Back to Journals » OncoTargets and Therapy » Volume 13
Effect of Ect2 Expression on the Growth of Triple-Negative Breast Cancer Cells with Paclitaxel Intervention
Authors Wang H ![]() , Liu H, Li J, Wei S, Liu X, Wan H, Zheng P, Zheng H
, Liu H, Li J, Wei S, Liu X, Wan H, Zheng P, Zheng H
Received 6 August 2020
Accepted for publication 4 December 2020
Published 16 December 2020 Volume 2020:13 Pages 12905—12918
DOI https://doi.org/10.2147/OTT.S275725
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Sanjay Singh
Hongkun Wang,1,2 Honggang Liu,1 Jun Li,2 Shuanyu Wei,2 Xiaojun Liu,2 Huili Wan,2 Peiming Zheng,2 Huixia Zheng2
1Department of Pathology, Beijing Tongren Hospital, Capital Medical University, Beijing, People’s Republic of China; 2Department of Pathology, First Clinical Medical College, Shanxi Medical University, Taiyuan City, Shanxi, People’s Republic of China
Correspondence: Honggang Liu
Department of Pathology, Beijing Tongren Hospital, Capital Medical University, Beijing 100005, People’s Republic of China
Email [email protected]
Object: To identify the expression levels of ECT2 (epithelial cell transforming sequence 2) in triple-negative breast cancer (TNBC) before and after administration of paclitaxel (PTX) and explore the interaction between ECT2 and PTX in breast cancer treatment.
Methods: Lentiviral (LV) packaging ECT2 overexpression and interference plasmids were constructed for in vitro assays. The effects of ECT2 expression on the TNBC cell line (HCC1806), particularly its roles in the proliferation, invasion, migration and apoptosis and cell cycle, were evaluated using the CCK-8 and other methods before and after PTX treatment. In nude mouse xenograft settings were performed to detect cell apoptosis and Ki-67 expression levels by TUNEL and immunohistochemical staining, respectively.
Results: In the vitro assays, before and after the PTX treatment, comparison of the LV-ECT2 and sh-ECT2 groups and the remaining three groups (control, LV-NC, sh-NC) showed statistically significant differences in terms of cell proliferation, invasion and migration and apoptosis and changes in the cell cycle. In the vivo assays, the control, LV-ECT2 and sh-ECT2 groups markedly outweighed the corresponding PTX-treated groups. The LV-ECT2, PTX, sh-ECT2 and sh-ECT2-PTX were all significantly different from the control group in terms of body weight and tumour size changes. Cell apoptosis occurred in the PTX, sh-ECT2 and sh-ECT2-PTX groups. About the Ki-67 proliferation index, the PTX, LV-ECT2-PTX, sh-ECT2 and sh-ECT2-PTX groups were significantly different from the control group.
Conclusion: ECT2, which is a major driving factor in the growth of breast cancer cells, plays an important role in regulating TNBC growth. PTX therapy had significantly improved efficacy after silencing ECT2. This finding indicates that the inhibition of ECT2 expression may facilitate the treatment of breast cancer as a new regimen and provide a theoretical basis for the development of new targeted drugs as a replacement for PTX in breast cancer treatment.
Keywords: ECT2, TNBC, paclitaxel
Introduction
Triple-negative breast cancer (TNBC) accounts for approximately 20% of all breast cancer cases and is common in younger women before menopause. In addition to the risk of recurrence and metastasis and the limited treatment options, TNBC remains a radical challenge in the field given the lack of targeted drugs and effective biomarkers for pre-treatment evaluation and prognostic assessment. National Comprehensive Cancer Network and other guidelines suggest that PTX is more efficacious than anthracyclines in breast cancer treatment. A preliminary experiment showed that ECT2 had an increased expression level in breast cancer, and its expression level was relatively high in TNBC. A survival analysis revealed that ECT2 and TNBC were independent prognostic factors in breast cancer patients.1 ECT2 mainly promotes cell proliferation during the G2/M phase, whereas PTX inhibits the proliferation of cancer cells by inducing a mitotic arrest. In the present study, in vivo and in vitro assays were performed to investigate the effects of ECT2 on TNBC cell proliferation, invasion, migration and apoptosis. Additionally, in the case of ECT2 overexpression and interference, cancer cells were treated with PTX to discuss how PTX inhibits the growth of cancer cells in vivo and in vitro and explore the interaction between ECT2 and PTX in TNBC, providing a theoretical basis for TNBC treatment and prognosis.
Materials and Methods
Cell Culture and Treatment
The frozen basaloid TNBC (B-TNBC) cell line HCC1806 (KeyGen Biotech, China) was quickly removed from liquid nitrogen and thawed at 37°C. The cells were then added to a medium containing 90% RPMI-1640 and 10% FBS using a pipette and placed in an incubator maintained at 37°C, 5% CO2, and saturated at 100% relative humidity. The following day, the medium was replaced, and a fresh medium was added for exponential growth of cells.
Constructing Lentiviral (LV) Vectors for ECT2 Overexpression
The complete genomic sequence of ECT2 was obtained from the National Centre for Biotechnology Information GenBank. LV packaging plasmids were constructed with the assistance of Nanjing KeyGen Biotech (Jiangsu, China). T4 DNA ligase was used for the construction of plasmid vectors for ECT2 overexpression (ie, FV115-ECT2-OE), which was examined by gel electrophoresis. Further, a negative control (NC) (FV115-NC) was established. HCC1806 cells were grown in adherent culture until transfection 24 h later. Transfection efficiency was determined by measuring the number of green fluorescent protein/red fluorescent protein (GFP/RFP)-transfected cells under a microscope. If the GFP/RFP-transfected cells accounted for at least 70% of all cells, the viruses were harvested, whereas the infected cells were subjected to reverse transcription polymerase chain reaction (RT-PCR). The LV plasmid vector Lenti-ECT2-3FLAG-OE was successfully constructed (Figure 1A and B).
