")
Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Effect of particle size on oral absorption of carvedilol nanosuspensions: in vitro and in vivo evaluation
Authors Liu D, Pan H, He F, Wang X, Li J, Yang X, Pan W
Received 22 April 2015
Accepted for publication 22 July 2015
Published 13 October 2015 Volume 2015:10(1) Pages 6425—6434
DOI https://doi.org/10.2147/IJN.S87143
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Carlos Rinaldi
Dandan Liu,1 Hao Pan,2 Fengwei He,1 Xiaoyu Wang,3 Jinyu Li,3 Xinggang Yang,3 Weisan Pan3
1Department of Pharmaceutical Engineering, School of Biomedical and Chemical Engineering, Liaoning Institute of Science and Technology, Benxi, People’s Republic of China; 2School of Pharmacy, Queen’s University Belfast, Belfast, Northern Ireland, UK; 3Department of Pharmaceutics, School of Pharmacy, Shenyang Pharmaceutical University, Shenyang, People’s Republic of China
Abstract: The purpose of this work was to explore the particle size reduction effect of carvedilol on dissolution and absorption. Three suspensions containing different sized particles were prepared by antisolvent precipitation method or in combination with an ultrasonication process. The suspensions were characterized for particle size, surface morphology, and crystalline state. The crystalline form of carvedilol was changed into amorphous form after antisolvent precipitation. The dissolution rate of carvedilol was significantly accelerated by a reduction in particle size. The intestinal absorption of carvedilol nanosuspensions was greatly improved in comparison with microsuspensions and solution in the in situ single-pass perfusion experiment. The in vivo evaluation demonstrated that carvedilol nanosuspensions and microsuspensions exhibited markedly increased Cmax (2.09- and 1.48-fold) and AUC0–t (2.11- and 1.51-fold), and decreased Tmax (0.34- and 0.48-fold) in contrast with carvedilol coarse suspensions. Moreover, carvedilol nanosuspensions showed good biocompatibility with the rat gastric mucosa in in vivo gastrointestinal irritation test. The entire results implicated that the dissolution rate and the oral absorption of carvedilol were significantly affected by the particle size. Particle size reduction to form nanosized particles was found to be an efficient method for improving the oral bioavailability of carvedilol.
Keywords: carvedilol, nanosuspensions, intestinal absorption, oral bioavailability, gastrointestinal irritation
Introduction
Oral delivery is regarded as the most widely used and readily accepted form of drug administration. However, an increasing number of new active pharmaceutical ingredients (APIs) that emerge as a consequence of combinatorial chemistry are poorly water-soluble and lipophilic, which result in poor and erratic absorption in the gastrointestinal tract.1,2 Since the poorly soluble drugs must be dissolved in the gastrointestinal lumen contents before oral absorption, improving the solubility and dissolution rate of poorly soluble APIs is rapidly becoming the leading hurdle for formulation scientists.3,4 Consequently, a variety of strategies have been developed to overcome this obstacle for poorly water-soluble APIs, such as solid dispersions,5,6 nanoemulsion,7 salt formation,8 melt extrusion,9,10 liposomes,11,12 nanostructured lipid carriers,13,14 and micellar systems.15,16
During the past several years, the use of particle size reduction methods to form stable nanosized particles has been proven to be an effective strategy to address this thorny problem. Nanosuspensions can be defined as liquid dispersion consisting of solid drug nanoparticles, which are stabilized by polymer and/or surfactant.17 According to the Noyes–Whitney equation,18 the reduction of particle size leads to a significant increase in the dissolution rate of the API, which in turn can lead to substantial increases in bioavailability.19,20 The nanoparticles can also adhere to the gastrointestinal mucosa, prolonging the transit time of the drug and consequently bringing a better absorption.
Previous studies have reported the particle size effect of nanosuspensions on drug dissolution and bioavailability.21,22 However, the preparations used were stabilized by different types or ratios of stabilizers, which may not truly reflect the effects of particle size. Until now, research studies of particle size effect using carvedilol particles stabilized with the same kind and content of stabilizers have scarcely been reported.
Carvedilol is an antihypertensive agent with nonselective β-blocking and α1-blocking activities. It has been extensively used in the treatment of hypertension, myocardial infarction, and congestive heart failure.23 Carvedilol is sparingly soluble in water with a logP of 4.115. Its oral bioavailability in humans as well as in animals is quite low due to extensive first-pass metabolism in liver and the slow dissolution rate in the intestinal tract.24,25 This study aims at developing carvedilol nanosuspensions by antisolvent precipitation–ultrasonication method and investigating the influence of particle size reduction on dissolution rate as well as oral bioavailability of carvedilol.
Materials and methods
Materials
Carvedilol was purchased from Shandong Qilu Pharmaceutical Co., Ltd. (Jinan, Shandong, People’s Republic of China). The commercially available carvedilol tablet (Luode®, 10 mg/tablet) was purchased from Beijing Juneng Pharmaceutical Co. Ltd. (Daxing, Beijing, People’s Republic of China). α-tocopherol succinate (VES) was obtained from Jiangsu Xixin Vitamin Co., Ltd. (Suqian, Jiangsu, People’s Republic of China). Sodium dodecyl sulfate (SDS) was purchased from TianJin Bodi Chemical Holding Co., Ltd. (Hexi, Tianjin, People’s Republic of China). Propranolol was purchased from Changzhou Yabang Pharmaceutical Co., Ltd. (Changzhou, Jiangsu, People’s Republic of China). Acetone was purchased from Yuwang Chemical Reagent Co. (Shandong, People’s Republic of China). All other chemicals and reagents used were of analytical grade or better.
