Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 21
Effective-Component Compatibility of Bufei Yishen Formula Alleviates Alveolar Epithelial Barrier Damage in COPD Through Inhibition of p38 MAPK Phosphorylation
Authors Shao X, Zhang L ![]() , Guo M, Wang C, Chu H, Mo Y, Lu R, Zhao P, Tian Y
, Guo M, Wang C, Chu H, Mo Y, Lu R, Zhao P, Tian Y ![]()
Received 12 January 2026
Accepted for publication 16 June 2026
Published 25 June 2026 Volume 2026:21 589282
DOI https://doi.org/10.2147/COPD.S589282
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Zijing Zhou
Xuejie Shao,1– 3,* Lanxi Zhang,3,* Mingyi Guo,1,2 Chen Wang,1,2 Haotian Chu,1,2 Yangqian Mo,1– 3 Ruilong Lu,1– 3 Peng Zhao,1– 3 Yange Tian1– 3
1Collaborative Innovation Center for Chinese Medicine and Respiratory Diseases Co-Constructed by Henan Province & Education Ministry of People’s Republic of China, Henan University of Chinese Medicine, Zhengzhou, People’s Republic of China; 2Henan Key Laboratory of Chinese Medicine for Respiratory Disease, Henan University of Chinese Medicine, Zhengzhou, People’s Republic of China; 3Academy of Chinese Medical Sciences, Henan University of Chinese Medicine, Zhengzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yange Tian, Email [email protected]
Purpose: Alveolar epithelial barrier (AEB) dysfunction drives the development of chronic obstructive pulmonary disease (COPD). This study aimed to investigate whether the effective-component compatibility of the Bufei Yishen formula (ECC-BYF) protects the AEB in COPD via anti-inflammatory mechanisms, and to elucidate the underlying pathways.
Methods: In vivo, a rat model of COPD was established via repeated bacterial infection and cigarette smoke exposure. Following treatment, we analyzed lung function, histopathology, epithelial ultrastructure, inflammatory factors, and AEB-related protein expression. In vitro, MLE-12 cells stimulated with TNF-α were pretreated with ECC-BYF to assess cell viability, inflammatory responses, and barrier protein expression. Additionally, network pharmacology was utilized to predict key therapeutic targets, and the involvement of the p38 MAPK signaling pathway was validated via Western blotting.
Results: In vivo, ECC-BYF improved pulmonary function and attenuated histopathological damage in COPD rats. Ultrastructural analysis showed that ECC-BYF improved the morphology of alveolar type 1 (AT1) and type 2 (AT2) cells and preserved intercellular junctions. Furthermore, ECC-BYF upregulated the expression of ZO-1, Occludin, SP-C, and SP-D proteins, increased Interleukin (IL)-10 in bronchoalveolar lavage fluid (BALF), and suppressed BALF levels of IL-6, IL-1β, and TNF-α. Network pharmacology identified TNF/IL-6 and MAPK signaling as key targets. In vitro, ECC-BYF reversed TNF-α-induced elevations in IL-6 and TNF-α mRNA, increased ZO-1, Occludin, and SP-C protein levels, and inhibited p38 MAPK phosphorylation. This reduction in p-p38 MAPK was concordantly observed in rat lung tissues. Critically, asiatic acid abrogated ECC-BYF’s suppressive effects, demonstrating that p38 MAPK inhibition is central to its barrier-protective efficacy.
Conclusion: ECC-BYF mitigates AEB disruption in COPD by inhibiting the p38 MAPK-mediated inflammatory response, highlighting a crucial mechanism for protecting barrier integrity.
Keywords: COPD, P38 MAPK pathway, alveolar epithelial barrier
Introduction
Chronic Obstructive Pulmonary Disease (COPD) is a heterogeneous disorder marked by irreversible airflow limitation. This obstruction results from abnormalities in the airways (such as bronchitis or bronchiolitis) and/or alveoli (emphysema).1 COPD is a primary contributor to global mortality, imposing a substantial socioeconomic burden. Current epidemiological estimates indicate approximately 210 million affected individuals worldwide, with 3.3 million annual deaths attributable to COPD.2,3 While COPD is characterized by a complex etiology, prolonged exposure to tobacco smoke remains the undisputed primary environmental risk factor driving its pathogenesis. Current COPD management primarily relies on smoking cessation, bronchodilators, and inhaled corticosteroids. Although these interventions partially alleviate clinical symptoms, they fail to effectively slow the progression of the disease.1,4 Therefore, it is critical to advance research on effective strategies for managing COPD.
The progressive decline in lung function, which presents the primary clinical challenge in COPD management, is fundamentally driven by the continuous disruption of the alveolar epithelial barrier (AEB). The AEB forms a natural physical defense against invading microbes and harmful substances, while activating immune cells to initiate innate immunity.5,6 Cigarette smoke, particulate matter, and microbial infections can breach this barrier, triggering inflammation and AEB damage (Supplementary Figure S1). During barrier impairment, inflammatory cells infiltrate the alveolar space, exacerbating epithelial integrity loss. Chronic injury induces compensatory structural destruction—manifested as alveolar rupture and bullae formation—which progressively disrupts gas exchange and accelerates emphysema development, ultimately driving COPD progression.7,8
Crucially, inflammatory dysregulation is central to AEB damage.9 Compromised epithelial cells release inflammatory mediators and recruit immune cells, thereby establishing a self-perpetuating inflammatory cascade.10,11 These mediators degrade tight junctions by downregulating ZO-1 and occludin (Supplementary Figure S1), destabilizing the AEB and promoting parenchymal destruction.12,13 Given this pathophysiology, synergistic strategies that concurrently suppress inflammation and restore alveolar barrier function hold significant therapeutic promise for COPD.14 Consequently, the preservation of the AEB from inflammation-induced damage has been identified as an effective strategy for COPD.
Emerging evidence suggests that the adjunctive use of Traditional Chinese medicine (TCM) alongside conventional therapy may offer beneficial effects for patients with COPD, particularly in alleviating clinical symptoms and improving overall quality of life.15 Bufei Yishen formula (BYF) serves as an efficacious formula for treating COPD (Patent ZL.201110117578.1). Clinical research has verified that BYF can mitigate the risk of acute exacerbation and promote patients’ exercise ability and functional status.16 Basic evidence supports the view that BYF can attenuate inflammation in COPD rats, regulate alveolar surfactant protein secretion, and maintain alveolar structure.17 Despite demonstrating efficacy in reducing systemic inflammation and alveolar destruction in a COPD rat model, the therapeutic mechanisms of BYF remain difficult to elucidate due to the complexity of its active components. To address this, we integrated systems pharmacology with in vitro and in vivo validation to screen and optimize the key active components.18,19 Finally, the Effective-Component Compatibility of BYF (ECC-BYF), which demonstrated efficacy comparable to that of the original BYF,20 was obtained. Previous fundamental research has revealed that ECC-BYF can reduce the thickening of the respiratory membrane and attenuate inflammation in COPD rats, and exerts a beneficial intervention effect,21 but the factors that regulate AEB during this process remain elusive.
To evaluate this, an in vivo COPD rat model was established through cigarette smoke inhalation combined with repetitive bacterial infection, alongside an in vitro model of TNF-α-stimulated MLE-12 cells. Using these models, this study aimed to investigate whether the ECC-BYF protects the AEB in COPD via anti-inflammatory mechanisms, and to elucidate the underlying pathways.
Materials and Methods
Chemicals and Reagents
Cigarettes (Hongqi Canal® Filter tip) were sourced from Henan Tobacco Industry Co., Ltd. (Anyang, China). Klebsiella pneumoniae (46114) was procured from the National Center for Medical Culture Collection (Beijing, China). N-acetylcysteine (NAC) was purchased from Zambon (S.p.A, Italia). The ECC-BYF formulation listed in Table 1 was supplied by Chengdu Must Bio-technology Co., Ltd. (Chengdu, China). All compounds were of >98% purity. TNF-α (L2880) was acquired from Sigma-Aldrich (St. Louis, MO, USA). ELISA kits (E-EL-R0012c, E-EL-R0015c, E-EL-R0016, E-EL-R2856c) were obtained from Elabscience (Wuhan, China). These antibodies were used: Surfactant protein D Antibody (Affinity Bioscience, DF13601), Anti-Prosurfactant Protein C antibody (Abcam, ab90716), IL-6 Polyclonal Antibody (Elabscience, E-AB-40073), ZO-1 (Affinity Bioscience, AF5145), Occludin Polyclonal antibody (Proteintech, 13409-1-AP), and Podoplanin Monoclonal Antibody (Invitrogen, 14–5381-82).