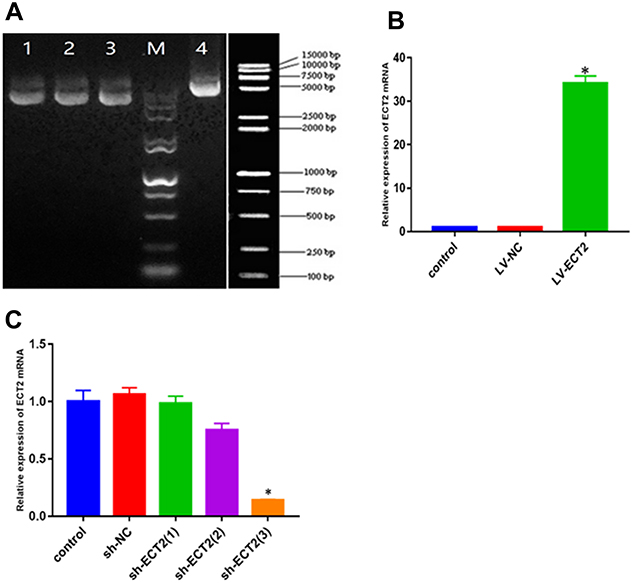 |
Figure 1 Plasmid gel electrophoresis and QPCR histogram. (A) 1–3 for ECT2 siRNA (sh-ECT2) gel electrophoresis, 4 for ECT2 overexpression (LV-ECT2) gel electrophoresis. (B) ECT2 overexpression group showed that there were significant differences between LV-ECT2 group and control and LV-NC group, and they were shown as mean ± SD. (C) sh-ECT2 group showed that there was significant difference between sh-ECT2(3) group and control group, and there was no significant difference between the other two groups and the control group. They were shown as mean ± SD. * P<0.05. |
Constructing LV Vectors for ECT2 Interference
Following the instructions for use, siControl and siRNA of ECT2 were provided by KeyGen Biotech (Jiangsu, China). Three target sequences were generated, and T4 DNA ligase was used for double digestion to construct plasmid vectors for ECT2 interference (ie, FV055-ECT2-shRNA), which was also examined by gel electrophoresis. Finally, the following interference target sequence was 5ʹ-GCTGAGCATTCCCTTTCCATA-3ʹ. Another NC (FV055-NC) was set up. HCC1806 cells grew under close observation within 6 h following transfection. If all observed cells were in good condition, no replacement of medium would be necessary. After continuous culture for another 48 h, quantitative RT-PCR was performed to confirm whether the target mRNAs were reduced, which indicated that the LV plasmid vector Lenti-ECT2-shRNA was successfully constructed (Figure 1A and C).
Experimental group including the control, LV-negative control (NC), LV-ECT2, sh-NC and sh-ECT2 groups, and Paclitaxel intervene in each group.
Cell Counting Kit (CCK)-8 Cell Proliferation Assay
After digestion and counting, the infected cells were used for the preparation of cell suspensions at the concentration of 5 × 104/L. Then, the cell suspensions were added to a 96-well culture plate at the density of 100 µL/well, and the plate was placed in a 5% CO2 incubator maintained at 37°C for cell culture. Cells in the culture plate were treated with CCK-8 at 24, 48 and 72 h after transfection with the ECT2 plasmids. Complete media were used for dilution of PTX to different concentrations, and the cells in each well were then mixed with 100 µL complete medium containing PTX at a given concentration. In the meantime, NCs were established. Cells were stained by adding 10 µL CCK-8 reagent (DOJINDO Laboratories, Japan) to each well. After continuous culture for another 3 h in an incubator, the optical density (OD) at 450 nm, PTX efficacy and inhibitory concentration of 50% (IC50) were measured. Experiments were performed in triplicate.
Transwell Invasion and Migration Assays
Cells under serum starvation were cultured in a serum-free medium for 24 h. Matrigel was thawed overnight at 4°C and diluted with a minimal medium. Then, 30 µL diluted Matrigel was added to the upper transwell chamber before incubation at 37°C for 120 min (this step was skipped in migration assay). Cells were digested and counted, and the cell densities were adjusted to 1 × 105/mL (invasion) and 5 × 104/mL (migration). After PTX treatment for 48 h, cells in the exponential phase of growth were used for experiments. Assays were performed following the given protocols. Cells on the lower surface of the polyethylene terephthalate membrane were visualised by microscopy and counted under 400x magnification in five different vision fields to average the results. All experiments were performed in triplicate.
Cell Apoptosis Assay
The cell apoptosis assay was performed using the Annexin V-APC apoptosis detection kit with 7-amino-actinomycin D (7-AAD). Cells in the exponential phase of growth were digested and inoculated into a six-well culture plate. The next day, upon adherence to the wall of the plate, the cells were divided into different groups and treated with the corresponding media, and NCs were established. The cells were harvested after PTX treatment for 48 h and digestion with 0.25% trypsin (without ethylenediaminetetraacetic acid (EDTA)). Then, the cells were washed twice with phosphate-buffered saline (PBS), and 5 × 105 cells were added to 500 µL binding buffer for suspension. Subsequently, the cell suspensions were stained with Annexin V-APC and mixed thoroughly with 7-AAD, followed by reaction for 5–15 min at room temperature in the dark. Finally, cell apoptosis was detected using a flow cytometer. All experiments were performed in triplicate.
