Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 16
Effects of Atmospheric Fine Particulate Matter and Its Carrier Microbes on Pulmonary Microecology in Patients with COPD
Authors Che C ![]() , Sun X, Wu Y, Ma L, Hu Y
, Sun X, Wu Y, Ma L, Hu Y ![]() , Yang W, Qi H
, Yang W, Qi H ![]() , Zhou Y
, Zhou Y
Received 9 April 2021
Accepted for publication 19 June 2021
Published 12 July 2021 Volume 2021:16 Pages 2049—2063
DOI https://doi.org/10.2147/COPD.S314265
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Chunli Che, 1 Xiazhong Sun, 2 Yuhan Wu, 1 Lixin Ma, 2 Yueying Hu, 3 Weiyan Yang, 3 Hong Qi, 2 Yumin Zhou 4
1Harbin Medical University, Harbin, 150001, Heilongjiang, People’s Republic of China; 2State Key Laboratory of Urban Water Resource and Environment, Harbin Institute of Technology, Harbin, 150090, People’s Republic of China; 3Department of Respiratory Medicine, The First Affiliated Hospital of Harbin Medical University, Harbin, 150001, Heilongjiang, People’s Republic of China; 4State Key Laboratory of Respiratory Disease, National Center for Respiratory Diseases, Guangzhou Institute of Respiratory Diseases, The First Affiliated Hospital, Guangzhou Medical University, Guangdong, Guangdong Sheng, People’s Republic of China
Correspondence: Hong Qi; Yumin Zhou Email [email protected]; [email protected]
Objective: The aim of this paper was to analyse the influence of atmospheric fine particulate matter (AFPM) and atmospheric microorganisms on the pulmonary microecology of chronic obstructive pulmonary disease (COPD) patients in northeast China.
Methods: Collected bronchoalveolar lavage fluid (BALF) of COPD patients in the high-risk period (group A) and low-risk period (group B) of AFPM inhalation and samples of AFPM in the same time range (group C) were collected. DNA sample sequencing, the bacterial abundance, and diversity bioinformatics of BALFs were performed by methods of Illumina MiSeq™ platform and Mothur and Uclust.
Results: A total of 58 samples were sequenced, including 22 samples from group A, 26 samples from group B and 10 samples from group C. A total of 2,005,790 bacterial sequences and 34,256 bacterial numbers were detected. Group B had the highest bacterial diversity of the three groups. Group B also had the highest bacterial abundance index value. There were differences in the classification of bacterial colonies for the three groups at the genus level. The types of bacteria in group C were more numerous than other groups, and group B was higher than group A, which indicates that there were more bacteria in BALF during the high-risk period of AFPM inhalation. The detection rates of Streptococcus, Mycoplasma, Roche, Pushia, Chlamydia trachomatis and Brucella for group C were significantly higher than group A. The COG and KEGG databases’ difference analysis results for the bacterial gene function abundance of group A and group B were 40.7% in group A and 38.9% in group B (R=0.098, P=0.006). The human disease abundance in group A and group B was 1.16% and 1.12%, respectively (P> 0.05).
Conclusion: The increase in the concentration of AFPM can increase the diversity and abundance of bacteria in the BALF of stable COPD patients.
Clinical Trial Registration Number: 2020XS04-02.
Keywords: COPD, atmospheric fine particulate matter, bronchoalveolar lavage fluid, microbe, pulmonary microecology
Introduction
Atmospheric fine particulate matter (PM2.5) has been proven to be the main air pollutant. It has a complex composition and is significantly harmful to the human body.1 Studies at home and abroad have confirmed that both short-term sharp exposure and long-term chronic exposure to atmospheric PM2.5 can shorten lifespan and increase the mortality rates of a number of acute and chronic diseases.1,2 Recently, Aziz et al3 confirmed that atmospheric fine particulate matter can carry more than 1300 kinds of microorganisms and speculated that certain kinds of microorganisms may be pathogenic to humans. The respiratory system is directly exposed to the atmospheric environment, which is affected by atmospheric fine particles directly. It has been confirmed that the incidence and progression of respiratory diseases such as COPD are closely related to fine particle inhalation.4 Current studies focus mainly on immunomodulatory mechanism changes5 and lung function injury6 caused by organic and inorganic aromatic components of fine particulate matter; the effects of microbes carrying fine particulate matter on COPD have rarely been reported. Developments in gene sequencing technology have shown that the human lower respiratory tract is a microecological environment containing bacteria in both healthy and sick conditions.7 Changes in pulmonary microecology, including changes in microbial load, bacterial diversity, species and abundance, may be an important reason for the occurrence, progression and deterioration of a number of diseases.8,9 Based on our previous studies of the composition of fine particulate matter, we conducted microbiological metagenomic detection of the pulmonary alveolar lavage fluid (BALF) of COPD patients at different stages of atmospheric fine particulate concentration in Harbin, northeast China. The objective was to analyse the influence of fine particulate matter and carrier microbes in Harbin on the pulmonary microecology of COPD patients; analyse the functional characteristics of bacterial genes in BALF; and explore the role of microecological changes related to fine particulate matter in the pathogenesis and progression of COPD.
Materials and Methods
Patients and BALF Collection
Approved by the Institutional Ethics Committee of the First Affiliated Hospital of Harbin Medical University, this study collected BALF from 48 enrolled patients with COPD in 2019. All methods used during the sampling process followed the relevant guidelines and regulations. The selected participants met all of the following criterias: (1) gave written informed consent; (2) were over 40 years old; (3) had been living in Harbin for more than 3 successive years; (4) had no history of smoking; (5) had no chronic airway diseases other than COPD; (6) were in a stable COPD condition. This study collected a 5 mL BALF sample from each selected patient, and then frozen samples (at −80°C) for further analysis. According to the air pollution data obtained from the China Environmental Air Online Monitoring and Analysis Platform (https://www.aqistudy.cn/historydata/about.php) and the official website of the Department of Ecology and Environment of Heilongjiang province (http://www.hljdep.gov.cn/), dense haze engulfs Harbin from every November to the following March. Thus, this study identified the period before March 2019 and after November 2019 as a high-risk period for particulate matter (PM) inhalation, and the rest of the year as a low-risk period for PM inhalation. According to the pollution conditions, BALF samples were given to group A (low-risk period of PM inhalation, 22 samples) and group B (high-risk period of PM inhalation, 26 samples).
PM2.5 Collection
PM sampling was conducted at the rooftop of the Harbin Institute of Technology (45°45′14″N, 126°40′54″E, ~15 m above the ground). The institute is centrally located, with commercial, educational, and living areas and no major stationary sources nearby, and may well reflect the PM conditions of urban Harbin. The PM samples were collected by high-volume samplers (Laoying, Qingdao, China; Tianhong, Wuhan, China) and quartz filters (PALL, NY, US) at a flow rate of 1.05 m3 min−1. Each sampling session lasted 24 hours (from 7:00 a.m. to 7:00 a.m. the following day). During the sampling process, all apparatuses were sterilized with 75% alcohol. After high temperature sterilization, the sampling film was wrapped in aluminium foil for later use. PM2.5 samples were collected and allocated to group C. The sampled filters were wrapped in aluminium foil and stored at −20 °C for the follow-up analysis.
