Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Effect of hydroxyapatite-containing microspheres embedded into three-dimensional magnesium phosphate scaffolds on the controlled release of lysozyme and in vitro biodegradation
Received 21 May 2014
Accepted for publication 28 June 2014
Published 1 September 2014 Volume 2014:9(1) Pages 4177—4189
DOI https://doi.org/10.2147/IJN.S68143
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Jongman Lee, Hui-suk Yun
Powder and Ceramics Division, Korea Institute of Materials Science, Changwon, Republic of Korea
Abstract: The functionality of porous three-dimensional (3D) magnesium phosphate (MgP) scaffold was investigated for the development of a novel protein delivery system and biomimetic bone tissue engineering scaffold. This enhancement can be achieved by incorporation of hydroxyapatite (HA)-containing polymeric microspheres (MSs) into a bulk MgP matrix, and a paste-extruding deposition (PED) system. In this work, the amount of MS and HA was precisely controlled when manufacturing MS-embedded MgP (MS/MgP) composite scaffolds. The main influence was researched in terms of in vitro lysozyme-release, in vitro biodegradation, mechanical properties, and in vitro calcification. The controlled release of lysozyme was indicated, while showing graded release patterns according to HA content. The composite scaffolds degraded gradually with MS content and degradation time. Due to the effect of HA inclusion, the higher HA-containing MS/MgP scaffolds could, not only delay the biodegradation process but also, compensate for the possible loss of mechanical properties. In this regard, it is reasonable to confirm the inverse relationship between biodegradation and corresponding compressive properties. In order to encourage bioactivity and osteoconductivity, the MS/MgP composite scaffolds were subjected to simulated body fluid treatment. Calcium deposition was, in turn, improved with increasing MS and HA content over time. This quantitative result was also proved using morphological and elemental analysis. In summary, a significant transformation of a monolithic MgP scaffold was directed toward a multifunctional bone tissue engineering scaffold equipped with controlled protein delivery, biodegradability, and bioactivity.
Keywords: protein delivery, bone tissue engineering
Introduction
The usefulness of microspheres (MSs) has been extended to combination with a continuous bulk matrix: solid polymer, hydrogel, and calcium phosphate cement (CPC).1 To this end, they can affect physicochemical and biological characteristics, in terms of porosity,2 mechanical properties,2 biodegradation,3 the release kinetics of bioactive agents,4 and bioactivity.3 One of the greatest advantages of introducing MS into a bulk matrix is to allow final composite scaffolds controlled delivery of biomolecules in a spatiotemporal manner. This is mainly because the direct incorporation of bioactive agents into bulk scaffolds might be vulnerable to denaturation when exposed to harsh preparation conditions (organic solvent, acidic byproduct, hydrophobicity, and so on).5
First, a composite system of MS-embedding solid polymers was developed for the sustained release of biomolecules and promoted tissue regeneration compared with MS-free scaffolds.3,6 Porous nano-hydroxyapatite (HA)/collagen/poly(L-lactic acid) (PLLA) composite scaffolds containing chitosan MSs were prepared using a thermally induced phase separation method.3 The compressive properties increased as the chitosan MS content increased, but the porosity decreased. The in vitro biodegradation rate increased with the enhancement of chitosan MSs, which in turn led to temporally controlled release of synthetic peptide. Second, a composite system of MS-embedding hydrogel was devised to serve as a controlled drug delivery carrier,4 reinforcement component,7 and cell delivery vehicle.8 The biodegradable gelatin MS/hydrogel composites were prepared to accomplish a simultaneous delivery of transforming growth factor-β1 (TGF-β1) and chondrocytes, respectively, for cartilage tissue engineering applications.4 As a result, a much slower release of TGF-β1 to the gelatin MS-encapsulating hydrogel composites was seen over the course of 28 days. Injectable alginate hydrogel composites combined with β-tricalcium phosphate (β-TCP) MSs were invented to overcome the lack of initial mechanical strength of the hydrogel constructs for bone tissue engineering.7 Third, a composite system of MS-embedding CPC was researched extensively to solve the major drawbacks of CPC: slow degradation rate attributed to a lack of macroporosity and poor drug-release capacity.1 In this regard, many types of biodegradable polymeric MSs were homogeneously incorporated into bone cements, not only to facilitate in vitro/in vivo biodegradation and the resultant formation of well-interconnected macroporous structures, but also, to obtain the controlled release of bioactive agents for a prolonged period of time, without an initial drug burst.9–11
In our previous studies, we developed a novel room-temperature process of three-dimensional (3D) magnesium phosphate (MgP) scaffold fabrication, using a paste-extruding deposition (PED) system.12 This technique enabled us to directly blend lysozyme, as a model bioactive substance, into MgP powder, to introduce homogeneous distribution in the scaffold. This was completely ascribed to a simple cementation process at room temperature, instead of the typical sintering process at high temperature. Both the drug-loading efficiency and in vitro release performance were, in turn, substantially enhanced compared with physical adsorption using lysozyme-containing solution. Nevertheless, we wished to develop a more advanced delivery system with a high efficacy. We recently investigated the manufacture of HA-containing polymeric MSs as a novel protein delivery carrier.13 High protein-loading capacity and controlled release kinetics were achieved by strong electrostatic interactions between nanosized HA and the lysozyme protein. For this reason, we combined the MgP scaffold with HA-containing polymeric MSs to produce the 3D bone tissue engineering composite scaffold.
In the present work, our aim was to develop MS-embedding MgP (MS/MgP) composite scaffolds that could contribute to in vitro lysozyme-release characteristics, in vitro biodegradation/corresponding mechanical properties, and in vitro calcification, in contrast to the bare MgP scaffold. For this purpose, MS/MgP composite scaffolds were prepared into two groups: (1) controlled MS content in MS/MgP scaffolds, ranging from 0% to 20% (w/w), and (2) controlled HA content in MSs, ranging from 0% to 55%. A synergistic combination of MgP scaffold and HA-containing polymeric MSs will be useful to create an efficient protein delivery carrier and bioactive physical platform for bone tissue engineering.
Materials and methods
Preparation of MSs and MgP powder
The development of gelatin/chitosan MSs, consisting of nanosized HA (Sigma-Aldrich Corp., St Louis, MO, USA), was reported in our previous publication.13 Briefly, 2.5% gelatin type B/chitosan with a weight ratio of 2 to 1 (Sigma-Aldrich Corp.) was dissolved in 50 mL of 0.1 M acetic acid (Sigma-Aldrich Corp.). Different amounts of HA (0%, 1.5%, and 3.0% [w/v]) were added to this solution and ultrasonicated for 5 minutes, for homogeneous dispersion of the HA. MSs with no HA were made for comparison following the same protocol. In this work, the theoretical percentage of HA in the total solid content was 38 wt% for a mixture containing 1.5% (w/v) HA and was 55 wt% for 3.0% (w/v) HA. For this reason, the final composite MSs with different HA content (0%, 38%, and 55%) were denoted as MS (0% HA), MS (38% HA), and MS (55% HA), respectively. The mixture was added dropwise into 5% cellulose acetate butyrate (Eastman Chemical Co., Kingsport, TN, USA) dissolved in 200 mL of n-butyl acetate (Samchun Pure Chemical Co., Ltd., Seoul, Republic of Korea) while stirring at 200 rpm for 30 minutes. The gelatin/chitosan MSs were subsequently cross-linked by adding glutaraldehyde (Merck & Co., Inc., Whitehouse Station, NJ, USA) to the emulsion at a final concentration of 0.25%, and stirred overnight. The composite MSs were then washed with acetone, rinsed in deionized (DI) water, and then collected using sieves with 150 μm and 250 μm pores. The residual glutaraldehyde from the cross-linking process was neutralized by placing the MSs in 2.0% sodium bisulfide for 1 day and then washing with DI water. The washed MSs were lyophilized for 1 day and stored in a desiccator until further use.