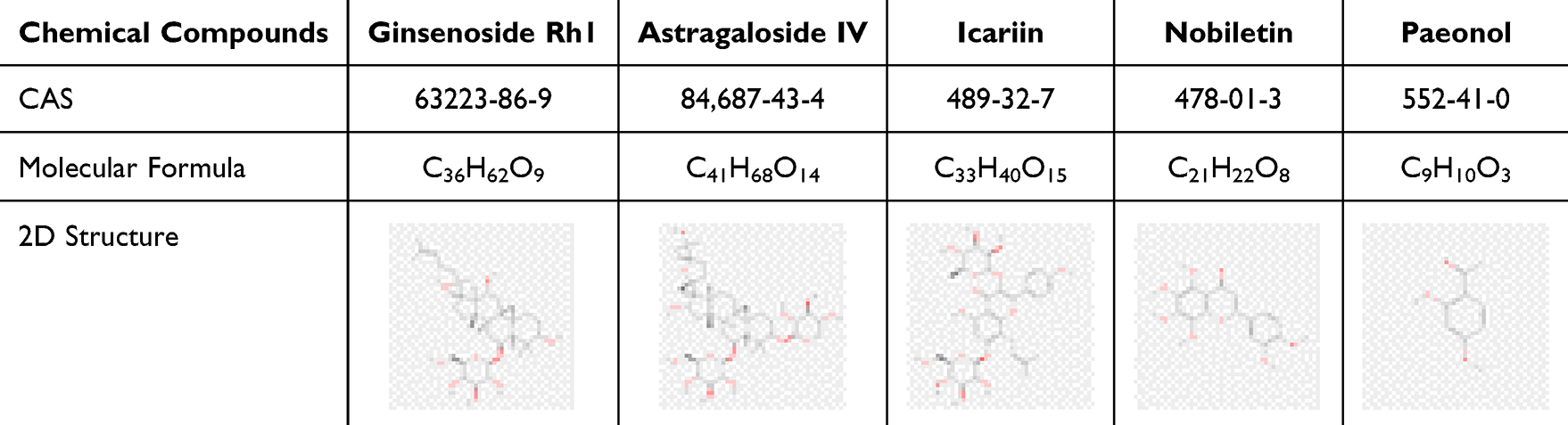 |
Table 1 The Compounds of ECC-BYF. The CAS Numbers, Molecular Formulas, and 2D Structures of ECC-BYF |
Study Design and Animal Model
This was a randomized, controlled animal experiment. The entire study lasted 16 weeks, comprising a COPD induction phase (weeks 1–8) and a treatment phase (weeks 9–16). The primary outcome was alveolar epithelial barrier integrity assessed by pulmonary pathology and junctional protein expression; secondary outcomes included inflammation and p38 MAPK phosphorylation. Seventy-two male Sprague-Dawley rats (age/weight) were obtained from Beijing HFK Bioscience Co., Ltd. The animal experiments received approval from the Experimental Animal Care and Ethics Committee of Henan University of Chinese Medicine (No. DWLL202003261) and were performed in strict accordance with the Laboratory animal-Guideline for ethical review of animal welfare (GB/T 35892–2018, National Standard of the People’s Republic of China). Sample size was based on our previous experience and published studies of COPD rat models, which showed that 10–12 animals per group were sufficient to detect meaningful differences in pulmonary function and histological parameters. With six groups, we allocated 12 rats per group to account for possible mortality during the modeling period. Randomization was performed using a computer-generated random number list. Rats were randomly assigned to six groups (n=12 each): Normal, Model, ECC-BYF H (ECC-H), ECC-BYF M (ECC-M), ECC-BYF L (ECC-L), and NAC. The allocation sequence was concealed from the investigators conducting the daily treatments and outcome assessments. The procedure for establishing the COPD model has been previously described.22 From weeks 1–8, COPD was induced by infection with Klebsiella pneumoniae and cigarette smoke. The Normal group received ambient air and saline instead of cigarette smoke and bacterial challenge. From weeks 9 to 16, the NAC, ECC-M, ECC-L, and ECC-H groups received NAC (54 mg/kg/d) and ECC-BYF (11.0, 5.5, and 1.75 mg/kg/d, respectively) by oral gavage. During the experimental period, mental state, activity level, and food and water intake was observed daily and body weight was recorded once per week. At the end of week 16, all rats were euthanized with sodium pentobarbital (40 mg/kg, i.p). After confirming anesthesia, pulmonary function was evaluated using a dedicated system.
Pulmonary Function and Histological Analysis
Pulmonary function parameters, including 50% expiratory flow (EF50), peak expiratory flow (PEF), and tidal volume (TV), were quantified four weeks via instrument and equipment (Buxco Inc., Wilmington, NC, USA), forced vital capacity (FVC), functional residual capacity (FRC), dynamic compliance (Cydn), and maximum expiratory flow (MMEF)were measured using FinePointe (PFT).
The lung tissue slices were processed with HE staining for morphological analysis using a light microscope (Olympus, Tokyo, Japan). Six views were captured from each slice, and the mean linear intercept (MLI)/ mean alveolar number (MAN) in a fixed area of the visual field were quantified. The wall thickness of the bronchial tube (Wt) and the ratio of the bronchial wall area (s) to the inner circumference (Pi) were utilized to indicate pathological changes in the bronchus. The short diameter of the bronchus should fall within the range of 100 to 300 µm to accurately determine its grade. Measurements included the three different long diameters (a1, a2, a3) and the different short diameters (b1, b2, b3), as well as external bronchial area (S1), internal bronchial area (S2), and internal perimeter (Pi) passing through the center of each bronchus. Wt(μm) = [(a1 - b1) + (a2 - b2) + (a3 - b3)] / (3 × 2), (s/Pi) = (S1 - S2) / Pi (Supplementary Figure S2).
Ultrastructure of Lung
The lung tissue was cut into 1 mm3 pieces, prefixed, dehydrated in an ethanol series, embedded in Epon812, and cut into ultra-thin sections. The ultrastructural changes of AEB were observed.
ELISA
Concentrations of IL-6, IL-10, IL-1β, and TNF-α in the BALF samples and IL-6, TNF-α levels in cell supernatant were quantified by ELISA kits in accordance with the manufacturer’s instructions.
Cell Culture and Treatment
The MLE-12 mouse alveolar epithelial cell line was purchased from the cell bank of the Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). MLE-12 was cultivated at 37 °C in DMEM/F12 (Cytiva, SH30023.01B) containing 2% FBS (EXCELL Bio, FSP500), 1% double antibody (penicillin-streptomycin, pricella, PB180120) and 1% ITS-X Media Supplement (Beyotime, C0345). MLE-12 cells were plated (3.5× 105 cells/mL), incubated with ECC-BYF (17, 35, and 70μg/mL), and stimulated with 10 ng/mL TNF-α for 24 hours.
Immunofluorescence
Lung tissues were incubated overnight at 4 °C with antibodies ZO-1, occludin, and double-stained PDPN/IL-6. A 12-well plate was utilized, and a specialized cell culture coverslip was placed within it. MLE-12 cells were seeded onto the cell climbing tablet. Intervention was performed after the cells successfully attached to the culture vessel. Primary antibodies for ZO-1, occludin, and SP-C were incubated overnight at 4 °C. The lung tissues and cell culture coverslip were subsequently treated with secondary antibody under dark conditions and re-stained with DAPI.
Immunohistochemistry
The processing steps for the lung tissues were roughly the same as those for immunofluorescence. Subsequently, the quantitative analysis of the integral optical density was determined using Image-Pro Plus 6.0 software.
Quantitative Real-Time PCR (qPCR) Assay
Total RNA was extracted and converted into complementary DNA. The mRNA levels were assessed by qPCR. The primers are listed in Table 2.
 |
Table 2 Mouse Primer Sequence |
Western Blot (WB) Assay
Following lysis in RIPA and protein concentration determination with a bicinchoninic acid assay, samples were resolved by SDS-PAGE and electroblotted onto PVDF (Millipore, ISEQ00010). Subsequently, PVDF were blocked with 5% non-fat milk, incubated overnight withZO-1, occludin, and GAPDH antibodies, and then labeled with chemiluminescent-linked secondary antibodies.
Integrated Analysis of Network Pharmacology
Prediction of Potential Targets of ECC-BYF
The canonical SMILES23 of these five compounds (Table 1) were used to mine potential targets from STITCH database,24 SwissTargetPrediction database,25 SEA database,26 PharmMapper database27 and TCMSP database.
Identification of COPD-Related Genes
The potential genes of inflammation and COPD were identified using the indexing terms “inflammation” and “chronic obstructive pulmonary disease” in the website with GeneCards,28 DrugBank29 and DisGeNet.30 All targets were submitted to UniProtKB31 to acquire the standardized gene symbols.
Generation and Analysis of the Protein–Protein Interaction (PPI) Network
The PPI was established using Cytoscape and STRING. Data were obtained from String database.32 Multiple Proteins were selected, and the common target Gene Symbol was entered. Homo sapiens was selected for Organisms. The task was submitted and the built PPI network file was downloaded. The top 100 targets were selected according to the degree value for further analysis.
Enrichment of KEGG Pathways
The functional enrichment analysis of KEGG Pathways of the key targets was in DAVID.33
Molecular Docking
The initial structures of proteins and compounds were retrieved from the PubChem and RCSB PDB. These protein structures were then subjected to preprocessing in PyMOL 2.3.4 to remove non-essential water and ligand molecules. Following preprocessing, the receptor proteins were prepared for docking (hydrogen addition and charge balancing) using AutoDock. The final docking simulations between the prepared receptors and ligands were executed. The outcome is quantified by the docking affinity score; a more negative value implies a stronger and more stable predicted binding complex.
Statistical Analysis
Statistical analyses were performed using SPSS (v24.0). Data are presented as mean ± SEM. Inter-group differences were assessed by one-way ANOVA. For data with homogeneous variance, Fisher’s LSD test was used for post-hoc comparisons; for data violating this assumption, Dunnett’s T3 test was implemented. Statistical significance was set at P < 0.05.
Results
ECC-BYF Treatment Ameliorated the Severity of COPD Rats
Over the 16-week experimental period, the Normal group rats exhibited good health. Model group rats developed progressive deterioration, and by week 8, significant weight loss was observed (P<0.01). At week 16, the ECC-H, ECC-M, ECC-L, and NAC groups showed improved mental status and significant weight recovery (P<0.05, P<0.01) (Supplementary Figure S3 A).