Animals
Wistar rats (male, weighted 200–250 g) and beagle dogs weighing 10–13 kg were provided by the Lab Animal Center of Shenyang Pharmaceutical University (Beijing, People’s Republic of China). All the experimental protocols were carried out in accordance with the guidelines for animal experimentation of Shenyang Pharmaceutical University (Shenyang, People’s Republic of China) and approved by the animal ethics committee of the institution.
Preparation of the formulations
The colloidal dispersions were prepared by an antisolvent precipitation method or in combination with an ultrasonication process.
Microsuspensions
Briefly, carvedilol (296 mg/mL) and VES (195 mg/mL) were completely dissolved in acetone to prepare the organic phase. Then, the organic phase was passed through a 0.45 μm filter (Shanghai Huan’ao Trading Company, Shanghai, People’s Republic of China) to remove the possible impurities. At 10°C, 1 mL of the organic solution was instantaneously injected at a stirring rate of 10,000 rpm for 1 minute using a B25 high shear homogenizer (BRT Equipment Technology Co. Ltd., Shanghai, People’s Republic of China) into 50 mL of water phase containing 0.47% (w/v) SDS. Then, the microsuspensions were kept under vacuum at room temperature for 24 hours to remove the acetone.
Nanosuspensions
The nanoparticles were obtained following the immediately ultrasonication of microsuspensions with an ultrasonic processor (20–25 kHz, Ningbo Scientz Biotechnology Co., Ltd., Zhejiang, People’s Republic of China) for 15 minutes (active every 3 seconds for a 3-second duration, 400 W) at 4°C–8°C. To remove the acetone, the nanosuspensions were then kept under vacuum at room temperature for 24 hours.
In order to improve the stability of carvedilol suspensions, the aqueous microsuspensions and nanosuspensions were freeze-dried with cryoprotectant (ie, maltose) at a concentration of 3% w/v. The suspensions were prefrozen in the refrigerator at −75°C for 12 hours and subsequently freeze-dried in a FD-1C-50 freeze-drier (Boyikang Laboratory Instruments Co., Ltd., Beijing, People’s Republic of China) at −25°C for 12 hours, followed by a secondary drying phase at 20°C for 4 hours.
Particle size analysis
Particle size was determined by laser diffraction using a Coulter LS 230 analyzer (Beckman-Coulter Co., Ltd., Brea, CA, USA) at room temperature. The particle size was volume-weighted and expressed as mean diameter. Each sample was measured three times. The Span value is a statistical parameter used to evaluate the particle size distribution and calculated using the following equation:
Span = (D90 − D10)/D50 | (1) |
where D10, D50, and D90 represent 10%, 50%, and 90%, respectively, of the cumulative particle size distribution at the given size.
Scanning electron microscopy
Scanning electron microscopy (SEM, SUPRA 35; Zeiss, Oberkochen, Germany) was employed to evaluate the morphology of carvedilol microsuspensions and nanosuspensions. Prior to observation, the samples were mounted onto metal stubs using a double-sided adhesive tape and sputtered with a thin layer of gold under vacuum.
Solid-state characterization
The solid state of carvedilol in different preparations was evaluated by differential scanning calorimetry (DSC) and powder X-ray diffractometry (PXRD). DSC analysis was performed using a DSC 1 calorimeter (Mettler Toledo, ZH, Schwerzenbach, Switzerland). The samples were scanned at a heating rate of 10°C/min over a temperature range of 50°C–250°C in an aluminum pan and sealed hermetically, using an empty pan as the reference. PXRD was performed using a powder X-ray diffractometer (D/max 2500, Rigaku, Tokyo, Japan) with a copper anode (Cu Kα radiation, K =0.15405 nm, 30 kV, 30 mA). The data were recorded over the 2θ range from 3° to 45° at a scan rate of 0.04°.
In vitro dissolution test
Dissolution tests of the preparations (all equivalent to 25 mg of carvedilol) were evaluated in a dissolution apparatus (ZRS-6G, Tianda Tianfa Technology Co., Ltd, Tianjin, People’s Republic of China) according to the USP paddle method. The samples were introduced directly into the vessels. The tests were performed at 37°C±0.5°C in 900 mL of dissolution medium with paddle speed set at 100 rpm. Five milliliters samples were drawn, and the same volume of fresh dissolution medium was added at 5, 15, 30, and 60 minutes, respectively. Then, the samples were filtered through a 0.1 μm syringe filter (Shanghai Huan’ao Trading Company, Shanghai, People’s Republic of China) immediately before dilution, when necessary. Drug content was determined using a ultraviolet spectrophotometer at 242 nm.