DNA Extraction and 16S rRNA Gene Amplicon
Before DNA extraction, sample pretreatments were conducted as follows: (1) The film of collected PM2.5 samples was cut into pieces with high-temperature sterilizing scissors so that the bacteria in the particulate matter could be in full contact with the lysate. This was done to ensure that the cell walls were fully lysed for DNA extraction, according to the operating instructions of the Mag-Bind Soil DNA Kit (OMEGA, Georgia, US). (2) Thawed 1 mL BALF samples were taken and sterilized in a 1 × PBS buffer solution in 5 mL tubes. The mixtures were vortexed and then centrifugated at 4 ° C in 200 g portions for two hours; the supernatant was discarded; and the remaining deposits were used for DNA extraction in the same process as described above.
In this study, the V3-V4 region was selected for 16S rRNA Gene Amplicon. Moreover, 341F (CCCTACACGACGCTCTTCCGATCTG[barcode]CCTACGGGNGGCWGCAG) and 805R (GACTGGAGTTCCTTGGCACCCGAGAATTCCA GACTACHVGGGTATCTAATCC) were selected as the universal primers. The extracted DNA was first beaked into several 500 bp fragments, and were then amplified by added universal primers for polymerase chain reactions (PCRs). PCR products with fragments amplified over 400 bp were further purified and recovered by 0.6 times magnetic beads (AgenCourt AMPure XP). After that, Qubit 3.0 (Thermo Fisher Scientific, MA, USA) was used to check for accurate quantification of the recovered DNA. The recovered products were eventually pooled to 20 pmol concentrations for DNA sequencing in an Illumina MiSeq (Shanghai Sangon Biotech).
Sequence Analysis
Following MiSeq sequencing, the barcode sequence, primer sequences and joint sequences were processed. Firstly, the primers were removed (Cutadapt, version 1.2.1). Then, the pair-end reads were merged into one sequence according to their overlaps (PEAR, version 0.9.6). Subsequently, samples were identified and distinguished by barcodes. Finally, the quality of the sample dataset was screened by quality control to obtain valid sequences (Prinseq, version 0.20.4). These sequences were further clustered into operational taxonomic units (OTUs) in QIIME (version 1.17), using 97% similarity.
Statistical Methods
Analysis of the relationship between OTU number and cluster similarity value: The relationship between changes in the number of OTU and the cluster similarity value was plotted, and the best similarity value was selected for OTU analysis and taxonomic analysis. The relationship between number of OTU and the similarity value was 0.86 −0.99, which was considered a sequence of high-quality abundance.
Analysis of bacterial colony abundance and diversity in samples: Alpha_diversity indices (Coverage, ACE, Chao1, Shannon and Simpson) were calculated by Mothur (version 1.30.1).
Taxonomy and difference analysis of bacterial colonies: To obtain taxonomy annotations, the RDP database (http://rdp.cme.msu.edu/misc/resources.jsp) was employed, coupled with BLAST (version 2.28). The corresponding significant differences of Alpha_diversity indices and taxonomy annotations among different samples were made by Analysis of Variance (ANOVA) using SPSS software (version 22).
Colony gene function analysis: The Clusters of Orthologous Groups of proteins (COG) and Kyoto Encyclopedia of Gene and Genomes (KEGG) databases were used to analyse gene function difference. The difference analysis of predicted bacterial function was conducted by STAMP (version 2.1.3). The PICRUST program (version 1.0.0) was used to predict the metabolic functions of bacterial communities.
Results