Cell Cycle Assay
Propidium iodide (PI) was used for cell cycle analysis. The cells were digested with trypsin-free EDTA, and 5 × 106 cells were harvested after centrifugation to prepare single-cell suspensions. Afterward, the suspensions were fixed with prechilled ethanol (70% vol) for 2 h (or overnight) and stored at 4°C. Before staining, the suspensions were washed with PBS to remove the fixation fluid. Then, 100 µL RNase A was added, followed by a water bath at 37°C for 30 min. The mixtures were stained with 400 µL PI in a dark place at 4°C for 30 min. Finally, cell cycle analysis was performed using a flow cytometer and data fitting software. All experiments were performed in triplicate.
Experimental Design of in vivo Assays
Nude Mouse Xenograft Assay
The in vivo assays involved 36 four- to five-week-old nude mice with severe combined immunodeficiency. Animal experiments were approved by the ethics committee of Shanxi Medical University. Each group cell suspension (1 × 107/mL) was harvested and was later used to inoculate the corresponding nude mice (0.1 mL each) subcutaneously from the right armpit after grouping. The diameter of each tumour xenograft was measured with a Vernier caliper. At 21 days following the inoculation, the nude mice were randomly divided into six groups (n = 6) when the tumours reached 70 mm3. The PTX, LV-ECT2-PTX and sh-ECT2-PTX groups received tail-vein injections of PTX at the dose of 6 mg/kg every two days and were closely observed for 21 days. During the same period, the control, LV-ECT2 and sh-ECT2 groups were intravenously injected with normal saline at the same dose and interval. The diameters of the tumours in nude mice were measured to assess the antitumor efficiency. Upon completion of the in vivo assays, the mice were euthanised to harvest tumours from their bodies for weight measurement. Antitumor efficiency was calculated using relevant parameters. All animal procedures were performed and approved by the Medical ethics committee of Shanxi Medical University, and followed the guidelines of welfare ethics committee of the laboratory animals of Shanxi Medical University.
Cell Apoptosis Assay
The tumour tissues removed from the mice were fully dewaxed and hydrated before cell apoptosis analysis based on the terminal deoxynucleotidyl transferase dUTP nick end labelling assay. Paraffin sections (3–4 um) were prepared. All samples were treated with a fresh mixture made from 90 µL 1X PBS and 10 µL 10X proteinase K. The reaction at 37°C was not terminated until 15–20 min, followed by the addition of 100 µL DNase I reaction buffer in varying Kunitz units and 1% Triton X-100. After standing at room temperature, the samples were washed and treated with 3% H2O2 methanol. Subsequently, every sample was mixed with 100 µL terminal transferase. The reaction occurred in a dark, humid environment at 37°C and was terminated 1 h later. Then, the samples were washed before the addition of enzyme-labelled avidins, stained with DAB reagent, counterstained and mounted.
Immunohistochemical (IHC) Analysis of Ki-67 Expression
The monoclonal antibody Ki-67, IHC assay kit and DAB staining kit were purchased from Fuzhou Maxim Biotechnologies Co., Ltd. IHC analysis was conducted using the two-step EnVision method. The assay was performed as directed by the manufacturer, with the known positive section as the positive control and PBS as the NC.
Statistical Analysis
IC50 was calculated using the Bliss method. Mean values were expressed by ‘mean ± standard deviation’. Group comparison was examined by t-test, and p values <0.05 were considered statistically significant. All results were analysed using SPSS 24.0.
Results
Effects of ECT2 Overexpression and Interference and PTX Therapy on Breast Cancer Cell Proliferation
According to the CCK-8 cell proliferation assay, before PTX treatment, the LV-ECT2 group had an OD value significantly higher than that of the control and LV-NC groups at 48 h (P < 0.05), indicating a remarkable improvement in the proliferation ability. On the other hand, the sh-ECT2 group had an OD value significantly lower than that of the control and sh-NC groups (P < 0.05), suggesting the inhibited cell proliferation in the sh-ECT2 group (Figure 2A).
 |
Figure 2 CCK-8 cell proliferation assay. (A) Before PTX treatment, (B) After treatment with PTX: LV-ECT2 group had an higher OD value at 48 h, and the sh-ECT2 group had an lower OD value at 48 h. (C) After PTX treatment, the inhibitory rate of each group was compared in the histogram.*P <0.05. |
Subsequently, the five groups were treated with PTX at different concentrations (3.91 to 250, 500 and 1000 nM). Cell proliferation was monitored at 48 h, and the PTX IC50 was 50 nM. The cell culture was continued after the addition of PTX (50 nM) to the five groups, and cell proliferation was monitored at 24, 48 and 72 h. At 48 h, the LV-ECT2 group had an inhibitory rate (IR) significantly lower than that of the control and LV-NC groups (P < 0.05), whereas the IR of the sh-ECT2 group was significantly higher than those of the control and sh-NC groups (P < 0.05) (Figure 2B and C).
Effects of ECT2 Overexpression and Interference and PTX Therapy on Migration and Invasion of Breast Cancer Cells
Effect on Migration of PTX-Treated Breast Cancer Cells
In terms of cell migration, marked changes were noted in the LV-ECT2 group. Compared with the control and LV-NC groups, the LV-ECT2 group had a notably higher cell migration rate, and the difference was statistically significant (P < 0.05). In the sh-ECT2 group, the cell migration rate dropped sharply and was significantly different from that in the control and sh-NC groups (P < 0.05) (Figure 3A and B).
 |
Figure 3 Cell migration assay. (A) The pictures of cell migration in each group. (B) The number of cell migration in 5 groups was compared in the histogram. *P <0.05. |
Following PTX treatment, all five groups exhibited decreased cell migration at varying degrees. The cell migration rate of the LV-ECT2 group did not reduce as drastically as those of the control and LV-NC groups, but the differences showed statistical significance (P < 0.05). In the sh-ECT2 group, the cell migration rate dropped sharply and was significantly different from that in the control and sh-NC groups (P < 0.05) (Figure 4A and B).
