDL
Preliminary experiments using 100 μg/mL of LDL showed limited toxicity and no apoptosis (data not shown). In order to be able to study the stress and death processes, the LDL concentration was doubled (200 μg/mL). Cells were incubated in the presence of 200 μg/mL of the different types of LDL for 24, 48, and 72 hours under hypoxia or normoxia. Analysis of cell morphology by phase contrast microscopy after ORO staining showed that LDL induced lipid accumulation within the cells, which seemed to be more important in cells incubated with Ox-LDL and Mox-LDL than in cells incubated with CM-LDL (Figure 2A). However, marked toxicity was also evidenced in these two conditions. Hypoxia increased the lipid loading, especially in Ox-LDL and Mox-LDL incubated cells. Quantification of the lipid loading was performed (Figure 2B). The results confirmed what was observed: an increase in lipid load in the presence of oxidized LDL, which was significantly enhanced when cells were exposed to hypoxia. It must be noted that cell death observed mainly in cells incubated with Ox-LDL and Mox-LDL under normoxia, inferred from propidium iodine fluorescence values, may lead to underestimation of lipid accumulation.
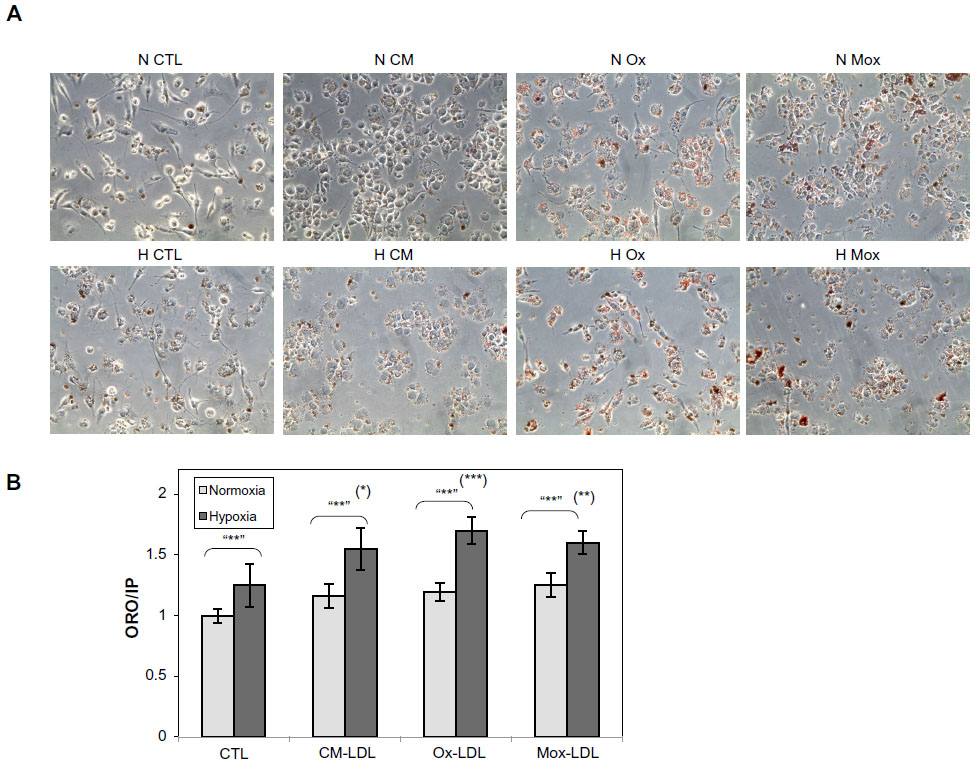 | Figure 2 Effect of hypoxia on lipid loading. |
The viability was then assessed using an MTT assay. Figure 3 and Table S1 show that under normoxia, the number of viable cells significantly decreased as the incubation time increased for the cells incubated in the presence of Ox-LDL and more importantly with Mox-LDL. Hypoxia markedly influences cell viability. First, the toxic effect of Mox-LDL was lower than under normoxia. Hypoxia could thus provide some protection against Mox-LDL-induced cell death. Second, after 48 and 72 hours of incubation in the presence of CM-LDL, the viability of macrophages incubated under normoxia was higher than hypoxia, which is the opposite of what was observed for both types of oxidized LDL. The same trend was also observed for untreated macrophages, ie, incubated without LDL. Both effects were statistically significant. The reason for this is unclear. Hypoxia per se induces stressful conditions that may affect cell viability. One example could be the depletion in glucose, pyruvate, and/or glutamine, which are substrates used in high amounts due to the inhibition of the oxidative phosphorylation. In the presence of oxidized LDL, these metabolic changes may lead to a lesser production of reactive oxygen species within the cells, hence the lower toxicity of these oxidized LDL under hypoxia.