MgP powder (farringtonite [Mg3(PO4)2]) was prepared following the methods described in our previous publication.12 Briefly, Mg(OH)2 (Junsei Chemical Co., Ltd., Tokyo, Japan) and H3PO4 (DC Chemical Co., Ltd., Seoul, Korea) were reacted with a molar ratio of 3 M to 2 M. Then, 2 M of H3PO4 was added to a 3 M of Mg(OH)2 suspension in a dropwise manner. The solution was mixed uniformly for 10 hours and left for 24 hours to age. The precipitate was filtered and dried at 80°C for 2 days and heat-treated at 850°C for 6 hours. The final product was ground in a planetary ball mill for 2 hours at a speed of 250 rpm.
Preparation of MS/MgP composite scaffold
In this work, the preparation of MS/MgP composite scaffolds could be categorized into two groups: (1) controlled MS content in MS/MgP scaffolds and (2) controlled HA content in MS. The MS content ranged from 0% to 20% (w/w) in the 55% HA-containing MS/MgP scaffolds; the HA content ranged from 0% to 55% in the 10% MS/MgP scaffolds. The HA-containing composite microspheres and MgP powder were mixed together using a 1% hydroxypropyl methyl cellulose solution (HPMC) (Sigma-Aldrich Corp.), with a powder/liquid ratio of 3 to 2–3 (wt:vol). A uniform MS/MgP green paste was formed for the PED system. In order to visualize the MSs in the MS/MgP composite scaffolds, as shown in Figure 1, MS (55% HA) was stained with 0.4% trypan blue solution (Gibco®; Life Technologies, Carlsbad, CA, USA), washed in DI water vigorously, and lyophilized for 1 day, prior to mixing with MgP powder. The MS/MgP paste, housed in a syringe, was mounted to a gantry robotic deposition apparatus, and equipped with specially-altered systems, such as an actuator, to control the position of the deposition nozzle for the fabrication of 3D structures. A nozzle size of 21G was used to fabricate the 3D MS/MgP scaffolds (10×10×4 mm) with a center-to-center distance of 1 mm. The MS/MgP scaffold green bodies were dried at room temperature, followed by immersion in 3.5 M diammonium hydrogen phosphate (DAHP) (Junsei Chemical Co., Ltd.) for 1 day, with gentle shaking to induce a cement reaction. They were then washed in DI water three times and dried completely by lyophilization. The MgP scaffold without the inclusion of MSs was also prepared for comparison. The final MS/MgP scaffolds were observed under a stereomicroscope (SMZ 1,500; Nikon Corp., Tokyo, Japan) to examine the morphology, strut thickness, and pore size (n=5). The apparent porosity of MS/MgP scaffolds (n=5) was calculated according to the following equation:
 | Figure 1 Gross images of MS/MgP composite scaffolds. |
Porosity = [1− (Dscaffold/Dbulk)] ×100, | (1) |
where Dscaffold means density of scaffold and Dbulk is density of bulk.
Morphological analysis of MS/MgP composite scaffold
For the morphological analysis, MS/MgP composite scaffolds were first sputter-coated with gold to minimize sample charging problems and evaluated by scanning electron microscope (SEM) (JSM-6610LV; JEOL Ltd., Tokyo, Japan) at an operating voltage of 15 kV. Surface and cross-sectional SEM micrographs were taken for each MS/MgP composite scaffold, at magnifications of ×50 and ×100.
Water absorption
For this test, the various MS (55% HA) content samples, ranging from 0% to 20% (w/w), were selected to constitute MS/MgP composite scaffolds. The water absorption of the MS/MgP composite scaffolds was determined by measuring hydrated weight in DI water at 37°C for 3 hours, with gentle shaking (n=5). MS/MgP scaffolds were then carefully patted with tissue to remove excess water and weighed to measure the hydrated weight (Wh). The percentage increase in water absorption can be calculated in contrast to the dried weight (Wd) of the MS/MgP scaffolds, according to the following equation:
Water absorption (%) = ([Wh − Wd]/Wh) ×100 | (2) |
Compressive properties
Based on the MS/MgP scaffold fabrication method described above, MS/MgP composite scaffolds with different MS- and HA-content were produced, while controlling the strut distance of 1.0 mm and nozzle size of 21 G. The dimension of the MS/MgP scaffolds was 10×10×4 mm. The compressive strength and modulus were measured using a uniaxial testing machine, with a 2 kN load cell at a cross-head rate of 1 mm/min (RB Model 302 ML™; R&B Inc., Daejon, Korea) (n=5).
In vitro biodegradation and compressive properties
MS/MgP composite scaffolds, with a dimension of 10×10×4 mm, were measured for dried weight prior to beginning this test. In order to provide the accelerated conditions for an in vitro biodegradation test, 10 mL of collagenase solution (50 μg/mL) (Sigma-Aldrich Corp.) containing 0.1% NaN3 (Sigma-Aldrich Corp.) was transferred, to submerge the MS/MgP scaffolds fully during the whole incubation time. Samples were placed in an incubator temperature-controlled at 37°C, while providing gentle shaking. At each predetermined time (days 5, 15, and 30), samples were collected, washed with DI water three times, and lyophilized to obtain the dried weight. The percentage of weight reduction was then calculated in comparison with the initial dried weight (n=5). The corresponding compressive properties at each biodegradation time (n=5) were evaluated as well, in terms of compressive strength and modulus, to investigate the relationship between in vitro biodegradation and compressive properties.