Bacterial Metagenomic Sequencing Results
A total of 58 samples were sequenced, including 22 samples from Group A, 26 samples from group B and 10 samples from Group C. A total of 2,005,790 bacterial sequences and 34,256 bacterial OTU numbers were detected. The sequence numbers and OTU numbers of each group are shown in Table 1. There were 2733 OTU coincidences between groups A and B; 1013 coincidences between groups A and C; and 2390 coincidences between groups B and C. The sequence length distribution of the QC samples for each group before and after QC processing is shown in Figure 1. Here, it can be seen that the BP length of each group’s QC sample after QC processing was within the range of 400–440, which is uniform and consistent and therefore indicates that the obtained data was of high accuracy. The correlation analysis between the detected number of OTU and the cluster similarity value is shown in Figure 2.
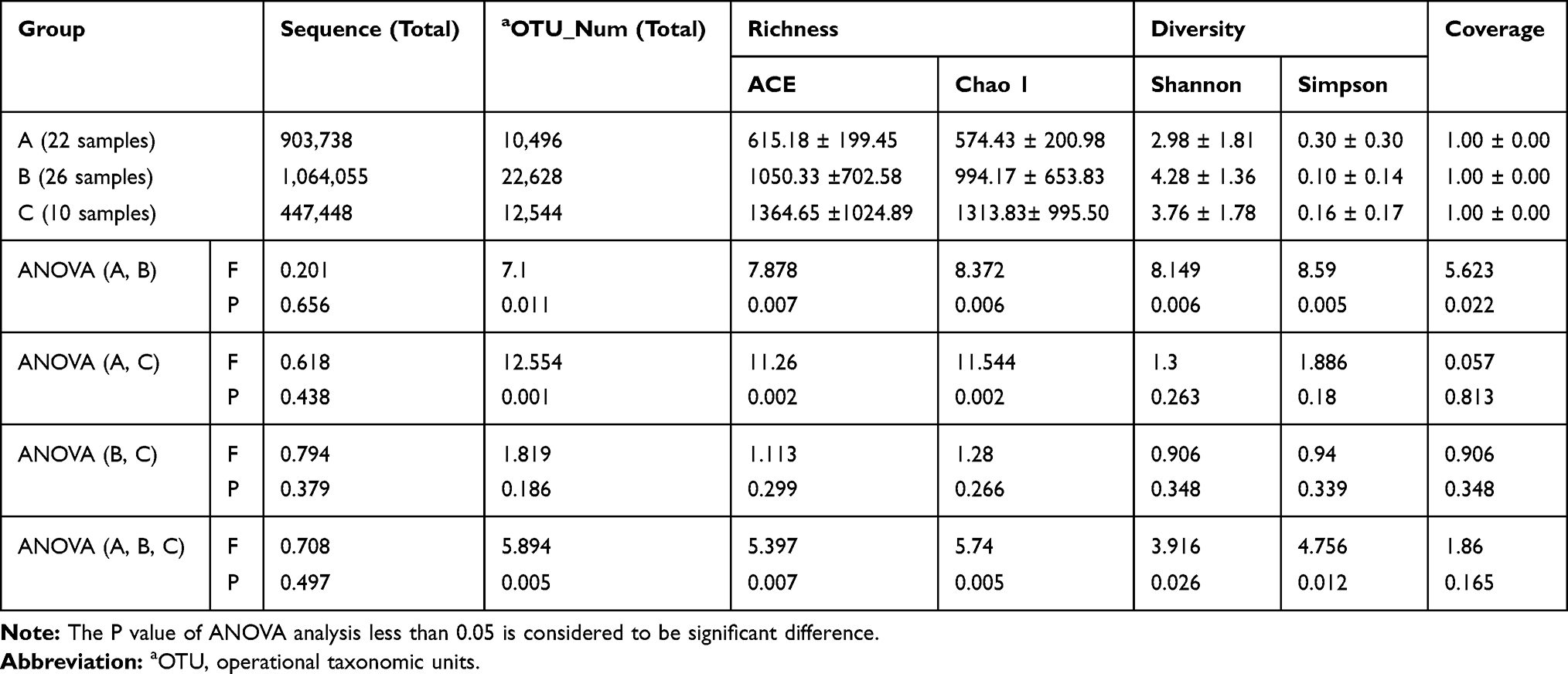 |
Table 1 Statistical Analysis of Species Richness and Community Diversity |
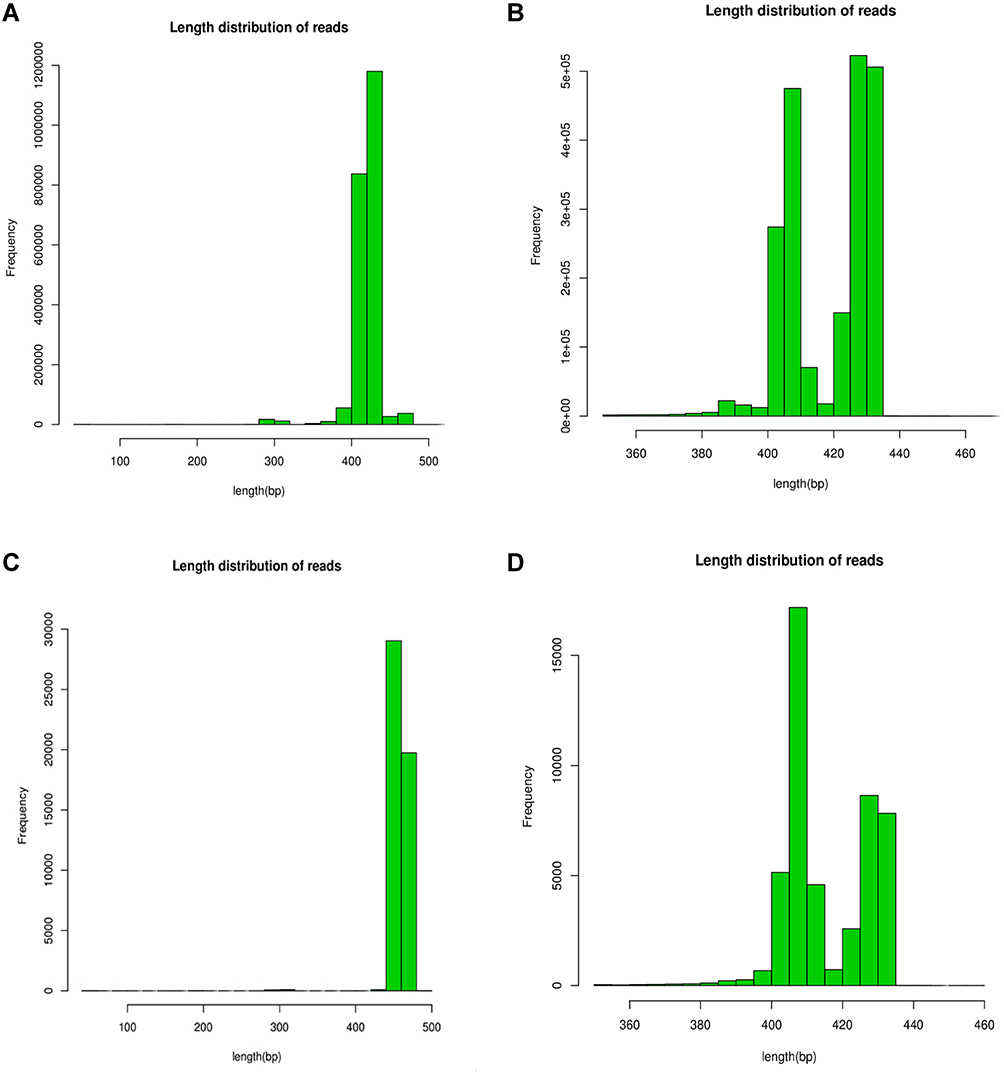 |
Figure 1 Sequence length distribution before QC processing for each group, the similarity value > 0.97 indicates that all the selected sequences are of high quality. (A) The sequence length of bacteria before QC in BALF. (B) Sequence length after QC of bacteria in BALF. (C) The sequence length before QC of bacteria in atmospheric fine particles. (D) Sequence length of bacteria after QC in atmospheric fine particles. |
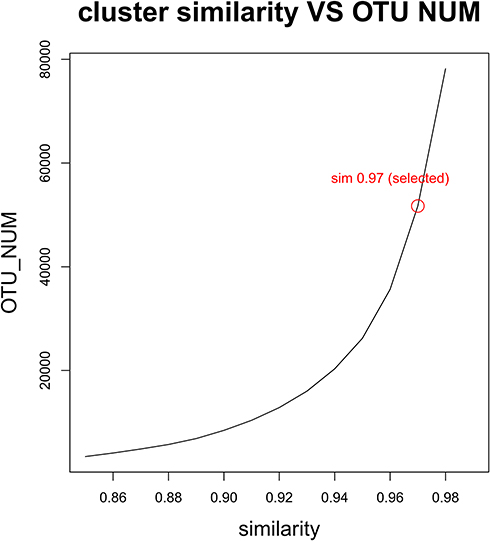 |
Figure 2 Relationship between OTU number and cluster similarity value. |
Bacterial Richness and Diversity
In total, approximately 2,005,790 bacterial sequences and 34,256 OTUs were obtained from 58 samples (Table 1). The average sequence reads of groups A, B and C were 41,079; 40,925; and 44,745, respectively. The average OTU numbers of groups A, B and C were 477; 807; and 1254, respectively. These results indicate that the number of bacterial sequences and the OTU numbers detected in inhalable PM were higher than those detected in BALF.
Coverage value evaluates the sequencing depth; values closer to 1 usually represent a lower probability of undetected sequences. The average coverage values of the three groups all tended toward 1, which proved the reliability of the sequences. Higher Chao1 and ACE values were used to reflect higher community richness.
Shannon and Simpson indices are commonly used to estimate microbial diversity. A greater Shannon value or lesser Simpson value reflects higher community diversity. From the calculated indices, the highest species richness value was in group C, the next in group B and the lowest in group A. Group C’s community diversity value was lower than group B’s, and group A’s was the lowest. The ANOVA analysis showed significant differences in species richness and community diversity between group A and Group B. However, there were no significant differences between groups B and C. Stated thus, it would appear that bacterial richness and diversity from BALF during the high-risk period of PM inhalation were much higher than they were during the low-risk period of PM inhalation, with significant differences. Bacterial richness and diversity during the high-risk period of PM inhalation approximated the bacterial richness and diversity in inhalable particles, and no significant differences were found. Collectively, the findings indicate that the abundant bacterial richness and diversity of BALF during the high-risk period of PM inhalation was most likely influenced by high concentrations of inhalable particle pollution.