By week 8, the COPD model rats exhibited a progressive decline in lung function, with non-invasive parameters (TV, EF50, and PEF) showing significant reductions relative to the Normal group (P<0.01). By week 16, these parameters remained significantly reduced, while invasive parameters including Cydn, FVC, and MMEF decreased significantly, and FRC increased significantly (P<0.01). All treatments reversed these deficits, improving TV, EF50, and PEF and normalizing Cydn, FVC, MMEF, and FRC (P<0.05, P<0.01) (Supplementary Figure S3 B–H).
Histologically, while Normal rats showed intact alveoli without inflammation, the Model group displayed severe alveolar fusion, inflammatory infiltration, and airway wall thickening. The ECC-BYF and NAC reduced alveolar fracture and inflammation (Figure 1), with quantitative improvements: increased MAN and decreased MLI, WT, and S/Pi (P<0.05, P<0.01).
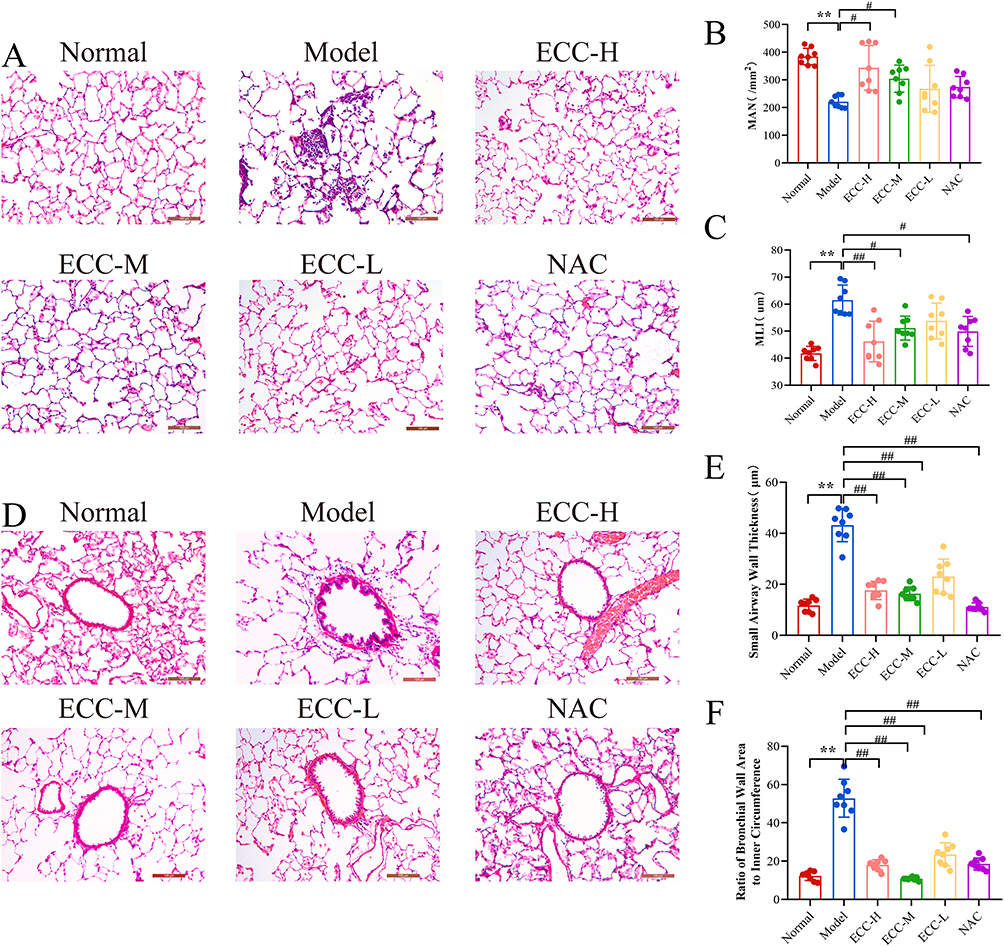 |
Figure 1 The effect of the ECC-BYF on the COPD rats. (A) HE staining of lung tissue in rats (×200). (B) Mean alveolar number (MAN). (C) Mean linear intercept (MLI). (D) HE staining of lung tissue in rats (×200). (E) wall thickness of the bronchial tube (Wt). (F) he ratio of the bronchial wall area (s) to the inner circumference (Pi). **P < 0.01, versus the normal group. ##P < 0.01, #P < 0.05, versus the model group. |
ECC-BYF Protected the AEB Structure and Function in Rats with COPD
Building upon the observed pulmonary dysfunction and histopathological damage, electron microscopy revealed critical ultrastructural alterations. In the Normal group, Alveolar type 1 (AT1) epithelial cells showed intact structure with smooth surfaces, abundant pinocytotic vesicles, and characteristic thin cytoplasmic extensions. Alveolar type 2 (AT2) epithelial cells exhibited well-organized microvilli, prominent lamellar bodies, and normal mitochondria. Tight intercellular junctions displayed high electron density without gaps. Conversely, in the Model group, both AT1 (blurred edges, reduced vesicles) and AT2 (vacuolated lamellar bodies, swollen/shrunken mitochondria) exhibited significant damage. Intercellular junctions showed widened gaps with decreased electron density. In comparison to the Model group, ECC-BYF and NAC restored AT1/AT2 ultrastructure, increasing pinocytotic vesicles and improving lamellar body/mitochondrial integrity. Intercellular junctions demonstrated notable recovery (Figure 2). Consistent with structural findings, ZO-1 and Occludin fluorescence intensity in Model group significantly decreased versus Normal (P<0.01), which was reversed by ECC-BYF and NAC treatments (Figure 3). SP-C and SP-D expression followed the same pattern: Model group showed marked reduction versus Normal (P<0.01), while treatments significantly upregulated expression versus Model (P<0.01) (Figure 4). Collectively, ECC-BYF preserved AEB integrity by simultaneously improving cellular ultrastructure and enhancing junctional/surfactant protein expression.
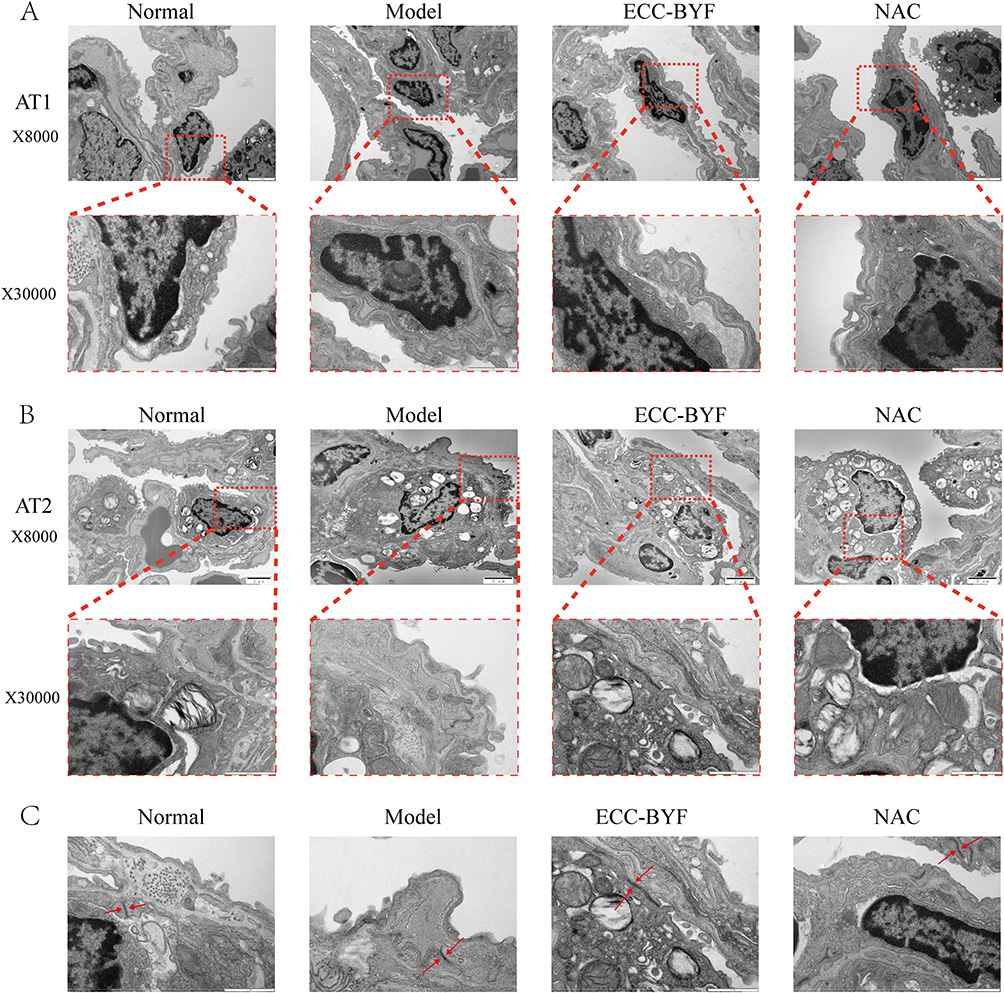 |
Figure 2 ECC-BYF improves AEB structure. (A) the ultrastructural changes observed in AT1 cells. (B) the ultrastructural changes observed in AT2 cells. The upper images of each group are magnified at ×8000, while the lower images, framed by red dotted lines, are magnified at ×30000. (C) Shows the changes in connections between alveolar epithelial cells, highlighting the tight connections indicated by the two red arrows at ×30000. |
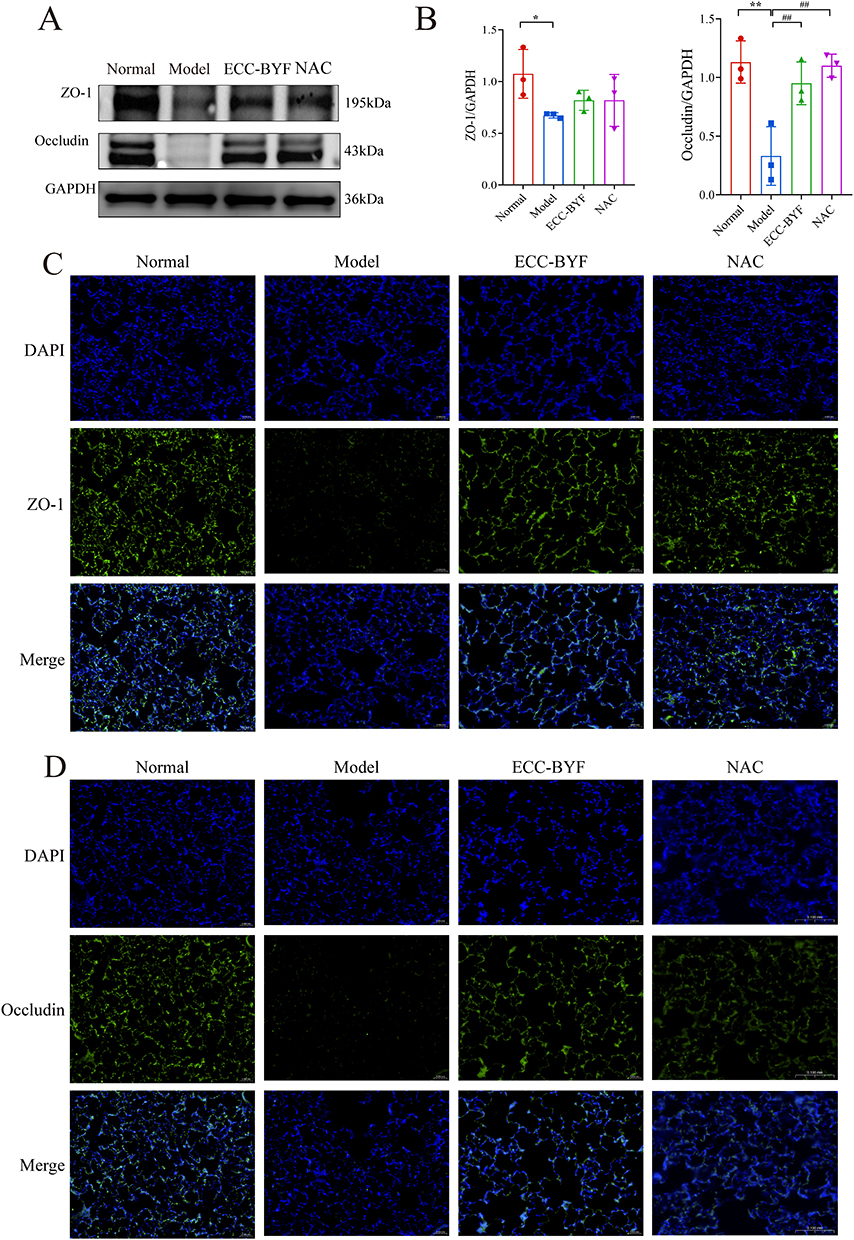 |
Figure 3 ECC-BYF improves AEB structure. (A and B) ZO-1 and Occludin expression in lung tissue was analyzed by Western blot. (C) ZO-1 immunofluorescence images of rats in each group (×400). (D) Immunofluorescence images of Occludin in each group (×400). DAPI blue fluorescent label; ZO-1, Occludin green fluorescent marks; Merge Blue and green merge tags. **P < 0.01, *P < 0.05, versus the normal group. ##P < 0.01, versus the model group. |
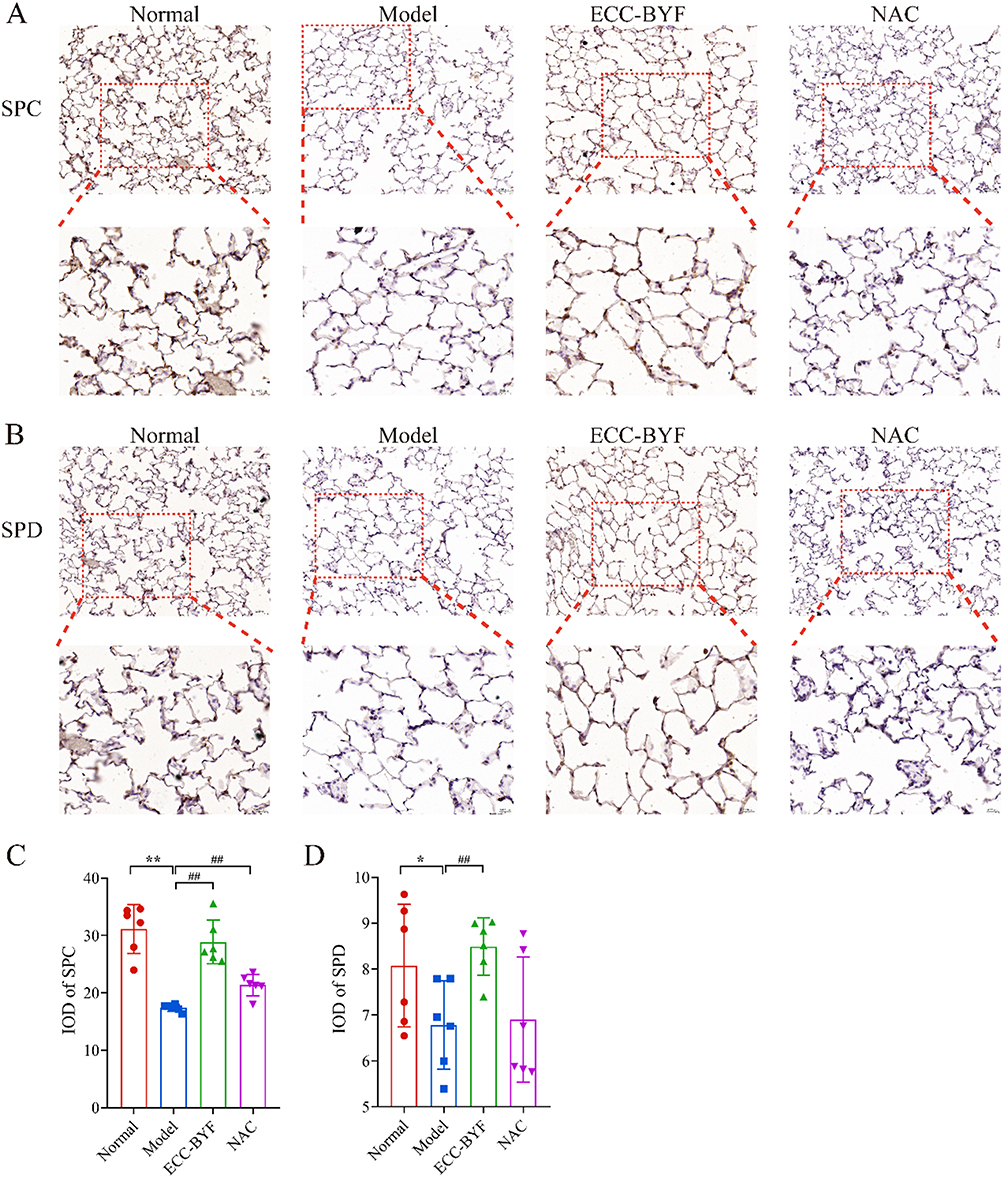 |
Figure 4 ECC-BYF improves AEB function. (A) SP-C immunohistochemical images of rats. (B) SP-D immunohistochemical images of rats. The upper images of each group are magnified at ×200, while the lower images, framed by red dotted lines, are magnified at ×400. (C) SP-C IOD value in rat lung tissue. (D) SP-D IOD value in rat lung tissue. IOD: Integrated optical density value. **P < 0.01, *P < 0.05, versus the normal group. ##P < 0.01, versus the model group. |
ECC-BYF Attenuated the Inflammatory Response in COPD Rats
The BALF of COPD model rats exhibited significantly increased levels of IL-6, IL-1β, and TNF-α (P<0.01). Conversely, IL-10 was markedly reduced. In contrast, ECC-BYF and NAC groups were significantly attenuated inflammation (P<0.01) (Figure 5A–D). In addition, no detectable expression of IL-6 was observed in Normal lung tissue, and the fluorescence of AT1 marker PDPN was stronger. The fluorescence of PDPN decreased significantly, while the fluorescence of IL-6 increased significantly in COPD rat model. The ECC-BYF and NAC significantly suppressed inflammatory responses, and enhanced fluorescence intensity of PDPN (Figure 5E).