In vitro lysozyme-release kinetics
In order to investigate the release kinetics of lysozyme from the MS/MgP composite scaffolds, 10% MS with different HA content (0% to 55%) was incubated overnight with 2 mL of lysozyme solution in phosphate-buffered saline (PBS) (0.5 mg/mL). The supernatant was then collected to measure ultraviolet (UV) absorbance at 280 nm, and the amount of lysozyme loaded into the composite MSs was calculated based on a standard curve, prepared using a series of lysozyme concentrations. The composite MSs were then washed once with DI water to eliminate nonspecifically bound lysozyme and lyophilized, prior to blending with MgP powder. Subsequently, 10% MS/MgP composite scaffolds were made following the same method shown above. For comparison, a direct addition of lysozyme into MgP scaffold was also prepared without using MSs as a lysozyme carrier. The amount of lysozyme for this sample was the same as that loaded into MS (55% HA), which was the largest lysozyme content in all the MS samples with different HA content. Briefly, lysozyme, dissolved in 1% HPMC solution, was uniformly blended with MgP powder to achieve a direct incorporation of lysozyme inside MgP scaffold. After lyophilizing the lysozyme-loading scaffolds, the dried weight was measured to calculate the lysozyme-loading content per scaffold; 0.37 g for MgP, 0.27 g for MS (0% HA)/MgP, 0.32 g for MS (38% HA)/MgP, and 0.32 g for MS (55% HA)/MgP (n=5). The lysozyme-release test began by placing MS/MgP composite MSs into 5 mL of PBS containing 0.1% NaN3 at 37°C, with gentle shaking (150 rpm). At each predetermined time, a sample of the medium was collected and replaced with an equal volume of fresh medium. The lysozyme content was analyzed quantitatively using a micro BCA™ protein assay kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA) according to the manufacturer’s instructions (n=5).
In vitro calcification
The capacity of MS/MgP composite scaffolds (10×10×4 mm) to absorb calcium ions was estimated by adding each MS/MgP scaffold to 10 mL of simulated body fluid (SBF) and incubating at 37°C for 5, 10, and 20 days, with gentle shaking. The SBF was prepared according to previously reported studies.14 At each predetermined time (day 5, 10, and 20), SBF was collected and replaced with fresh fluid. The calcium ion content deposited on the MS/MgP composite scaffolds was then determined qualitatively using a commercially available assay kit (QuantiChrom™ Calcium Assay Kit, BioAssay Systems, Hayward, CA, USA) (n=5). The initial calcium ion concentration of SBF was compared with the retrieved SBF solution at each time noted, to calculate the calcium ion deposition. In brief, 5 μL of retrieved samples were reacted with 200 μL of working solution and incubated for 3 minutes at room temperature, followed by measurement of UV absorbance at 612 nm, using a UV-Visible spectrophotometer (UVmini-1240; Shimadzu Corp., Kyoto, Japan). After completing the calcium ion deposition test in SBF, the samples at day 20 were washed carefully with DI water and dried thoroughly to carry out a morphological and elemental analysis by SEM and energy dispersive spectroscopy (EDS) (Oxford Instruments, Abingdon, UK), respectively.
Statistics
All quantitative results were expressed as means ± standard deviation for number n of samples analyzed. Statistical analysis was carried out using one-way analysis of variance (ANOVA), followed by the Tukey test for post hoc multicomparisons to determine whether the differences were significant. A value of P<0.01 or P<0.05 was accepted as statistically significant.
Results and discussion
Microscopic characterization of MS/MgP composite scaffolds
HA-containing composite MS was prepared according to the preparation method described above. As described in our previous publication, the mean diameters for the composite MS in the dried state increased slightly with increasing HA content, from 85±11 μm to 95±12 μm to 101±13 μm.13
The gross images of MS/MgP composite scaffolds are shown in Figure 1A. The dimension of the MS/MgP scaffolds (10×10×4 mm) was well maintained without any expansion or shrinkage during the whole manufacturing process. Due to the advantage of the PED system, the structures could be accurately controlled so as to fabricate well-interconnected porous 3D scaffolds. A gradual increase in blue color could be seen while adding the blue-stained MSs into MS/MgP composite scaffolds ranging from 0% to 20% (w/w). This was more evident when observing the MS/MgP composite scaffolds under a stereomicroscope (Figure 1B). Moreover, it seemed that the surface of the composite scaffolds became rougher as more MS content was added to MS/MgP scaffolds. The strut thickness and pore size of all MS/MgP composite scaffolds were similar, at 540–580 μm and 420–440 μm, respectively (n=5). In addition, the apparent porosity was found to be 50%–52% (n=5).
In the SEM micrographs (Figure 2), the existence of MS was clearly seen, either in the core or on the surface of the struts. As was shown in Figure 1, more MSs could be found in the MS/MgP composite scaffolds while increasing MS content from 2% to 20% (w/w). As a result, the 20% MS/MgP scaffold exhibited the highest distribution of MS content, whereas others contained relatively few MSs.
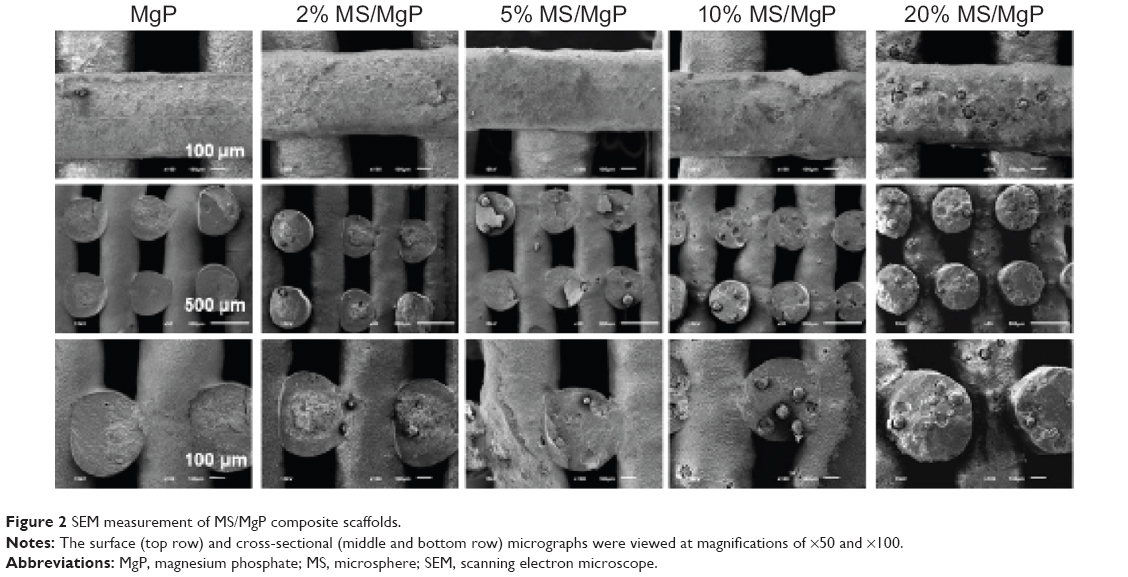 | Figure 2 SEM measurement of MS/MgP composite scaffolds. |
Water absorption
The capacity of water absorption (%) by the MS/MgP scaffolds was explained in Figure 3. Bare MgP scaffold showed the lowest water absorption, of 39%, after soaking in DI water for 3 hours. However, the water uptake increased gradually with increasing MS content from 2% to 20%. The 10% and 20% MS/MgP composite scaffolds showed a dramatic increase in water uptake, to 48% and 53%, respectively.