Results of Bacterial Community Characteristics Analysis and Differences Among Samples
Results of Diversity of Bacterial Community Composition Analysis
In this study, the OTU was classified as a bacterial species based on the RDP classifier method of Bergey’s taxonomy. Since generation-level classification is of more value to clinical applications, this paper reports only the probability values of each sequence obtained by the Naive Bayesian assignment algorithm in generation-level rank; the results are shown in Table 2. There were significant differences among groups A, B and C at the genus level, with a higher number of bacterial species in group C than in groups A and B, which indicated that the bacterial species in PM2.5 were more abundant. The number of bacterial species in group B’s BALF was higher than that in group A’s, suggesting that the increase of bacterial species in the BALF of COPD patients was related to the high concentration of fine particulate inhalation in the high-risk period of atmospheric fine particulate pollution.
 |
Table 2 Statistical Analysis of Bacterial Taxonomy Annotations in Three Groups |
Comparison of Abundance Values of Bacterial Colony Species in Samples
Bacterial species with relative abundance, ie greater than 1%, were reported in this study, as shown in Figures 3 and 4. The bacteria with the highest abundance in group A’s BALF were Phyllobacterium (16.40%), followed by Stenotrophomonas (10.32%), Mesorhizobium (7.64%), Pseudomonas (6.50%) and Streptococcus (5.60%). Among these, Stenotrophomonas and Pseudomonas contain common pathogenic bacteria in clinic. Moreover, the abundance of Acinetobacter (1.91%) in group A was higher than that of other groups, considering that it was related to the bacterium’s colonization of the airway. Group B had the highest abundance of Streptococcus (6.60%), followed by Mesorhizobium (5.54%), Prevotella (3.88%), Methylobacterium (3.69%) and Mycoplasma (3.04%). Streptococcus and Mycoplasma were the main pathogenic bacteria of community-acquired infection, and the detection rate of Streptococcus and Mycoplasma in group B was higher than that in group A. This suggests that these two bacteria were related to the inhalation of fine particulate matter. The bacterial species in group C were significantly more abundant than those in groups A and B, and the dominant ones were significantly different from those present in the BALF. In group C, the bacterial genera were in descending order: Lactococcus (20.44%), Pseudomonas (15.32%), Serratia (6.76%), Arcobacter (3.91%), Psychrobacter (2.88%), Sphingomonas (2.46%) and Lactobacillus (1.15%). Most strains of Lactococcus are beneficial bacteria isolated from dairy products and plant products, although some pathogenic bacteria are also found in Lactococcus. For example, Lactococcus lactis, Lactococcus garvieae and Streptococcus lactarius can cause septicemia, endocarditis, osteomyelitis, coxitis, liver abscesses, etc. Pseudomonas, which is present in soil, fresh water and sea water, has been found to contain 29 species, of which at least 3 are pathogenic to animals or humans. Serratia was once considered a harmless environmental contaminant, but it has become an important opportunistic pathogen due to its invasiveness and resistance to many commonly used antimicrobial agents. Psychrobacter can resist extreme cold and has not been found to contain pathogenic bacteria.
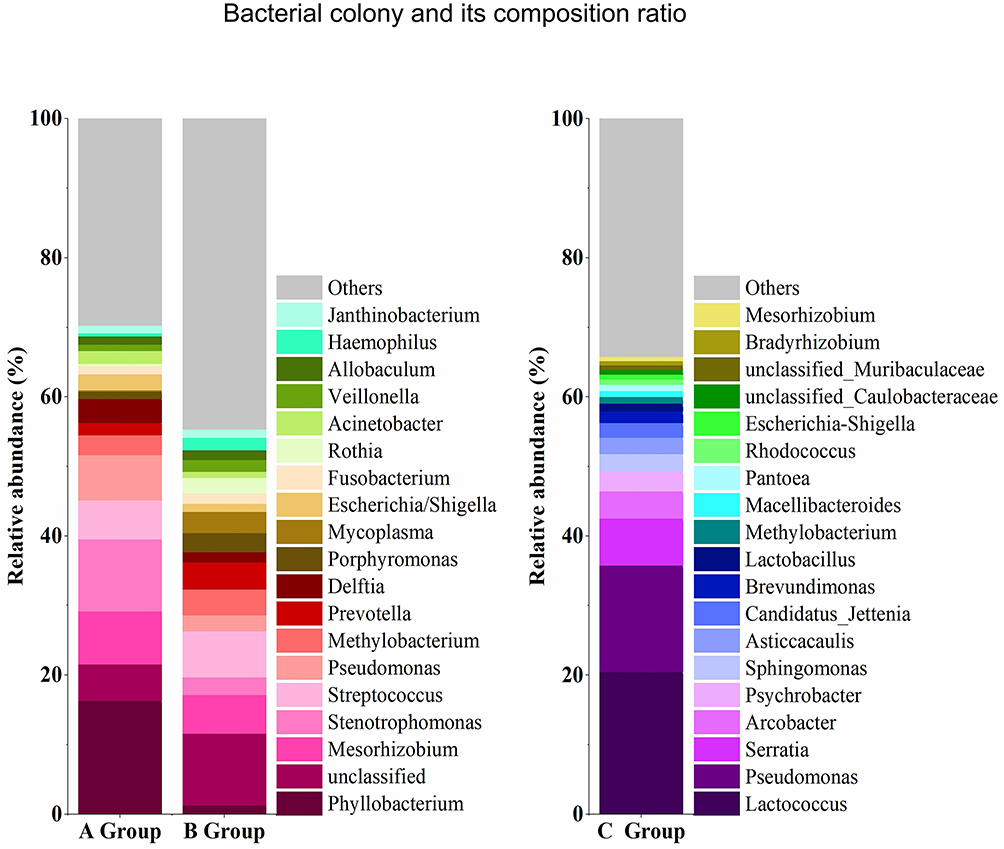 |
Figure 3 Name and composition ratio of the top 20 bacterial colonies in BALF and PM2.5 samples. |
 |
Figure 4 Name and composition ratio of bacterial colonies with abundance greater than 1% in BALF and PM2.5 samples. |
The Abundance Distribution Characteristics of Pathogenic Bacteria Carried by PM2.5 in BALF
According to the bacterial species specified in the Description of Pathogenic Bacteria,7 this study further analysed the abundance distribution characteristics of pathogenic bacteria carried by PM2.5 in the BALF of COPD patients; the results are shown in Table 3. All bacteria except Fusobacterium necrophorum were detected in Group B, and Streptococcus, Mycoplasma, Rothia, Prevotella, Rickettsiaceae, Chlamydia trachomatis and Brucella were significantly more abundant in group B than in group A. This indicates that the pathogenic bacteria detected in group B’s BALF were related to fine particulate inhalation. Therefore, it is necessary to prevent the above bacterial infections during periods of fine particulate pollution. Chlamydia trachomatis and Brucella, in particular, are the main pathogenic bacteria of two infectious diseases in clinic.
