Back to Journals » Journal of Inflammation Research » Volume 19
Effective Component Compatibility of Bufei Yishen Formula III Alleviates Pulmonary Vascular Inflammation in COPD: Via VEGF165/P38 MAPK Pathway
Authors Song Q ![]() , Cui L, Lu R, Shao X, Xu K, Tian Y
, Cui L, Lu R, Shao X, Xu K, Tian Y ![]()
Received 17 February 2025
Accepted for publication 8 January 2026
Published 23 January 2026 Volume 2026:19 523270
DOI https://doi.org/10.2147/JIR.S523270
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Cynthia Koziol-White
Qinghua Song,1,2 Lili Cui,1,2 Ruilong Lu,1,2 Xuejie Shao,1,2 Kexin Xu,1– 3 Yange Tian1,2,4
1Henan University of Chinese Medicine, Zhengzhou, Henan, 450046, People’s Republic of China; 2Collaborative Innovation Center for Chinese Medicine and Respiratory Diseases Co-Constructed by Henan Province and Education Ministry of People’s Republic of China, Zhengzhou, Henan, 450046, People’s Republic of China; 3First Affiliated Hospital of Henan University of Chinese Medicine, Zhengzhou, Henan, 450000, People’s Republic of China; 4Academy of Chinese Medical Sciences, Henan University of Chinese Medicine, Zhengzhou, Henan, 450046, People’s Republic of China
Correspondence: Yange Tian, Henan University of Chinese Medicine, Zhengzhou, Henan, 450046, People’s Republic of China, Tel +86-13783656761, Email [email protected]
Purpose: Chronic inflammation of the lungs can affect pulmonary vascular remodeling in chronic obstructive pulmonary disease (COPD). The Bufei Yishen formula (BYF) and Effective-compound combination of BYF III (ECC-BYF III) ameliorate lung histopathological injury and remodeling, but the mechanism remains unclear. This study aimed to observe the effects of ECC-BYF III on pulmonary vascular inflammation in COPD and to elucidate its detailed mechanism.
Methods: In vivo, COPD rat model was established through cigarette smoke exposure (CSE) combined with repeated infections of Klebsiella pneumoniae. Rats were randomly treated with ECC-BYF III (5.5 mg/kg, once a day) or doxofylline (36 mg/kg, once a day) for eight weeks. In vitro, Human umbilical vein endothelial cells (HUVECs) and human monocyte leukemia cells (THP-1) were induced with 10 μg/mL LPS for 24h. The pulmonary function, histopathology, inflammatory factor levels, immunoblotting results were evaluated.
Results: Compared with the model group, ECC-BYF III significantly improved the lung function, alleviated pulmonary artery inflammation and relieved pulmonary vascular remodeling in COPD rats. At the molecular level, ECC-BYF III down-regulated VEGF165/P38 MAPK signaling pathway. In the inflammatory model of HUVEC induced by LPS, 35 and 70μg/mL ECC-BYF III significantly decreased the levels of tumor necrosis factor -α (TNF-α), interleukin-1β (IL-1β) and Endothelin-1 (ET-1) mRNA, and increased the expression of endothelial nitric oxide synthase (eNOS) mRNA. In addition, ECC-BYF III also inhibited VEGF165/P38 MAPK pathway in LPS-induced HUVEC and THP-1/HUVEC co-cultured inflammatory models.
Conclusion: Our findings demonstrate that ECC-BYF III can improve pulmonary vascular remodeling in COPD rats, and its key pharmacodynamic mechanism involves the inhibition of the VEGF165/P38 MAPK pathway, thereby reducing inflammatory infiltration.
Keywords: chronic obstructive pulmonary disease, Chinese medicine, pulmonary vascular inflammation, VEGF165/P38 MAPK pathway
Introduction
Chronic obstructive pulmonary disease (COPD) is a complex condition characterized by chronic inflammation-induced tissue damage and irreversible airflow limitation. It typically results from prolonged exposure to noxious gases or particles. Common symptoms include cough, expectoration and dyspnea, with a significant risk of disease progression. 1 Globally, COPD is among the leading causes of death, accounting for approximately three million deaths each year,2,3 and this number is projected to rise to over 5.4 million annual deaths from COPD and related conditions by 2060.4 The economic and social burden of COPD is substantial with direct medical expenses in China ranging from 33.33% to 118.09% of the local average annual income.5 Therefore, there is an urgent need to intensify research efforts toward effective prevention and treatment strategies for COPD.
The inhalation of noxious gases and particles induces chronic inflammation in patients with COPD, leading to the accumulation of macrophages, neutrophils, T lymphocytes, and other inflammatory cells in the airways. These cells subsequently release inflammatory mediators such as interleukin-6 (IL-6) and interleukin-1β (IL-1β) which contribute to the destruction of alveolar structures, airway mucosal edema, increased mucus secretion, and subsequent airway narrowing and obstruction, ultimately resulting in dyspnea. Chronic inflammation can also affect the pulmonary vascular system, causing pathological vascular remodeling in COPD.6 Inflammatory stimuli activate smooth muscle cells in pulmonary vessels to proliferate and stimulate resting endothelial cells through cytokines such as vascular endothelial growth factor (VEGF). This process induces vascular wall thickening and muscularization,7 increases pulmonary vascular resistance, and gradually elevates pulmonary artery pressure.8
Moreover, activated vascular endothelial cells release a variety of inflammatory mediators, such as adhesion molecules, chemokines, cytokines. These inflammatory mediators stimulate macrophages to migrate to endothelial cells, mediating the systemic dissemination of tumor necrosis factor -α (TNF-α), IL-1β and other factors, thereby aggravating the inflammation.9 Therefore, inhibiting vascular inflammation and improving vascular remodeling are essential to delaying the progress of COPD.
VEGF is a critical activator that mediates abnormal vascular growth and permeability, stimulating the overexpression of pre-existing blood vessels and promoting endothelial proliferation.10 Among the four subtypes of VEGF, VEGF-a165 (VEGF165) is the most potent pro-angiogenic factor and the predominant form, promoting endothelial cell proliferation and increasing the permeability of the endothelial layer.11,12 P38 mitogen-activated protein kinase (P38 MAPK) is a key component of the MAPK pathway, regulating inflammatory responses and playing a role in cell proliferation, cytokine production, cytoskeletal remodeling, and other cellular processes.13 Research indicates that VEGF165 levels increase under inflammatory conditions in rats, synergistically activating the P38 MAPK signaling pathway through VEGFR2. Inhibition of VEGF expression reduces VEGFR2 activity, leading to downregulation of the P38 MAPK pathway, thereby mitigating damage to vascular endothelial cell adhesion and effectively alleviating both angiogenesis and inflammatory responses. Thus, targeting the VEGF165/P38 MAPK pathway presents a promising approach for reducing vascular inflammation.14
The treatment of COPD primarily relies on bronchodilators, mucolytic agents, and immunomodulatory drugs to relieve symptoms and prevent further decline in lung function.1 Long-term management is usually required; however, prolonged use of inhaled bronchodilators may cause adverse reactions such as arrhythmia and tremor.15 In recent years, Traditional Chinese Medicine (TCM) has shown considerable promise in COPD management due to its multi-component and multi-target characteristics.16,17 The Bufei Yishen formula (BYF) has been recognized as an effective treatment for COPD.18 Previous studies have demonstrated that the BYF can effectively improve the clinical symptoms of COPD patients, reduce acute exacerbations, and improve the quality of life,19 with no significant difference in the incidence of adverse events compared with the control group. Additionally, it has been shown to mitigate pulmonary inflammation in COPD rats, inhibit the thickening of pulmonary arteriolar walls and smooth muscle, and improve right ventricular remodeling.20–22 Following extensive screening and repeated verification of its effective-compound combination (ECC), the ECC-BYF III (ZL. 201811115372.3) was developed, including ginsenoside Rh1, astragaloside IV, icariin, paeonol, and nobiletin, which retains the efficacy of BYF.23–26 Although existing research indicated that ECC-BYF III can ameliorate lung histopathological damage and small pulmonary vascular remodeling in COPD rats,27 its potential mechanism is still unclear.