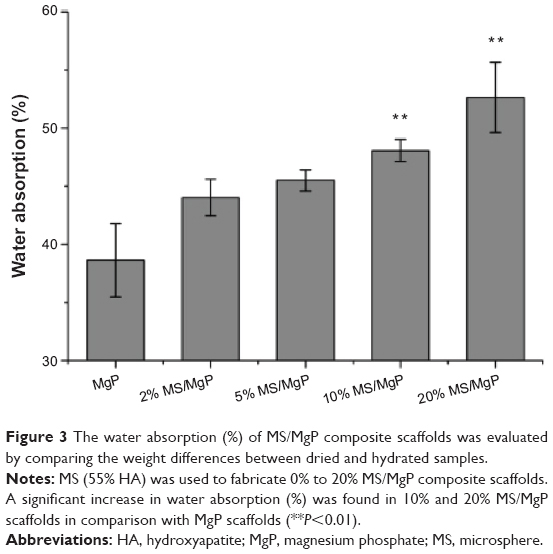 | Figure 3 The water absorption (%) of MS/MgP composite scaffolds was evaluated by comparing the weight differences between dried and hydrated samples. |
According to our previous work on HA-containing composite MSs,13 the water content (%) of the composite MSs varied from 64.5% to 81.1%, depending on the HA content embedded in polymeric MSs. In this work, the experiments were mostly conducted using 55% HA-containing MSs. This MS has the lowest water content (%), of 64.5%, but still had excellent hydrophilic capacity. Due to the significant influence of MSs, it was possible to enhance the hydrophilic properties of bare MgP scaffold by simple incorporation of MS into the MgP matrix. Thus, 10% and 20% MS/MgP scaffolds were able to achieve improvement in water absorption (%) relative to the MgP scaffold (**P<0.01). The proportional increase in water absorption (%) could be attributed to the increasing amount of hydrophilic composite MSs enveloped in the MS/MgP composite scaffolds.
Compressive properties
The compressive properties of MS/MgP composite scaffolds are depicted in Figure 4. The compressive properties of MgP scaffolds are indicated using a dotted line for comparison. The HA-containing MS/MgP composite scaffolds maintained the compressive strength and modulus, both similar to the level of MgP scaffold throughout all MS content samples (2% to 20%). However, 0% HA-containing MS/MgP scaffolds resulted in a gradual decrease in compressive strength, which finally reached its lowest, at 20% MS/MgP scaffolds (P<0.01). Regarding the compressive modulus, most of the MS/MgP composite scaffolds exhibited similar values to the MgP scaffold, except for 20% MS/MgP scaffolds. In this case, a significant reduction was only seen in the 0% HA-containing MS/MgP scaffold (P<0.05). The inclusion of HA into MS was therefore advantageous to the MS/MgP composite scaffolds because the HA addition would be able to preserve the original mechanical characteristics of the MgP scaffold and also, prevent a substantial decrease in compressive properties.
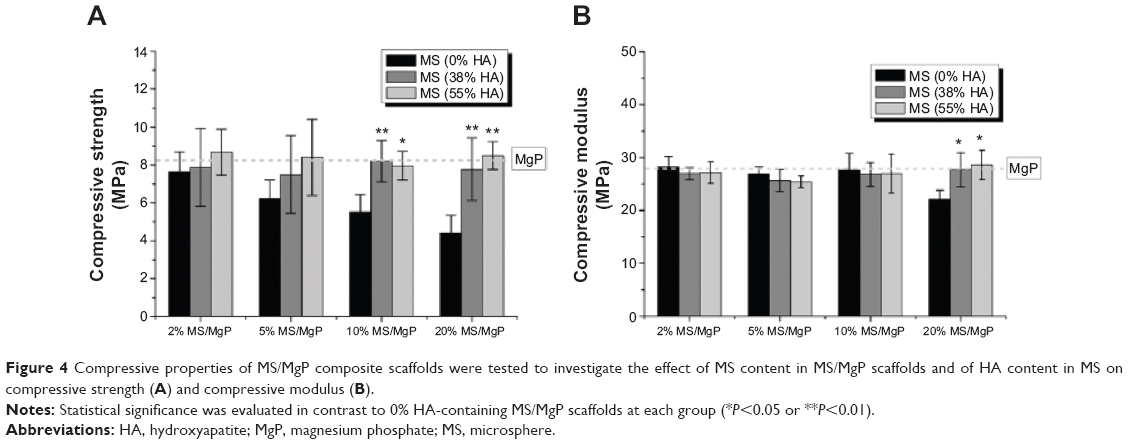 | Figure 4 Compressive properties of MS/MgP composite scaffolds were tested to investigate the effect of MS content in MS/MgP scaffolds and of HA content in MS on compressive strength (A) and compressive modulus (B). |
In vitro biodegradation and compressive properties
The in vitro biodegradation of MS/MgP composite scaffolds were evaluated in two different aspects: the effect of MS content in MS/MgP scaffolds (Figure 5A) and of HA content in MS (Figure 5B). The steady increase in the in vitro biodegradation was monitored over time. This meant that all MS/MgP composite scaffolds would be degraded gradually in proportion to the incubation time. The in vitro biodegradation was facilitated with increasing MS content, from 0% to 20%. The greatest weight reduction occurred with the 20% MS/MgP composite scaffolds throughout the whole incubation time (day 5 and day 15 P<0.01; day 30 P<0.05). With regard to the influence of HA content in MS, a substantial increase in biodegradation (%) was shown with the reduction of HA content from 55% to 0% (P<0.01).
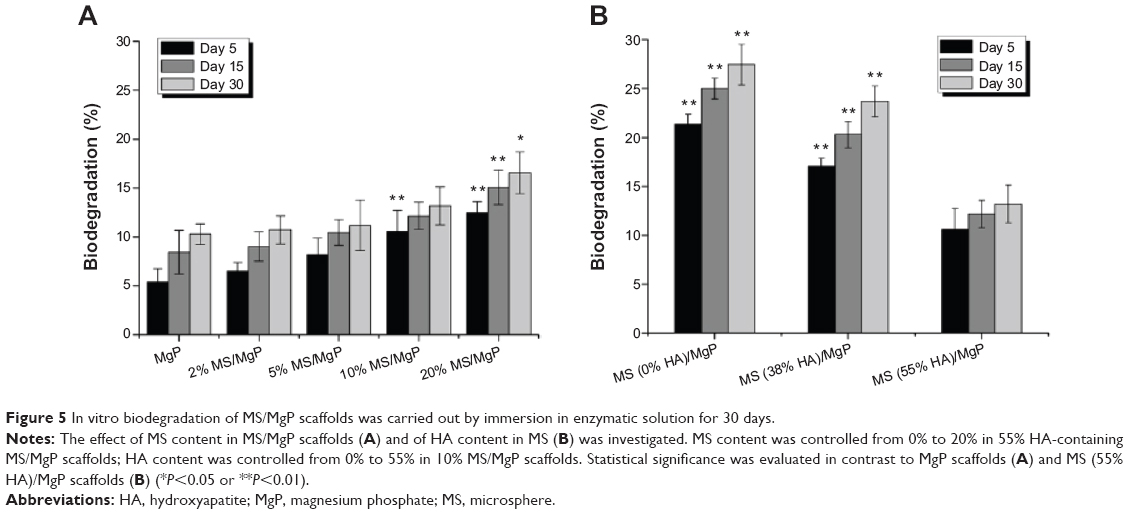 | Figure 5 In vitro biodegradation of MS/MgP scaffolds was carried out by immersion in enzymatic solution for 30 days. |
The corresponding compressive properties at each biodegradation time are shown in Figure 6, and this revealed a close relationship to the in vitro biodegradation. The compressive strength of MS/MgP composite scaffolds began to decrease after being biodegraded over time (Figure 6A). Contrary to the in vitro biodegradation result (Figure 5), the more the MS/MgP composite scaffolds were degraded, the fewer compressive properties were obtained as a function of biodegradation time. For this reason, the lowest compressive strength was found with 20% MS/MgP composite scaffolds from day 5 to day 30. Due to the superior biodegradability of the HA-containing 10% MS/MgP composite scaffolds, significant decrease in compressive properties was found with 0% and 38% HA-containing 10% MS/MgP scaffolds (Figure 6B). However, the 55% HA-containing 10% MS/MgP scaffolds were able to prevent this significant loss of mechanical properties to some extent.