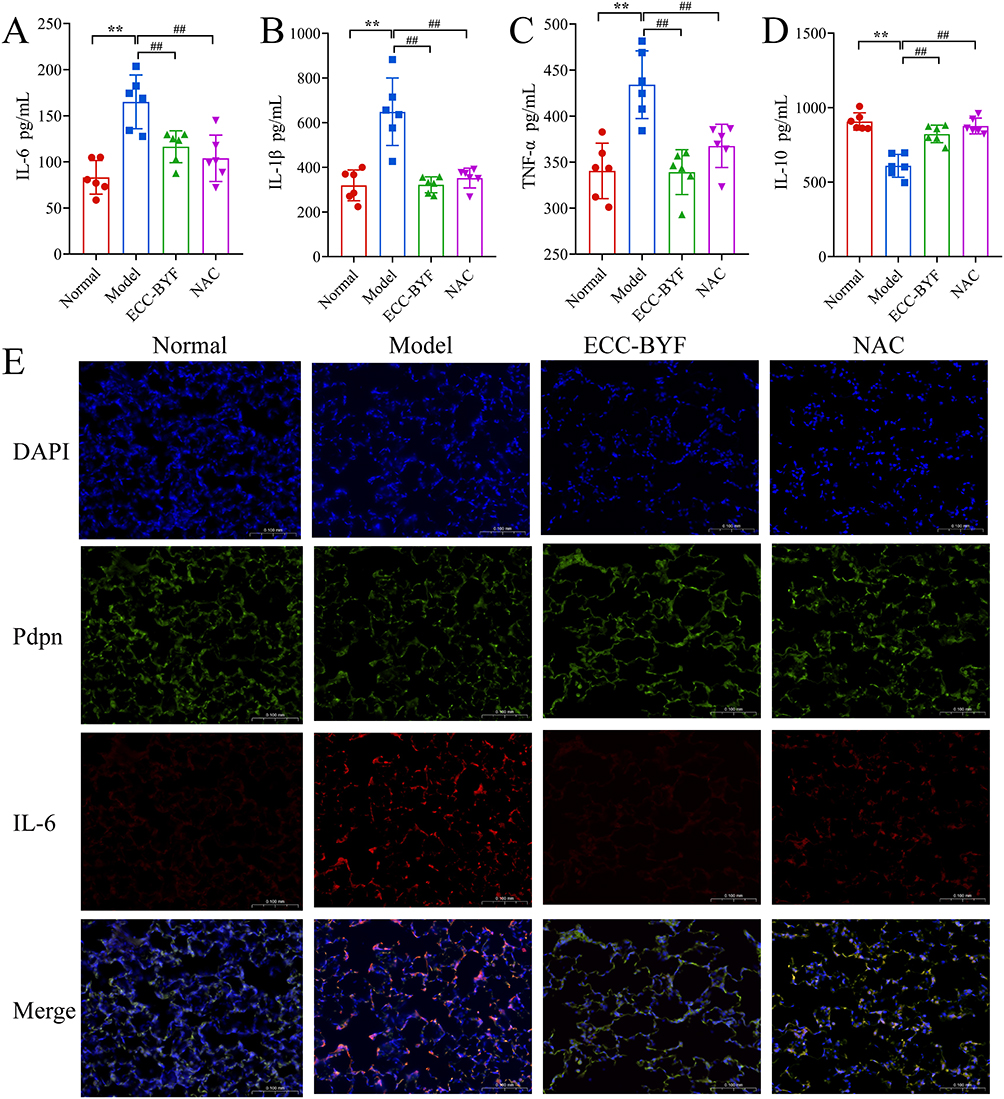 |
Figure 5 ECC-BYF attenuated the inflammatory response in rats with COPD. (A–D) IL-6, IL-1β, TNF-α, IL-10 level in BALF samples. (E) DAPI blue fluorescent label; PDPN green fluorescent label; IL-6 red fluorescent label; Merge Blue, green, and red merge tags (×200). **P < 0.01, versus the normal group. ##P < 0.01, versus the model group. |
ECC-BYF Protects TNF-α‑Induced AEB
To investigate the cellular mechanisms underlying the effects of ECC-BYF, we established a TNF-α-induced MLE-12 cell model. CCK-8 results indicated that concentrations of TNF-α less than 10 ng/mL showed no apparent impact on the viability of MLE-12 after 24 h of treatment (Supplementary Figure S4 A). To further determine the optimal concentration of TNF-α for the alveolar epithelial cell barrier injury model, qPCR assay results revealed that the 10 ng/mL TNF-α exhibited the pronounced effect on the expression of epithelial injury indicators and inflammatory factors (P<0.01) (Supplementary Figure S4 B and C). Thus, the optimal dose for inducing epithelial inflammation injury in the model was determined to be 10 ng/mL.
The CCK-8 assay showed that cell viability was maintained at ECC-BYF concentrations ranging from 0 to 70 µg/mL (Supplementary Figure S4 D). Subsequent experiments utilized ECC-BYF at concentrations of 17.5, 35, and 70 μg/mL. The qPCR results showed that IL-6 and TNF-α mRNA expression was significantly augmented in the TNF-α induced MLE-12. The levels of IL-6 and TNF-α genes in the ECC-BYF groups were significantly diminished (P<0.01) (Supplementary Figure S4 E and F). These results validated that ECC-BYF can alleviate the inflammatory damage to alveolar epithelial cells.
ZO-1 and Occludin expression decreased in the Model group and increased to different degrees in 17.5, 35 and 70 μg/mL ECC-BYF groups (Figure 6A–C). ZO-1 in Normal group showed green fluorescence, distributed continuously along the cell membrane, appearing oval intact, with high fluorescence intensity. TNF-α downregulated the positive expression of ZO-1 protein. The fluorescence of ZO-1 protein in 17.5, 35, 70 μg/mL ECC-BYF was elevated, while the ZO-1 in 70 μg/mL ECC-BYF was significantly restored to a nearly normal state. Occludin exhibited a similar relative expression pattern to ZO-1 across all groups (Figure 6D and E). SP-C fluorescence intensity was lower in the Model group compared to the Normal group. Following treatment with 35 and 70 μg/mL ECC-BYF, SP-C fluorescence intensity was higher than in the Model group, with the highest intensity observed in the 70 μg/mL group (Supplementary Figure S4 G).
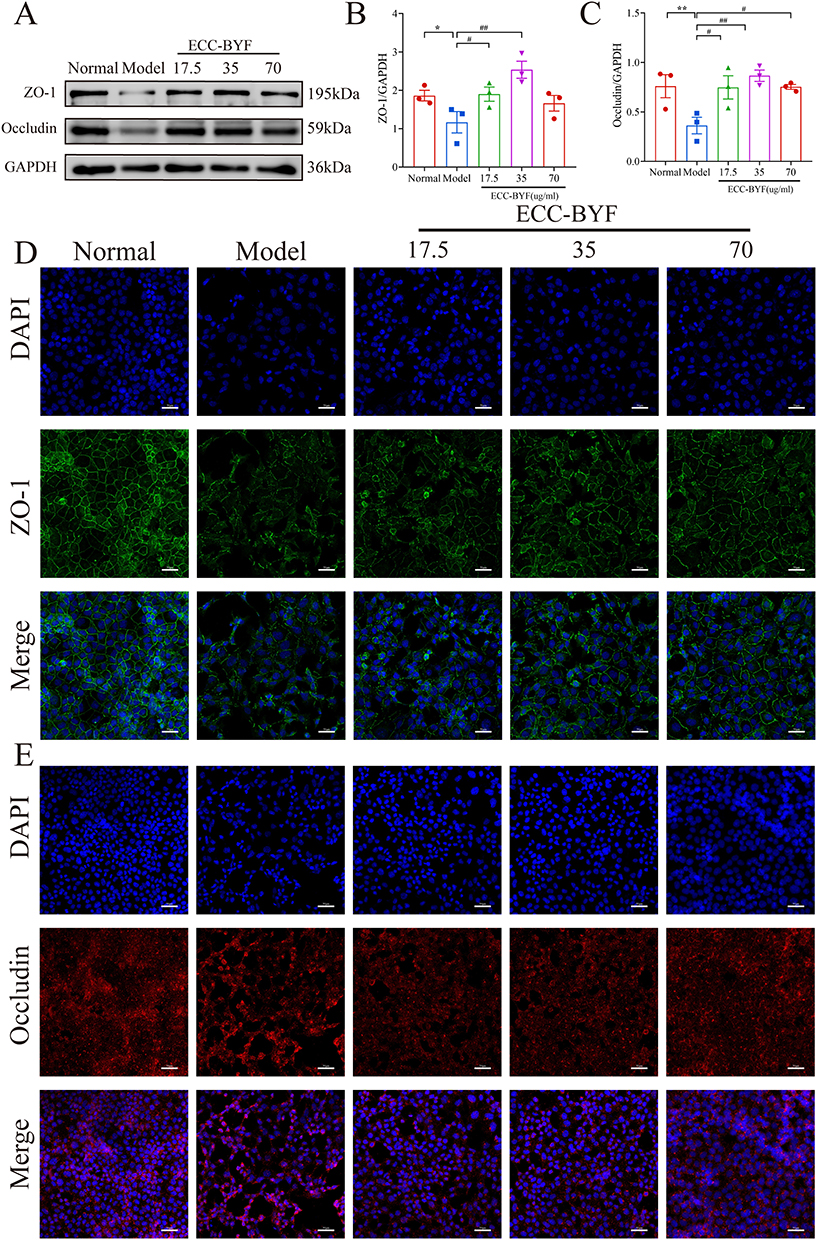 |
Figure 6 ECC-BYF improved the structure of AEB induced by TNF-α. (A–C) Western blotting results of the expression of ZO-1 and Occludin. (D and E) The expression of ZO-1 and Occludin was measured using IF. DAPI blue fluorescent label; ZO-1 green fluorescent label; Occludin red fluorescent label. **P < 0.01, *P < 0.05, versus the normal group. ##P < 0.01, #P < 0.05, versus the model group. |
Network Pharmacology Analysis of ECC-BYF in COPD
We identified the relevant targets of astragaloside, icariin, tangerine, paeonol, and ginsenoside from five public databases. After removing duplicate entries, we identified 295, 321, 121, 175, and 279 potential targets for astragaloside, icariin, tangerine, paeonol, and ginsenoside, respectively. Furthermore, we screened for COPD and inflammation-related targets across three public databases, resulting in 2785 unique COPD-related targets and 1802 unique inflammation-related targets after duplicates were removed. Based on the Venny, 247 targets overlapped between the ECC-BYF and COPD and inflammation-related targets (Figure 7A). These 247 common targets were submitted to STRING public database to map PPI (Figure 7B). The PPI network was further analyzed to identify the top 100 targets, including TNF, IL6, and ALB. The constructed PPI network contained 100 nodes and 3020 edges (Figure 7C), and these 100 targets were subsequently analyzed using the DAVID public database. The KEGG demonstrated that the pathways mainly centered on the processes of inflammatory response. Among them, MAPK signaling pathway was significantly enriched (Figure 7D). To further validate the binding potential of the five compounds towards p38 MAPK, molecular docking was performed for each component. The results indicated that all five monomers possessed favorable binding activities (Figure 7E–I).