As the VEGF165/P38 MAPK signaling axis plays a crucial role in endothelial dysfunction and vascular remodeling, investigation of its modulation by ECC-BYF III may provide new insights into the molecular basis of its therapeutic actions. In this study, a COPD rat model was established through chronic exposure to cigarette smoke combined with repeated Klebsiella pneumoniae infection. In addition, an LPS-induced inflammatory injury model in human umbilical vein endothelial cells (HUVECs) and a THP-1/HUVEC co-culture inflammation model were employed to further elucidate the cellular mechanisms involved. These experiments were designed to investigate the regulatory effects of ECC-BYF III on pulmonary vascular inflammation and the VEGF165/P38 MAPK axis, providing mechanistic insight into how ECC-BYF III modulates inflammatory responses and alleviates pulmonary microvascular remodeling in COPD.
Materials and Methods
Animals
Male SD rats (250±20 g) were purchased from Beijing Weitong Lihua Experimental Animal Technology Co. Ltd (China, certificate number: 110011211105823815). The study was approved by the Experimental Animal Ethics Committee of Henan University of Traditional Chinese Medicine (DWLL202003261), and all procedures were conducted in accordance with the principles of the 3Rs (Replacement, Reduction, and Refinement) to ensure the welfare of laboratory animals.
Drugs and Reagents
The ECC-BYF III is composed of ginsenoside Rh1, astragaloside IV, icariin, paeonol, and nobiletin. Ginsenoside Rh1 (lot: CHB180608) was purchased from Chengdu Chroma Biotechnology Co. Ltd (China). Astragaloside IV (lot: MUST-17022804), icariin (lot: MUST-16111710), and paeonol (lot: MUST-16071405) were purchased from Chengdu Must Biotechnology Co. Ltd (China). Nobiletin (lot: HL-20170312) was purchased from Xi’an Huilin Biotechnology Co. Ltd (China). The purity of the compound was confirmed to be no less than 98% via high-performance liquid chromatography (HPLC). Each component was dissolved in 0.5% Carboxymethyl Cellulose-Na (CMC-Na) in specific proportions to form a suspension. Doxofylline (State Drug Approval Number H19991048) was purchased from Heilongjiang Fuhe Pharmaceutical Group Co. Ltd.
Filter cigarettes containing13 mg tar, 1.1 mg nicotine, and 13 mg NO were provided by China Tabacco Henan Industrial Co. Ltd. Klebsiella pneumoniae (strain number: 46117) was selected from the China Medical Bacteria Preservation Management Center of China Institute for Drug and Biological Products Verification.
VEGF (Cat: GTX21316) antibody and endothelin-1 (ET-1, Cat: GTX116033) antibody were purchased from Gene Tex (USA), the cluster of differentiation 68 (CD68, Cat: DF7518) antibody was purchased from Affinity (USA). The hematoxylin-eosin (HE) staining kit were purchased from Servicebio biotechnology Co. Ltd (Wuhan, China, G1005). Rat Tumor Necrosis Factor Alpha (TNF-α) ELISA Kit and interleukin-1β (IL-1β) ELISA Kit were purchased from Elabscience Biotechnology Co., Ltd. (Wuhan, China; E-EL-R2856c, E-EL-R0012c). Interleukin-6 (IL-6) ELISA Kit was purchased from BD Biosciences Pharmingen (USA, 550319).
COPD Modeling, Grouping, and Drug Administration
From week 1 to week 8, the rat model of COPD was induced by cigarette smoke exposure (CSE) combined with repeated bacterial infection.28 CSE conducted at a concentration of 3000 ± 500 ppm for 40 minutes, twice daily, using a smoke generator (Yuyan Instruments, Co., Ltd). Bacterial infection was induced by intranasal instillation of Klebsiella pneumoniae suspension (6 × 108 CFU/mL, 0.1 mL per rat) once every 5 days throughout the modeling period.
The rats were randomly divided into four groups (n=12): Normal group, model group, doxofylline group, and ECC-BYF III group. Except for the normal group, the other three groups were modeled using the same method as described previously. From weeks 9 to 16, the normal and model groups were orally administered with 0.5% CMC-Na (0.5 mL/100 g); The ECC-BYF III group received ECC-BYF III orally (5.5 mg/(kg·d), 0.5 mL/100 g), and the doxofylline group received doxofylline orally (36 mg/(kg·d), 0.5 mL/100 g). Dose of doxofylline was calculated according the following formula (D: dose; K: body shape index, K = A/W2/3, A: surface area in m2, W: weight in kg):
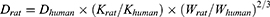
The treatment lasted for 8 weeks, at the 16th week, lung tissue, blood and bronchoalveolar lavage fluid (BALF) of rats were collected for subsequent experiments.
Cell Culture and Treatment
Human umbilical vein endothelial cells (HUVECs) were purchased from the Cell Resource Center of Shanghai Academy of Life Sciences, and human monocyte leukemia cells (THP-1) were purchased from Wuhan Procell Life Technology Co. Ltd. (China).
In order to screen the optimal modeling concentration of LPS and the optimal intervention concentration of ECC-BYF III. The HUVECs were cultured in a 1640 complete medium containing 10% fetal bovine serum (FBS) at 37°C with 5% CO2. The cells were then exposed to 0–20 μg/mL LPS (0.625 μg/mL, 1.25 μg/mL, 2.5 μg/mL, 5 μg/mL, 10μg/mL and 20 μg/mL), 0–280 μg/mL ECC-BYF III (35 μg/mL, 70 μg/mL, 140 µg/mL, 280 µg/mL) and 0–0.8 μg/mL VEGF-IN (0.025 μg/mL 0.05μg/Ml, 0.1 μg/mL, 0.2 μg/mL, 0.4 μg/mL and 0.8 μg/mL) for 24 h. MTT reagent was added and incubated for 4 h before the absorbance was measured at 570 nm using a microplate reader. CCK-8 reagent was added and incubated for 4 h before measuring absorbance at 450 nm using a microplate reader.
The HUVECs were placed in 6-well plates (1 × 106 cells/well). The cells were divided into four groups: control group (control), LPS group (10μg/mL, model), LPS + 35 ECC-BYF III group (35 μg/mL), and LPS + 70 ECC-BYF III group (70 μg/mL). Following a 3-hour exposure to ECC-BYF III for the 35 μg/mL and 70 μg/mL groups, all groups except the control group were induced with 10 μg/mL LPS for 24h.
The HUVECs were uniformly inoculated in 12-well plate and cultured in a 37°C incubator for 2 h. Meanwhile, THP-1 cells were treated with 0.05 μg/mL PMA for 2 hours. The 12-well plate was then removed, and 1.5 mL of complete medium (RPMI 1640 medium + 10% FBS) was added to each well. Subsequently, 0.5 mL of PMA-treated THP-1 cells were added to transwell inserts, which were incubated in a 37°C incubator for 24 h before being exposed to 10 μg/mL LPS for 24 h. The co-culture cells were divided into six groups: control group (control), LPS group (10 μg/mL, model), LPS + 70 ECC-BYF III group (70 μg/mL), VEGF-IN group (0.1 μg/mL), LPS+ VEGF-IN group and LPS+ECC-BYF III + VEGF-IN group. All groups except the control group were induced with 10 μg/mL LPS for 24 h.
Pulmonary Function
At the end of week 16, tidal volume (TV), 50% tidal volume expiratory flow (EF50), and Peak expiratory flow (PEF) were measured using whole body plethysmography (WBP) system (Buxco, NC, USA).