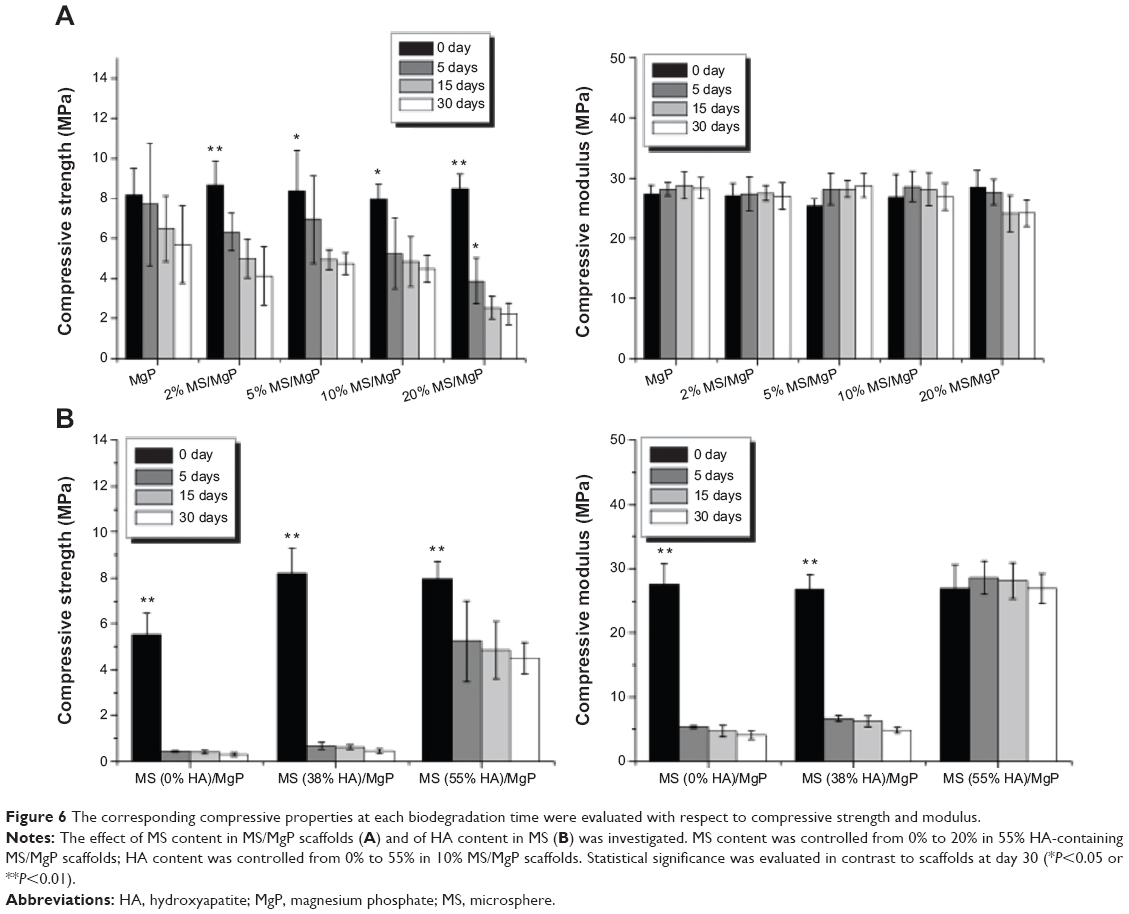 | Figure 6 The corresponding compressive properties at each biodegradation time were evaluated with respect to compressive strength and modulus. |
In Figure 5, the in vitro biodegradation pattern of MS/MgP composite scaffolds was highly associated with MS content (%), HA content (%), and degradation time (day). In this work, we recognized that MS/MgP composite scaffolds would be susceptible to degrade in enzymatic solutions because the MSs were mainly constituted of biodegradable natural polymers, such as gelatin and chitosan. As a result, the largest weight reduction took place in the 20% MS/MgP composite scaffolds at day 30. The biodegradation of MS-embedding bioceramic scaffolds has been extensively studied under in vitro and in vivo circumstances.9,10 In most cases, the biodegradable polymeric MS was impregnated into bioceramic matrix in a certain ratio and kept under physiological conditions for a prolonged period of time. Then, the result was usually evaluated in terms of the morphology, porosity (%), weight loss (%), mechanical properties, and so on. For example, a composite scaffold of gelatin MS/CPC was prepared to create a macroporous structure after being degraded in PBS for 5 weeks.15 A higher porosity was obtained with the degradation of gelatin MS. This could subsequently lead to a gradual decrease in compressive strength with MS content and degradation time, which is very consistent with our results as shown in Figures 5 and 6. As well, the bulk erosion of gelatin MSs began after 3 weeks degradation so that many macropores with interconnectivity was spread out everywhere.
Given the influence of HA content in MS, the inclusion of HA into the MSs was able to, not only retard the biodegradation process but also, compensate for the potential loss of compressive properties in a sustained manner. In comparison with the biodegradation (%) at day 30, 0% HA-containing 10% MS/MgP scaffold was 27.5%, which was twice as large as 13.2% (55% HA-containing 10% MS/MgP scaffold; Figure 5B). However, the corresponding compressive strength of these two samples was measured as 0.3 MPa and 4.5 MPa, respectively. It is therefore straightforward that the improved compressive properties of the 55% HA-containing MS/MgP composite scaffolds were mostly attributed to the significant effect of HA inclusion. In order to reinforce the mechanical properties of bone tissue engineering scaffolds, HA has been typically incorporated into a bulk polymeric phase, with or without surface modification: PLLA,16 poly (ε-caprolactone),17 and chitosan.18 In most cases, the compressive properties of HA-containing composite scaffolds would improve with the increasing weight ratio and remain stabilized until a certain point. This is mainly ascribed to the enhanced interaction between HA and the bulk matrix and good dispersion of HA inside the matrix.17 Furthermore, it was reported that the electrostatic interactions of gelatin and chitosan with HA nanoparticles could slow down the in vitro biodegradation and mechanical properties.18,19 As with the previous studies, we also confirmed the inverse relationship between the in vitro biodegradation and corresponding compressive properties of MS/MgP composite scaffolds. The critical importance of the HA addition into MSs was emphasized, so as to prevent a severe reduction of compressive properties with in vitro biodegradation time.
