Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 13
Effect of background long-acting beta2-agonist therapy on the efficacy and safety of a novel, nebulized glycopyrrolate in subjects with moderate-to-very-severe COPD
Authors Kerwin EM ![]() , Tosiello R
, Tosiello R ![]() , Price B
, Price B ![]() , Sanjar S
, Sanjar S ![]() , Goodin T
, Goodin T
Received 26 April 2018
Accepted for publication 30 July 2018
Published 19 September 2018 Volume 2018:13 Pages 2917—2929
DOI https://doi.org/10.2147/COPD.S172408
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Edward M Kerwin,1 Robert Tosiello,2 Barry Price,2 Shahin Sanjar,2 Thomas Goodin2
1Clinical Research Institute of Southern Oregon, Inc., Medford, OR, USA; 2Sunovion Pharmaceuticals Inc., Marlborough, MA, USA
Background: Phase III studies demonstrated efficacy and safety of nebulized glycopyrrolate inhalation solution (GLY) in subjects with COPD. Secondary analyses were performed to examine the effect of background long-acting beta2-agonist (LABA) use on the efficacy and safety of nebulized GLY.
Methods: In two 12-week placebo-controlled studies (GOLDEN 3 and GOLDEN 4) and one 48-week, open-label active-controlled study (GOLDEN 5), a total of 2,379 subjects were stratified by background LABA use (LABA-yes: n=861; LABA-no: n=1,518) and randomized to placebo vs GLY 25 or 50 µg twice daily, or GLY 50 µg twice daily vs tiotropium (TIO) 18 µg once daily. Lung function, patient-reported outcomes, exacerbations, and safety were assessed.
Results: Compared with placebo, pooled data from the 12-week studies showed significant improvements from baseline with GLY 25 and 50 µg across LABA subgroups in trough FEV1 (LABA-yes: 0.101 and 0.110 L; LABA-no: 0.092 and 0.101 L, respectively; P<0.001) and St George’s Respiratory Questionnaire total score (SGRQ; LABA-yes: -2.957 and -3.888; LABA-no: -3.301 and -2.073, respectively; P<0.05). Incidence of treatment-emergent adverse events (TEAEs) was similar in LABA subgroups, and lower in GLY 25 µg vs placebo. In the 48-week active-controlled study, GLY and TIO both showed improvement from baseline across LABA subgroups in FEV1 (LABA-yes: 0.106 and 0.092 L; LABA-no: 0.096 and 0.096 L, respectively) and in SGRQ total score (LABA-yes: -5.190 and -3.094; LABA-no: -4.368 and -4.821, respectively). Incidence of TEAEs was similar between GLY and TIO, and across LABA subgroups. Exacerbation rates were similar across treatments and LABA subgroups, and cardiovascular events of special interest were more frequent in the LABA-no subgroup. Nebulized GLY, combined with LABA, did not generate any additional safety signals.
Conclusion: Nebulized GLY demonstrated efficacy and was well tolerated up to 48 weeks in subjects with COPD with/without background LABA.
Keywords: COPD, nebulized glycopyrrolate, eFlow® CS, LAMA, background LABA
Plain language summary
COPD is the third leading cause of death in the US. Patients with COPD have limited airflow to the lungs and experience symptoms such as breathlessness and cough. COPD limits the ability to work, exercise, and carry out everyday activities and can reduce quality of life. Treatment of COPD with bronchodilator therapy is important for disease management. Bronchodilators improve health status, reduce symptoms, and can decrease the frequency of COPD exacerbations. Different types of bronchodilators can be used alone, or in combination, to manage COPD; the treatment depends on an individual patient’s needs. Nebulized glycopyrrolate inhalation solution (GLY) is the first nebulized long-acting muscarinic antagonist (LAMA) approved in the US by the Food and Drug Administration for the treatment of patients with COPD. This analysis examines whether the continued use of another bronchodilator, a long-acting beta2-agonist (LABA) – with or without inhaled corticosteroid (ICS) treatment – affects the efficacy and safety of nebulized GLY in patients with moderate-to-very-severe COPD. Data from three Phase III trials were analyzed: two 12-week, placebo-controlled studies and one 48-week, open-label active-controlled study. Approximately one-third of patients were using combination LAMA/LABA (±ICS) therapy. We found that lung function and patient-reported outcomes were significantly improved and the overall and cardiovascular safety profiles were acceptable in patients treated with nebulized GLY with and without background LABA (±ICS). Nebulized GLY benefits COPD patients receiving LABA (±ICS), as well as those on rescue medication only.