HE Staining
The left lung tissue was fixed, sliced, stained with hematoxylin and eosin and examined under an optical microscope to observe pathological injury in pulmonary arterioles with diameters ranging from 100 μm to 200 μm. The number of white blood cells per unit area of the pulmonary arteriolar wall was counted. Pathological measurement of pulmonary vessels included pulmonary vessel diameter (VD), wall thickness (WT), total area (TA), wall area (WA=TA-LA), and luminal area (LA). Wall thickness to vessel diameter ratio was calculated as WT%=(WT/VD) × 100%, wall area to total area ratio as WA%=(WA/TA) × 100%, and luminal area to total area ratio as LA%=(LA/TA) × 100%. The pathological injury of lung airway was measured. Six visual fields were randomly selected from each HE staining section, mean alveolar numbers (MAN) and mean linear intercept (MLI) in the field of vision were measured. Draw a “+” in the field of vision under ×200 microscope, measure the length (L), and record the number of alveolar septa (Ns) and the number of alveoli (Na), alveolar area (A), calculated MLI and MAN according to the formula MLI (μm) = L / Ns, MAN (/mm2) = Na / A. Airway wall thickness (AWT) is used to evaluate the degree of small airway stenosis, select a small airway with a diameter of 100~300 μm under the ×200 microscope, measure three inner diameters (d1, d2, d3) and three corresponding outer diameters (c1, c2, c3), and calculate the airway wall thickness according to the formula AWT = ((c1-d1) + (c2-d2) + (c3-d3)/3)/2.
Immunohistochemistry
The lung tissue sections were subjected to immunohistochemical(IHC) staining to detect VEGF (1:1000), ET-1 (1:1000,), CD68 (1:1000). Image Pro Plus 6.0 professional image acquisition and analysis system (Media Cybernetics Inc.) was used to calculate the integral optical density of the images.
Enzyme Linked Immunoassay Assay ELISA
The levels of IL-6, TNF-α, IL-1β in serum and BALF were measured using a Rat ELISA Kit, according to the manufacturer’s instructions.
Endothelial Permeability Detection
Endothelial barrier permeability was evaluated using a Transwell system. Evans blue–albumin working solution was added to the upper chamber, while 4% BSA solution was added to the lower chamber. The cells were incubated for 1 hour, after which the medium from the lower chamber was collected. The absorbance at 620 nm was measured using a microplate reader, and the concentration of leaked Evans blue was calculated based on a standard curve to quantify endothelial permeability.
Western Blotting
Total protein extracts were prepared from cells or lung tissues, quantified using a bicinchoninic acid (BCA) protein assay kit, and separated by 10% SDS-PAGE. Proteins were transferred to PVDF membranes. The following antibodies were used: VEGF (1:1000, Gene Tex, USA), VEGFR2 (1:1000, CST, USA), P38 (1:1000, CST, USA), p-P38 (1:1000, CST, USA), and an anti-rabbit secondary antibody (1:5000, Gene Tex, USA). Images were acquired using a gel imaging analysis system (ChemiDoc MP, USA), and the bands were analyzed using Image J software.
Quantitative Polymerase Chain Reaction Assay (qPCR)
Total RNA was extracted using the QIAzol Lysis Reagent (QIAGEN) and reverse transcribed using HiScript® II Q RT SuperMix for qPCR kit (Vazyme). qPCR was performed on a QuantStudio 6 system (Life Technologies) using PowerUpTM SYBRTM Green Master Mix (ABI). All primers are listed in the Table 1.
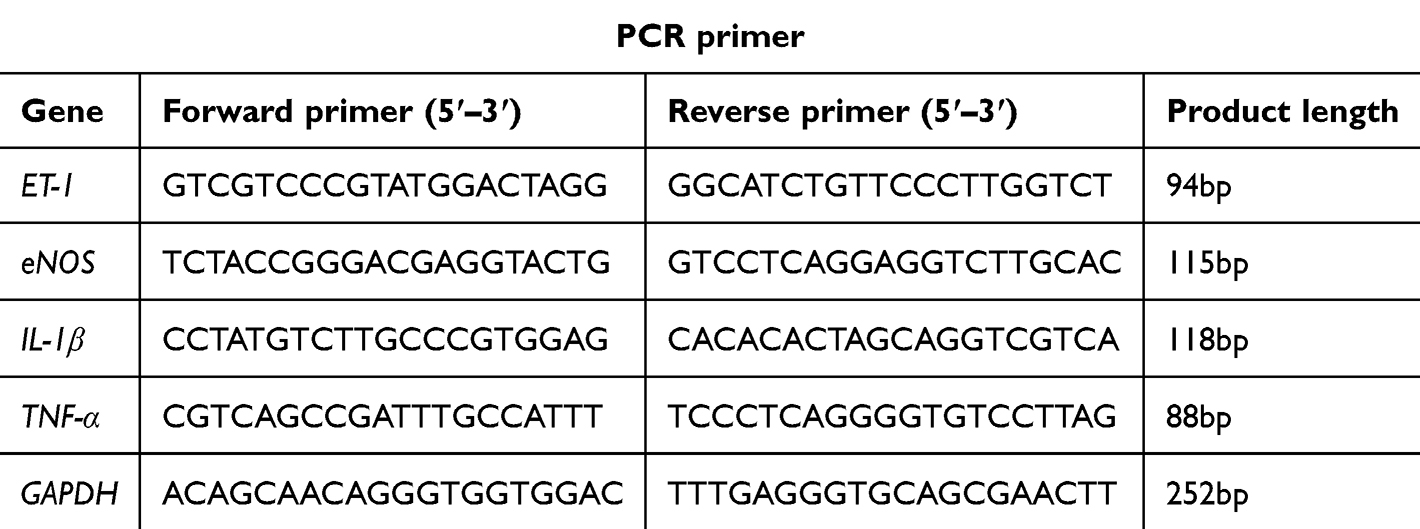 |
Table 1 qPCR primer sequences |
Immunofluorescence
Immunofluorescence was used to detect the expression changes of platelet endothelial cell adhesion molecule-1 (CD31) (1:1000)/IL-6 (1:1000) and CD31 (1:1000) /IL-1β (1:1000) proteins in lung tissue sections and VEGF (1:100), VEGFR2 (1:100) and p-P38 (1:100) proteins in THP-1 cells and HUVECs. A laser confocal microscope (Carl Zeiss AG, Germany) was used to observe and collect images.
Statistical Analysis
All values were presented as means ± standard errors. Statistical data was analyzed using the Statistical Package for the Social Sciences (SPSS 26.0, Chicago, USA), and multiple sample groups were compared using one-way analysis of variance (ANOVA), and P < 0.05 was considered statistically significant.
Results
ECC-BYF III Ameliorated the Lung Function and Lung Histopathological Injury of COPD Rats
Pulmonary function was measured to monitor the degree of airflow restriction in the rats. As shown in Figure 1A, CSE and Klebsiella pneumoniae infection decreased lung function in rats including TV, EF50, and PEF, while ECC-BYF III and doxofylline treatment significantly improved these parameters. HE staining revealed that lung tissues from the Model group displayed obvious structural damage, including alveolar enlargement, decreased alveolar number, rupture and fusion of alveolar walls, and thickening of airway walls compared with the Normal group. Treatment with ECC-BYF III or doxofylline markedly alleviated these pathological alterations, resulting in more intact alveolar architecture and reduced airway wall thickness (Figure 1B). Quantitative analysis further showed that the MLI and AWT were significantly increased, while MAN was significantly decreased in the Model group relative to Normal. Administration of ECC-BYF III or doxofylline significantly reduced MLI and AWT and increased MAN compared with the Model group (Figure 1C). These results suggested that ECC-BYF III significantly improved pulmonary function and ameliorated lung histopathological structure in COPD rats.