 |
Figure 4 Cell migration assay after PTX treatment. (A) The pictures of cell migration in each group. (B) The number of cell migration in 5 groups was compared in the histogram. *P <0.05. |
Effect on Invasion of PTX-Treated Breast Cancer Cells
Results from the transwell invasion assay showed that the LV-ECT2 group exhibited a significantly higher invasiveness than the control and LV-NC groups, and statistical analysis indicated that the differences were statistically significant (P < 0.05). Compared with the control and sh-NC groups, the sh-ECT2 group showed a significantly lower level of invasiveness (P < 0.05) (Figure 5A and B).
 |
Figure 5 Cell invasion assay. (A) The pictures of cell invasion in each group. (B) The number of cell invasion in 5 groups was compared in the histogram. *P <0.05. |
PTX was later administered to the five groups accordingly. The LV-ECT2 group experienced a drop in the number of invasive cells, although they still outnumbered the invasive cells in the control and LV-NC groups, and the differences had statistical significance (P < 0.05). In the sh-ECT2 group, the number of invasive cells was reduced substantially, and compared with the control and sh-NC groups, the differences were statistically significant (P < 0.05) (Figure 6A and B).
 |
Figure 6 Cell invasion assay after PTX treatment. (A) The pictures of cell invasion in each group. (B) The number of cell invasion in 5 groups was compared in the histogram. *P <0.05. |
Effects of ECT2 Overexpression and Interference and PTX Therapy on Breast Cancer Cell Apoptosis
Flow cytometry (FCM) was performed to analyse the apoptotic rates of breast cancer cells in the five groups. Comparison of the LV-ECT2 group and its counterparts (ie, control and LV-NC) showed that the apoptotic rate of the LV-ECT2 group was markedly reduced, and the differences were statistically significant (P < 0.05). On the other hand, the sh-ECT2 group showed a considerable increase in the apoptotic rate, and the differences between the sh-ECT2 group and the control and sh-NC groups showed statistical significance (P < 0.05) (Figure 7A and B). After the PTX treatment, cell apoptosis in the five groups increased at varying rates. However, compared with the control and LV-NC groups, the change in the apoptotic rate of the LV-ECT2 group was minimal, but the differences were statistically significant (P < 0.001). The sh-ECT2 group had an apoptotic rate significantly higher than that of the remaining four groups, and the differences between the sh-ECT2 group and the control and sh-NC groups exhibited statistical significance (P < 0.001) (Figure 8A and B).
 |
Figure 7 Cell apoptosis assay. (A) Changes of apoptosis rate in each group. (B) The cell apoptosis rate in 5 groups was compared in the histogram. *P <0.05. |
 |
Figure 8 Cell apoptosis assay after PTX treatment. (A) Changes of apoptosis rate in each group. (B) The cell apoptosis rate in 5 groups was compared in the histogram. **P <0.001. |
Effects of ECT2 Overexpression and Interference and PTX Therapy on Cell Cycle of Breast Cancer Cells
In the FCM, the cell cycle was divided into three compartments, including G0/G1, S and G2/M phases, based in the intracellular DNA content. The cell cycle analysis showed that compared with the control and LV-NC groups, the LV-ECT2 group exhibited marginal changes in the percentage (%) of cells and DNA content in the G1 and S phases but underwent substantial changes in both respects during the G2/M phase. Likewise, the sh-ECT2 group, in comparison with the control and sh-NC groups, also experienced small changes in the G1 and S phases but had major changes in the G2/M phase, in which concerning values for the percentage (%) of cells and DNA content were observed (Figure 9A and B). After PTX was given, significant differences were observed between the LV-ECT2 group and the control and LV-NC groups in terms of the percentage (%) of cells and DNA content during the G1 and G2/M phases, whereas the sh-ECT2 group was significantly different from the control and sh-NC groups in terms of the percentage (%) of cells and DNA content during the G2/M phase (Figure 10A and B).
 |
Figure 9 Cell cycle assay. (A) Cell cycle outcome diagram in each group. (B) A comparison of the cell cycle phases is shown in the histogram. *P <0.05. |
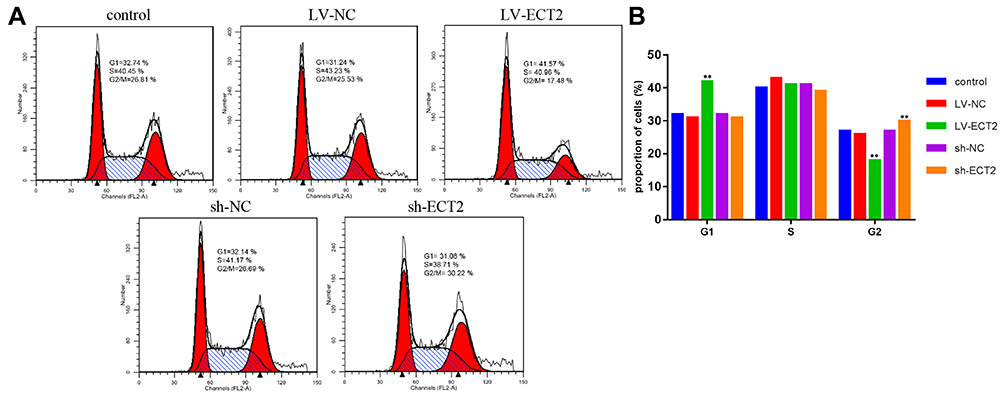 |
Figure 10 Cell cycle assay after PTX treatment. (A) Cell cycle outcome diagram in each group. (B) A comparison of the cell cycle phases is shown in the histogram. **P <0.001. |
Effects of ECT2 Overexpression and Interference and PTX Therapy on Nude Mouse Xenograft Tumour Growth
During PTX treatment, each nude mouse was weighed every two days, and the ratio of long/short diameter of each tumour was measured using a Vernier caliper. Experimental results showed that during the assay, the control, LV-ECT2 and sh-ECT2 groups became substantially heavier than the PTX, LV-ECT2-PTX and sh-ECT2-PTX groups, and the differences were statistically significant (P < 0.05). In addition, in the sh-ECT2-PTX group, the body weight and tumour size changes in the nude mice were sluggish and stagnant. Except for the LV-ECT2-PTX group, the remaining groups were all significantly different from the control group in terms of body weight and tumour size changes (P < 0.05) (Figure 11A–D).