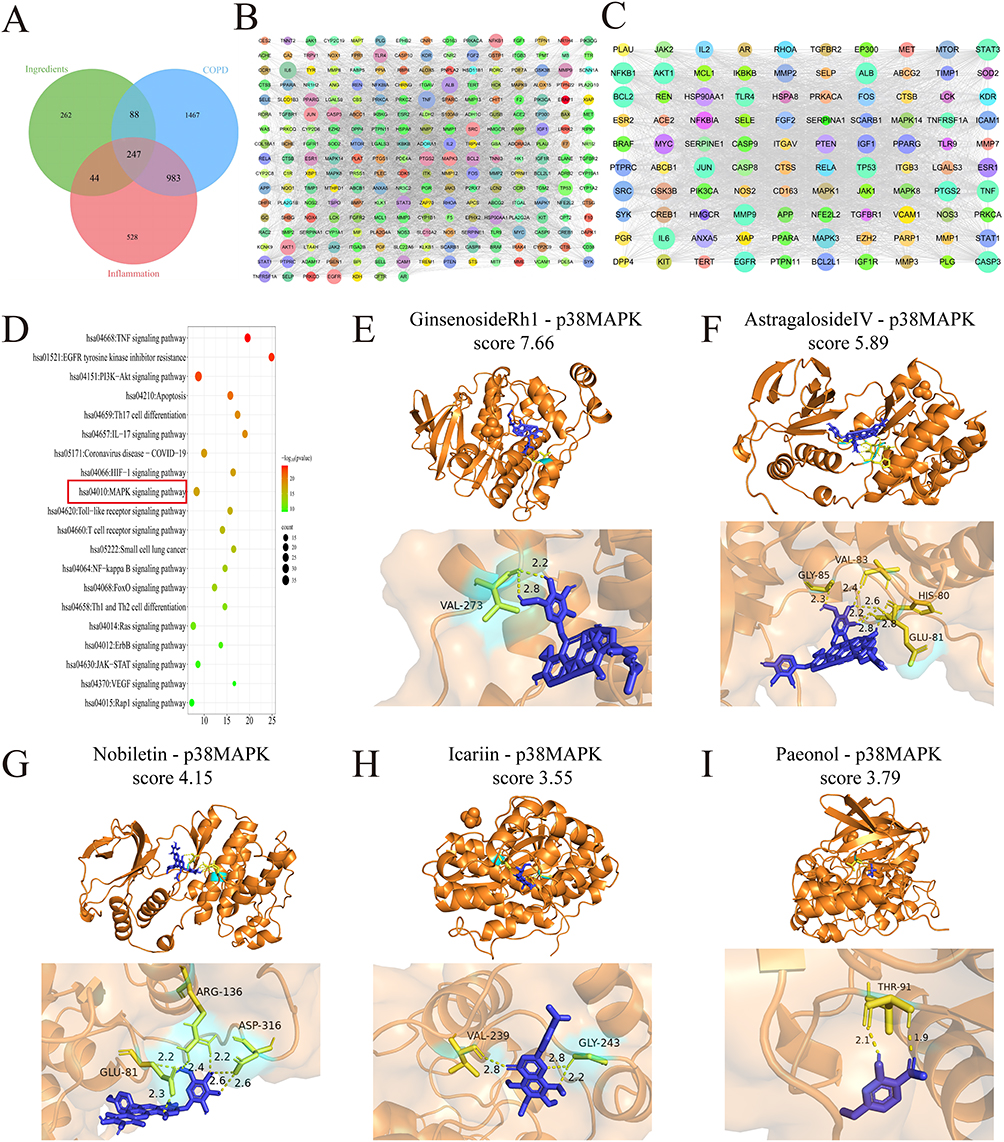 |
Figure 7 Effect of ECC-BYF on p38 MAPK of alveolar epithelial injury cell model (A) The COPD-related targets of ECC-BYF indicated by Venn diagram. (B) PPI network of ECC-BYF preventing COPD. (C) According to the scores of Dgree, 100 targets were selected. (D) KEGG pathway. The MAPK signaling pathway (red box) was identified as a key pathway. (E–I) Molecular docking of p38MAPK and ECC-BYF. |
The ECC-BYF Prevents AEB Dysfunction and Activates p38 MAPK
WB revealed that the expression of phosphorylated p38 MAPK (p-p38) in the Model group was statistically upregulated (P < 0.01). Critically, ECC-BYF (17.5–70 μg/mL) dose-dependently suppressed p-p38 expression, with maximal inhibition at 70 μg/mL (Figure 8A and B). Consistent with in vitro findings, ECC-BYF significantly reduced p-p38 levels in COPD rat lungs (P < 0.01) (Figure 8C and D). To assess the effect of p38 MAPK activation, cells were treated with the p38 agonist asiatic acid. The addition of asiatic acid alongside ECC-BYF resulted in higher p-p38 levels and increased IL-6 expression compared to cells treated with ECC-BYF alone (Figure 8E–G). Collectively, these data demonstrate that ECC-BYF suppresses p38 MAPK activation in TNF-α-stimulated epithelial cells and lung tissues, suggesting this pathway contributes to its barrier-protective effects.
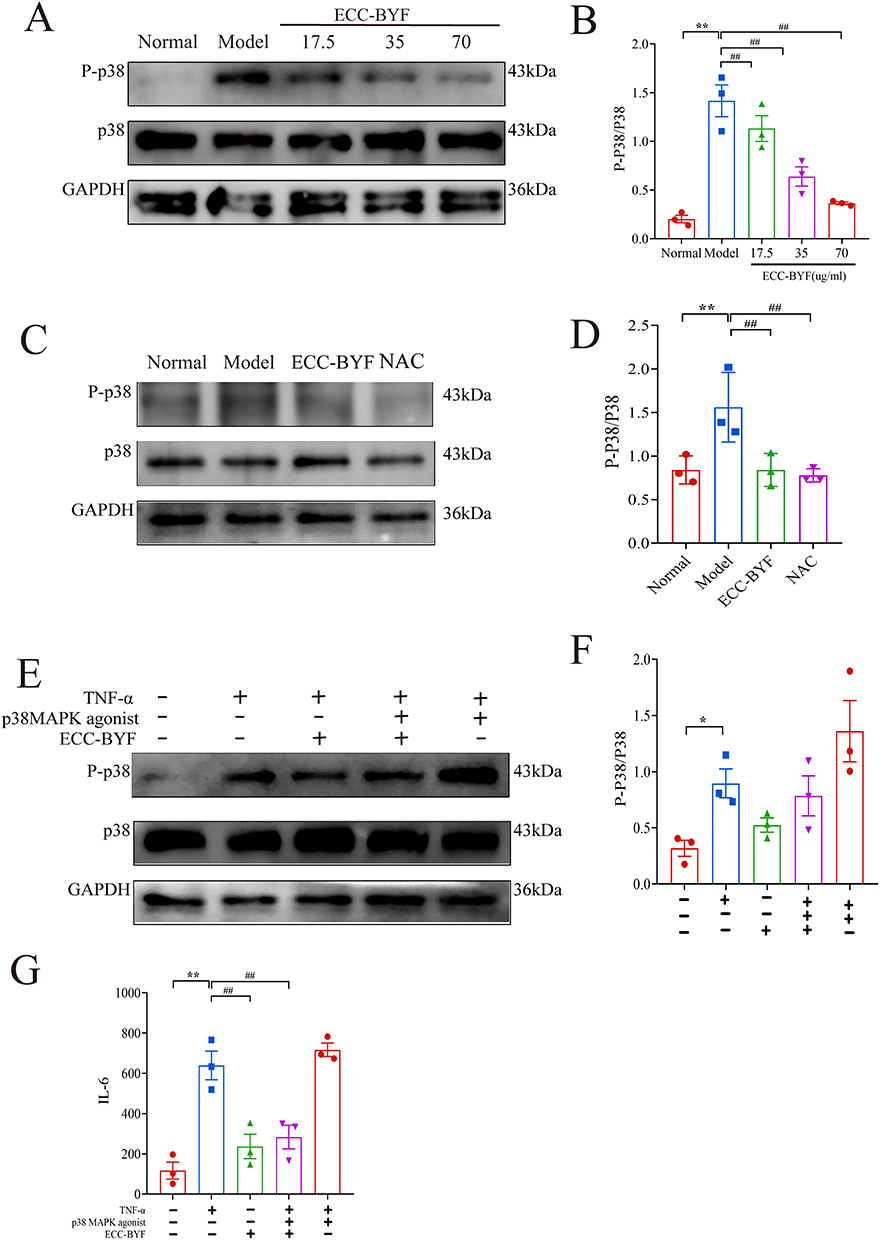 |
Figure 8 The ECC-BYF prevent barrier dysfunction and activate p38 MAPK (A and B) The expression of p-p38 MAPK in MLE-12 were determined by Western blotting. (C and D) The expression of p-p38 MAPK in COPD rats were determined by Western blotting. (E and F) The expression of p-p38 MAPK were determined in MLE-12 by Western blotting. (G) ECC-BYF in MLE-12 on the expression mRNA of IL-6. **P < 0.01, *P < 0.05, versus the normal group. ##P < 0.01, versus the model group. |
Discussions
In this study, we demonstrated that ECC-BYF significantly restored the structure and function of the AEB in COPD. We found that ECC-BYF mitigated pathological lung injury and attenuated the abnormal inflammatory response. Furthermore, our combined network analysis and experimental validation established that the cytoprotective efficacy of ECC-BYF is centrally mediated through the inhibition of the p38 MAPK signaling pathway. These findings reveal a novel mechanistic link between ECC-BYF-mediated p38 MAPK inhibition and the preservation of AEB function in COPD.