 |
Figure 1 ECC-BYF III inhibited CSE combined with Klebsiella pneumoniae-induced COPD in rats. (A) Changes in TV, EF50, and PEF pulmonary function in rats. (B) HE staining of lung tissue of rats (×200). (C) Changes in MLI, MAN, and AWT in rats. Values are presented as the mean ± SD. n=6~8. *P < 0.05, **P< 0.01. |
ECC-BYF III Attenuated Inflammation in COPD
Under the stimulation of CSE, macrophages, neutrophils and other white blood cells were recruited into the lungs, aggravating inflammatory infiltration and releasing a large number of inflammatory mediators, including IL-6 and IL-1β.29 These cytokines serve as markers for inflammation. Macrophages, which are the predominant immune cells in the lung, were identified using CD68 labeling.30 CD31, a marker expressed by vascular endothelial cells, was used to label these cells.31 HE staining revealed marked inflammatory cell infiltration in the lung tissues of COPD rats, characterized by a significant increase in the number of white blood cells and macrophages around the airways and alveolar septa compared with the Normal group (Figure 2A). Treatment with ECC-BYF III or doxofylline markedly reduced inflammatory cell accumulation, indicating a clear attenuation of pulmonary inflammation. IHC staining for CD68 further demonstrated a pronounced elevation of CD68-positive macrophages in the Model group, whereas their numbers were significantly reduced following treatment with ECC-BYF III or doxofylline (Figure 2B). Quantitative analysis of fluorescence intensity revealed that the integrated optical density (IOD) of IL-6 and IL-1β co-localized with CD31 was significantly increased in the Model group compared with the Normal group, whereas ECC-BYF III and doxofylline treatments markedly reduced their expression levels (Figure 2C). In addition, the levels of IL-6 and IL-1β in serum and BALF were markedly elevated (Figure 2D), while treatment with ECC-BYF III and doxofylline significantly reduced these inflammatory responses.
 |
Figure 2 Effect of ECC-BYF III on inflammation in rats with COPD. (A) HE staining of rat pulmonary vessels (×400) and mean leukocyte values of lung tissues. (B) IHC analysis of CD68 (×400). (C) Immunofluorescence assay for the expression of CD31 with IL-1β and IL-6 in rat lung tissue (IF, ×200). (D) IL-6 level in serum and bronchial alveolar lavage fluid (BALF) samples. (E) Effect of ECC-BYF III on TNF-α and IL-1β mRNA in human umbilical vein endothelial cells (HUVECs). Values are presented as the mean ± SD. n=6~8. *P < 0.05, **P< 0.01. |
LPS (0.625~20 μg/mL) and ECC-BYF III (35~280 µg/mL) were used to induce HUVEC cells, but neither had significant effects on the survival rate of HUVECs (Figure S1A). Therefore, 10 μg/mL LPS with the strongest inflammatory reaction was selected to induce HUVECs, 35 µg/mL and 70 µg/mL ECC-BYF III with the highest cell survival rate were selected for next treatment. Under the stimulation of 10 μg/mL LPS, the mRNA of TNF-α and IL-1β in HUVEC cells were significantly increased (Figure S1B), and the mRNA of TNF-α and IL-1β were significantly decreased after the intervention of 35 µg/mL and 70 µg/mL ECC-BYF III (Figure 2E).
ECC-BYF III Relieved Pulmonary Vascular Remodeling in COPD
Pulmonary vascular wall thickening was observed via HE staining, and the degree of vascular remodeling was quantitatively analyzed using WT%, WA% and LA%. ET-1 is a polypeptide produced by vascular endothelium, which promotes pulmonary vascular endothelial proliferation, thereby aggravating pulmonary vascular remodeling.32 Endothelin receptor B (ETB) is a receptor for ET-1, which can be activated and coupled with endothelial nitric oxide synthase (eNOS) on the surface of endothelial cells, leading to vascular injury.33 HE staining revealed significant pulmonary vascular remodeling in COPD rats after CSE combined with Klebsiella pneumoniae exposure, as evidenced by vascular wall thickening and luminal narrowing (Figure 3A). Quantitative analysis showed increased WT% and WA% and decreased LA% in the Model group, which were markedly improved by ECC-BYF III and doxofylline treatment (Figure 3B). Immunofluorescence staining of alpha-smooth muscle actin (α-SMA) and CD31 was performed to further evaluate vascular remodeling and endothelial integrity. In the Model group, α-SMA expression was markedly enhanced and extended outward, indicating smooth muscle proliferation and wall