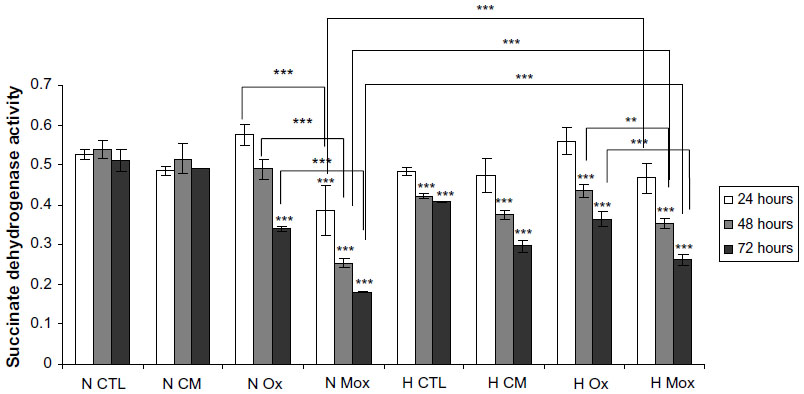 | Figure 3 Effects of hypoxia on low-density lipoprotein-induced cytotoxicity. |
UPR
Cholesterol overload is well known to disturb the endoplasmic reticulum (ER) membrane and to generate ER stress, which induces the UPR – a stress response well known to occur in atheromatous plaques.30,31 A growing body of evidence indicates that the ER plays a key role in the death of plaque macrophages and is a central regulator of death pathways.32
Thus, this study aimed to characterize ER stress in the foam cells as well as to study the possible effect of hypoxia on this stress. The abundance of phosphorylated eIF2α was assessed by Western blot analysis. Thapsigargin, a powerful sarco/endoplasmic reticulum Ca2+ adenosine triphosphatase inhibitor, was used as a positive control for UPR (200 nM for 20 hours under normoxia). A marked increase in the level of eIF2α phosphorylation was indeed observed (Figure 4A). The results shows that, under normoxia, incubation in the presence of Ox-LDL increased the abundance of phosphorylated eIF2α. Surprisingly, after 72 hours of incubation, the cells incubated in the presence of Mox-LDL, which displayed the highest lipid load, only presented a moderate increase in eIF2α phosphorylation (Figure 4A). It has to be noted that after 24 and 48 hours of incubation, Mox-LDL induced a higher level of eIF2α phosphorylation (Figure S1). After 24 hours, the abundance of phosphorylated eIF2α in the cells incubated with Mox-LDL and with Ox-LDL was comparable. However, while the level of eIF2α phosphorylation remained high throughout the 72 hours of incubation in the cells incubated with Ox-LDL, it began to decrease at 48 hours in the cells incubated with Mox-LDL. After 72 hours of incubation under hypoxia, the amount of phosphorylated eIF2α shows the same profile as under normoxia.
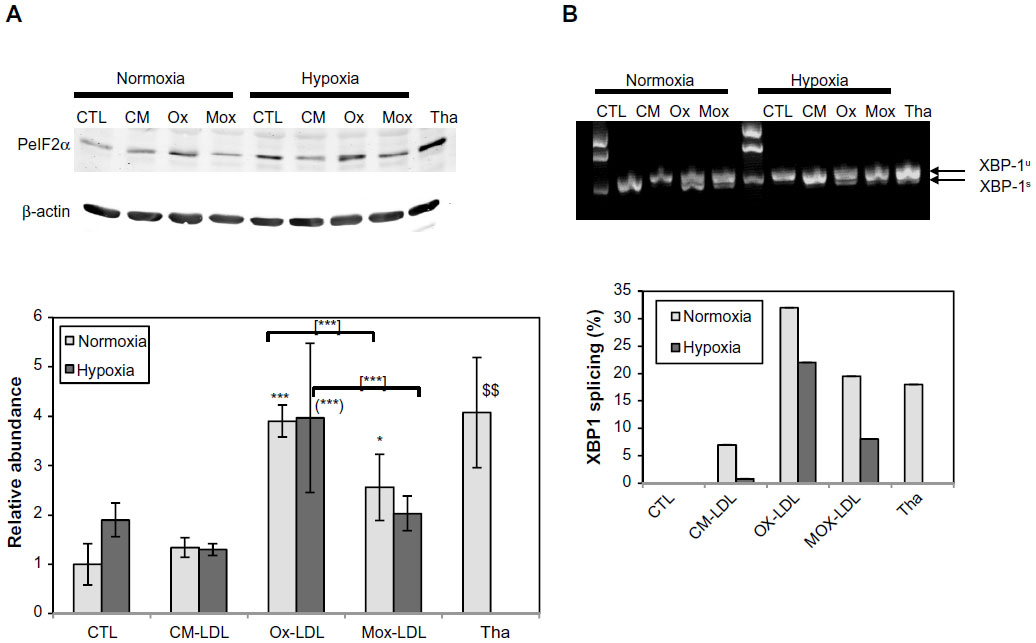 | Figure 4 Effects of hypoxia on low-density lipoprotein-induced unfolded protein response. |
The amount of spliced XBP1 messenger RNA was studied by polymerase chain reaction using primers designed on both sides of the spliced intron. Figure 4B shows that for the cells incubated under normoxia, the level of the spliced XBP1 messenger RNA was higher in the cells incubated with Mox-LDL and to a larger extent in the cells incubated with Ox-LDL, with a slight inhibitory effect of hypoxia. Thapsigargin, used as a positive control, also induced XBP1 splicing.
Altogether, these results indicate that the UPR is triggered when the macrophages are incubated with Ox-LDL and Mox-LDL, but following different kinetics. CM-LDL does not seem to induce ER stress. Hypoxia does not seem to influence the induction of UPR.
Autophagy
Since autophagy can be triggered by ER stress, this process was studied. One of the most commonly used markers of this pathway is the LC3 II protein, formed by the covalent binding of phosphatidylethanolamine with LC3 I during the formation of autophagosomal vesicles.33,34 The abundance of LC3 II was assessed by Western blot analysis in cells incubated for 72 hours in the presence of 200 μg/mL LDL. Figure 5A shows that the level of LC3 II was slightly increased in cells incubated in the presence of Mox-LDL and markedly increased in cells incubated with Ox-LDL under normoxia. Hypoxia alone did not induce LC3 conversion. On the other hand, hypoxia seemed to prevent the effect of Ox-LDL. Similar results were obtained after 24 and 48 hours of incubation (data not shown). The abundance of the LC3 I form was increased in parallel to the LC3 II form. Thapsigargin, used as a positive control for ER stress, also markedly induced the accumulation of the phosphatidylethanolamine-conjugated form of LC3.