In situ single-pass intestinal perfusion in rats
Briefly, Wistar rats were fasted overnight prior to the experiments, but had free access to water. Each rat was anesthetized with an intraperitoneal injection of urethane (1 g/kg). The anesthetized rats were restrained in a supine position and kept under infrared lamps to maintain body temperature. Following midline incision of the abdomen, approximately 10 cm of the duodenum, jejunum, ileum, and colon were simultaneously exposed, and small incisions were made in both sides of each segments. The segment was rinsed with physiological saline at 37°C, moistened, and covered with absorbent cotton, and then the selected segments were cannulated with plastic tubes, ligated at both sides. The freshly prepared preparations were dispersed in Krebs Ringer’s solution (7.8 g NaCl, 0.35 g KCl, 0.37 g CaCl2, 1.37 g NaHCO3, 0.02 g MgCl2, 0.32 g NaH2PO4, and 1.4 g glucose in 1,000 mL purified water), maintained at 37°C, and then perfused to the selected segments at a flow rate of 0.2 mL/min. Thereafter, samples were collected every 15 minutes, and the experiments lasted for 105 minutes. Microtubes containing the ingoing perfusion solution and the perfusate samples were weighed and recorded. Meanwhile, the length and diameter of the selected segments were determined. Then, all the collected samples and the perfusate solution were diluted with methanol, centrifuged at 13000 rpm for 10 minutes, and stored at −20°C until analysis.
The concentration of carvedilol in these samples was analyzed by high-performance liquid chromatography (HPLC). The HPLC system was composed of a model LC-20A pump (Shimadzu, Kyoto, Japan) and a model SPDM20A programmable photodiode array detector (Shimadzu, Kyoto, Japan) with Diamonsil C18 column (200×4.6 mm, 5 μm) (Dikma, Lake Forest, CA, USA). The mobile phase consisted of methanol, NaH2PO4/Na2HPO4 (5.375:1 M ratio) at 33 mM total concentration in water and glacial acetic acid (62:38:0.3, v/v/v). The flow rate was 1.0 mL/minutes, and the effluent was detected at 242 nm.
The perfusion of the free carvedilol solution (carvedilol dissolved in ethanol and then diluted in Krebs Ringer’s solution, 10 μg/mL) was performed as control. The effective permeability (Peff) of drug in the intestinal segments was calculated according to the following equation:
|
|
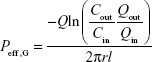
where Cin and Cout are the concentration of the perfusate solution and collected samples, respectively, Qin and Qout are their respective volumes, Q is the flow rate, r and l are the radius and length of the perfused intestinal segments, respectively.
Pharmacokinetic study
Preparation of the formulations
For the convenience of administration in in vivo studies, carvedilol microsuspension and nanosuspension capsules (containing 12.5 mg of carvedilol) were prepared, respectively. Briefly, freeze-dried carvedilol nanosuspensions and microsuspensions were passed through a 60-mesh screen, respectively, and precisely weighed using an electronic balance (Shanghai Minqiao Precise Science Instrument Co., Shanghai, People’s Republic of China). The powders were then filled into Coni-Snap® hard gelatin capsule shells (Size 1, CAPSUGEL, Suzhou, People’s Republic of China).
In vivo studies in beagle dogs
Six healthy male beagle dogs were divided into three groups comprising two animals in each. A randomized, three-period crossover single-dose study was conducted, and the washout period was 1 week. The dogs were fasted overnight with free access to water. Three types of carvedilol formulations at a dose of 50 mg/body weight were orally administrated to three groups of dogs, ie, carvedilol microsuspension capsules, nanosuspension capsules, and the commercial tablets (the reference preparation). Blood samples (3 mL) were collected before administration and at 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 14, and 24 hours after administration. All the blood samples were separated immediately by centrifugation at 5,800 rpm for 10 minutes and stored at −20°C until determination. The concentration of carvedilol in plasma was determined using a validated HPLC assay. The HPLC conditions for carvedilol analysis and plasma sample preparation method conformed to the former research studies.26
Pharmacokinetic data analysis
Pharmacokinetic data analysis was carried out using DAS® 2.0 software (Mathematical Pharmacology Professional Committee of China, Shanghai, People’s Republic of China). The various pharmacokinetic parameters that were analyzed included peak concentration (Cmax), the time to reach Cmax (Tmax), and the area under the concentration–time curve (AUC0–t). All results were expressed as mean ± SD values. Analysis of variance test was performed to demonstrate statistical differences using SPSS 12.0 software (SPSS, Chicago, IL, USA). P-value less than 0.05 was considered significant.
In vivo irritation test
Wistar rats were used to evaluate the oral tolerance to carvedilol nanosuspensions and the coarse suspensions (prepared by dispersing commercial carvedilol tablets in 0.5% sodium carboxymethylcellulose solution and sonicated for 10 minutes). The rats were randomly divided into three groups comprising three animals in each with free access to water and were fasted 12 hours before euthanasia. Carvedilol coarse suspensions and nanosuspensions, 10 mg/body weight, were orally administered to rats in two groups once a day for 7 days, respectively. The control group only received the same amount of saline. Two hours after the last administration, the rats were euthanized, and the tissue samples (5 mm of small intestine) were fixed with 4% formaldehyde, embedded in paraffin, and made into histological sections for histopathology assessments.
Results and discussion
Particle size and SEM analysis
From the preparation and formulation screening processes previously studied,26 the microsuspensions and nanosuspensions with mean particle size of 4,895 nm and 212 nm were prepared (Table 1), respectively. Following lyophilization, the microsuspensions and nanosuspensions could be gently redispersed without obvious change in particle size (Table 1). Carvedilol nanosuspensions showed narrow size distribution, whereas the microsuspensions were widely distributed (based on the Span value). These data were corresponding to the information gained from SEM analysis. Figure 1 presents the morphologies of raw carvedilol, microsuspensions and nanosuspensions obtained by drying at room temperature. These particle size data and SEM images indicated that particles in the two kind of suspensions were different not only in size, but also in shape, which would influence their dissolution behavior and bioavailability.27 Carvedilol coarse powders had an irregular shape, while the microsuspensions were globular cluster like, and the nanosuspensions exhibited a flaky shape.