 |
Figure 11 Nude mice experiment. (A) The tumor size in 6 groups. (B) The weight curves of nude mice in 6 groups at 21 days. (C) The changes of tumor weight of 6 groups after 21 days in the histogram. (D) The volume curves of tumor in 6 groups at 21 days. ** P <0.001. |
Effects of ECT2 Overexpression and Interference and PTX Therapy on Apoptosis and Proliferation of Nude Mouse Xenograft Tumour Tissues
In the control, LV-ECT2 and LV-ECT2-PTX groups, the tumour tissues presented very limited numbers of apoptotic cells. By contrast, apoptosis occurred often in the PTX, sh-ECT2 and sh-ECT2-PTX groups. Thus, cell apoptosis was promoted in the PTX-treated groups, in which ECT2 was underexpressed. Particularly, apoptosis occurred in over 50% of the tumour cells in the sh-ECT2-PTX group (Figure 12A).
 |
Figure 12 (A) Cell apoptosis assay by tunnel. The pictures of tumor cell apoptosis in each group. (B) Immunohistochemical expression of Ki-67 protein in each group. |
Ki-67 IHC staining was performed to assess cell proliferation in each group. If a section was stained evenly, 200 cells were counted at 20x magnification in five different vision fields to average the results. Otherwise, cells in cold and hot zones were counted to average the results.2,3 Comparison of the proliferation indexes of the control group and the PTX, LV-ECT2-PTX, sh-ECT2 and sh-ECT2-PTX groups showed statistically significant differences (P < 0.05). Although the LV-ECT2 group had the highest proliferation index, in this respect, it showed no significant difference compared with the control group (P > 0.05) (Figure 12B).
Discussion
ECT2 refers to the epithelial cell transforming sequence 2. As a guanine nucleotide exchange factor for Rho GTPases, ECT2 is associated with diverse malignant phenotypes of human cancers.4 ECT2 resides on chromosome 3q26, a highly conserved proto-oncogene. It was isolated from the cDNA expression library of keratinocytes.5 The DH-PH-C domain of ECT2 lacks N-terminal sequence, which can regulate the activity and nuclear localization of Ect2. When the nuclear membrane breaks down, in the process of mitosis, Ect2 mutates from the nucleus to the whole cytoplasm. Ect2 localized in the cytoplasm has constitutive GEF activity and can transform fibroblasts and induce cell division.6,7 This wrong localization of Ect2 in the cytoplasm plays a critical role in the oncogenic function of Ect2.5 When the cells lacking in Ect2, the formation of cell cleavage furrow and the assembly of contraction ring appear defects and cause cytokinesis arrest. But, the transformation activity of Ect2 was activated when a large amount of Ect2 accumulates in the cytoplasm, which promoted cell division and transformation. This is also the reason why Ect2 is located in the cytoplasm in some cancer cells during the interphase period.8 The expression level of ECT2 is strongly associated with cell cycle control and cell division.9 In the G2/M phase, ECT2 is phosphorylated and promotes cell division by binding with different factors.10 Up to now, ECT2 expression is upregulated in a range of cancers, including lung cancer,11 ovarian cancer,12 bile duct cancer13 and gliomas.14,15 Thus, ECT2 mainly promotes the proliferation of cancer cells at the G2/M phase.
ECT2 has a relatively high expression level in different breast cancer cells and promotes tumour progression.16,17 Mansour et al16 reported that expression level of Ect2 may be a marker of TNBC, which can predict the invasion and metastasis of tumour. In our preliminary experiment, ECT2 was highly expressed in TNBC and associated with the prognosis of the disease.1 TNBC is the most invasive breast cancer that lacks known specific molecular targets and targeted therapy, with lumpectomy, radiotherapy and cytotoxic chemotherapy as the mainstays of treatment options. Given that B-TNBC accounts for over 50% of all TNBC cases, in this study, the highly proliferative and invasive HCC1806 breast cancer cells were used for in vivo and in vitro assays.18 The in vitro assays showed that ECT2 overexpression yielded a remarkable improvement in the abilities of proliferation, invasion and migration, whereas silencing ECT2 meant decreased proliferation, invasion and migration of cancer cells. Moreover, the apoptotic rate in the LV-ECT2 group was reduced significantly, whereas the sh-ECT2 group saw a marked increase in the number of apoptotic cells. All these findings indicate that ECT2 expression is, to a certain extent, related to the growth of cancer cells. Overexpressed ECT2 promotes proliferation, invasion and migration of TNBC cells and inhibits cell apoptosis. On the contrary, silencing ECT2 can inhibit TNBC cell proliferation, invasion and migration and accelerate cell apoptosis. After PTX took effect, the proliferation, invasion and migration of TNBC cells were significantly inhibited, and the number of apoptotic cells was elevated substantially. In the sh-ECT2 group, the apoptotic rate climbed sharply. According to the assay results, malignant progression was strongly affected by ECT2, regardless of whether it was in overexpressed or silenced form. In combination with the use of PTX, ECT2 inhibited malignant transformation, which is probably associated with certain signalling pathways at the molecular level.