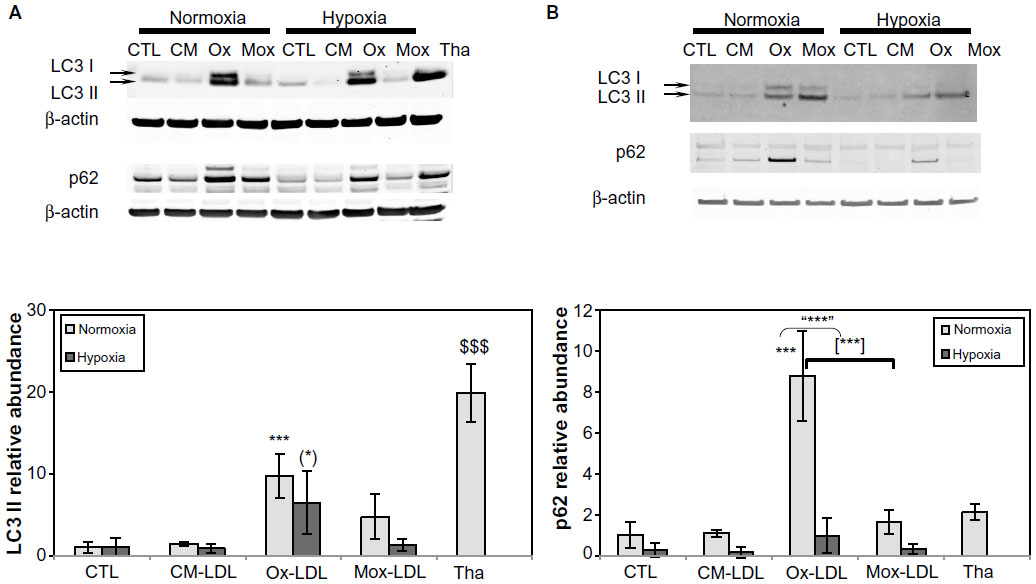 | Figure 5 Effects of hypoxia on low-density lipoprotein-induced autophagy. |
In order to ascertain the possible triggering of autophagy by oxidized LDL, the amount of p62, described to be degraded in cells undergoing autophagy, was assessed. It associates with oxidized proteins and ubiquitinated protein aggregates and targets them to the autophagosome by interaction with LC3 II.35–37 In doing so, p62 is also degraded and its amount decreases as the autophagic process goes on. Under normoxia, the abundance of the protein was increased in the presence of Mox-LDL and to a larger extent in the presence of Ox-LDL (Figure 5A). Under hypoxia, the abundance of this protein was decreased. It has to be noted that PBMC-derived macrophages show a similar profile of p62 and LC3 abundance when incubated in the presence of the different LDL (Figure 5B).
To further study the process of autophagy, TEM analysis was performed. Control cells displayed normal cellular morphology while cells incubated in the presence of Ox-LDL or Mox-LDL displayed large lipid droplets and typical features of autophagy such as double membrane vesicles and multilamellar bodies (Figure 6). These structures (Figures 6.1 and 6.2) were observed in cells incubated under normoxia with Ox-LDL. They correspond to engulfed cytosol by ER or ER self-engulfment, which is described to happen in ER stress-associated autophagy. Structures corresponding to myelin figures, also called multilamellar bodies, and resulting from the degradation of a membranous organelle (Figures 6.3 and 6.4), were observed in cells incubated under normoxia in the presence of Mox-LDL.
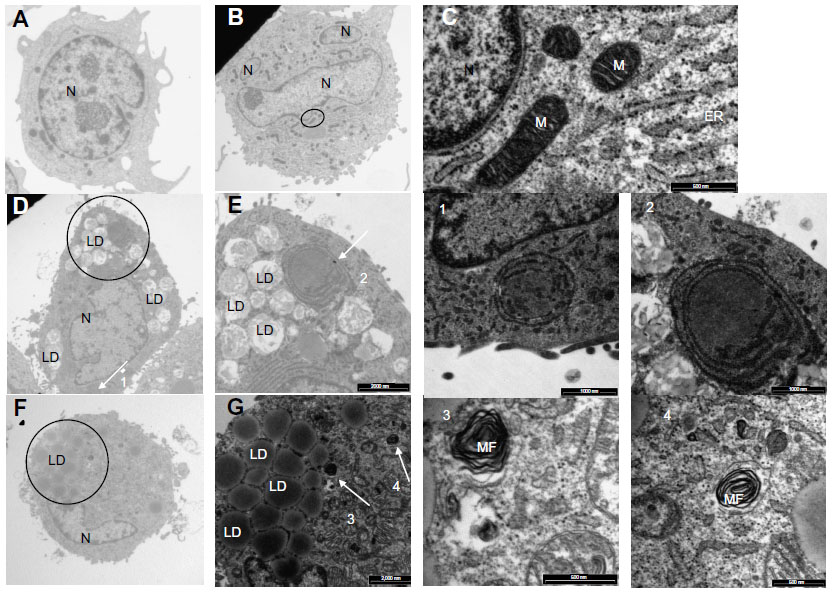 | Figure 6 Effects of low-density lipoproteins on cell morphology. |
A large number of cells incubated in the presence of Mox-LDL especially under normoxia were dying with an important number of vesicles supporting the possibility of death by excessive autophagy (Figure 7A). Some cells incubated in the presence of Ox-LDL under normoxia also displayed vesicularization but to a lesser extent than cells incubated with Mox-LDL (Figure 7B). Necrosis cannot been excluded, indeed rupture plasma membrane was observed. However, in this case, there should be no vesicularization of the cells. Apoptotic features were also observed (Figure 7B): chromatin was condensed; the nucleus was no longer round, suggesting the formation of nucleus-derived vesicles; and the cell was blebbing, indicating apoptotic body formation. While the cells incubated in the presence of Mox-LDL only displayed moderate markers of macroautophagy, TEM analysis showed characteristics of pronounced macroautophagy and of cell dismantling, indicating that these cells may be dying by excessive autophagy.