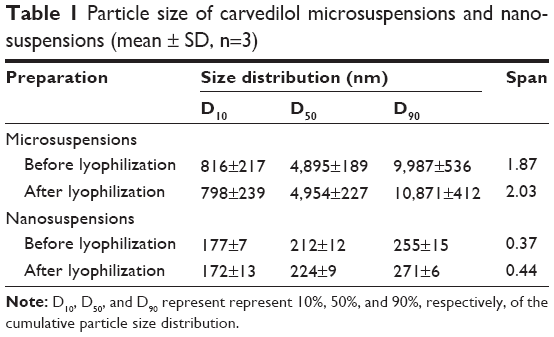 | Table 1 Particle size of carvedilol microsuspensions and nanosuspensions (mean ± SD, n=3) |
 | Figure 1 SEM micrographs. |
Solid-state characterization by DSC and PXRD
To study potential changes in crystalline state of different sized drug particles, DSC and PXRD measurements were performed. As shown in the DSC patterns (Figure 2), pure carvedilol shows a single sharp endothermic melting peak at 116.47°C, indicative of crystalline characteristics of carvedilol. For SDS and VES, sharp endothermic peaks were observed at 193.38°C and 76.91°C due to their crystalline state nature. In the physical mixture, the melting peaks of all components appeared but drifted slightly because of mixing. After incorporation into nanosuspensions and microsuspensions, the characteristic peak of carvedilol disappeared completely, irrespective of the particle size, implying the noncrystalline state of the drug. To further confirm the physical state of carvedilol, PXRD measurement was performed. Figure 3 shows the PXRD curves of pure carvedilol, VES, SDS powder, physical mixtures, and the freeze-dried carvedilol microsuspensions and nanosuspensions powder. As shown in Figure 3, pure carvedilol, VES, and SDS showed intense and characteristic diffraction peaks, respectively. However, no trace of the typical crystalline peaks of carvedilol was observed for both nanosuspensions and microsuspensions, indicating the noncrystalline state of carvedilol. The results generated from DSC and PXRD in combination illustrated that carvedilol freeze-dried nanosuspension and microsuspension powders were all in the amorphous form state. And the transition from the crystalline state to the amorphous state probably took place during the antisolvent precipitation step.
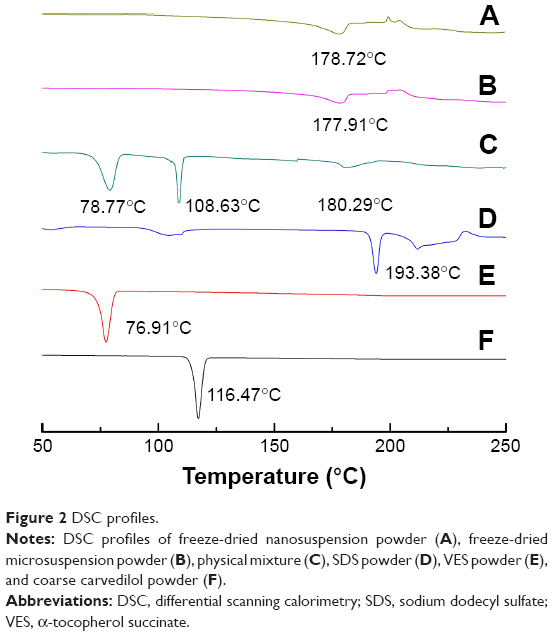 | Figure 2 DSC profiles. |
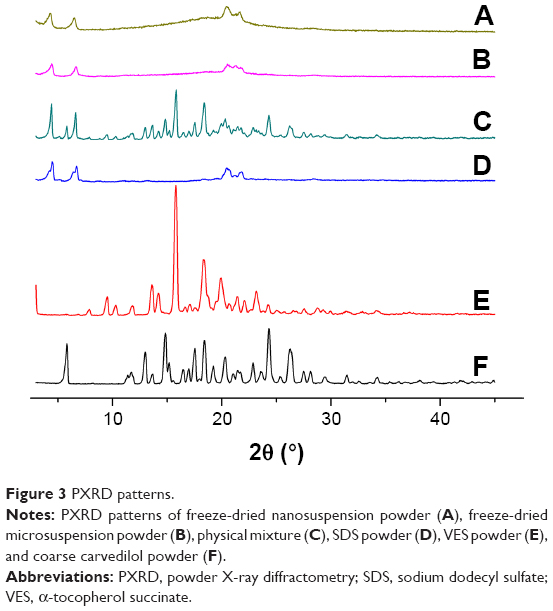 | Figure 3 PXRD patterns. |
In vitro dissolution studies
The dissolution profiles of bulk carvedilol, the commercial tablet, microsuspensions, and nanosuspensions are shown in Figure 4. Buffer medium with pH 1.0 was used to ensure sink condition during the dissolution rate testing. The dissolution rates of the four preparations were 20.63%, 38.16%, 78.90%, and 91.94% in 5 minutes, respectively (Figure 4A). Another medium with pH 6.8 was used to compare the drug dissolution rate at a more physiological pH. The dissolution rates of the four preparations were 8.13%, 23.89%, 69.03%, and 90.15% in 60 minutes, respectively (Figure 4B). The results indicated that the dissolution rate of carvedilol from the preparations followed the Noyes–Whitney equation:
|
|
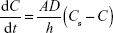
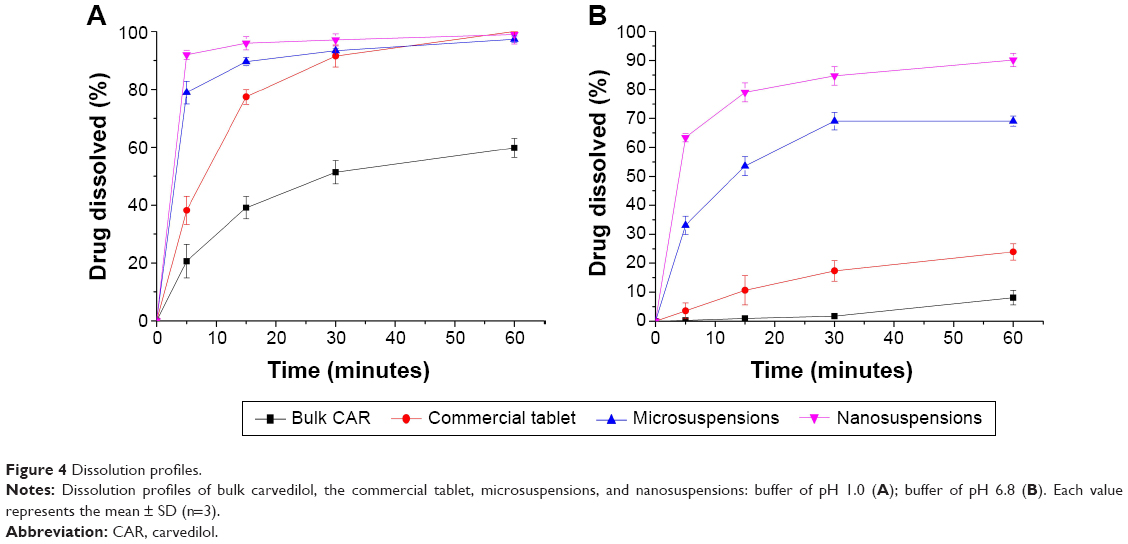 | Figure 4 Dissolution profiles. |