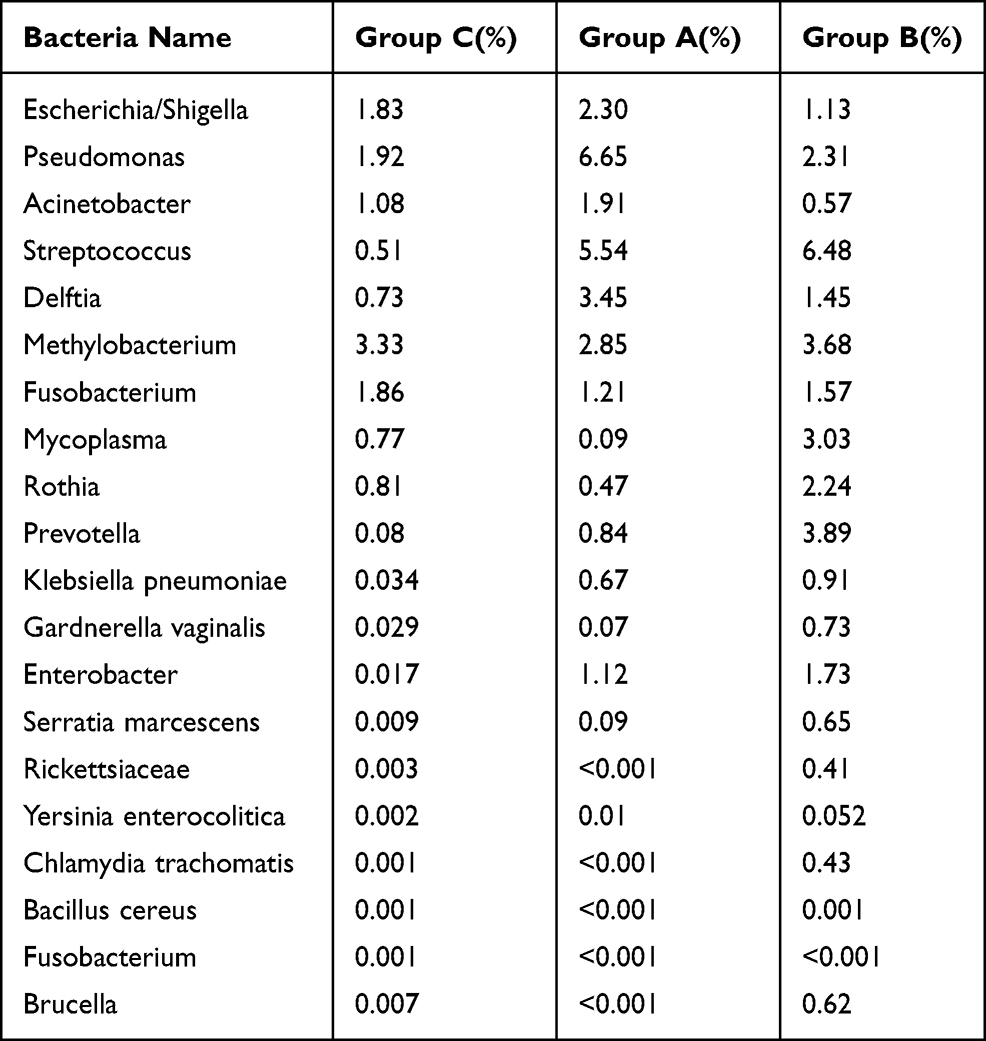 |
Table 3 Abundance Distribution Characteristics of Pathogenic Bacteria in PM2.5 Carrier Bands in BALF of COPD Patients |
Difference Analysis of Bacterial Gene Function in the BALF of Groups A and B
More and more studies have confirmed that changes in bacterial microecology are an important factor in the occurrence and development of diseases. Therefore, according to the bacterial genome sequencing results in the BALF of groups A and B (Figure 5), the COG and KEGG databases were used to analyse the gene abundance and functional differences in the BALF of the two groups (Figure 6). Furthermore, the role of bacterial microecology in the progression of COPD and its possible mechanism for action were explored in this study.
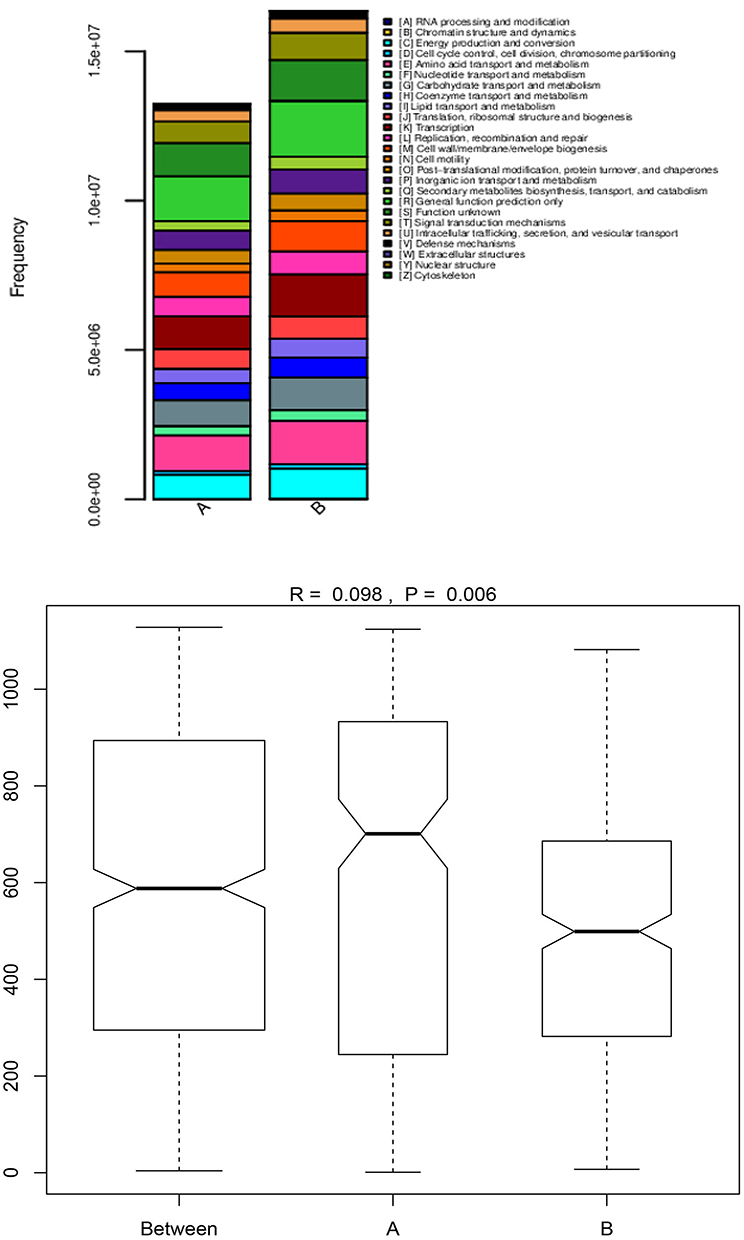 |
Figure 5 The abundance and difference analysis of bacterial gene functional characteristics are in group A and group B. |
 |
Figure 6 Function category difference graph. The left side of the figure shows the abundance ratio of different functional genes in the two groups of samples, the middle side shows the difference ratio of functional genes in the 95% confidence interval, and the right side shows the P value. Functional genes with P < 0.05 were marked in red, indicating significant differences. Only the 25 functional genes with the lowest P values are listed in the figure. |
The results of the functional analysis showed that there were significant differences between the functional abundance of bacterial genes in groups A and B (P=0.006) (Figure 5). The expression abundance of metabolism-regulating genes was 40.7% in group A and 38.9% in group B. In terms of amino acids, inorganic ions and carbohydrate transport and metabolism, the expression abundance of genes in both groups was >22.0%, higher than the metabolic regulation of other substances. This suggests either that the bacteria present in BALF participated in the damage and repair processes of the airways and lung tissues, or that normal physiological and metabolic functions were active.