thickening, while CD31 expression was significantly reduced and discontinuous, reflecting endothelial injury. Treatment with ECC-BYF III or doxofylline restored CD31 expression and reduced α-SMA fluorescence intensity, suggesting alleviation of vascular remodeling and preservation of vascular structure (Figure 3C). Immunohistochemistry showed strong ET-1 staining in the vascular endothelium of the Model group, while ECC-BYF III and doxofylline significantly reduced ET-1 expression (Figure 3D and E). In HUVECs induced by LPS, ECC-BYF III dose-dependently decreased ET-1 and increased eNOS mRNA expression (Figure 3F), indicating its potential to alleviate pulmonary vascular remodeling in COPD. Furthermore, the Evans blue permeability assay revealed a significant increase in EB concentration in the Model group, reflecting endothelial barrier dysfunction. Treatment with ECC-BYF III significantly reduced EB leakage in a dose-dependent manner, suggesting that ECC-BYF III can restore endothelial integrity and improve vascular permeability (Figure 3G).
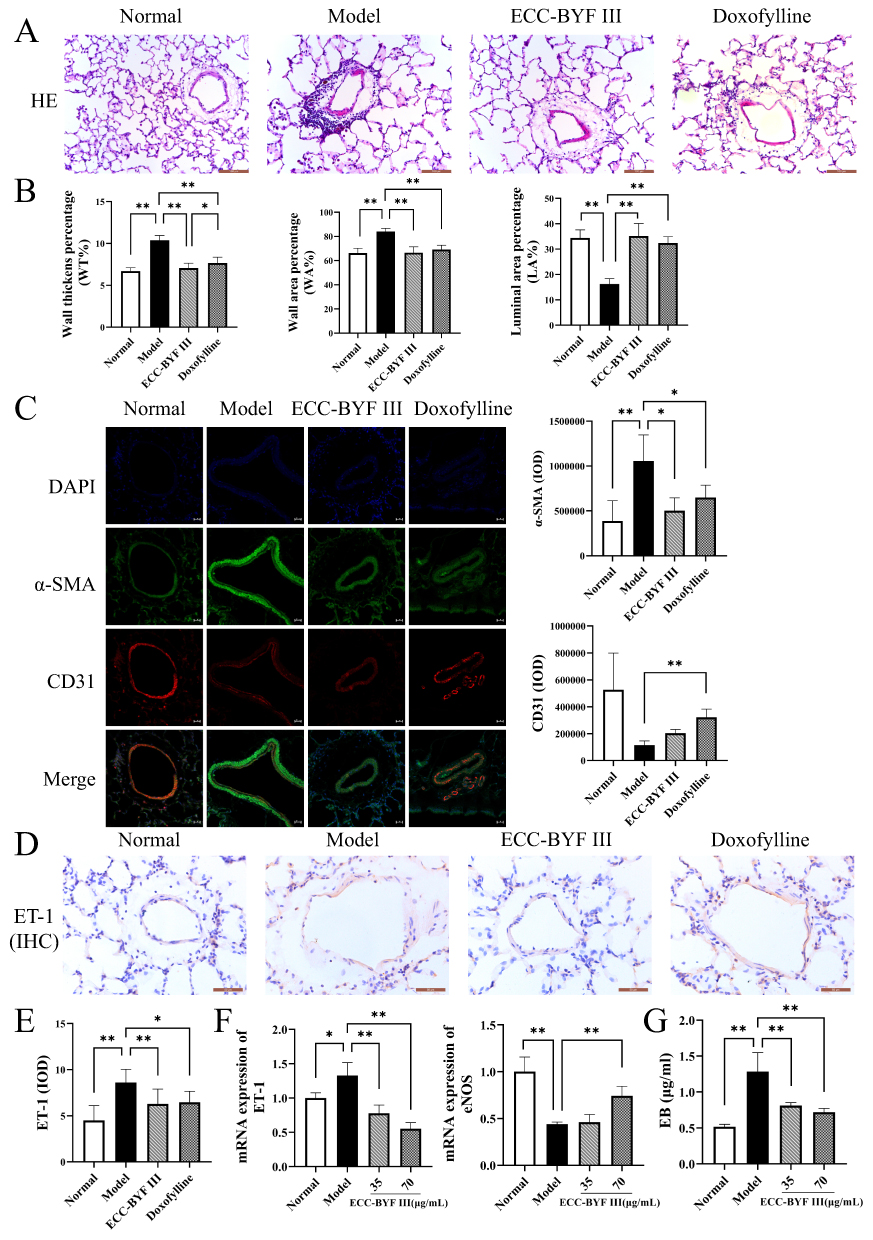 |
Figure 3 Effect of ECC-BYF III on pulmonary vascular remodeling in rats with COPD. (A) Changes in vascular wall thickness in rats (HE, ×400). (B) Wall thickens percentage (WT%), wall area percentage (WA%), luminal area percentage (LA%) structural changes in pulmonary arterioles of rats. (C) Immunofluorescence images showing the localization of α-SMA and CD31 in rat lung tissues (IF, ×400). (D) IHC analysis of Endothelin-1 (ET-1, ×400). (E) Integral optical density (IOD) changes of ET-1 in rat lung tissue. (F) Effect of ECC-BYF III on ET-1 and endothelial nitric oxide synthase (eNOS) mRNA in HUVECs. (G) Evans blue albumin extravasation assay. Values are presented as the mean ± SD. n=6~8. *P < 0.05, **P< 0.01. |
ECC-BYF III Inhibited Activation of the VEGF165/P38 MAPK Pathway in COPD Rats
VEGF165/ P38 MAPK pathway promotes the secretion of inflammatory factors and is involved in the prognosis of pulmonary vascular diseases in COPD. CSE stimulates vascular smooth muscle cells to release VEGF, causing the Y1214 site of tyrosine kinase VEGF receptor 2(VEGFR2) with GTPase. This initiates signaling to the downstream P38 MAPK pathway, ultimately leading to vascular endothelial thickening and extensive inflammatory responses.34,35 In this study, IHC analysis revealed a significant increase in VEGF expression in the pulmonary vascular endothelium of COPD rats, indicating the activation of angiogenic and inflammatory signaling (Figure 4A). Western blot analysis further demonstrated that, compared with the normal group, the model group showed marked upregulation of VEGF, VEGFR2, and phosphorylated P38 (Figure 4B and C), along with a notable increase in the phosphorylation of the downstream effector heat shock protein 27 (HSP27) in the P38 MAPK pathway (Figure 4D). These findings confirmed the activation of the VEGF165/P38 MAPK signaling cascade. Importantly, treatment with ECC-BYF III and doxofylline significantly reduced the expression of these proteins, suggesting effective inhibition of this signaling axis.
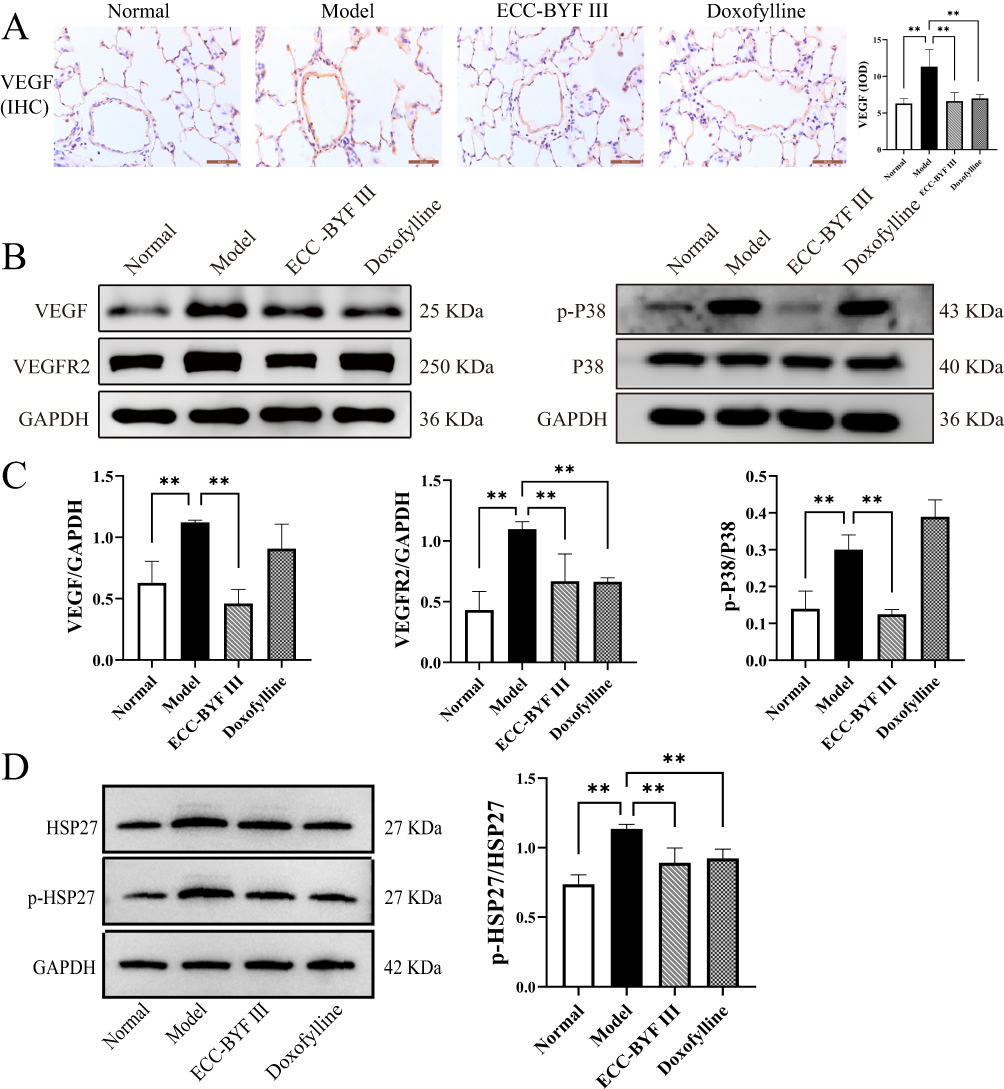 |
Figure 4 Inhibitory effect of ECC-BYF III on VEGF165/P38 MAPK pathway in COPD rats. (A) IHC analysis of VEGF in rat lung tissue (×400). (B) VEGF, VEGFR2, P38 MAPK protein expression in lung tissues of rat. (C) Protein expression of VEGF, VEGFR2 and p-P38 in rat lung tissue. (D) Protein expression of p-HSP27 and HSP27 in rat lung tissue. n=6~8. Data are expressed as the mean ± SD of three independent experiments. **P< 0.01. |
Inhibition of ECC-BYF III on VEGF165/P38 MAPK Pathway in HUVECs and THP-1 Cells Induced by LPS
The inhibitory effect of ECC-BYF III on the VEGF165/P38 MAPK pathway was verified in vitro. Immunofluorescence results demonstrated a significant up-regulation of VEGF and VEGFR2 expression in HUVECs following LPS treatment. Conversely, the expression of both VEGF and VEGFR2 was down-regulated after treatment with 35 and 70 μg/mL ECC-BYF III, with the 70 μg/mL concentration showing a more pronounced effect (Figure 5A). WB results revealed a significant increase in p-P38 MAPK protein expression in the model group. However, treatment with 35 μg/mL and 70 μg/mL ECC-BYF III effectively inhibited p-P38 MAPK and p-HSP27 expression, with the higher concentration demonstrating a more substantial inhibition (Figure 5B and C).