The cell cycle analysis showed that ECT2 mainly promoted the proliferation of cancer cells during the G2/M phase. When a cell proceeds to the G2 phase, ie, the gap between DNA synthesis and mitosis, large numbers of RNA, proteins (including MTPs) and other substances required for mitosis will be synthesised. During this period, ECT2 binds with diverse molecules, thereby promoting cell division and proliferation.19,20 In non-small cell lung cancer, ECT2 promotes cell proliferation during the G2/M phase. In this phase, ECT2 requires phosphorylation of the T328 residue by PKCι to achieve the proliferation-promoting effect.21,22 Hara et al23 discovered that in HEK 293T cells, T341 served as the phosphorylation site; with increased activity, phosphorylated ECT2, together with other relevant factors, regulated the expression of ECT2, ensuring ECT2’s function as a cell division promoter. All these events occurred in the G2/M phase. Therefore, ECT2 expression is elevated as DNA synthesis proceeds during the G2/M phase, which in turn promotes DNA synthesis and cell division.24 In the present study, we inferred that ECT2 in the breast cancer cells was also phosphorylated, but further study is needed to identify the phosphorylation site. In the case of ECT2 overexpression, enhanced proliferation and reduced apoptosis were observed in TNBC cells, that is, exogenous ECT2 played an important role in the proliferation of TNBC cells, which primarily occurred in the G2/M phase. When ECT2 in the TNBC cells was silenced, decreased proliferation, invasion and migration were detected; the apoptotic rate and percentage (%) of cells in the G2/M phase were elevated significantly. These results indicate a cell division arrest during the G2/M phase following the downregulation of ECT2, which resulted in an inhibitory effect on the proliferation of cancer cells and a significant increase in the apoptotic rate. On this basis, the proliferation of breast cancer cells may be effectively inhibited by downregulating endogenous ECT2 expression, offering a real breakthrough in the treatment of breast cancer.
High-dose PTX inhibits tumour growth by blocking cell proliferation and promoting massive cell death at the G2/M phase. However, if a lower dose is administered, PTX may only induce MTP accumulation resulting in the inhibition of spindle formation, thereby arresting the cell cycle at the G2/M phase.25,26 This study used a concentration gradient of PTX to determine the PTX IC50 for the HCC1806 cell line in relevant assays. When treated with PTX, the LV-ECT2 group significantly differed from the control and LV-NC groups in terms of cell proliferation, invasion, migration and apoptosis, whereas no such statistically significant differences were observed between the control and LV-NC groups. Additionally, the LV-ECT2 group significantly differed from the control and LV-NC groups regarding changes in the percentage (%) of cells and DNA content in the G2/M-phase. Thus, PTX mainly induced cell apoptosis at the G2/M phase. In cancer cells with overexpressed ECT2, PTX not only inhibited exogenous ECT2 but also reasonably downregulated the expression of endogenous ECT2. Therefore, an antagonistic effect existed between ECT2 and PTX in these cells. We know that a large number of microtubules are accumulated in the cells after paclitaxel contacts cells and can not form spindle body and spindle filament, so cell division is arrested in G2 and M phases. But the role of Ect2 is to promote the spindle to pull chromosomes and then complete cell division. Viewed from this perspective, can we infer from our experimental results that Ect2 can only act on the spindle, but not depolymerize microtubule of formed spindle, so it can not fundamentally antagonize the effect of paclitaxel. Of course, further studies should be undertaken to investigate the underlying mechanism. In nasopharynx cancer cells, dishevelled-associated antagonist of β-catenin homolog 2 is an effective inhibitor that induces the G2/M phase arrest, inhibits cell proliferation and promotes cell apoptosis, making the cancer cells highly sensitive to PTX.27 In this study, cell proliferation in the sh-ECT2 group was significantly reduced following PTX treatment, whereas its apoptotic rate was markedly increased, especially at the G2/M phase. These findings indicate that the cancer cells exhibited a significantly higher level of sensitivity to PTX after ECT2 has been silenced. Thus, the downregulation of ECT2 expression is an effective approach to further improve the sensitivity of cancer cells to PTX, inhibit cancer cell proliferation and promote cell apoptosis. The combination of PTX and a given TCM extract has a synergistic effect on inhibiting the proliferation and promoting the apoptosis of prostate cancer cells through G2/M phase arrest.28 Reportedly, the extract of Trametes robiniophila Murr. (Huaier) and PTX has a synergistic effect on inhibiting the growth of two breast cancer cell lines (MCF-7 and MDA-MB-231) in vitro. In vivo studies have demonstrated that these medicines, especially when used in combination, can significantly inhibit the growth of xenograft tumours, although they induce cell cycle arrests in different phases, with the TCM extract mainly taking effect during the G0/G1 phase.29
In this study, each nude mouse in the LV-ECT2 group showed negligible changes in body volume. However, the tumour size and weight were significantly higher than those of the nude mice in other groups. Further, the apoptotic rate of the LV-ECT2 group approached zero, whereas the Ki-67 proliferation index was up to 90%. This condition indicates that ECT2 overexpression can significantly promote the proliferation of cancer cells. After PTX treatment, the LV-ECT2 group slightly exceeded the control group in terms of tumour size and weight. However, the apoptotic rates of the two groups were close to each other. Meanwhile, the Ki-67 proliferation index of the LV-ECT2 group was significantly lower than that of the control group probably because PTX produced a strong antagonistic effect on ECT2 expression. As a result, this condition might have failed to fully inhibit tumour growth but effectively inhibited the tumour proliferative activity. PTX can possibly inhibit ECT2 expression in breast cancer cells, but the underlying mechanism is still unclear. No significant difference was observed in the tumour growth between the sh-ECT2 and PTX groups. Although tumour size and weight in the PTX group grew slightly compared with those of the control group, the nude mice continually gained weight following PTX treatment. Further evidence is needed to confirm whether tumour growth can be inhibited in this case to keep the nude mice alive with tumour permanently. The tumour growth in the sh-ECT2 group was almost stagnant at 21 days after PTX treatment, the apoptotic rate exceeded 50%, and the Ki-67 proliferation index dropped to the lowest level. Still, no significant changes were noted in the body weight and tumour size compared with the early stage of PTX treatment. Analysis revealed that silencing endogenous ECT2 and the use of PTX may produce a therapeutic effect. When endogenous ECT2 was silenced, and PTX was administered as a combination, the therapeutic effect was multiplied, and the nude mice almost stopped growing. These results indicate that silencing ECT2 also improved the sensitivity to PTX.