In terms of cell processes and signal regulation, the expression abundance of biofilm synthesis genes in cell walls was 20.7% in group A and 22.0% in group B (P<0.01), suggesting that the bacteria present in BALF were involved in or accelerated the biofilm formation process. Biofilm formation is one of the mechanisms of bacterial resistance, and bacteria are more likely to be resistant during periods of air pollution. This further suggests that atmospheric fine particles may affect the biofilm formation of COPD patients’ pulmonary microecological bacteria, thereby causing or aggravating the generation of bacterial resistance. Regarding the functional characteristics of information storage and processing, the abundance of genes function involved in replication, recombination and repair regulation was 15.1% in group A and 19.8% in group B (P<0.01), suggesting strong genetic functions such as regulation of epithelial injury, repair, airway reconstruction and hyperplasia of airway mucus cells following injury by COPD lesions. These regulated pathological processes are the main pathophysiological processes in the progression of COPD. Moreover, the fact that group B’s abundance of genes function and information storage processes were greater than group A’s indicates that the atmospheric fine particles influenced the progression of COPD during periods of serious atmospheric pollution. The mechanism of influence may be to accelerate the process of airway damage repair and airway remodelling by changing the abundance of pulmonary microecological genes in patients with COPD, thereby leading to disease progression.Where human disease regulation was concerned, the abundance of genes functions of group A and group B were 1.16% and 1.12%, respectively (P>0.05). With regards to the regulation of infectious diseases, immune system diseases, urinary system diseases and nervous system diseases, there was no significant difference in the functional abundance of the two groups. However, the excessive abundance of genes indicated that the genes involved in the aforementioned disease processes were more active. The abundance of genes related to the regulation of cardiovascular system diseases, cancers and other tumour diseases was greater in group A than in group B (P<0.05), while the abundance of genes related to the regulation of metabolic diseases, allergic diseases and digestive diseases was greater in group B than in group A (P<0.05). The above results showed that the gene function of group B was relatively high in regulating the metabolism of carbohydrates, metabolic diseases, allergic diseases, etc., suggesting that inhalation of atmospheric fine particles may have adverse effects on metabolic diseases, such as diabetes, and allergic diseases, such as bronchial asthma and allergic rhinitis. The gene function of group A was higher in the regulation of cardiovascular system diseases, cancers and other tumour diseases, suggesting that airway microecology has an impact not only on pulmonary diseases but also on other systemic diseases, especially the incidence of malignant tumours.
Discussion
Harbin area, located in the northeast of China, has a cold temperate climate with long winters, and has a high incidence of COPD. Every year, the transitions from autumn to winter and winter to spring are the periods of high incidence of AECOPD. The causes of AECOPD mainly include infection of various pathogenic microorganisms10–12 and seasonal changes. Climate change has certain characteristics in this area. First, particulate matter aerosols are hard to conductive in winters with low temperatures.13,14 Second, heating time is long, and fuel combustion releases an enduring, high concentration of waste into the air.15,16 Furthermore, the rapid increase in the number of urban motor vehicles in recent years has led to an increase in exhaust emissions. Due to air pressure and other factors, the presence of these harmful substances in the air has increased, which in turn leads to a rise in the concentration of inhaled fine particles. In the past few years, ATS/RES17,18 guidelines have repeatedly proposed that atmospheric fine particulate matter is an important cause of the onset of COPD onset AECOPD. At present, it has been confirmed that the composition of fine particulate matter is complex; it contains a variety of organic aromatic hydrocarbons and inorganic elements. We have confirmed with previous studies that these inorganic elements and organic aromatic hydrocarbons can be detected in COPD patients’ BALF, and that their concentration is many times higher than it is in normal blood.19 The results of Cao et al20 study on microbes in fine particulate areas in Beijing confirmed that fine particulate matter can carry a large number of microbes. Given the prevalence of COPD and the particularities of the situation of atmospheric particulate matter in Harbin, we performed metagenomic detection of microorganisms in the alveolar lavage fluids of COPD patients during high and low particulate pollution periods. At the same time, we detected the microbes in atmospheric fine particulate matter during the high pollution period by metagenomics in order to compare the correlations among bacterial colonies. Thus, we can identify inhalation of microorganisms in particulate matter carriers and their influence on airway microecology. All the patients included in our study presented stable cases of COPD, and we excluded AECOPD. On one hand, infection in AECOPD patients can interfere with the diversity and abundance classification of bacterial colonies. On the other hand, bronchoalveolar lavage in AECOPD patients, whereas airway microecology in stable patients more accurately reflects correlations with particulate inhalation. According to our results, in terms of bacterial diversity, the Shannon value of group B in the high pollution period was the highest, at 4.28 ± 1.36, followed by the fine particulate matter group, and finally the low pollution period group. In order to ensure more accurate results, we also verified the results using the Simpson index. The verification results were consistent, but this result was inconsistent with relevant reports.21 The difference analysis of bacterial abundance and bacterial taxonomy also showed that group B had the highest index.
According to the bacterial sequencing and the detected OTU number, group B’s sequence number and OTU number were 1,064,055 and 22,628, respectively. Therefore, we believe that the results of this study accurately reflect the true bacterial colony in the BALF of COPD patients in this region. Moreover, the results also showed that there were 2390 overlapping sequences in groups B and C, and 1013 overlapping sequences in groups A and C. This indicates that the bacterial colony composition in BALF during the high pollution period is more obviously affected by the bacterial colonization of atmospheric fine particles. It also indicates that microbes carried by atmospheric fine particulate matter influence the airway microecology of patients with COPD.
Research has shown that there is no significant difference in microorganism species at the phylum and genus levels in particulate matter with different particle sizes.22 Some studies have indicated that classification at the genus level varies greatly at the species level.23,24 In this study, metagenomic sequencing analysis can only achieve genus-level classification. However, based on the authors’ experience in clinical diagnosis and the treatment of bacterial infections, the results of gene-level sequencing are of great clinical significance. Therefore, this study reports the sequencing results of bacterial taxonomy at the genus level.
The results show that there were significant differences between the abundance of dominant bacterial genera in the BALF of COPD patients during the period of high fine particulate matter pollution and the period of low fine particulate matter pollution (F=5.29, P=0.026). We confirmed that the bacterial colony composition was different in the high pollution period and the low pollution period. We further verified the differences among the three sample groups, showing that their bacterial colony composition was also different (F=14.986, P<0.001). The distribution of bacterial colonies and abundance in the atmosphere was significantly different from that in the BALF. To determine the pathogenicity of bacteria carried by fine particulate matter, we further analysed the pathogen species of the fine particulate matter carried25 in the BALF. The result was that the abundance of these species was different in group B, and the group’s detectable rates of the Streptococcus, Mycoplasma, Rothia, Pushtun, Rickettsia, Chlamydia trachomatis and Brucella genera were obviously higher than those of group A.