 |
Figure 5 Inhibitory effect of ECC-BYF III on VEGF165/P38 MAPK pathway in HUVECs and Human monocytic-leukemia (THP-1) cells. (A) Effect of ECC-BYF III on VEGF and VEGFR2 in HUVECs (IF, ×400). (B) Effect of ECC-BYF III on P38 MAPK in HUVECs. (C) Effect of ECC-BYF III on HSP27 in HUVECs. (D) Immunofluorescence images of VEGF protein expression in HUVECs and THP-1 cells (IF, ×400). (E) Immunofluorescence images of p-P38 protein expression in HUVECs and THP-1 cells (IF, ×400). n=3. Data are expressed as the mean ± SD of three independent experiments. *P < 0.05, **P< 0.01. |
THP-1 monocytes can be induced into macrophages through specific treatments, which serve as the primary source of macrophages. Hypoxia-induced THP-1 cells and HUVEC cells were found to promote VEGF secretion from THP-1 cells, accelerating angiogenesis in endothelial cells through coupling with VEGFR2 receptors in HUVECs.36 In addition, LPS-induced THP-1 cells and HUVEC cells can mediate the phosphorylation of P38, c-Jun N-terminal kinase (JNK) and extracellular regulated protein kinases (ERK), thereby amplifying the inflammatory response.37 The study’s findings also confirmed these observations. In the co-culture system of THP-1 cells and HUVEC cells induced by LPS, VEGF and p-P38 protein fluorescence signals were prominent. CCK-8 assay results showed that 0.1 μg/mL of VEGF inhibitor (VEGF-IN) had no significant cytotoxicity (Figure S1C). After VEGF-IN treatment, fluorescence weakened, and ECC-BYF III treatment further reduced the expression of VEGF and p-P38 protein. Additionally, co-treatment of VEGF-IN and ECC-BYF III further reduced protein expression levels (Figure 5D and E).
Discussion
COPD is a chronic respiratory disease characterized by persistent and progressive airflow obstruction. The primary driving factor is chronic inflammation resulting from prolonged exposure to harmful particles or gases, such as CSE. This inflammatory response not only leads to alveolar structural damage but also contributes to pathological changes in the airways and pulmonary vessels through complex molecular mechanisms.
In response to harmful particles, immune cells like macrophages, neutrophils, and T cells are activated, releasing pro-inflammatory cytokines, such as IL-6, IL-1β, and TNF-α. These pro-inflammatory mediators further recruit additional immune cells, amplifying the inflammatory response.9 Long-term inflammation leads to excessive mucus secretion causing airway stenosis and obstruction.38,39 Protease released by inflammatory cells degrade the elastic fibers of alveolar wall, leading to the rupture and fusion of alveoli, which further exacerbates airflow limitation and manifests as a severe decline in pulmonary function.40,41 ECC-BYF III is a multi-component formulation consisting of ginsenoside Rh1, astragaloside IV, icariin, paeonol, and nobiletin. Ginsenoside Rh1 has been shown to reduce allergic airway inflammation by regulating the NF-κB and AKT pathways.42 Astragaloside IV can prevent lung injury and inflammation through the MAPK pathway. Growing evidence suggests that these compounds have anti-inflammatory effects and may act together to suppress inflammatory signaling.43 ECC-BYF III, composed of these five compounds in defined proportions, reduced the decline of lung function induced by CSE combined with Klebsiella pneumoniae in rats, as well as alleviate alveolar destruction and pathological airway thickening. Additionally, we observed a significant accumulation of leukocytes in the lungs of COPD rats, along with abnormally high expression levels of IL-6 and IL-1β in both the serum and pulmonary vasculature. ECC-BYF III effectively reduced this inflammatory response.
In the pathological process of COPD, lung inflammation not only affects airway, but also affects pulmonary vascular remodeling. This process involves several signal pathways, including the VEGF165/P38 MAPK,44 TLR4/NF-κB pathway45 and ERK/Akt pathway.46 Under inflammatory conditions, an imbalance between ET-1 and eNOS secreted by vascular endothelial cells disrupts endothelial homeostasis and leads to vascular dysfunction. ET-1 acts as a potent vasoconstrictive peptide that promotes smooth muscle proliferation and extracellular matrix deposition, thereby contributing to vascular remodeling. In contrast, NO derived from eNOS maintains vascular integrity by inducing vasodilation and suppressing inflammation.47 Meanwhile, pro-inflammatory factors such as IL-1β, TNF-α and IL-6 activates vascular smooth muscle cells to release VEGF165. This factor binds to its receptor VEGFR2 which, via the Y1214 site, couples with the signaling molecule RhoGTP, transmitting signals to the downstream P38 MAPK pathway. The activation of this pathway promotes vascular endothelial thickening, vascular remodeling, and further induces angiogenesis48(Figure 6). In this study, the co-localization of CD31 and α-SMA revealed endothelial injury and loss of vascular integrity in COPD rats. Concurrently, ET-1 expression was increased while eNOS expression was markedly decreased, indicating endothelial dysfunction and disruption of the vasoconstriction/vasodilation balance. Treatment with ECC-BYF III effectively reversed these changes. Moreover, the observed downregulation of the VEGF165/P38 MAPK signaling pathway further highlights the ability of ECC-BYF III to attenuate vascular remodeling through modulation of the VEGF165/P38 MAPK axis.
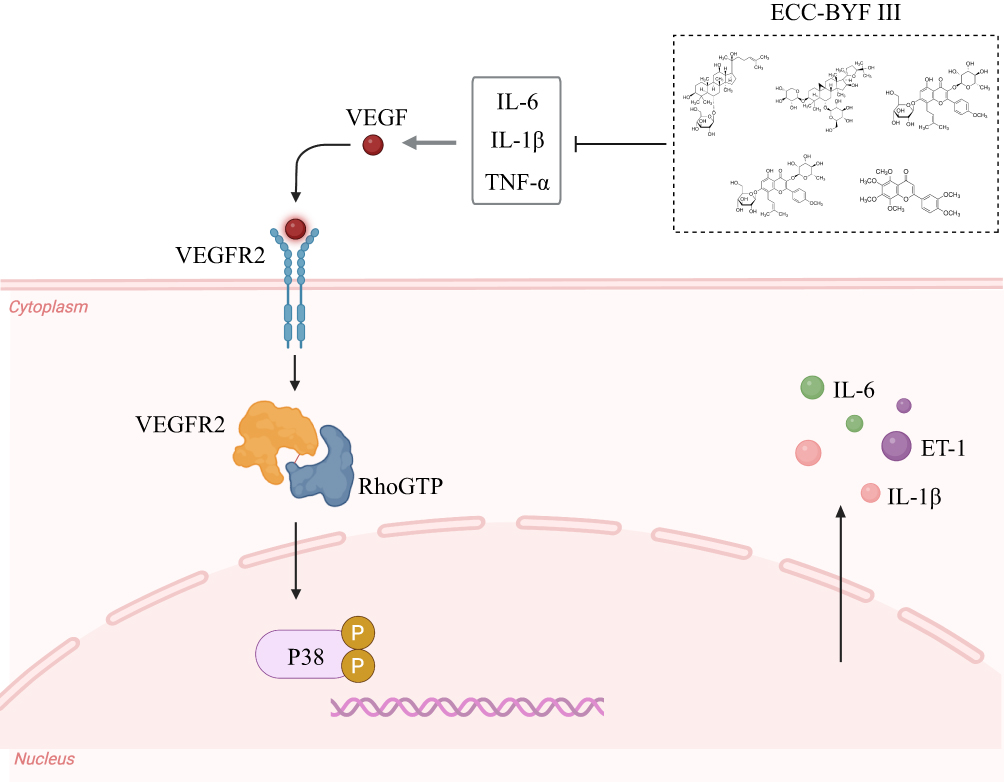 |
Figure 6 Diagram showing the potential mechanism of VEGF165/P38 MAPK signaling involved in pulmonary vascular remodeling. |
Furthermore, to verify the downstream regulatory effects of this pathway, we examined the p-HSP27, a well-recognized substrate of P38 MAPK that regulates cytoskeletal dynamics and endothelial permeability.49 The results showed that p-HSP27 was markedly elevated in COPD rats, reflecting enhanced P38 MAPK activity and endothelial stress. ECC-BYF III administration significantly decreased the p-HSP27/HSP27 ratio, indicating that ECC-BYF III attenuates P38 MAPK–mediated HSP27 phosphorylation, thereby stabilizing endothelial structure and reducing vascular inflammation. Collectively, these findings demonstrate that ECC-BYF III mitigates pulmonary vascular remodeling in COPD by suppressing the VEGF165/P38 MAPK signaling cascade.