where dC/dt is the dissolution rate, D is the diffusion coefficient of the solute, A is the thickness of the dissolution boundary layer, S is the surface area, Cs is the saturation solubility, and C is the bulk concentration.28 It is widely accepted that the improvement in dissolution of drug can be ascribed to the particle size reduction, especially to the nanometer range, providing a significant increase in surface area available to dissolve. According to the Prandtl equation, the diffusion distance h decreases for very small particles.29 The reduction in diffusion distance h and the simultaneous increase in saturation solubility Cs can also lead to an increased dissolution velocity of the nanosuspensions. Consequently, dissolution studies of the four preparations clearly showed that particle size reduction is a practical way to improve the dissolution rate of carvedilol.
Absorption of carvedilol in rat intestine
The absorption of free carvedilol, microsuspensions, and nanosuspensions in the rat intestine was performed by in situ single-pass intestine perfusion method. The results indicated that the absorption of carvedilol was changed with the site of the intestine segments (Figure 5). Briefly, Peff of carvedilol solution was the highest in the jejunum, and decreased in the duodenum, ileum, and colon, which could attribute to the different physiological characteristics of selected intestinal segments. Compared with carvedilol solution, Peff of carvedilol nanosuspensions was remarkably increased by 1.74-, 1.42-, 1.89-, and 1.82-fold in the duodenum, jejunum, ileum, and colon, respectively. For microsuspensions, Peff increased by 1.38-, 1.12-, 1.42-, and 1.29-fold in the selected intestinal segments, respectively. The Peff of nanosuspensions was significantly higher than that of microsuspensions in all of the intestinal segments (P<0.05). It is well known that the diffusion across the unstirred water layer is a crucial barrier to the absorption of poorly water soluble drugs from the intestinal fluid.30 The reduction of carvedilol particle size could promote its diffusion across the unstirred water layer and access the brush border membrane of enterocytes. Moreover, the nanometer-sized particles of carvedilol nanosuspensions could enable the interfacial interaction with the mucin layer, thereby further enhancing the absorption of carvedilol from the intestinal lumen.21 Meanwhile, SDS used in the nanosuspensions and microsuspensions could improve the intestinal absorption of carvedilol from the intestinal epithelium by the paracellular pathway.31 The presence of SDS and VES in the suspensions could also contribute to the increased saturation solubility of carvedilol, and thus improve the absorption of the drug.