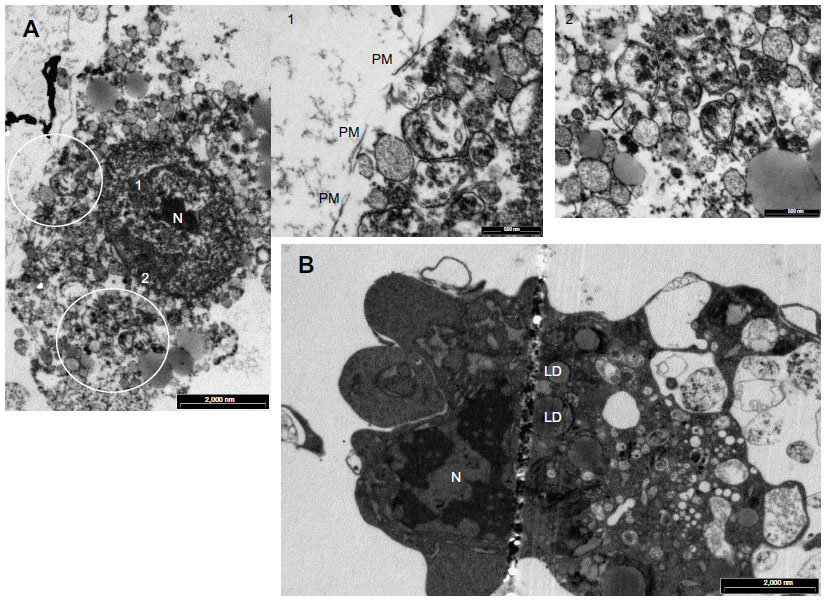 | Figure 7 Effects of oxidized low-density lipoproteins on cell morphology under normoxia. |
Figure 8 shows cells incubated under hypoxia either with Ox-LDL (Figure 8A) or with Mox-LDL (Figure 8B). Figure 8.1 shows a structure looking like ER engulfed material and Figure 8.2 shows a structure looking like a double membrane vesicle, which could also be a mitochondrion. Figure 8.3 and 8.4 both show autophagic-like vesicles, which are surrounded by a double membrane.
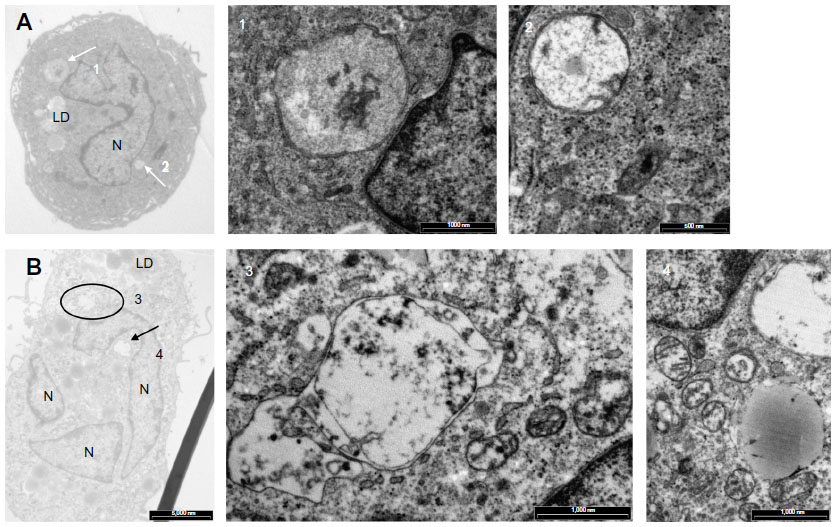 | Figure 8 Effects of oxidized low-density lipoproteins on cell morphology under hypoxia. |
Apoptosis
As apoptosis could account for the diminished cell viability observed in the cells incubated in the presence of Ox-LDL or Mox-LDL under normoxia, the activation of caspases was assessed by Western blot analysis. Upon activation, inactive precursor procaspases are cleaved, generating small and large subunits that dimerize. Two of these dimers interact to form the active caspase.
Western blot analyses show that under normoxia, the abundance of the cleaved form of executor caspases 3 and 7 was increased in the cells incubated in the presence of Ox-LDL and to a higher extent in the cells incubated in the presence of Mox-LDL. This indicates that these enzymes were activated. This was not observed under hypoxia (Figure 9A), suggesting that hypoxia prevented caspase activation in cells incubated in the presence of oxidized LDL. It has to be noted that PBMC-derived macrophages displayed a profile of cleaved caspase 3 similar to the one observed for THP-1-derived macrophages (Figure 9C). Western blot analyses also showed a higher level of PARP cleavage in THP-1-derived macrophages incubated with Mox-LDL under normoxia, which was prevented by hypoxia (data not shown).
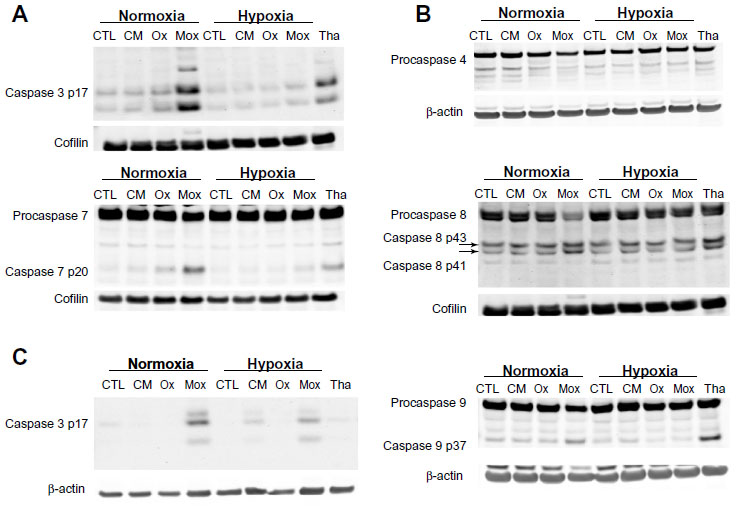 | Figure 9 Effects of low-density lipoproteins and hypoxia on apoptosis. |
The activation of executor caspases results from the activity of initiator caspases. The amount and profile of cleavage of initiator caspases that could be involved here were assessed by Western blot analysis.