E3 ubiquitin ligase E6-associated protein (E6AP) can inhibit the metastasis of breast cancer by regulating the activity of ECT2 and Rho GTPase, with the E6AP-ECT2 axis serving as the therapeutic target.16 In vivo and in vitro assays and positive and negative validation showed that ECT2 can accelerate breast cancer, and tumour growth was significantly inhibited by downregulating ECT2. Additionally, PTX lowered the overall proliferation ability and increase the apoptotic rate of cancer cells in vivo and in vitro. In accordance with the changes in cell proliferation and apoptosis in the sh-ECT2-PTX group, this assumption was especially true when ECT2 was silenced. ECT2 in TNBC plays an important regulatory role in cell proliferation, invasion, migration and apoptosis. However, concrete evidence is still needed to fully understand how ECT2 promotes the development and progression of breast cancer and identify relevant signalling pathways involved in this process. In the study of esophageal squamous cell carcinoma, it was found that Ect2 can regulate the expression of VEGF and MMP9 through RhoA-ERK signaling pathway to inhibit tumour cells progression.30 It is also found that Ect2 is closely related to the activation of Rho/ERK signal transduction axis in hepatocellular carcinoma. Knockdown of ECT2 can significantly inhibit the activity of Rho GTPases and the activation of ERK, reduce the ability of cell proliferation and migration.31 It has been reported that the targeted inhibition of Ect2 activity may lead to the blocking of Rac1/RhoA/Cdc42 downstream signaling pathway, and then inhibit the progress of breast cancer.16 So, Rho/ERK or Rac1/RhoA/Cdc42 or other signaling pathways are related to the role of Ect2 in breast cancer, which needs further study. Thus, ECT2 is probably a key driving factor in the growth of breast cancer cells. Silencing ECT2 causes a remarkable improvement in the therapeutic effect of PTX, which offers breast cancer patients a new treatment option. A study reported that ECT2 is targeted during the S phase and is deactivated when protease-dependent degradation occurs following cell division in the G2/M phase.32 This condition proves the specificity of ECT2 and provides strong evidence for its use in a future potential targeted therapy that may outperform PTX in treating breast cancer.
Funding
Beijing Municipal Education Commission (KZ201910025033), Beijing, China. Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding (ZYLX101814), Beijing, China.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Wang HK, Liang JF, Zheng HX, Xiao H. Expression and prognostic significance of ECT2 in invasive breast cancer. J Clin Pathol. 2018;71(5):442–445. doi:10.1136/jclinpath-2017-204569
2. Jain P, Doval DC, Batra U, et al. Ki-67 labeling index as a predictor of response to neoadjuvant chemotherapy in breast cancer. Jpn J Clin Oncol. 2019;49(4):329–338. doi:10.1093/jjco/hyz012
3. Del Rosario Taco Sanchez M, Soler-Monsó T, Petit A, et al. Digital quantification of KI-67 in breast cancer. Virchows Arch. 2019;474(2):169–176. doi:10.1007/s00428-018-2481-3
4. Jin Y, Yu Y, Shao Q, et al. Up-regulation of ECT2 is associated with poor prognosis in gastric cancer patients. Int J Clin Exp Pathol. 2014;7(12):8724–8731.
5. Miki T, Smith CL, Long JE, et al. Oncogene ect2 is related to regulators of small GTP-binding proteins. Nature. 1993;362(6419):462–465. doi:10.1038/362462a0
6. Saito S, Tatsumoto T, Lorenzi MV, et al. Rho exchange factor ECT2 is induced by growth factors and regulates cytokinesis through the N-terminal cell cycle regulator-related domains. J Cell Biochem. 2003;90(4):819–836. doi:10.1002/jcb.10688
7. Chen M, Pan H, Sun L, et al. Structure and regulation of human epithelial cell transforming 2 protein. Proc Natl Acad Sci U S A. 2020;117(2):1027–1035. doi:10.1073/pnas.1913054117
8. Justilien V, Lewis KC, Murray NR, Fields AP. Oncogenic Ect2 signaling regulates rRNA synthesis in NSCLC. Small GTPases. 2019;10(5):388–394. doi:10.1080/21541248.2017.1335274
9. Tan H, Wang X, Yang X, Li H, Liu B, Pan P. Oncogenic role of epithelial cell transforming sequence 2 in lung adenocarcinoma cells. Exp Ther Med. 2016;12(4):2088–2094. doi:10.3892/etm.2016.3584
10. Niiya F, Tatsumoto T, Lee KS, Miki T. Phosphorylation of the cytokinesis regulator ECT2 at G2/M phase stimulates association of the mitotic kinase Plk1 and accumulation of GTP-bound RhoA. Oncogene. 2006;25(6):827–837. doi:10.1038/sj.onc.1209124
11. Ulke HM, Mutze K, Lehmann M, et al. The oncogene ECT2 contributes to a hyperplastic, proliferative lung epithelial cell phenotype in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2019;61(6):713–726. doi:10.1165/rcmb.2019-0047OC
12. Zhou S, Wang P, Su X, et al. High ECT2 expression is an independent prognostic factor for poor overall survival and recurrence-free survival in non-small cell lung adenocarcinoma. PLoS One. 2017;12(10):e0187356. doi:10.1371/journal.pone.0187356
13. Gao J, Dai C, Yu X, Yin XB, Zhou F. Upregulated microRNA-194 impairs stemness of cholangiocarcinoma cells through the Rho pathway via inhibition of ECT2. J Cell Biochem. 2020. doi:10.1002/jcb.29648
14. Cheng YS, Lin C, Cheng YP, Yu YL, Tang CT, Hueng DY. Epithelial cell transformation sequence 2 is a potential biomarker of unfavorable survival in human gliomas. Neurol India. 2014;62(4):406–409. doi:10.4103/0028-3886.141278
15. Sano M, Genkai N, Yajima N, et al. Expression level of ECT2 proto-oncogene correlates with prognosis in glioma patients. Oncol Rep. 2006;16(5):1093–1098.