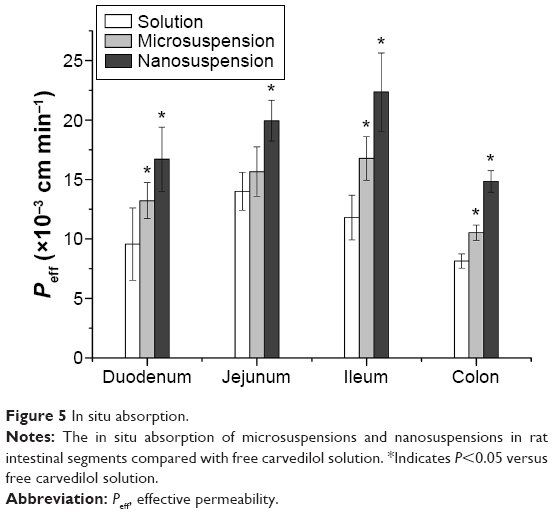 | Figure 5 In situ absorption. |
Pharmacokinetic study
A pharmacokinetic study was performed in beagle dogs to assess the absorption efficiency of carvedilol suspensions. Following oral administration, there were significant differences in pharmacokinetic profiles among the commercial tablets, the microsuspension and nanosuspension capsules. The plasma concentration–time profiles of carvedilol are presented in Figure 6, and the pharmacokinetic parameters are displayed in Table 2. As expected, significant increase in absorption was observed with the nano- and microsuspensions. In contrast with carvedilol coarse suspensions (the commercial tablets), the nanosuspensions and microsuspensions had higher Cmax (2.09- and 1.48-fold), AUC0–t (2.11- and 1.51-fold), and shorter Tmax (0.34- and 0.48-fold) (all P<0.05). Furthermore, enhanced absorption was observed with the nanosuspensions as compared with the microsuspensions. These findings were in accordance with the results from the in vitro dissolution and the in situ single-pass perfusion tests, indicating that the differences in carvedilol absorption are primarily attributed to the dissolution and intestinal absorption behavior of carvedilol with different particle sizes. Meanwhile, the direct uptake of the nanoparticles by mechanisms involving M-cells in Peyer’s patches of the gastrointestinal lymphoid tissue, thus avoid the first-pass metabolism effect, which is primarily responsible for the low bioavailability of carvedilol. Furthermore, the rapid absorption could attribute to the increased aqueous solubility and the size effect of carvedilol.
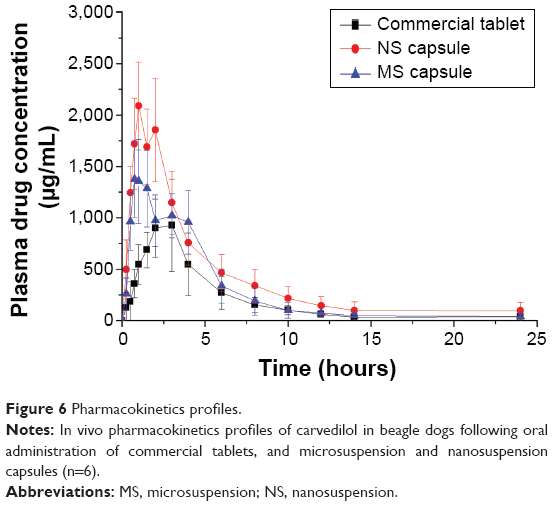 | Figure 6 Pharmacokinetics profiles. |
Although the results (Figure 6 and Table 2) showed that the Cmax and AUC0–t were increased with the reduction of particle size, the increase in these parameters can also give rise to pronounced fluctuations in plasma concentration and the more intensified adverse effects such as arrhythmia, hypotension, and so on. To address this issue, we have developed a novel controlled delivery system of carvedilol nanosuspensions.32 The in vivo test in beagle dogs demonstrated that the novel system could significantly improve the bioavailability of carvedilol and that the plasma concentrations were more stable than that of the marketed tablets. It was a promising strategy in improving the oral bioavailability, minimizing the frequency of drug administration, and lowering the average peak plasma concentration of poorly soluble drugs.
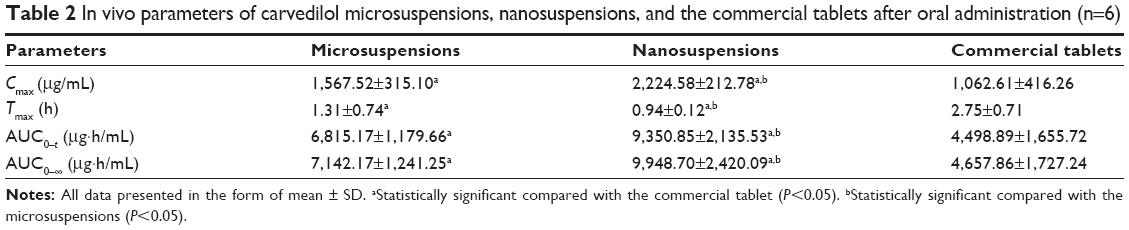 | Table 2 In vivo parameters of carvedilol microsuspensions, nanosuspensions, and the commercial tablets after oral administration (n=6) |
In vivo gastrointestinal irritation test
The long-term gastrointestinal irritation of carvedilol nanosuspensions was further studied using Wistar rats to exam the irritation effect after oral administration. Figure 7 shows the histopathology of the rat gastric mucosa treated with various formulations to observe their effect on tissue integrity and cell structure. Pathology of the rat gastric mucosa demonstrated that there was hardly any evidence of hemorrhagic mucosal erosions and disruption of intestinal villi. The results indicated that there was a good biocompatibility between the preparations and the rat gastric mucosa.