This study adopted randomised controlled research methods, chose stable COPD patients and ruled out factors that may interfere with the composition and abundance of bacterial colonies in the BALF of COPD patients—such as infection factors, underlying diseases, age, etc.—and took the high concentration of fine particulate inhalation as the main influencing factor. Therefore, we believe that the results of this study are credible. We confirmed that the pathogenic bacteria detected in group B’s BALF were related to the inhalation of fine particulate matter and that the pathogenic microorganisms could be inhaled into the airway. We further confirmed that the pathogenic microorganisms could stay in the airway for a long time, leading to bacterial infection and other adverse events.
Another important finding of this study was that the abundance detection of Chlamydia trachomatis and Brucella genus was high in group B (Chlamydia trachomatis 0.43%, Brucella genus 0.62%), but very low in group A. This indicates that these two infective pathogenic microorganisms26,27 may be transmitted by airborne fine particulate matter. This finding suggests the possible infectious routes of these two diseases, which is of great importance to their prevention and treatment.
More and more studies have confirmed that changes in bacterial microecology are an important factor in the occurrence and development of diseases.28 Therefore, according to the results of bacterial metagenomic sequencing in the BALF of group A and Group B using COG and KEGG databases, the gene abundance value and functional difference in BALF of two groups were analysed to explore the role of bacterial microecology in the disease progression of COPD patients and the possible pathogenesis. The gene function predictions of the COG and KEGG databases, in particular the KEGG database’s predictive method of correlating system function at the cell, species and ecosystem levels, have yielded important information for this study’s gene function analysis. This information may be of great value to subsequent research on the pathogenesis of atmospheric fine particulate matter leading to COPD and other related diseases. The functional analysis results of the two databases showed that microbial flora in the airway had active metabolic functions in both high and low pollution periods, especially in the transport and metabolism of amino acids, inorganic ions and carbohydrates. This active metabolic function leads to an increase in nutrient consumption, which further leads to a decrease in immune function and an increase in the risk of infection, contraction of other diseases and disease progression.29
In terms of regulating the repair function, the functional abundance values of genes involved in the regulation of replication, recombination, and repair were 15.1% in group A and 19.8% in group B (P<0.01), suggesting that the functions of genes involved in the regulation of epithelial injury, repair, airway reconstruction and proliferation following airway mucous cell injury in the formation of COPD lesions are relatively strong. These regulated pathological processes are the main pathophysiological processes in the progression of COPD. The bacterial abundance value of group B was higher than that of group A, suggesting that atmospheric fine particles in the period of high pollution have an impact on the progression of COPD. The pathogenesis of COPD progression may be caused by the acceleration of airway damage, airway repair and airway remodelling processes due to changes in the abundance of pulmonary microecological genes in COPD patients.
An equally important finding was that gene abundance in the genetic expression and signal regulation of cell wall biofilm synthesis was greater in groups A and B than group C. Additionally, gene abundance in the genetic expression and signal regulation of cell wall biofilm synthesis was relatively high in both groups, ie 20.7% in group A and 22.0% in group B (P<0.01). Biofilm formation is an important mechanism of bacterial resistance.30,31 The results of this study suggest that the bacterial microecology in the BALF of COPD patients is involved in the formation of bacterial resistance in vivo, and preliminarily reveal that drug resistance may be caused by the formation of biofilm. This finding will provide important insights into the mechanisms of drug resistance in infectious diseases. Additionally, the greater abundance of group B indicates that atmospheric fine particles may affect the proportion of abundance and diversity of bacterial microecological colonies in the airway. A greater abundance may accelerate the formation of bacterial biofilm; lead to the emergence of bacterial resistance; and accelerate the occurrence of resistant diseases.
The bacterial microecology of airways plays an important role in the regulation of human diseases, as demonstrated by the genetic analysis of disease regulatory function by the KEGG database. The gene function abundance values of group A and group B were 1.16% and 1.12% (P>0.05) respectively, suggesting that the bacterial microecology of airways has a regulatory effect on infectious diseases, contagions, immune system diseases, urinary system diseases, nervous system diseases, cancer, etc. The role of group B in the regulation of metabolic diseases, allergic diseases, and digestive system diseases was significantly enhanced, ie group B>group A (P<0.05). We suggest that the related components of fine particulate matter may be involved in the occurrence and development of the aforementioned diseases, and of special interest to research concerned with the relationship between metabolic diseases, such as diabetes, and particulate matter.
Conclusion
A total of 58 samples were sequenced, including 22 samples from Group A, 26 samples from group B and 10 samples from Group C. A total of 2,005,790 bacterial sequences and 34,256 bacterial numbers were detected. Group B had the highest bacterial diversity of the three groups. Group B also had the highest bacterial abundance index value. There were differences in the classification of bacterial colonies for the three groups at the genus level. The types of bacteria in group C were more numerous than other groups, and group B was higher than group A, which indicates that there were more bacteria in BALF during the high-risk period of AFPM inhalation. The detection rates of Streptococcus, Mycoplasma, Roche, Pushia, Chlamydia trachomatis and Brucella for group C were significantly higher than group A. The COG and KEGG databases’ difference analysis results for the bacterial gene function abundance of group A and Group B were 40.7% in Group A and 38.9% in group B (R=0.098, P=0.006). The human disease abundance in group A and group B was 1.16% and 1.12%, respectively (P>0.05). The increase in the concentration of PM2.5 can increase the diversity and abundance of bacteria in the BALF of stable COPD patients, which participates in the mechanism of airway remodelling and drug resistance in patients with stable COPD.
Data Sharing Statement
Availability of data and material need to be approved by the respiratory medicine department of the Fourth Affiliated Hospital of Harbin Medical University.
Ethics Approval and Consent to Participate
This study has been reviewed and approved by the Research Ethics Committee of the Harbin Medical University (no. 2020XS04-02). The clinical samples were specifically isolated for this research, all of the patients provided written informed consent. All relevant ethical safeguards have been met in line with the Declaration of Helsinki.
Consent for Publication
All authors have been personally and actively involved in substantive work leading to the report, and will hold themselves jointly and individually responsible for its content.
Acknowledgments
We would like to acknowledge the work of State Key Laboratory of Urban Water Resource and Environment, Harbin Institute of Technology.
Funding
The National Key Laboratory of Sponsored by Open Project of State Key Laboratory of Respiratory Disease (Project Funding Number: SKLRD-OP-201902). The National Key Laboratory of Urban Water Resources and Water, Environment Full Funds. (Project Funding Number: ESK201602).
Disclosure
The authors have no conflicts of interest to declare.
References
1. Han C, Oh J, Lim YH, et al. Long-term exposure to fine particulate matter and development of chronic obstructive pulmonary disease in the elderly. Environ Int. 2020;143:105895. doi:10.1016/j.envint.2020.105895
2. Tsai DH, Riediker M, Berchet A, et al. Effects of short- and long-term exposures to particulate matter on inflammatory marker levels in the general population. Environ Sci Pollut Res Int. 2019;26(19):19697–19704. doi:10.1007/s11356-019-05194-y
3. Abd Aziz A, Lee K, Park B, et al. Comparative study of the airborne microbial communities and their functional composition in fine particulate matter (PM2.5) under non-extreme and extreme PM2.5 conditions. Atmos Environ. 2018;194:82–92. doi:10.1016/j.atmosenv.2018.09.027
4. Zhang Q, Zhang PW, Cai YD. The Use of Protein-Protein Interactions for the Analysis of theAssociations between PM2.5 and Some Diseases.Biomed Res Int. 2016;2016(2016–5–8):1–7. doi:10.1155/2016/4895476
5. Guo L, Zhang K, Liu J T, et al. Mechanism of lung injury caused by high temperature and PM_(2.5) in COPD rats. J Environ Occup Med. 2018.