AT1 and AT2 collectively maintain the structural and functional integrity of the AEB, with disruption of this barrier constituting a pathological feature in COPD.34–36 Our analyses demonstrate that ECC-BYF directly counteracts this pathology by restoring AT1/AT2 cell morphology, reducing vacuolation in AT2 lamellar bodies, and enhancing junctional integrity. Concurrently, ECC-BYF upregulated critical tight junction proteins and surfactant components in vivo. This restoration mechanistically reverses the core drivers of barrier failure—compromised tight junction integrity and surfactant deficiency—which collectively increase alveolar surface tension, impair bacterial clearance, and destabilize alveolar architecture.37–42 Furthermore, our in vitro studies demonstrated that ECC-BYF effectively reversed TNF-α-induced barrier dysfunction. Specifically, ECC-BYF upregulated the expression of ZO-1, occludin, and SP-C, which successfully restored critical junctional proteins and surfactants. This highlights the protection of the AEB as a viable therapeutic strategy for COPD. Collectively, these findings show that ECC-BYF preserves alveolar ultrastructure and barrier competence, thereby mitigating epithelial remodeling and functional decline.
The significance of barrier repair extends beyond structural recovery, as AEB disruption directly fuels pulmonary inflammation. Inflammation is a primary pathogenic mechanism in COPD, with repeated stimulation leading to a continuous decline in lung function and airflow limitation, significantly affecting patient prognosis.43 When barrier integrity is compromised, alveolar epithelial cells aberrantly secrete inflammatory mediators, which induce immune cells that further release proteases and oxidants, exacerbating epithelial damage.44 TNF-α serves as a pivotal orchestrator in this process: it is markedly elevated in COPD patients’ airways and lung tissue,11 where it not only perpetuates inflammation but also directly impairs junctional integrity and surfactant function. In this context, ECC-BYF demonstrates dual efficacy: it significantly suppresses TNF-α-induced release of inflammatory factors in alveolar epithelial cells, while also reducing expression of IL-6, TNF-α, and IL-1β in COPD model rats. Collectively, by coordinately preserving alveolar ultrastructure and disrupting TNF-α-driven inflammatory cascades, ECC-BYF mitigates the intertwined pathologies of epithelial remodeling, functional decline, and chronic inflammation that define COPD.
Network pharmacology analysis showed that MAPK was an important signaling pathways of the ECC-BYF in the treatment of COPD. The MAPK pathway consists of a family of serine/threonine protein kinases, including signaling pathways such as p38 MAPK, which can modulate inflammatory mediators, thereby exacerbating the inflammatory cascade in the lung.45,46 In recent years, p38 MAPK has arisen as a novel molecular intervention point for COPD,47 numerous p38 MAPK inhibitors have exhibited efficacy in attenuating inflammatory injury in pulmonary disease, with its inhibitors demonstrating significant effects in the clinical management.48 Molecular docking results also suggested that the ECC-BYF could directly bind to p38 MAPK. The current data showed upregulation of p38 MAPK phosphorylation in MLE-12 cells induced by TNF-α. Notably, ECC-BYF was found to inhibit the increase in phosphorylated p38 MAPK. Furthermore, Asiatic acid (a p38 MAPK agonist)49 abolished the effect of ECC-BYF on AEB function. Similar to previous reports, our data confirm that p38 MAPK activation is a critical driver of inflammation and AEB dysfunction in COPD. These findings indicate that the protective effect of ECC-BYF on the AEB from inflammation-induced impairment may be attributable to the suppression of p38 MAPK signaling. While previous studies have established the general anti-inflammatory properties of ECC-BYF, the specific novelty of the present study lies in identifying the alveolar epithelial barrier as its primary structural target. To the best of our knowledge, this is the first demonstration that ECC-BYF preserves AEB integrity directly through the inhibition of p38 MAPK phosphorylation. This dual-validation approach—combining network analysis with in vivo and in vitro models—distinguishes our findings from prior research that assessed lung function in broader terms.
Despite the promising findings, this study has several limitations that should be considered. First, the protective effects of ECC‑BYF were demonstrated in a rat model of COPD and in vitro cellular assays, which do not fully recapitulate the complex pathogenesis of human disease. Future clinical trials are therefore required to validate the translational potential of these findings. Second, ECC‑BYF is a multi‑component herbal formulation, and the exact active monomer(s) responsible for p38 MAPK inhibition remain to be identified. Subsequent pharmacokinetic and pharmacodynamic studies will be needed to isolate and characterize the active constituents. Finally, while p38 MAPK was identified as a central mediator, COPD pathogenesis involves crosstalk among multiple signaling pathways; further studies exploring parallel networks will help to map the full mechanism of action.
Conclusions
This study elucidated the protective mechanism of ECC-BYF against AEB compromise in COPD. ECC-BYF mitigated pathological injury and inflammation, and preserved AEB integrity. Moreover, the inhibition of p38 MAPK phosphorylation mediated the protective effects of ECC-BYF in maintaining AEB function. In summary, the findings support the conclusion that ECC-BYF preserves AEB integrity in COPD via the specific targeting of p38 MAPK pathway. Future investigations are warranted to validate these findings in additional preclinical COPD models, to assess the long-term safety and efficacy of ECC-BYF, and to explore its translational potential in clinical settings.
Data Sharing Statement
Data for this study can be obtained from the corresponding author according to the rules.
Ethical Statement
The protocol of this study was approved by the Ethic Committee of Henan University of Traditional Chinese Medicine (Zhengzhou, Henan, China, No. DWLL202003261) and were performed in strict accordance with the Laboratory animal—Guideline for ethical review of animal welfare (GB/T 35892-2018, National Standard of the People’s Republic of China).
Acknowledgments
The authors gratefully acknowledge the Electron Microscope Science Henan University of Chinese Medicine for their technical assistance, and special thanks are extended to Professors Ning sun and Caili Zhang for their assistance.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research is funded by the National Natural Science Foundation of China (No.82405382), Program for Science & Technology Innovation Talents in Universities and Colleges of Henan Province (NO.242102311251), Special Scientific Research Project for Chinese Medicine in Henan Province’s “Double First-Class” Discipline Development Plan (NO. HSRP-DFCTCM-2023-7-19), and the National Key Research and Development Program of China (2023YFC3502600).
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management and prevention of chronic obstructive pulmonary disease (2026 report). Available from: https://www.goldcopd.org/2026-gold-report.