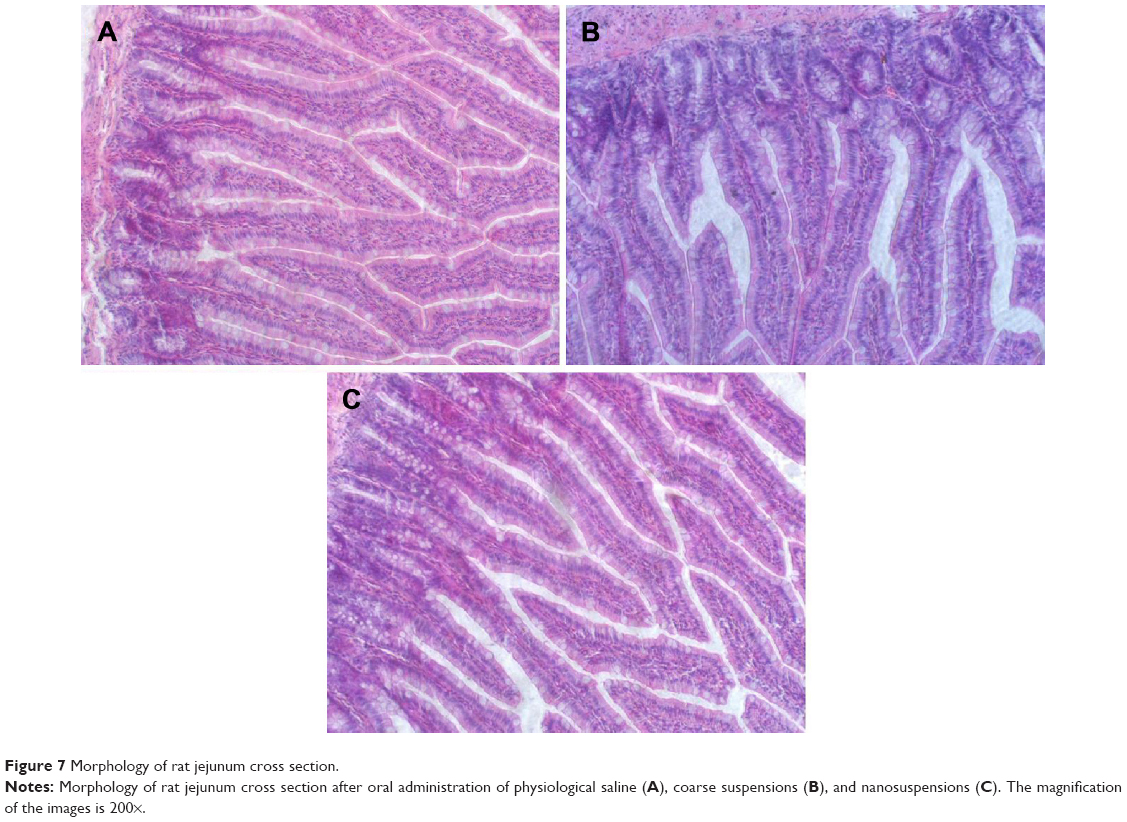 | Figure 7 Morphology of rat jejunum cross section. |
Conclusion
In this study, carvedilol microsuspensions and nanosuspensions were prepared by antisolvent precipitation method or in combination with an ultrasonication process. The solid form of carvedilol altered from the crystalline state to the amorphous state after incorporation into nanosuspensions or microsuspensions. The dissolution and oral absorption properties of the carvedilol suspensions were obviously particle size-dependent and were dramatically enhanced when the particle size was reduced to the nanometer range. Peff of nanosuspensions in all of the intestinal segments was greatly improved in the in situ single-pass perfusion experiment compared with carvedilol microsuspensions and solution. The in vivo study demonstrated that the nanosuspensions and microsuspensions had higher Cmax (2.09- and 1.48-fold), AUC0–t (2.11- and 1.51-fold), and shorter Tmax (0.34- and 0.48-fold) in contrast to carvedilol coarse suspensions. Furthermore, carvedilol nanosuspensions showed good biocompatibility with the rat gastric mucosa in in vivo gastrointestinal irritation test. The overall results indicated that particle size reduction to form nanosized particles was found to be an efficient method for improving the oral bioavailability of carvedilol.
Acknowledgments
This work was supported by the Foundation of Liaoning Educational Committee (number L2014504), by the Doctoral Scientific Research Foundation of Liaoning Institute of Science and Technology (number 1406B04), and by the State Key Laboratory of Long-acting and Targeting Drug Delivery System.
Disclosure
The authors report no conflicts of interest in this work.