Introduction
COPD is characterized by airflow limitation and persistent respiratory symptoms, the most common symptoms being dyspnea, cough, and sputum production.1 An estimated 15.7 million American adults have been diagnosed with COPD,2 and the disease is the third leading cause of death in the US.3 Patients with COPD have a considerably reduced quality of life,4–6 and the disease limits an individual’s ability to work, exercise, and participate in everyday social activities.4,5
Treatment of COPD with bronchodilator therapy is a key approach to disease management and can reduce symptoms, improve health status, and may reduce the frequency and severity of exacerbations.1 Current treatment paradigms include the use of a long-acting muscarinic antagonist (LAMA) as initial maintenance therapy for patients with low exacerbation risk, but high burden of symptoms.1 LAMAs are preferred to long-acting beta2-agonists (LABAs) in these cases due to reduced risk of exacerbation.7,8 An increase in exacerbation risk or symptom burden may warrant an escalation in treatment, for example, to a combination of a LAMA with a LABA or LABA/inhaled corticosteroid (ICS).1 The availability of a range of LAMA mono- and combination therapies enables clinicians to personalize a patient’s treatment over their COPD disease continuum. Patients at risk of frequent exacerbations may benefit from “triple therapy,” which can be achieved by combining LAMA with a separately dosed LABA/ICS.9
On the basis of the Phase III GOLDEN studies, the US Food and Drug Administration (FDA) approved GLY (LONHALA®, Sunovion Pharmaceuticals Inc., Marlborough, MA, USA; 25 μg twice daily [BID]), delivered by the innovative eFlow® Closed System (CS) nebulizer (MAGNAIR®, PARI Pharma GmbH, Starnberg, Germany), for the long-term maintenance treatment of airflow obstruction in patients with COPD.10 This is the first nebulized LAMA approved by the FDA for the maintenance treatment of COPD. Specifically, the efficacy and safety of this nebulized GLY were evaluated in three randomized, parallel-group, multicenter Phase III studies: two replicate, 12-week, placebo-controlled studies (GOLDEN 3 [NCT02347761] and GOLDEN 4 [NCT02347774]),11 and one 48-week, active-controlled, open-label study (GOLDEN 5 [NCT02276222]).12
Published findings from the placebo-controlled studies showed that nebulized GLY demonstrated statistically significant and clinically important improvements in lung function, improved patient-reported outcomes over 12 weeks, and was well tolerated.11 The long-term, active-controlled safety study established that nebulized GLY was well tolerated, with no overall and cardiovascular (CV) safety signals, with improvements in FEV1 and health status that persisted over 48 weeks.12
All three Phase III GOLDEN studies were prospectively designed to include stratification of subjects using background LABA or LABA/ICS therapy throughout the treatment period. The patient populations were representative of the general COPD population and included subjects with very severe COPD (FEV1 % predicted <30%),1 as well as those with CV risk factors. Subgroup analyses were performed to assess the effect of background LABA therapy on nebulized GLY efficacy and safety outcomes.
Methods
Study design and patients
The designs for the Phase III GOLDEN studies have been published previously (Figure 1).11,12 Briefly, in the 12-week, multicenter, placebo-controlled, double-blind studies (GOLDEN 3 and GOLDEN 4), subjects (N=1,294) were randomized in a 1:1:1 ratio to receive placebo or GLY (25 or 50 μg BID), via the eFlow CS nebulizer. In the 48-week, multicenter, active-controlled, open-label study (GOLDEN 5), subjects (N=1,086) were randomized in a 4:3 ratio to receive GLY 50 μg BID delivered via eFlow CS nebulizer or tiotropium (TIO) 18 μg once daily (QD) delivered via the HandiHaler® dry powder inhaler. Randomization in each of the studies was stratified by background LABA use (yes/no) and by CV risk (high/low). Approximately 30% of subjects (limited by protocol) in GOLDEN 3 and GOLDEN 4, and ~40% in GOLDEN 5 continued background LABA use (with or without a concomitant ICS) during the treatment period. Nearly two-thirds of the study populations had pre-existing CV risk factors. Ipratropium bromide, as supplemental medication, and albuterol (salbutamol), as rescue medication, were permitted.