In vitro lysozyme-release kinetics
For this experiment, 10% MSs with different HA content were homogeneously embedded into MgP scaffolds to investigate the effect of HA on the in vitro lysozyme-release kinetics. The in vitro release pattern was monitored for 30 days and analyzed in terms of cumulative release amount (μg) (Figure 7A) and cumulative release percentage (%) (Figure 7B). The lysozyme-loading content of MS and MS/MgP composite scaffolds could be obtained as follows. First, the lysozyme-loading content of 10% MS with different HA contents was: 810 μg for MS (0% HA), 889 μg for MS (38% HA), and 891 μg for MS (55% HA). Second, the final lysozyme-loading content of the 10% MS/MgP composite scaffolds was calculated based on the dried weight of each sample: 166 μg for MgP scaffold, 109 μg for MS (0% HA)/MgP scaffold, 140 μg for MS (38% HA)/MgP scaffold, and 140 μg for MS (55% HA)/MgP scaffold. In Figure 7A, the MgP scaffold, which had direct blending with lysozyme, showed an initial burst until 7 days and then continued to retain the highest release of lysozyme until 30 days. On the other hand, the MS/MgP composite scaffolds showed a controlled release rate without any serious loss and showed graded lysozyme-release patterns in accordance with HA content. The lysozyme release kinetics of composite scaffolds is shown in order of MS (38% HA)/MgP and MS (55% HA)/MgP. A slower release patten of MS (55% HA)/MgP is attributed to the strong affinity of HA to lysozyme as depicted in Figure 7A.
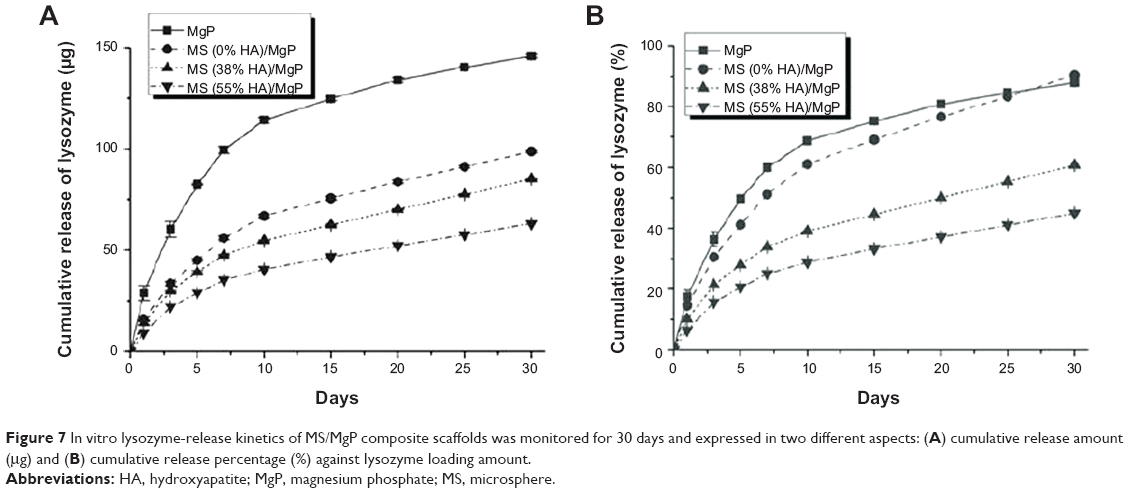 | Figure 7 In vitro lysozyme-release kinetics of MS/MgP composite scaffolds was monitored for 30 days and expressed in two different aspects: (A) cumulative release amount (μg) and (B) cumulative release percentage (%) against lysozyme loading amount. |
In our previous study, an HA-containing gelatin/chitosan MS was developed as an efficient protein delivery carrier that not only enables the loading of high quantities of lysozyme, but also, maintains a sustained release without an initial burst.13 The HA effect on lysozyme delivery was enhanced with increases in HA content from 38% to 55%. This outstanding feature could be explained by the increase in specific surface areas and strong electrostatic interactions of HA-containing composite MSs. For more advanced applications, we endeavored to combine the novel MS with MgP scaffolds, to allow for the higher loading capacity and prolonged release of lysozyme in a controlled manner. As presented in Figure 7, it was obvious that the incorporation of MS into MgP scaffold could result in a controllable lysozyme-release pattern without a severe initial burst. The slower release of lysozyme took place in the higher HA-containing MS/MgP composite scaffolds; in contrast, the fast release of lysozyme was seen on the bare MgP scaffold, due to the rapid diffusion.
Various types of MSs have been incorporated into a bulk matrix consisting of either biodegradable polymers or bioceramics.20,21 In most cases, the MS-containing composite structures were usually aimed to overcome the conventional limitations of tissue engineering scaffolds, especially poor drug delivery ability. Based on recent publications, it would be feasible to achieve a sustained release of a single biomolecule or sequential delivery of dual bioactive molecules for the tissue engineering application.3,22 In view of clinical applications, antibiotics (eg, gentamicin) or growth factors (eg, bone-morphogenetic protein-2) were encapsulated within poly(lactic-co-glycolic acid) MSs and subsequently mixed with calcium phosphate bone cement to generate a controlled drug delivery system for the effective treatment of bone infections and bone regeneration, respectively.11,23
In vitro calcification
The ability of MS/MgP composite scaffolds to initiate in vitro calcification by SBF treatment was evaluated in two different aspects: the effect of MS content in MS/MgP scaffolds (Table 1) and of HA content in MS (Table 2). MgP scaffolds contained the lowest calcium ion deposition throughout the entire incubation time. On the other hand, MS/MgP composite scaffolds steadily enhanced the in vitro calcium deposition, while increasing MS content from 2% to 20%. At day 20, the 10% and 20% MS/MgP composite scaffolds resulted in the highest quantity of calcium ion deposition (P<0.01) (Table 1). With respect to HA content in MS, 55% HA-containing MS/MgP composite scaffolds led to the highest level of calcium ion deposition (P<0.05), followed by 38% HA-containing MS/MgP scaffolds (Table 2).