6. Bi Z, Zhou H.Research progress of lung diseases caused by air fine particulate matter and its mechanism. Environmental Engineering. 2016.
7. Wang HY, Gu X, Weng Y. Quantitative analysis of pathogens in the lower respiratory tract of patients with chronic obstructive pulmonary disease. BMC Pulm Med. 2015;15(1):94. doi:10.1186/s12890-015-0094-z
8. Willis A, Bunge J, Whitman T, et al. Improved detection of changes in species richness in high diversity microbial communities. J Royal Stat Soc. 2016;963–977.
9. Duan X. Research progress on the relationship between chronic obstructive pulmonary disease and respiratory microecology. Adv Clin Med. 2020;10(6):884–889. doi:10.12677/ACM.2020.106135
10. Moghoofei M, Jamalkandi SA, Moein M, Salimian J, Ahmadi A. Bacterial infections in acute exacerbation of chronic obstructive pulmonary disease: a systematic review and meta-analysis. Infection. 2020;48(1):19–35. doi:10.1007/s15010-019-01350-1
11. Cui J, Yan W, Xie H. A retrospective seroepidemiologic survey of Chlamydia pneumoniae infection in patients in Beijing between 2008 and 2017. PLoS One. 2018;13(11):e0206995. doi:10.1371/journal.pone.0206995
12. Zhao CL, Huang JW, Zhang L. Respiratory virus infections and inflammatory cytokines in hospitalized patients with acute exacerbation of chronic obstructive pulmonary disease. ZhonghuaJie He He Hu Xi Za Zhi. 2018;41(12):942–948.
13. Shao SC, Chang YH, Cao F, et al. High-frequency evolution of urban atmospheric ammonia and ammonium and its gas-to-particle conversion mechanism in Nanjing City. Huan Jing Ke Xue. 2019;40(10):4355–4363.
14. Chen WW, Liu Y, Wu XW, et al. Spatial and temporal characteristics of air quality and cause analysis of heavy pollution in Northeast China. Huan Jing Ke Xue. 2019;40(11):4810–4823. doi:10.13227/j.hjkx.201807159
15. Zhao H, Che H, Wang Y, et al. Aerosol vertical distribution and typical air pollution episodes over northeastern China during 2016 analyzed by ground-based lidar. Aerosol Air Qual Res. 2018;18:918e937.
16. Zhao H, Ma Y, Wang Y, et al. Aerosol and gaseous pollutant characteristics during the heating season(winterespring transition) in the Harbin-Changchun megalopolis, northeastern China. J Atmos Sol Terr Phys. 2019;188:26e43.
17. Thurston GD, Balmes JR, Garcia E, Gilliland FD, Rice MB. Outdoor air pollution and new-onset airway disease. an official American Thoracic Society workshop report. Ann Am Thorac Soc. 2020;17(4):387–398. doi:10.1513/AnnalsATS.202001-046ST
18. Thurston GD, Kipen H, Annesi-Maesano I, et al. A joint ERS/ATS policy statement: what constitutes an adverse health effect of air pollution? An analytical framework. Eur Respir J. 2017;49(1):1600419. doi:10.1183/13993003.00419-2016
19. Che C, Li J, Dong F, et al. Seasonal characteristic composition of inorganic elements and polycyclicaromatic hydrocarbons in atmospheric fine particulate matter and bronchoalveolar lavage fluid of COPD patients in Northeast China. Respir Med. 2020;171:1–5.
20. Cao C, Jiang WJ, Wang BY, et al. Inhalable microorganisms in Beijing’s PM2.5 and PM10 pollutants during a severe smog event. Environ Sci Technol. 2014;48(3):1499–1507. doi:10.1021/es4048472
21. Liu T, Chen X, Xu Y, et al. Gut microbiota partially mediates the effects of fine particulate matter on type 2 diabetes: evidence from a Population-Based Epidemiological Study. Environ Int. 2019;130:104882. doi:10.1016/j.envint.2019.05.076
22. Wang BY, Lang JD, Zhang LN, et al.Characterizing Beijing's Airborne Bacterial Communities in PM2.5 and PM1 Samples During Haze Pollution Episodes Using 16S rRNA Gene Analysis Method. Huan Jing KeXue. 2015;36(8):2727–34.
23. Yan D, Zhang T, Su J, et al. Structural variation in the bacterial community associated with airborne particulate matter in Beijing, China, during hazy and nonhazy days. Appl Environ Microbiol. 2018;84(9):e00004–18. doi:10.1128/AEM.00004-18
24. Sun Y, Xu S, Zheng D, et al. Effects of haze pollution on microbial community changes and correlation with chemical components in atmospheric particulate matter. Sci Total Environ. 2018;637–638:507–516. doi:10.1016/j.scitotenv.2018.04.203
25. Li JL, Hu YR, Liu L, et al. PM2.5 exposure perturbs lung microbiome and its metabolic profile in mice. Sci Total Environ. 2020;721:137432. doi:10.1016/j.scitotenv.2020.137432
26. Chow EP, Fairley CK. The role of saliva in gonorrhoea and chlamydia transmission to extragenital sites among men who have sex with men: new insights into transmission. J Int AIDS Soc. 2019;22(SupplSuppl 6):e25354. doi:10.1002/jia2.25354
27. Liu Z, Shen TF, Wei DW, et al. Analysis of the epidemiological, clinical characteristics, treatment and prognosis of human brucellosis during 2014–2018 in Huludao, China. Infect Drug Resist. 2020;11(13):435–445. doi:10.2147/IDR.S236326
28. Sun ZH, Zhu QL, Shen Y, et al. Dynamic changes of gut and lung microorganisms during chronic obstructive pulmonary disease exacerbations. Kaohsiung J Med Sci. 2020;36(2):107–113. doi:10.1002/kjm2.12147
29. Coleman MF, Cozzo AJ, Pfeil AJ, et al. Cell intrinsic and systemic metabolism in tumor immunity and immunotherapy. Cancers (Basel). 2020;12(4):852. doi:10.3390/cancers12040852
30. Rahdar HA, Malekabad ES, Dadashi A-R. Correlation between biofilm formation and carbapenem resistance among clinical isolates of Klebsiella pneumoniae. Ethiop J Health Sci. 2019;29(6):745–750. doi:10.4314/ejhs.v29i6.11
31. Nguyen T, Argudín MA, Deplano A. Antibiotic resistance, biofilm formation, and intracellular survival as possible determinants of persistent or recurrent infections by staphylococcus aureus in a vietnamese tertiary hospital: focus on bacterial response to moxifloxacin. Microb Drug Resist. 2020;26(6):537–544. doi:10.1089/mdr.2019.0282
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.