Figure 9B shows that procaspase 4 was detected in the cells in all conditions, but its abundance was decreased in the cells incubated in the presence of Mox-LDL under normoxia. It is possible that the decrease in the abundance of the proenzyme is due to proteolytic cleavage due to its activation. In this case, this would suggest that Mox-LDL triggered caspase activation by the ER-associated caspase 4. Again, hypoxia protected the cells because the cleavage was not observed in the cells incubated under low oxygen partial pressure. However, as the cleaved fragments of the activated caspase could not be evidenced, it is not certain that the decrease in procaspase abundance was really due to activation.
Procaspase 8 was detected in the cells in all conditions. Again, as for caspase 4, the abundance was much lower in the cells incubated in the presence of Mox-LDL under normoxia. The cleaved fragments p41 and p43 were also detected in all conditions, and they were more abundant in the cells incubated in the presence of Mox-LDL under normoxia. This means that, in this case, the diminished abundance of the total form is actually associated with proteolytic activator cleavage. This was not observed under hypoxia. Similar results were obtained for caspase 9. It has to be noted that, for this caspase, the cleaved fragment evidenced corresponds to p37 and that p35 was never detected. This means that this caspase has been activated during the caspase cascade by caspase 3 and not by caspase 4 or by itself in the apoptosome.38,39 All together, these results suggest that Mox-LDL favored foam cell apoptosis, while Ox-LDL, often described as very toxic, only triggered a low level of apoptosis. Mox-LDL were the most potent inducer of apoptosis, and hypoxia protected the cells from apoptosis.
Discussion
Lipid loading
In this work, it was shown that foam cells were obtained by 72-hour incubation of THP-1-derived macrophages with 100 or 200 μg/mL of Ox-LDL and more markedly with Mox-LDL. Surprisingly, a foam cell phenotype at least as pronounced as the one observed with Ox-LDL was also observed in the cells incubated in the presence of CM-LDL. Although at first sight this was surprising, there are different hypotheses to explain this observation. First, other data support the fact that THP-1-derived macrophages can become foam cells when incubated with CM-LDL. Indeed, in the first research aimed at the characterization of the THP-1 cell line, Banka et al reported that THP-1 differentiated with PMA became foam cells when incubated with physiological concentrations of native LDL.40 It might be hypothesized that this lipid accumulation with THP-1-derived macrophage is an artifact due to the nature of the THP-1 cell line or due to the use of PMA, well known to increase macropinocytosis. However, the fact that PBMCs differentiated into macrophages with M-CSF and incubated for 72 hours with 200 μg/mL of CM-LDL also store important quantities of lipids invalidates this hypothesis.
It is well known that endothelial cells can oxidize native LDL, rendering them proinflammatory and prone to internalization.41 Macrophages may display a much more potent array of modifying/oxidizing activities and be more effective for lipoprotein oxidization/modification. Indeed, data shows that when incubated for 24 hours in the presence of native LDL, macrophages induce oxidization of LDL rendering them prone to internalization.42,43
Foam cells obtained with the different modified LDL displayed a high lipid load, which was the highest in cells incubated in the presence of Mox-LDL. This was observed with THP-1-derived macrophages as well as with PBMC-derived macrophages. In all cases, the lipid droplets were delineated by adipophilin, but the adipophilin border was clearly more pronounced in cells incubated in the presence of Mox-LDL (data not shown for PBMCs). This protein plays an important role in the lipid droplet formation and emerges as an atherosclerosis marker.27,44 The fact that the adipophilin edge always appeared thicker in cells incubated in the presence of Mox-LDL could account for more efficient lipid storage under these conditions.
It was shown that, in THP-1-derived macrophages, hypoxia enhanced lipid load in control cells as well as in cells incubated in the presence of oxidized LDL. Hypoxia-increased lipid load may be due to a decrease in the catabolism of fatty acids, to an increase in the synthesis of fatty acids, or to an upregulation of the scavenger receptor-dependent internalization. Bostrom et al28 showed that hypoxia caused an accumulation of lipid droplets in human macrophages incubated in the presence of Ox-LDL, which was correlated to an increase in triglyceride level but not in the level of cholesterol and cholesterol esters. Using DNA microarrays, they evidenced an increase in the transcription of ADRP and enzymes involved in the synthesis of fatty acid, such as SCD1, and a decrease in the transcription of enzymes involved in the β-oxidation of fatty acids such as acetyl–CoA synthase and acetyl–CoA dehydrogenase, leading to an actual decrease in the rate of β-oxidation.
On the other hand, Crucet et al29 demonstrated that increased membrane expression of some scavenger receptors could account for enhanced lipid load in RAW264.7 macrophages exposed to Ox-LDL under hypoxia. Hypoxia induced CD36 receptor relocalization to the plasma membrane and increased Lox-1 expression. Lox-1 seems to be important for Ox- LDL uptake since neutralizing anti-Lox-1 antibodies blocked this uptake.
Cytotoxicity
It was observed that 72 hours of incubation with 100 μg of LDL/mL induced neither toxicity nor apoptosis. Although surprising given the cytotoxicity generally associated with oxidized/modified LDL, these data are consistent with other observations from Vicca et al. Indeed, working on THP-1, U937, and PBMCs, they showed that once differentiated into macrophages, these cells were very resistant, withstanding oxidized LDL exposure that would kill monocytes.45,46 So, in order to study the death pathways, the concentration of LDL/mL had to be increased up to 200 μg. With these conditions, the cells incubated in the presence of Ox-LDL, and especially in the presence of Mox-LDL, displayed a diminished viability as assessed by the MTT assay. This reduction in cell viability was less pronounced when the cells were incubated at low oxygen partial pressure, which suggests a protective effect of hypoxia.