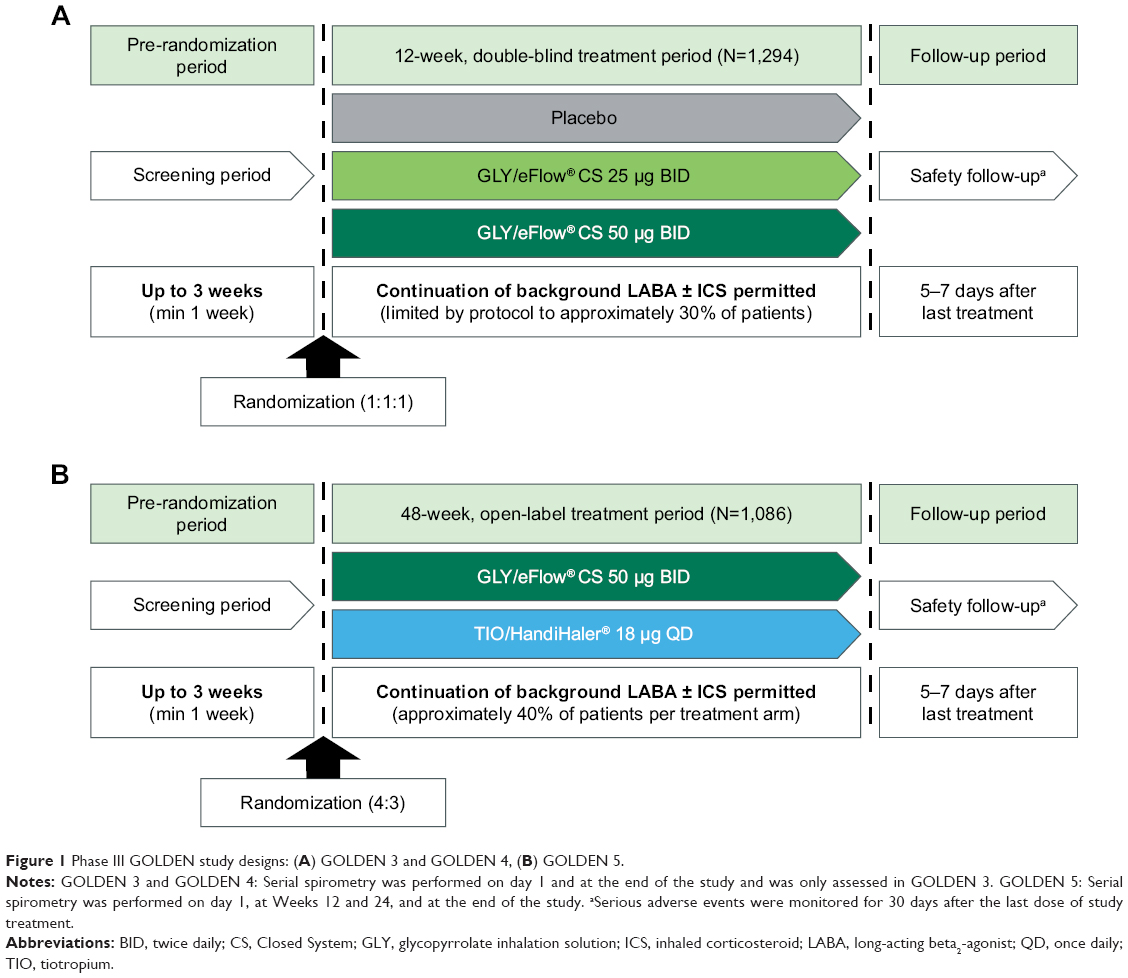 | Figure 1 Phase III GOLDEN study designs: (A) GOLDEN 3 and GOLDEN 4, (B) GOLDEN 5. |
In all Phase III studies, key eligibility criteria included males or females aged ≥40 years, current or ex-smokers with ≥10 pack-year smoking history, a clinical diagnosis of moderate-to-very-severe COPD (as defined by the Global Initiative for Chronic Obstructive Lung Disease [GOLD] 2014 criteria),1 and qualifying post-bronchodilator (ipratropium 68 μg) spirometry (FEV1 ≤80% of predicted normal, FEV1 >0.7 L, and FEV1/forced vital capacity ratio <0.70).