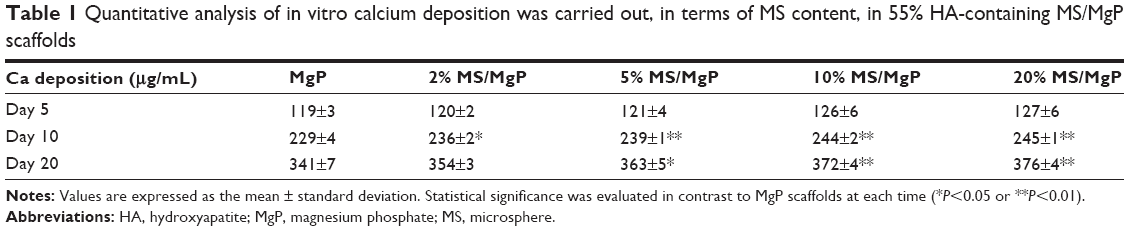 | Table 1 Quantitative analysis of in vitro calcium deposition was carried out, in terms of MS content, in 55% HA-containing MS/MgP scaffolds |
 | Table 2 Quantitative analysis of in vitro calcium deposition was carried out, in terms of HA content, in 10% MS/MgP scaffolds |
In the SEM morphological analysis (Figure 8), the flake-like apatite crystals were newly produced and distributed on the surface of all MS/MgP composite scaffolds. As with the quantitative result of calcium ion deposition (Table 1), 20% MS/MgP composite scaffolds could generate relatively more apatite crystals than other MS/MgP scaffolds (Figure 8A). Nevertheless, it was difficult to find morphological variations between the different HA-containing MS/MgP scaffolds (Figure 8B). The elemental analysis of the neoformation of apatite crystals was also carried out by means of EDS. A representative EDS result is illustrated, in Figure 8A, to confirm the noticeable peaks for calcium and phosphate. The EDS patterns were comparable with each other, regardless of different MS and HA content.
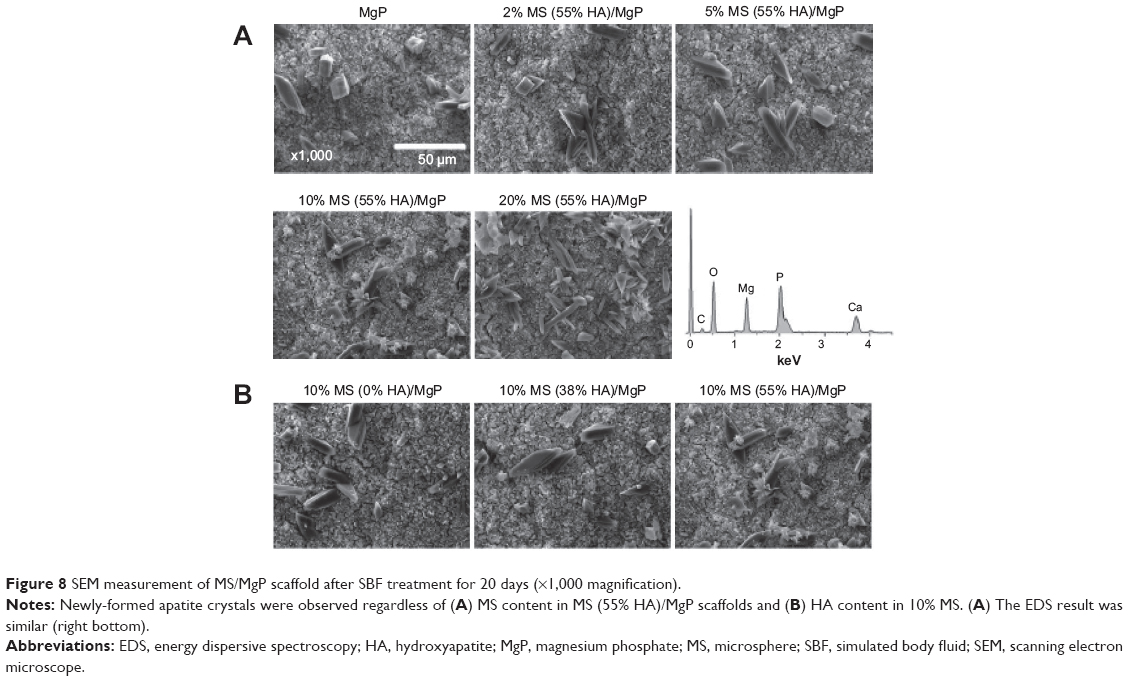 | Figure 8 SEM measurement of MS/MgP scaffold after SBF treatment for 20 days (×1,000 magnification). |
The effect of HA addition on in vitro calcification of HA-containing MSs was already investigated in our previous study, by soaking them in SBF at physiological conditions.13 We found that the calcium ion deposition was facilitated with increasing incubation time and HA content in MSs. This result is consistent with the present work using MS/MgP composite scaffolds (Tables 1 and 2). The in vitro calcium deposition was proportional to MS content in MS/MgP scaffold, HA content in MSs, and the incubation time. Overall, the important role of nanosized HA would be able to accelerate the in vitro apatite-forming ability of MS/MgP composite scaffolds by SBF treatment. Using morphological and elemental analysis, flake-like apatite crystals and noticeable calcium/phosphate peaks were obviously detected in all SBF-treated scaffolds, respectively (Figure 8). However, there was no evidence of apatite formation in bare MgP scaffold kept in DI water for the same period of time (Figure S1). To date, SBF solution has been widely utilized for the functionalization of biomaterials as it could mimic the actual physiological conditions and consequently, improve both in vitro bioactivity and in vivo performance. To this end, many types of biomaterials would be able to create the bone-like apatite layers on the surface and integrate with surrounding host bony tissues, without forming fibrous interfacial tissue.24
Conclusion
A synergistic combination of MgP scaffold and MS was carried out in this work to fulfill the requirements for bone tissue engineering scaffold. Porous 3D structures with good interconnectivity were fabricated using a PED system. Controlled lysozyme-release kinetics were exhibited in accordance with HA content. In vitro biodegradation was directly affected by MS and HA content throughout the entire incubation period, which led to an inverse relationship with corresponding compressive properties. In vitro calcium deposition was initiated by SBF treatment so that the biomimetic apatite crystals could be formed on the MS/MgP composite scaffolds. Taken together, these results suggest that an MS/MgP composite scaffold is a promising candidate material to achieve sustained drug-release capacity, biodegradability, and bioactivity for effective bone regeneration.
Acknowledgments
This work was supported by the Mid-Career Researcher Program, through a National Research Foundation of Korea (NRF) grant funded by the Korea Ministry of Education, Science, and Technology (MEST) (number 2011-0017572).
Disclosure
The authors report no conflicts of interest in this work.