Macrophage inflammation,50 LPS,51 and cigarette smoke can induce pulmonary inflammation, infiltrate endothelial cell damage, and activate vascular smooth muscle cells to release VEGF165. In subsequent in vitro experiment, we simulated the inflammatory injury environment of HUVECs induced by LPS and THP-1/HUVECs co-culture system. The results indicated that ECC-BYF III significantly reduced the expression of TNF-α and IL-1β in LPS-induced HUVECs. Additionally, it inhibited the expression of VEGF165, VEGFR2, and p-P38 in a dose-dependent manner, further confirming that ECC-BYF III could alleviate the inflammatory response and reduce the expression of VEGF165. Moreover, the combination of ECC-BYF III with VEGF-IN resulted in more pronounce deduction in the levels of VEGF165 and its downstream target p-P38. These findings suggest that ECC-BYF III can inhibit the VEGF165/P38 MAPK pathway and may synergistically interact with other signaling pathways to mitigate vascular remodeling in COPD while exerting anti-inflammatory effects.
This study demonstrated the therapeutic potential of ECC-BYF III in alleviating pulmonary vascular inflammation and remodeling by inhibiting the VEGF165/P38 MAPK signaling cascade and explored its underlying mechanisms. However, the LPS-induced THP-1/HUVEC co-culture model used in vitro cannot fully replicate the complex pathological features of COPD-related vascular remodeling. In future studies, we plan to employ lung organoid or organ-on-chip models to better simulate the vascular microenvironment and further elucidate the mechanisms through which ECC-BYF III exerts its regulatory effects.
Conclusion
These findings indicate that ECC-BYF III effectively treats COPD by improving lung function, alleviating pulmonary vascular inflammation and remodeling, and exert its effects by inhibiting the activation of the VEGF165/P38 MAPK pathway. This could provide future directions for studying the anti-inflammatory properties of ECC-VYF III as well as a better understanding of the role of VEGF165/P38 MAPK in inflammatory disorders.
Data Sharing Statement
The original contributions presented in the study are included in the article. Further inquiries can be directed to the corresponding authors.
Funding
This study was financially supported by the National Key Research and Development Program of China (2023YFC3502605) and National Natural Science Foundation of China (82074406 and 81973822).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. 2025. Available from: https://goldcopd.org.
2. World Health Organization. Chronic obstructive pulmonary disease. 2024. Available from: https://www.who.int/news-room/fact-sheets/detail/chronic-obstructive-pulmonary-disease-(copd).
3. Li HY, Gao TY, Fang W, et al. Global, regional and national burden of chronic obstructive pulmonary disease over a 30-year period: estimates from the 1990 to 2019 global burden of disease study. Respirology. 2023;28(1):29–16. doi:10.1111/resp.14349
4. World Health Organization. Projections of mortality and causes of death, 2016 and 2060. Available from: https://colinmathers.com/2022/05/10/projections-of-global-deaths-from-2016-to-2060/f.
5. Zhu B, Wang Y, Ming J, et al. Disease burden of COPD in China: a systematic review. Int J Chron Obstruct Pulmon Dis. 2018;13(1):1353–1364. doi:10.2147/COPD.S161555
6. Szucs B, Szucs C, Petrekanits M, et al. Molecular characteristics and treatment of endothelial dysfunction in patients with COPD: a review article. Int J Mol Sci. 2019;20(18):4329. doi:10.3390/ijms20184329
7. He S, Ma C, Zhang L, et al. GLI1-mediated pulmonary artery smooth muscle cell pyroptosis contributes to hypoxia-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2020;318(3):L472–L482. doi:10.1152/ajplung.00405.2019
8. Shlobin OA, Adir Y, Barbera JA, et al. Pulmonary hypertension associated with lung diseases. Eur Respir J. 2024;64(4):2401200. doi:10.1183/13993003.01200-2024
9. Polverino F, Celli BR, Owen CA. COPD as an endothelial disorder: endothelial injury linking lesions in the lungs and other organs? (2017 Grover Conference Series). Pulm Circ. 2018;8(1):767789184. doi:10.1177/2045894018758528
10. Apte RS, Chen DS, Ferrara N. VEGF in signaling and disease: beyond discovery and development. Cell. 2019;176(6):1248–1264. doi:10.1016/j.cell.2019.01.021
11. Zanini A, Chetta A, Imperatori AS, et al. The role of the bronchial microvasculature in the airway remodelling in asthma and COPD. Respir Res. 2010;11(1):132. doi:10.1186/1465-9921-11-132
12. Westergren-Thorsson G, Bagher M, Andersson‐Sjöland A, et al. VEGF synthesis is induced by prostacyclin and TGF -β in distal lung fibroblasts from COPD patients and control subjects: implications for pulmonary vascular remodelling. Respirology. 2018;23(1):68–75. doi:10.1111/resp.13142
13. Bai J, Bai Y, Wang X, et al. Carbon monoxide-releasing molecule-3 ameliorates acute lung injury in a model of hemorrhagic shock and resuscitation: roles of P38 MAPK signaling pathway. Shock. 2021;55(6):816–826. doi:10.1097/SHK.0000000000001684
14. Liu C, He L, Wang J, et al. Anti-angiogenic effect of Shikonin in rheumatoid arthritis by downregulating PI3K/AKT and MAPKs signaling pathways. J Ethnopharmacol. 2020;260:113039. doi:10.1016/j.jep.2020.113039
15. Wang C, Zhou J, Wang J, et al. Progress in the mechanism and targeted drug therapy for COPD. Signal Transduct Target Ther. 2020;5(1):248. doi:10.1038/s41392-020-00345-x
16. Shi F, Cao J, Zhou D, et al. Revealing the clinical effect and biological mechanism of acupuncture in COPD: a review. Biomed Pharmacother. 2024;170:115926. doi:10.1016/j.biopha.2023.115926
17. Chen T, Ding L, Zhao M, et al. Recent advances in the potential effects of natural products from traditional Chinese medicine against respiratory diseases targeting ferroptosis. Chin Med. 2024;19(1):49. doi:10.1186/s13020-024-00918-w
18. Li SY, Li JS, Wang MH, et al. Effects of comprehensive therapy based on traditional Chinese medicine patterns in stable chronic obstructive pulmonary disease: a four-center, open-label, randomized, controlled study. BMC Complement Altern Med. 2012;12:197. doi:10.1186/1472-6882-12-197
19. Li J, Xie Y, Zhao P, et al. A Chinese herbal formula ameliorates COPD by inhibiting the inflammatory response via downregulation of p65, JNK, and P38. Phytomedicine. 2021;83:153475. doi:10.1016/j.phymed.2021.153475
20. Qin YQ, Chen YL, Zhao P, et al. Tiaobu Feishen therapy inhibits inflammation induced by cigarette smoke extracts in a human monocyte/macrophage cell line. J Traditional Chin Med. 2021;41(3):360–366. doi:10.19852/j.cnki.jtcm.2021.03.003