UPR
Further analysis showed that the levels of UPR markers were increased in the cells incubated in the presence of Mox-LDL and to a larger extent in the cells incubated in the presence of Ox-LDL, especially under normoxia. These data suggest that hypoxia protects the macrophages from lipid load-induced cell death. This protection could be provided by the chaperone overexpression driven by HIF-1. Grp78 and ERO1 were both overexpressed in the THP-1-derived macrophages incubated under hypoxia, in the presence or not of LDL (data not shown). Both chaperones have been described to protect many cell types. Indeed, hypoxia-induced Grp78 overexpression has been described to protect tumor cells against proapoptotic drugs47 and cardiomyocytes against different stresses.48 Moreover in astrocytes, hypoxia-induced Grp78 overexpression contributes to the secretion of IL-6 which protects these cells.49 ERO1 displays the same type of cytoprotective effects. This needs to be further investigated.
Autophagy
Autophagy markers for the cells incubated in the presence of Mox-LDL were weak compared to the ones observed for the cells incubated in the presence of Ox-LDL. However, the cells incubated in the presence of Mox-LDL under normoxia displayed an important increase in the amount of p62. In this case, as LC3 II was not increased, the possibility of autophagy blockade was ruled out. Like Ox-LDL, Mox-LDL probably induced an oxidative stress that would have been more pronounced under normoxia and would have resulted in Nrf2 activation and p62 overexpression. Indeed, performing a comparative study of Ox-LDL and Mox-LDL, Calay et al showed that both types of LDL induced oxidative stress with Nrf2 activation.23
TEM analysis revealed that the cells incubated in the presence of Ox-LDL, whatever the oxygen concentration, displayed structures typical of macroautophagy and more precisely of ER-phagy. Surprisingly, despite low levels of molecular markers of autophagy, the cells incubated in the presence of Mox-LDL also displayed structures typical of autophagy such as myelin figures and double membrane vesicles. Moreover, a lot of cells incubated in the presence of Mox-LDL, especially under normoxia, were very damaged and featured a lot of vesicle-like structures. This suggests that despite showing obvious features of apoptosis, these cells also die with or by excessive autophagy.
Apoptosis
The study of apoptosis revealed caspase cleavage was particularly pronounced in the cells incubated in the presence of Ox-LDL and even more pronounced in the cells incubated with Mox-LDL. The mechanism initiating apoptosis may involve the mitochondrial pathway as shown by Aflaki et al.50 Mox-LDL-induced apoptosis was markedly reduced under hypoxia. Numerous studies have demonstrated that hypoxia protects against apoptosis induced by different stresses. It is particularly the case in cancer cells51–53 but may also apply to macrophages.14
If macrophages incubated in the presence of Mox-LDL die, partly by apoptosis, the cells incubated in the presence of Ox-LDL do not die or die by another death pathway. Such a low level of apoptosis in these cells that displayed pronounced UPR markers with marked eIF2α phosphorylation is surprising because long-term eIF2α phosphorylation is well known to trigger ER-associated apoptosis by both CHOP induction and decreased protein synthesis.54 Paradoxically, UPR and especially long-lasting eIF2α phosphorylation are also described to trigger autophagy.55,56 This process could protect the cells incubated in the presence of Ox-LDL and delay apoptosis. Western blot analysis revealed that the cells incubated in the presence of Ox-LDL, whatever the oxygen concentration, displayed elevated levels of both LC3 II and p62. This could mean two things, either blockade of autophagic vacuole degradation or strong induction of the process with increased abundance of both LC3 and p62. As p62 is normally degraded during macroautophagy, one explanation for its increased amount has been long disturbances in the autophagic flux.33 Such disturbances could also account for the increased abundance of LC3 protein. However, p62 can also increase in response to different stresses such as oxidative stress. In this case, increase of p62 is due to transcriptional upregulation by Nrf2, of which p62 is a target gene.57 As Ox-LDL are well known to induce oxidative stress, it is likely that they activate Nrf2, which would in turn increase the amount of p62. This phenomenon needs to be experimentally confirmed. Combined with autophagy induction by the UPR, it could explain the results here.
Conclusion
A better understanding and characterization of the interplay between death processes in foam cells obtained with the different types of LDL is of great interest. Because autophagy and apoptosis are often associated in dying cells in atherosclerosis, it would be very interesting to understand how these pathways crosstalk in order to manipulate them for therapeutic goals. Indeed, the death of foam cells by apoptosis leads to destabilization of the plaque and to death by autophagy, which could be beneficial. The results clearly showed that Ox-LDL and Mox-LDL induced different cell death pathways, which may trigger inflammation to a different extent. This is an important observation to take into account in order to design new therapeutic strategies.
Similarly, understanding the effect of hypoxia is also a priority. It has been shown that most macrophages are exposed to a low oxygen partial pressure in atherosclerotic plaques. As hypoxia can modify the response of foam cells to stresses as shown here, hypoxia should be more taken into account in the development of new therapeutic strategies for atherosclerosis.
Acknowledgments
B Vlaminck, D Calay, and M Genin were financed by a FRIA (Fonds de la Recherche pour l’Industrine et l’Agriculture) grant (Brussels, Belgium). This article presents results of the Belgian Programme on Interuniversity Poles of Attraction initiated by the Belgian State, Prime Minister’s Office, Science Policy Programming. The responsibility is assumed by its authors. The authors also thank the Fonds National de la Recherche Scientifique (FNRS) for financial support. The authors thank Corry Charlier and David Degleize for the electron microscopy service.
Disclosure
The authors report no conflicts of interest in this work.