References
Wang H, Leeuwenburgh SC, Li Y, Jansen JA. The use of micro- and nanospheres as functional components for bone tissue regeneration. Tissue Eng Part B Rev. 2012;18(1):24–39. | ||
Habraken WJ, Wolke JG, Mikos AG, Jansen JA. Injectable PLGA microsphere/calcium phosphate cements: physical properties and degradation characteristics. J Biomater Sci Polym Ed. 2006;17(9):1057–1074. | ||
Niu X, Feng Q, Wang M, Guo X, Zheng Q. Porous nano-HA/collagen/PLLA scaffold containing chitosan microspheres for controlled delivery of synthetic peptide derived from BMP-2. J Control Release. 2009; 134(2):111–117. | ||
Park H, Temenoff JS, Holland TA, Tabata Y, Mikos AG. Delivery of TGF-beta1 and chondrocytes via injectable, biodegradable hydrogels for cartilage tissue engineering applications. Biomaterials. 2005;26(34): 7095–7103. | ||
Zhu G, Mallery SR, Schwendeman SP. Stabilization of proteins encapsulated in injectable poly(lactide-co-glycolide). Nat Biotechnol. 2000; 18(1):52–57. | ||
Kempen DH, Kruyt MC, Lu L, et al. Effect of autologous bone marrow stromal cell seeding and bone morphogenetic protein-2 delivery on ectopic bone formation in a microsphere/poly(propylene fumarate) composite. Tissue Eng Part A. 2009;15(3):587–594. | ||
Matsuno T, Hashimoto Y, Adachi S, et al. Preparation of injectable 3D-formed beta-tricalcium phosphate bead/alginate composite for bone tissue engineering. Dent Mater J. 2008;27(6):827–834. | ||
Wang C, Gong Y, Zhong Y, Yao Y, Su K, Wang DA. The control of anchorage-dependent cell behavior within a hydrogel/microcarrier system in an osteogenic model. Biomaterials. 2009;30(12):2259–2269. | ||
Habraken WJ, Liao HB, Zhang Z, et al. In vivo degradation of calcium phosphate cement incorporated into biodegradable microspheres. Acta Biomater. 2010;6(6):2200–2211. | ||
Félix Lanao RP, Leeuwenburgh SC, Wolke JG, Jansen JA. In vitro degradation rate of apatitic calcium phosphate cement with incorporated PLGA microspheres. Acta Biomater. 2011;7(9):3459–3468. | ||
Schnieders J, Gbureck U, Thull R, Kissel T. Controlled release of gentamicin from calcium phosphate-poly(lactic acid-co-glycolic acid) composite bone cement. Biomaterials. 2006;27(23):4239–4249. | ||
Lee J, Farag MM, Park EK, Lim J, Yun HS. A simultaneous process of 3D magnesium phosphate scaffold fabrication and bioactive substance loading for hard tissue regeneration. Mater Sci Eng C Mater Biol Appl. 2014;36:252–260. | ||
Lee J, Yun HS. Hydroxyapatite-containing gelatin/chitosan microspheres for controlled release of lysozyme and enhanced cytocompatibility. J Mater Chem B. 2014;2(9):1255–1263. | ||
Kokubo T, Takadama H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials. 2006;27(15):2907–2915. | ||
Li M, Liu X, Liu X, Ge B, Chen K. Creation of macroporous calcium phosphate cements as bone substitutes by using genipin-crosslinked gelatin microspheres. J Mater Sci Mater Med. 2009;20(4):925–934. | ||
Fang Z, Feng Q. Improved mechanical properties of hydroxyapatite whisker-reinforced poly(L-lactic acid) scaffold by surface modification of hydroxyapatite. Mater Sci Eng C Mater Biol Appl. 2014;35:190–194. | ||
Wang Y, Dai J, Zhang Q, Xiao Y, Lang M. Improved mechanical properties of hydroxyapatite/poly(ε-caprolactone) scaffolds by surface modification of hydroxyapatite. Appl Surf Sci. 2010;256(20):6107–6112. | ||
Thein-Han WW, Misra RD. Biomimetic chitosan-nanohydroxyapatite composite scaffolds for bone tissue engineering. Acta Biomater. 2009; 5(4):1182–1197. | ||
Leeuwenburgh SC, Jo J, Wang H, Yamamoto M, Jansen JA, Tabata Y. Mineralization, biodegradation, and drug release behavior of gelatin/apatite composite microspheres for bone regeneration. Biomacromolecules. 2010;11(10):2653–2659. | ||
Tang G, Zhang H, Zhao Y, Li X, Yuan X, Wang M. Prolonged release from PLGA/HAp scaffolds containing drug-loaded PLGA/gelatin composite microspheres. J Mater Sci Mater Med. 2012;23(2):419–429. | ||
Girod Fullana S, Ternet H, Freche M, Lacout JL, Rodriguez F. Controlled release properties and final macroporosity of a pectin microspheres-calcium phosphate composite bone cement. Acta Biomater. 2010;6(6):2294–2300. | ||
Wang X, Wenk E, Zhang X, Meinel L, Vunjak-Novakovic G, Kaplan DL. Growth factor gradients via microsphere delivery in biopolymer scaffolds for osteochondral tissue engineering. J Control Release. 2009;134(2):81–90. | ||
Ruhé PQ, Boerman OC, Russel FG, Spauwen PH, Mikos AG, Jansen JA. Controlled release of rhBMP-2 loaded poly(dl-lactic-co-glycolic acid)/calcium phosphate cement composites in vivo. J Control Release. 2005;106(1–2):162–171. | ||
Zadpoor AA. Relationship between in vitro apatite-forming ability measured using simulated body fluid and in vivo bioactivity of biomaterials. Mater Sci Eng C Mater Biol Appl. 2014;35:134–143. |
Supplementary material
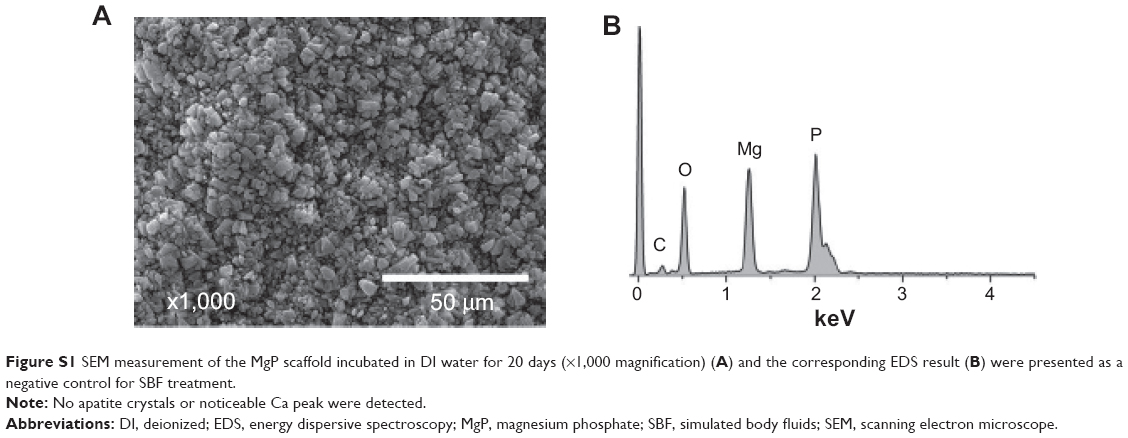 | Figure S1 SEM measurement of the MgP scaffold incubated in DI water for 20 days (×1,000 magnification) (A) and the corresponding EDS result (B) were presented as a negative control for SBF treatment. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.