References
Kawabata Y, Wada K, Nakatani M, Yamada S, Onoue S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: basic approaches and practical applications. Int J Pharm. 2011;420(1):1–10. | ||
Chen H, Khemtong C, Yang X, Chang X, Gao J. Nanonization strategies for poorly water-soluble drugs. Drug Discov Today. 2011;16(7):354–360. | ||
Lipinski CA. Poor aqueous solubility-an industry wide problem in ADME screening. Am Pharm Rev. 2002;5:82–85. | ||
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2012;64:4–17. | ||
Paudel A, Worku ZA, Meeus J, Guns S, Van den Mooter G. Manufacturing of solid dispersions of poorly water soluble drugs by spray drying: formulation and process considerations. Int J Pharm. 2013;453(1):253–284. | ||
Yan Y-D, Sung JH, Kim KK, et al. Novel valsartan-loaded solid dispersion with enhanced bioavailability and no crystalline changes. Int J Pharm. 2012;422(1):202–210. | ||
Shakeel F, Faisal MS. Nanoemulsion: a promising tool for solubility and dissolution enhancement of celecoxib. Pharm Dev Technol. 2010;15(1):53–56. | ||
Tao T, Zhao Y, Wu J, Zhou B. Preparation and evaluation of itraconazole dihydrochloride for the solubility and dissolution rate enhancement. Int J Pharm. 2009;367(1):109–114. | ||
Breitenbach J. Melt extrusion: from process to drug delivery technology. Eur J Pharm Biopharm. 2002;54(2):107–117. | ||
Lakshman JP, Cao Y, Kowalski J, Serajuddin AT. Application of melt extrusion in the development of a physically and chemically stable high-energy amorphous solid dispersion of a poorly water-soluble drug. Mol Pharm. 2008;5(6):994–1002. | ||
Fenske DB, Chonn A, Cullis PR. Liposomal nanomedicines: an emerging field. Toxicol Pathol. 2008;36(1):21–29. | ||
Mohammed A, Weston N, Coombes A, Fitzgerald M, Perrie Y. Liposome formulation of poorly water soluble drugs: optimisation of drug loading and ESEM analysis of stability. Int J Pharm. 2004;285(1):23–34. | ||
Zhuang CY, Li N, Wang M, et al. Preparation and characterization of vinpocetine loaded nanostructured lipid carriers (NLC) for improved oral bioavailability. Int J Pharm. 2010;394(1–2):179–185. | ||
Lin C, Chen F, Ye T, et al. A novel oral delivery system consisting in “drug-in cyclodextrin-in nanostructured lipid carriers” for poorly water-soluble drug: vinpocetine. Int J Pharm. 2014;465(1–2):90–96. | ||
Lukyanov AN, Torchilin VP. Micelles from lipid derivatives of water-soluble polymers as delivery systems for poorly soluble drugs. Adv Drug Deliv Rev. 2004;56(9):1273–1289. | ||
Lu Y, Park K. Polymeric micelles and alternative nanonized delivery vehicles for poorly soluble drugs. Int J Pharm. 2013;453(1):198–214. | ||
Patravale VB, Date AA, Kulkarni RM. Nanosuspensions: a promising drug delivery strategy. J Pharm Pharmacol. 2004;56(7):827–840. | ||
Noyes AA, Whitney WR. The rate of solution of solid substances in their own solutions. J Am Chem Soc. 1897;19(12):930–934. | ||
Scholz A, Abrahamsson B, Diebold SM, et al. Influence of hydrodynamics and particle size on the absorption of felodipine in labradors. Pharm Res. 2002;19(1):42–46. | ||
Hintz RJ, Johnson KC. The effect of particle size distribution on dissolution rate and oral absorption. Int J Pharm. 1989;51(1):9–17. | ||
Xia D, Cui F, Piao H, et al. Effect of crystal size on the in vitro dissolution and oral absorption of nitrendipine in rats. Pharm Res. 2010;27(9):1965–1976. | ||
Sun W, Mao S, Shi Y, Li LC, Fang L. Nanonization of itraconazole by high pressure homogenization: Stabilizer optimization and effect of particle size on oral absorption. J Pharm Sci. 2011;100(8):3365–3373. | ||
Wen H, Jiang H, Lu Z, et al. Carvedilol ameliorates sympathetic nerve sprouting and electrical remodeling after myocardial infarction in rats. Biomed Pharmacother. 2010;64(7):446–450. | ||
Morgan T. Clinical pharmacokinetics and pharmacodynamics of carvedilol. Clin Pharmacokinet. 1994;26(5):335–346. | ||
Hokama N, Hobara N, Kameya H, Ohshiro S, Sakanashi M. Rapid and simple micro-determination of carvedilol in rat plasma by high-performance liquid chromatography. J Chromatogr B Biomed Sci Appl. 1999;732(1):233–238. | ||
Liu D, Xu H, Tian B, et al. Fabrication of carvedilol nanosuspensions through the anti-solvent precipitation-ultrasonication method for the improvement of dissolution rate and oral bioavailability. AAPS Pharm Sci Tech. 2012;13(1):295–304. | ||
Mosharraf M, Nyström C. The effect of particle size and shape on the surface specific dissolution rate of microsized practically insoluble drugs. Int J Pharm. 1995;122(1):35–47. | ||
Junghanns J-UA, Müller RH. Nanocrystal technology, drug delivery and clinical applications. Int J Nanomed. 2008;3(3):295. | ||
Müller R, Jacobs C, Kayser O. Nanosuspensions as particulate drug formulations in therapy: rationale for development and what we can expect for the future. Adv Drug Deliv Rev. 2001;47(1):3–19. | ||
Porter CJ, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6(3):231–248. | ||
Anderberg EK, Artursson P. Epithelial transport of drugs in cell culture. VIII: effects of sodium dodecyl sulfate on cell membrane and tight junction permeability in human intestinal epithelial (Caco-2) cells. J Pharm Sci. 1993;82(4):392–398. | ||
Liu D, Yu S, Zhu Z, et al. Controlled delivery of carvedilol nanosuspension from osmotic pump capsule: in vitro and in vivo evaluation. Int J Pharm. 2014;475(1–2):496–503. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.