References
Tabas I, Williams KJ, Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116(16):1832–1844. | |
Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15(5):551–561. | |
Stocker R, Keaney JF Jr. Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84(4):1381–1478. | |
Greaves DR, Gordon S. The macrophage scavenger receptor at 30 years of age: current knowledge and future challenges. J Lipid Res. 2009; 50 Suppl:S282–S286. | |
Levitan I, Volkov S, Subbaiah PV. Oxidized LDL: diversity, patterns of recognition, and pathophysiology. Antioxid Redox Signal. 2010;13(1):39–75. | |
Schrijvers DM, De Meyer GR, Herman AG, Martinet W. Phagocytosis in atherosclerosis: molecular mechanisms and implications for plaque progression and stability. Cardiovasc Res. 2007;73(3):470–480. | |
Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol. 2005;25(11):2255–2264. | |
Van Vre EA, Ait-Oufella H, Tedgui A, Mallat Z. Apoptotic cell death and efferocytosis in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32(4):887–893. | |
Bjornheden T, Levin M, Evaldsson M, Wiklund O. Evidence of hypoxic areas within the arterial wall in vivo. Arterioscler Thromb Vasc Biol. 1999;19(4):870–876. | |
Hulten LM, Levin M. The role of hypoxia in atherosclerosis. Curr Opin Lipidol. 2009;20(5):409–414. | |
Sluimer JC, Gasc JM, van Wanroij JL, et al. Hypoxia, hypoxia-inducible transcription factor, and macrophages in human atherosclerotic plaques are correlated with intraplaque angiogenesis. J Am Coll Cardiol. 2008;51(13):1258–1265. | |
Mateo J, Izquierdo-Garcia D, Badimon JJ, Fayad ZA, Fuster V. Noninvasive assessment of hypoxia in rabbit advanced atherosclerosis using 18F-fluoromisonidazole positron emission tomographic imaging. Circ Cardiovasc Imaging. 2014;7(2):312–320. | |
Marsch E, Sluimer JC, Daemen MJ. Hypoxia in atherosclerosis and inflammation. Curr Opin Lipidol. 2013;24(5):393–400. | |
Parathath S, Yang Y, Mick S, Fisher EA. Hypoxia in murine atherosclerotic plaques and its adverse effects on macrophages. Trends Cardiovasc Med. 2013;23(3):80–84. | |
Daugherty A, Dunn JL, Rateri DL, Heinecke JW. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J Clin Invest. 1994;94(1):437–444. | |
Carr AC, McCall MR, Frei B. Oxidation of LDL by myeloperoxidase and reactive nitrogen species: reaction pathways and antioxidant protection. Arterioscler Thromb Vasc Biol. 2000;20(7):1716–1723. | |
Hazen SL, Heinecke JW. 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human atherosclerotic intima. J Clin Invest. 1997;99(9):2075–2081. | |
Malle E, Marsche G, Arnhold J, Davies MJ. Modification of low-density lipoprotein by myeloperoxidase-derived oxidants and reagent hypochlorous acid. Biochim Biophys Acta. 2006;1761(4):392–415. | |
Nicholls SJ, Hazen SL. Myeloperoxidase, modified lipoproteins, and atherogenesis. J Lipid Res. 2009;50 Suppl:S346–S351. | |
Yan LJ, Lodge JK, Traber MG, Matsugo S, Packer L. Comparison between copper-mediated and hypochlorite-mediated modifications of human low density lipoproteins evaluated by protein carbonyl formation. J Lipid Res. 1997;38(5):992–1001. | |
Zarev S, Bonnefont-Rousselot D, Jedidi I, et al. Extent of copper LDL oxidation depends on oxidation time and copper/LDL ratio: chemical characterization. Arch Biochem Biophys. 2003;420(1):68–78. | |
Boudjeltia KZ, Legssyer I, Van Antwerpen P, et al. Triggering of inflammatory response by myeloperoxidase-oxidized LDL. Biochem Cell Biol. 2006;84(5):805–812. | |
Calay D, Rousseau A, Mattart L, et al. Copper and myeloperoxidase-modified LDLs activate Nrf2 through different pathways of ROS production in macrophages. Antioxid Redox Signal. 2010;13(10):1491–1502. | |
Notte A, Ninane N, Arnould T, Michiels C. Hypoxia counteracts taxol-induced apoptosis in MDA-MB-231 breast cancer cells: role of autophagy and JNK activation. Cell Death Dis. 2013;4:e638. | |
DuRose JB, Tam AB, Niwa M. Intrinsic capacities of molecular sensors of the unfolded protein response to sense alternate forms of endoplasmic reticulum stress. Mol Biol Cell. 2006;17(7):3095–3107. | |
Bickel PE, Tansey JT, Welte MA. PAT proteins, an ancient family of lipid droplet proteins that regulate cellular lipid stores. Biochim Biophys Acta. 2009;1791(6):419–440. | |
Paul A, Chan L, Bickel PE. The PAT family of lipid droplet proteins in heart and vascular cells. Curr Hypertens Rep. 2008;10(6):461–466. | |
Bostrom P, Magnusson B, Svensson PA, et al. Hypoxia converts human macrophages into triglyceride-loaded foam cells. Arterioscler Thromb Vasc Biol. 2006;26(8):1871–1876. | |
Crucet M, Wust SJ, Spielmann P, Luscher TF, Wenger RH, Matter CM. Hypoxia enhances lipid uptake in macrophages: role of the scavenger receptors Lox1, SRA, and CD36. Atherosclerosis. 2013;229(1):110–117. | |
Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res. 2010;107(7):839–850. | |