16. Mansour M, Haupt S, Chan AL, et al. The E3-ligase E6AP represses breast cancer metastasis via regulation of ect2-rho signaling. Cancer Res. 2016;76(14):4236–4248. doi:10.1158/0008-5472.CAN-15-1553
17. Tanaka I, Chakraborty A, Saulnier O, et al. ZRANB2 and SYF2-mediated splicing programs converging on ect2 are involved in breast cancer cell resistance to doxorubicin. Nucleic Acids Res. 2020;48(5):2676–2693. doi:10.1093/nar/gkz1213
18. van Geldermalsen M, Wang Q, Nagarajah R, et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene. 2016;35(24):3201–3208. doi:10.1038/onc.2015.381
19. He D, Xiang J, Li B, Liu H. The dynamic behavior of Ect2 in response to DNA damage. Sci Rep. 2016;6:24504. doi:10.1038/srep24504
20. Cook DR, Solski PA, Bultman SJ, et al. The ect2 rho Guanine nucleotide exchange factor is essential for early mouse development and normal cell cytokinesis and migration. Genes Cancer. 2011;2(10):932–942. doi:10.1177/1947601912437035
21. Justilien V, Fields AP. Ect2 links the PKCiota-Par6alpha complex to Rac1 activation and cellular transformation. Oncogene. 2009;28(41):3597–3607. doi:10.1038/onc.2009.217
22. Justilien V, Jameison L, Der CJ, Rossman KL, Fields AP. Oncogenic activity of Ect2 is regulated through protein kinase C iota-mediated phosphorylation. J Biol Chem. 2011;286(10):8149–8157. doi:10.1074/jbc.M110.196113
23. Hara T, Abe M, Inoue H, et al. Cytokinesis regulator ECT2 changes its conformation through phosphorylation at Thr-341 in G2/M phase. Oncogene. 2006;25(4):566–578. doi:10.1038/sj.onc.1209078
24. Murata Y, Minami Y, Iwakawa R, et al. ECT2 amplification and overexpression as a new prognostic biomarker for early-stage lung adenocarcinoma. Cancer Sci. 2014;105(4):490–497. doi:10.1111/cas.12363
25. Yoo HJ, Park J, Yoon TH. High throughput cell cycle analysis using microfluidic image cytometry (μFIC). Cytometry A. 2013;83(4):356–362. doi:10.1002/cyto.a.22261
26. Torres K, Horwitz SB. Mechanisms of taxol-induced cell death are concentration dependent. Cancer Res. 1998;58(16):3620–3626.
27. Zhang Y, Fan J, Fan Y, et al. The new 6q27 tumor suppressor dact2, frequently silenced by CPG methylation, sensitizes nasopharyngeal cancer cells to paclitaxel and 5-fu toxicity via β-catenin/cdc25c signaling and g2/m arrest. Clin Epigenetics. 2018;10(1):26. doi:10.1186/s13148-018-0459-2
28. Doğan Şiğva ZÖ, Balci Okcanoğlu T, Biray Avci Ç, et al. Investigation of the synergistic effects of paclitaxel and herbal substances and endemic plant extracts on cell cycle and apoptosis signal pathways in prostate cancer cell lines. Gene. 2019;687:261–271. doi:10.1016/j.gene.2018.11.049
29. Yang L, Song Z, Wang X, Yang W, Wang M, Liu H. Huaier extract enhances the treatment efficacy of paclitaxel in breast cancer cells via the nf-κb/iκbα pathway. Oncol Rep. 2017;38(6):3455–3464. doi:10.3892/or.2017.6024
30. Sun BY, Wei QQ, Liu CX, et al. ECT2 promotes proliferation and metastasis of esophageal squamous cell carcinoma via the RhoA-ERK signaling pathway. Eur Rev Med Pharmacol Sci. 2020;24(15):7991–8000. doi:10.26355/eurrev_202008_22482
31. Chen J, Xia H, Zhang X, et al. ECT2 regulates the Rho/ERK signalling axis to promote early recurrence in human hepatocellular carcinoma. J Hepatol. 2015;62(6):1287–1295. doi:10.1016/j.jhep.2015.01.014
32. Liot C, Seguin L, Siret A, Crouin C, Schmidt S, Bertoglio J. APC (cdh1) mediates degradation of the oncogenic Rho-GEF Ect2 after mitosis. PLoS One. 2011;6(8):e23676. doi:10.1371/journal.pone.0023676
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.