2. Safiri S, Carson-Chahhoud K, Noori M, et al. Burden of chronic obstructive pulmonary disease and its attributable risk factors in 204 countries and territories, 1990-2019: results from the global burden of disease study 2019. BMJ. 2022;378:e069679. doi:10.1136/bmj-2021-069679
3. Adeloye D, Song P, Zhu Y, et al. Global, regional, and national prevalence of, and risk factors for, chronic obstructive pulmonary disease (COPD) in 2019: a systematic review and modelling analysis. Lancet Respir Med. 2022;10(5):447–18. doi:10.1016/S2213-2600(21)00511-7
4. Moll M, Silverman EK. Precision approaches to chronic obstructive pulmonary disease management. Annu Rev Med. 2024;75(1):247–262. doi:10.1146/annurev-med-060622-101239
5. Kageyama T, Ito T, Tanaka S, Nakajima H. Physiological and immunological barriers in the lung. Semin Immunopathol. 2024;45(4–6):533–547. doi:10.1007/s00281-024-01003-y
6. Herminghaus A, Kozlov AV, Szabó A, et al. A barrier to defend - models of pulmonary barrier to study acute inflammatory diseases. Front Immunol. 2022;13:895100. doi:10.3389/fimmu.2022.895100
7. Suki B, Sato S, Parameswaran H, Szabari MV, Takahashi A, Bartolák-Suki E. Emphysema and mechanical stress-induced lung remodeling. Physiology. 2013;28(6):404–413. doi:10.1152/physiol.00041.2013
8. Tuder RM, Petrache I. Pathogenesis of chronic obstructive pulmonary disease. J Clin Invest. 2012;122(8):2749–2755. doi:10.1172/JCI60324
9. Watanabe N, Fujita Y, Nakayama J, et al. Anomalous epithelial variations and ectopic inflammatory response in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2022;67(6):708–719. doi:10.1165/rcmb.2021-0555OC
10. Dong -L-L, Liu Z-Y, Chen K-J, et al. The persistent inflammation in COPD: is autoimmunity the core mechanism? Eur Respir Rev. 2024;33:230137. doi:10.1183/16000617.0137-2023
11. Singh S, Verma SK, Kumar S, et al. Correlation of severity of chronic obstructive pulmonary disease with potential biomarkers. Immunol Lett. 2018;196:1–10. doi:10.1016/j.imlet.2018.01.004
12. Yu W, Ye T, Ding J, et al. miR-4456/CCL3/CCR5 pathway in the pathogenesis of tight junction impairment in chronic obstructive pulmonary disease. Front Pharmacol. 2021;12:551839. doi:10.3389/fphar.2021.551839
13. Lee P-H, Park S, Lee Y-G, Choi S-M, An M-H, Jang A-S. The impact of environmental pollutants on barrier dysfunction in respiratory disease. Allergy Asthma Immunol Res. 2021;13(6):850–862. doi:10.4168/aair.2021.13.6.850
14. Sim TY, Harith HH, Tham CL, et al. The protective effects of a synthetic geranyl acetophenone in a cellular model of TNF-α-induced pulmonary epithelial barrier dysfunction. Mol. 2018;23:1355. doi:10.3390/molecules23061355
15. Chan KH, Tsoi YYS, McCall M. The effectiveness of traditional Chinese medicine (TCM) as an adjunct treatment on stable COPD patients: a systematic review and meta-analysis. Evid Based Complement Alternat Med. 2021;2021:5550332. doi:10.1155/2021/5550332
16. Li S, Li J, Wang M, et al. Effects of comprehensive therapy based on traditional Chinese medicine patterns in stable chronic obstructive pulmonary disease: a four-center, open-label, randomized, controlled study. BMC Complement Altern Med. 2012;12:197. doi:10.1186/1472-6882-12-197
17. Tian Y, Li J, Li Y, et al. Effects of Bufei Yishen granules combined with acupoint sticking therapy on pulmonary surfactant proteins in chronic obstructive pulmonary disease rats. BioMed Res Int. 2016;2016:8786235. doi:10.1155/2016/8786235
18. Li J-S, Liu X-F, Dong H-R, et al. Effective-constituent compatibility-based analysis of Bufei Yishen formula, a traditional herbal compound as an effective treatment for chronic obstructive pulmonary disease. J Integr Med. 2020;18(4):351–362. doi:10.1016/j.joim.2020.04.004
19. Li J, Ma J, Tian Y, et al. Effective-component compatibility of Bufei Yishen formula II inhibits mucus hypersecretion of chronic obstructive pulmonary disease rats by regulating EGFR/PI3K/mTOR signaling. J Ethnopharmacol. 2020;257:112796. doi:10.1016/j.jep.2020.112796
20. Li J, Xie Y, Zhao P, et al. A Chinese herbal formula ameliorates COPD by inhibiting the inflammatory response via downregulation of p65, JNK, and p38. Phytomedicine Int J Phytother Phytopharm. 2021;83:153475. doi:10.1016/j.phymed.2021.153475
21. Xu K, Shao X, Lu R, et al. Effective-component compatibility of Bufei Yishen formula III protects lung air-blood barrier by regulating the oxidative stress: via the nuclear factor-E2-Related factor 2 pathway. Int J Chron Obstruct Pulmon Dis. 2025;20:2211–2226. doi:10.2147/COPD.S513071
22. Li Y, Li S-Y, Li J-S, et al. A rat model for stable chronic obstructive pulmonary disease induced by cigarette smoke inhalation and repetitive bacterial infection. Biol Pharm Bull. 2012;35:1752–1760. doi:10.1248/bpb.b12-00407
23. Kim S, Chen J, Cheng T, et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2021;49:D1388–95. doi:10.1093/nar/gkaa971
24. STITCH 5: augmenting protein-chemical interaction networks with tissue and affinity data - PubMed. 2024. Available from: https://pubmed.ncbi.nlm.nih.gov/26590256/.
25. SwissTargetPrediction: updated data and new features for efficient prediction of protein targets of small molecules - PubMed. 2024. Available from: https://pubmed.ncbi.nlm.nih.gov/31106366/.
26. Relating protein pharmacology by ligand chemistry - PubMed. 2024. Available from: https://pubmed.ncbi.nlm.nih.gov/17287757/.
27. PharmMapper 2017 update: a web server for potential drug target identification with a comprehensive target pharmacophore database - PubMed. 2024. Available from: https://pubmed.ncbi.nlm.nih.gov/28472422/.
28. Zhang W, Bojorquez-Gomez A, Velez DO, et al. A global transcriptional network connecting noncoding mutations to changes in tumor gene expression. Nat Genet. 2018;50(4):613–620. doi:10.1038/s41588-018-0091-2
29. Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46(D1):D1074–82. doi:10.1093/nar/gkx1037
30. The DisGeNET cytoscape app: exploring and visualizing disease genomics data - PubMed. 2024. Available from: https://pubmed.ncbi.nlm.nih.gov/34136095/.
31. UniProt: the universal protein knowledgebase in 2021 - PubMed. 2024. Available from: https://pubmed.ncbi.nlm.nih.gov/33237286/.
32. Szklarczyk D, Kirsch R, Koutrouli M, et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023;51(D1):D638–46. doi:10.1093/nar/gkac1000
33. Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi:10.1038/nprot.2008.211
34. Chan M, Liu Y. Function of epithelial stem cell in the repair of alveolar injury. Stem Cell Res Ther. 2022;13(1):170. doi:10.1186/s13287-022-02847-7
35. Yang J, Hernandez BJ, Martinez Alanis D, et al. The development and plasticity of alveolar type 1 cells. Dev Camb Engl. 2016;143:54–65. doi:10.1242/dev.130005
36. Feng J, Liu L, He Y, Wang M, Zhou D, Wang J. Novel insights into the pathogenesis of virus-induced ARDS: review on the central role of the epithelial-endothelial barrier. Expert Rev Clin Immunol. 2021;17(9):991–1001. doi:10.1080/1744666X.2021.1951233
37. Casals C, Campanero-Rhodes MA, García-Fojeda B, Solís D. The role of collectins and galectins in lung innate immune defense. Front Immunol. 2018;9:1998. doi:10.3389/fimmu.2018.01998
38. Martin A, Tempra C, Yu Y, et al. Exposure to aldehyde cherry e-liquid flavoring and its vaping byproduct disrupt pulmonary surfactant biophysical function. Environ Sci Technol. 2024;58(3):1495–1508. doi:10.1021/acs.est.3c07874
39. Guagliardo R, Pérez-Gil J, De Smedt S, Raemdonck K. Pulmonary surfactant and drug delivery: focusing on the role of surfactant proteins. J Control Release. 2018;291:116–126. doi:10.1016/j.jconrel.2018.10.012
40. Garavaglia ML, Bodega F, Porta C, Milzani A, Sironi C, Dalle-Donne I. Molecular impact of conventional and electronic cigarettes on pulmonary surfactant. Int J Mol Sci. 2023;24(14):11702. doi:10.3390/ijms241411702
41. Beutel O, Maraspini R, Pombo-García K, Martin-Lemaitre C, Honigmann A. Phase separation of zonula occludens proteins drives formation of tight junctions. Cell. 2019;179(4):923–936.e11. doi:10.1016/j.cell.2019.10.011
42. Li J, Qi Z, Li D, et al. Alveolar epithelial glycocalyx shedding aggravates the epithelial barrier and disrupts epithelial tight junctions in acute respiratory distress syndrome. Biomed Pharmacother Biomedecine Pharmacother. 2021;133:111026. doi:10.1016/j.biopha.2020.111026
43. Xu J, Zeng Q, Li S, Su Q, Fan H. Inflammation mechanism and research progress of COPD. Front Immunol. 2024;15:1404615. doi:10.3389/fimmu.2024.1404615
44. Liang X, Wang J, Guan R, et al. Limax extract ameliorates cigarette smoke-induced chronic obstructive pulmonary disease in mice. Int Immunopharmacol. 2018;54:210–220. doi:10.1016/j.intimp.2017.11.004
45. Saleem S. Targeting MAPK signaling: a promising approach for treating inflammatory lung disease. Pathol Res Pract. 2024;254:155122. doi:10.1016/j.prp.2024.155122
46. Anjum J, Mitra S, Das R, et al. A renewed concept on the MAPK signaling pathway in cancers: polyphenols as a choice of therapeutics. Pharmacol Res. 2022;184:106398. doi:10.1016/j.phrs.2022.106398
47. Holtzman MJ, Zhang Y, Wu K, Romero AG. Mitogen-activated protein kinase-guided drug discovery for post-viral and related types of lung disease. Eur Respir Rev. 2024;33(171):230220. doi:10.1183/16000617.0220-2023
48. Charron CE, Russell P, Ito K, et al. RV568, a narrow-spectrum kinase inhibitor with p38 MAPK-α and -γ selectivity, suppresses COPD inflammation. Eur Respir J. 2017;50(4):1700188. doi:10.1183/13993003.00188-2017
49. Zhang H, Chen Y, Li F, et al. Elamipretide alleviates pyroptosis in traumatically injured spinal cord by inhibiting cPLA2-induced lysosomal membrane permeabilization. J Neuroinflammation. 2023;20(1):6. doi:10.1186/s12974-023-02690-4
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.