Zhou J, Lhotak S, Hilditch BA, Austin RC. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation. 2005;111(14):1814–1821. | |
Tabas I. Macrophage apoptosis in atherosclerosis: consequences on plaque progression and the role of endoplasmic reticulum stress. Antioxid Redox Signal. 2009;11(9):2333–2339. | |
Klionsky DJ, Abeliovich H, Agostinis P, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4(2):151–175. | |
Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3(6):542–545. | |
Bjorkoy G, Lamark T, Johansen T. p62/SQSTM1: a missing link between protein aggregates and the autophagy machinery. Autophagy. 2006;2(2):138–139. | |
Ichimura Y, Kominami E, Tanaka K, Komatsu M. Selective turnover of p62/A170/SQSTM1 by autophagy. Autophagy. 2008;4(8):1063–1066. | |
Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–24145. | |
Malladi S, Challa-Malladi M, Fearnhead HO, Bratton SB. The Apaf-1*procaspase-9 apoptosome complex functions as a proteolytic-based molecular timer. EMBO J. 2009;28(13):1916–1925. | |
Yamamuro A, Kishino T, Ohshima Y, et al. Caspase-4 directly activates caspase-9 in endoplasmic reticulum stress-induced apoptosis in SH-SY5Y cells. J Pharmacol Sci. 2011;115(2):239–243. | |
Banka CL, Black AS, Dyer CA, Curtiss LK. THP-1 cells form foam cells in response to coculture with lipoproteins but not platelets. J Lipid Res. 1991;32(1):35–43. | |
Henriksen T, Mahoney EM, Steinberg D. Enhanced macrophage degradation of low density lipoprotein previously incubated with cultured endothelial cells: recognition by receptors for acetylated low density lipoproteins. Proc Natl Acad Sci U S A. 1981;78(10):6499–6503. | |
Cathcart MK, Morel DW, Chisolm GM 3rd. Monocytes and neutrophils oxidize low density lipoprotein making it cytotoxic. J Leukoc Biol. 1985;38(2):341–350. | |
Parthasarathy S, Printz DJ, Boyd D, Joy L, Steinberg D. Macrophage oxidation of low density lipoprotein generates a modified form recognized by the scavenger receptor. Arteriosclerosis. 1986;6(5):505–510. | |
Larigauderie G, Cuaz-Perolin C, Younes AB, et al. Adipophilin increases triglyceride storage in human macrophages by stimulation of biosynthesis and inhibition of β-oxidation. FEBS J. 2006;273(15):3498–3510. | |
Ermak N, Lacour B, Drueke TB, Vicca S. Role of reactive oxygen species and Bax in oxidized low density lipoprotein-induced apoptosis of human monocytes. Atherosclerosis. 2008;200(2):247–256. | |
Vicca S, Hennequin C, Nguyen-Khoa T, et al. Caspase-dependent apoptosis in THP-1 cells exposed to oxidized low-density lipoproteins. Biochem Biophys Res Commun. 2000;273(3):948–954. | |
Fernandez PM, Tabbara SO, Jacobs LK, et al. Overexpression of the glucose-regulated stress gene GRP78 in malignant but not benign human breast lesions. Breast Cancer Res Treat. 2000;59(1):15–26. | |
Pan YX, Ren AJ, Zheng J, et al. Delayed cytoprotection induced by hypoxic preconditioning in cultured neonatal rat cardiomyocytes: role of GRP78. Life Sci. 2007;81(13):1042–1049. | |
Hori O, Matsumoto M, Kuwabara K, et al. Exposure of astrocytes to hypoxia/reoxygenation enhances expression of glucose-regulated protein 78 facilitating astrocyte release of the neuroprotective cytokine interleukin 6. J Neurochem. 1996;66(3):973–979. | |
Aflaki E, Radovic B, Chandak PG, et al. Triacylglycerol accumulation activates the mitochondrial apoptosis pathway in macrophages. J Biol Chem. 2011;286(9):7418–7428. | |
Rohwer N, Cramer T. Hypoxia-mediated drug resistance: novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist Updat. 2011;14(3):191–201. | |
Sermeus A, Genin M, Maincent A, et al. Hypoxia-induced modulation of apoptosis and BCL-2 family proteins in different cancer cell types. PLoS One. 2012;7(11):e47519. | |
Flamant L, Roegiers E, Pierre M, et al. TMEM45A is essential for hypoxia-induced chemoresistance in breast and liver cancer cells. BMC Cancer. 2012;12:391. | |
Marciniak SJ, Ron D. Endoplasmic reticulum stress signaling in disease. Physiol Rev. 2006;86(4):1133–1149. | |
Py BF, Boyce M, Yuan J. A critical role of eEF-2K in mediating autophagy in response to multiple cellular stresses. Autophagy. 2009;5(3):393–396. | |
Talloczy Z, Jiang W, Virgin HW 4th, et al. Regulation of starvation- and virus-induced autophagy by the eIF2α kinase signaling pathway. Proc Natl Acad Sci U S A. 2002;99(1):190–195. | |
Jain A, Lamark T, Sjottem E, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285(29):22576–22591. |
Supplementary materials
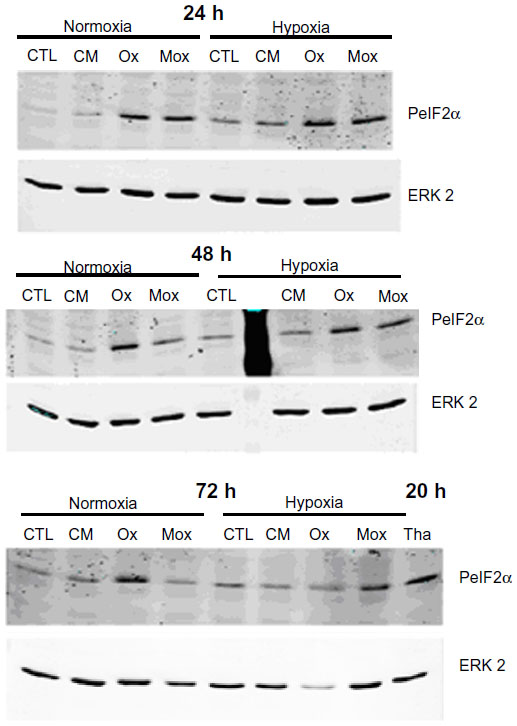 | Figure S1 Effects of hypoxia and low-density lipoproteins on the unfolded protein response. |
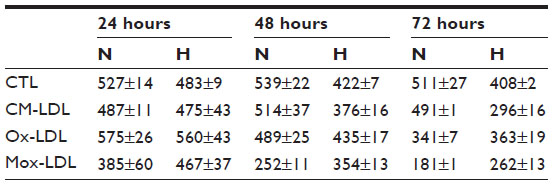 | Table S1 Effects of low-density lipoproteins and hypoxia on cytotoxicity |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.