21. Zhao P, Li J, Yang L, et al. Integration of transcriptomics, proteomics, metabolomics and systems pharmacology data to reveal the therapeutic mechanism underlying Chinese herbal Bufei Yishen formula for the treatment of chronic obstructive pulmonary disease. Mol Med Rep. 2018;17(4):5247–5257. doi:10.3892/mmr.2018.8480
22. Zhao P, Li J, Tian Y, et al. Restoring Th17/Treg balance via modulation of STAT3 and STAT5 activation contributes to the amelioration of chronic obstructive pulmonary disease by Bufei Yishen formula. J Ethnopharmacol. 2018;217:152–162. doi:10.1016/j.jep.2018.02.023
23. Li J, Zhao P, Yang L, et al. System biology analysis of long-term effect and mechanism of Bufei Yishen on COPD revealed by system pharmacology and 3-omics profiling. Sci Rep. 2016;6:25492. doi:10.1038/srep25492
24. Li JS, Liu XF, Dong HR, et al. Effective-constituent compatibility-based analysis of Bufei Yishen formula, a traditional herbal compound as an effective treatment for chronic obstructive pulmonary disease. J Integr Med. 2020;18(4):351–362. doi:10.1016/j.joim.2020.04.004
25. Li J, Ma J, Tian Y, et al. Effective-component compatibility of Bufei Yishen formula II inhibits mucus hypersecretion of chronic obstructive pulmonary disease rats by regulating EGFR/PI3K/mTOR signaling. J Ethnopharmacol. 2020;257:112796. doi:10.1016/j.jep.2020.112796
26. Liu L, Qin Y, Cai Z, et al. Effective-components combination improves airway remodeling in COPD rats by suppressing M2 macrophage polarization via the inhibition of mTORC2 activity. Phytomedicine. 2021;92:153759. doi:10.1016/j.phymed.2021.153759
27. Miao YF, Zhang LX, Jin FL, et al. Effect of effective-component compatibility of Bufei Yishen formula combined with acupuncture on small pulmonary vessels in COPD rats. Chin J Integr Tradit West Med. 2021;41(08):973–980. doi:10.7661/j.cjim.20210604.174
28. Li Y, Li SY, Li JS, et al. A rat model for stable chronic obstructive pulmonary disease induced by cigarette smoke inhalation and repetitive bacterial infection. Biol Pharm Bull. 2012;35(10):1752–1760. doi:10.1248/bpb.b12-00407
29. Poto R, Loffredo S, Palestra F, et al. Angiogenesis, lymphangiogenesis, and inflammation in Chronic Obstructive Pulmonary Disease (COPD): few certainties and many outstanding questions. Cells. 2022;11(10):1720. doi:10.3390/cells11101720
30. Jiang Y, Zhao Y, Zhu X, et al. Effects of autophagy on macrophage adhesion and migration in diabetic nephropathy. Ren Fail. 2019;41(1):682–690. doi:10.1080/0886022X.2019.1632209
31. Guo CR, Han R, Xue F, et al. Expression and clinical significance of CD31, CD34, and CD105 in pulmonary ground glass nodules with different vascular manifestations on CT. Front Oncol. 2022;12:956451. doi:10.3389/fonc.2022.956451
32. Zhao Y, Zhu H, Yang Y, et al. AQP1 suppression by ATF4 triggers trabecular meshwork tissue remodeling in ET-1-induced POAG. J Cell Mol Med. 2020;24(6):3469–3480. doi:10.1111/jcmm.15032
33. Gopalakrishna D, Pennington S, Karaa A, et al. ET-1 stimulates superoxide production by eNOS following exposure of vascular endothelial cells to endotoxin. Shock. 2016;46(1):60–66. doi:10.1097/SHK.0000000000000576
34. Heldin J, O’Callaghan P, Hernandez VR, et al. FGD5 sustains vascular endothelial growth factor A (VEGFA) signaling through inhibition of proteasome-mediated VEGF receptor 2 degradation. Cell Signal. 2017;40:125–132. doi:10.1016/j.cellsig.2017.09.009
35. Watterston C, Halabi R, McFarlane S, et al. Endothelial Semaphorin 3fb regulates Vegf pathway-mediated angiogenic sprouting. PLoS Genet. 2021;17(8):e1009769. doi:10.1371/journal.pgen.1009769
36. Zhang G, Tao X, Ji B, et al. Hypoxia-driven M2-polarized macrophages facilitate cancer aggressiveness and temozolomide resistance in glioblastoma. Oxid Med Cell Longev. 2022;2022(1):1614336. doi:10.1155/2022/1614336
37. Kim C, Sim H, Bae JS. Benzoylpaeoniflorin activates anti-inflammatory mechanisms to mitigate sepsis in cell-culture and mouse sepsis models. Int J Mol Sci. 2022;23(21). doi:10.3390/ijms232113130
38. Kesimer M, Ford AA, Ceppe A, et al. Airway mucin concentration as a marker of chronic bronchitis. N Engl J Med. 2017;377(10):911–922. doi:10.1056/NEJMoa1701632
39. Hill DB, Button B, Rubinstein M, et al. Physiology and pathophysiology of human airway mucus. Physiol Rev. 2022;102(4):1757–1836. doi:10.1152/physrev.00004.2021
40. Johnson SR. Untangling the protease web in COPD: metalloproteinases in the silent zone. Thora. 2016;71(2):105–106. doi:10.1136/thoraxjnl-2015-208204
41. Dey T, Kalita J, Weldon S, et al. Proteases and their inhibitors in chronic obstructive pulmonary disease. J Clin Med. 2018;7(9):244. doi:10.3390/jcm7090244
42. Jin Y, Tangchang W, Kwon OS, et al. Ginsenoside Rh1 ameliorates the asthma and allergic inflammation via inhibiting Akt, MAPK, and NF-κB signaling pathways in vitro and in vivo. Life Sci. 2023;321:121607. doi:10.1016/j.lfs.2023.121607
43. Wang Z, Wu Y, Pei C, et al. Astragaloside IV pre-treatment attenuates PM2.5-induced lung injury in rats: impact on autophagy, apoptosis and inflammation. Phytomedicine. 2022;96:153912. doi:10.1016/j.phymed.2021.153912
44. Maniatis NA, Kotanidou A, Catravas JD, et al. Endothelial pathomechanisms in acute lung injury. Vascul Pharmacol. 2008;49(4–6):119–133. doi:10.1016/j.vph.2008.06.009
45. Zhang S, Zou J, Li P, et al. Curcumin protects against atherosclerosis in apolipoprotein E-knockout mice by inhibiting toll-like receptor 4 expression. J Agric Food Chem. 2018;66(2):449–456. doi:10.1021/acs.jafc.7b04260
46. Jhun H, Baek S, Kim J, et al. Effect of Korean Magnolia obovata extract on platelet-derived growth factor-induced vascular smooth muscle cells. Chin J Integr Med. 2020;26(9):677–682. doi:10.1007/s11655-019-3171-y
47. Durán WN, Breslin JW, Sánchez FA. The NO cascade, eNOS location, and microvascular permeability. Cardiovasc Res. 2010;87(2):254–261. doi:10.1093/cvr/cvq139
48. Schermuly RT, Ghofrani HA, Wilkins MR, et al. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol. 2011;8(8):443–455. doi:10.1038/nrcardio.2011.87
49. Gallagher ER, Oloko PT, Fitch TC, et al. Lysosomal damage triggers a p38 MAPK-dependent phosphorylation cascade to promote lysophagy via the small heat shock protein HSP27. Curr Biol. 2024;34(24):5739–5757.e8. doi:10.1016/j.cub.2024.10.061
50. Harkness LM, Kanabar V, Sharma HS, et al. Pulmonary vascular changes in asthma and COPD. Pulm Pharmacol Ther. 2014;29(2):144–155. doi:10.1016/j.pupt.2014.09.003
51. Hou X, Yang S, Yin J. Blocking the REDD1/TXNIP axis ameliorates LPS-induced vascular endothelial cell injury through repressing oxidative stress and apoptosis. Am J Physiol Cell Physiol. 2019;316(1):C104–C110. doi:10.1152/ajpcell.00313.2018
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.