The GOLDEN 3 (SUN101-301: project approval number 28481) and GOLDEN 4 (SUN101-302: project approval number 28482) study protocols were approved by Quorum Review institutional review board (IRB) North American (US and Canadian) Board (Panel II), and GOLDEN 5 (SUN101-303: project approval number 28446) by Quorum Review IRB US Board (Panel IV), prior to patient enrollment and were conducted in accordance with the protocols, International Council for Harmonization Good Clinical Practice guidelines, and Declaration of Helsinki. All subjects provided written informed consent.
Statistical analysis
All statistical procedures were performed using SAS® v9.2 (SAS Institute Inc., Cary, NC, USA).
LABA subgroup analyses were pre-specified as secondary analyses. Reported P-values from hypothesis tests for differences between treatments within subgroups were not adjusted for multiplicity. Efficacy results are expressed as least squares (LS) mean ± standard error (SE).
Twelve-week, placebo-controlled studies
Data from the replicate GOLDEN 3 and GOLDEN 4 studies were pooled for analysis. The primary pooled efficacy endpoint was change from baseline in trough FEV1 at week 12. Other pooled efficacy endpoints included change from baseline in health status measured by St George’s Respiratory Questionnaire (SGRQ)13 total score at week 12, SGRQ responder rate (a responder was defined as a subject with a ≥4.0 unit reduction in SGRQ total score), change from baseline in Evaluating Respiratory Symptoms in COPD (E-RS™; Evidera Inc., Bethesda, MD, USA)14 total score, and incidence of exacerbations. A COPD exacerbation was defined as an increase in clinician-assessed COPD symptoms (eg, dyspnea, cough, sputum volume, or sputum purulence) during at least 2 consecutive days and was classified as mild (self-managed, with increased short-acting bronchodilator and/or ICS use), moderate (treated with antibiotics and/or systemic corticosteroids), or severe (requiring hospitalization). Any event occurring within 30 days of a prior event was considered a continuation of the index event.
FEV1 was analyzed using a mixed model for repeated measures (MMRM), with change from baseline in trough FEV1 as the response variable and including a factor for background LABA use.11 Change from baseline SGRQ total score at week 12 was analyzed by analysis of covariance (ANCOVA) with change from baseline in SGRQ total score as the response variable and including a factor for background LABA use. E-RS total scores were recorded daily by subjects using an electronic diary and were analyzed using the same MMRM model as for FEV1, with baseline E-RS total score being included as a covariate. No adjustments were made for multiple treatment comparisons in the pooled data set or secondary analyses.
The intent-to-treat (ITT) and safety populations consisted of all subjects receiving ≥1 dose of study drug and, for efficacy, one post-dose pulmonary function assessment. Changes from baseline in the efficacy assessments were evaluated for all subjects who received double-blind study drug, regardless of their completion status. Subjects who discontinued the randomized treatment before week 12, but continued to be followed, were analyzed using retrieved dropout data (all collected data). Efficacy and safety analyses were performed using on-treatment and all collected data, and the analyses indicated that the endpoint results were similar in both data sets.11 On-treatment data are presented.
Forty-eight-week, active-controlled study
Primary safety endpoints included discontinuations due to treatment-emergent adverse events (TEAEs), and other safety endpoints included incidence of major adverse CV events (MACE), including CV death, ischemia/infarction, and stroke. Efficacy endpoints included change from baseline at 48 weeks in trough FEV1, SGRQ total score, and E-RS total score and incidence of COPD exacerbations. An ANCOVA model and MMRM were used for the selected efficacy endpoints as per the 12-week studies.12
Safety analyses were conducted using the safety population and efficacy analyses using the ITT population, both consisting of all subjects randomized to treatment who received ≥1 dose of study drug.
Safety analyses
Safety data were analyzed using descriptive statistics.11,12 TEAEs were coded according to MedDRA Version 15.1 and summarized by treatment, system organ class, and preferred term. CV events of special interest were examined using Standardized MedDRA Query analysis and included cardiac arrhythmia, arrhythmia-related events, cardiac failure, ischemic heart disease, QT prolongation, and myocardial infarction (MI). Vital signs (including heart rate and blood pressure), 12-lead electrocardiogram (ECG), and clinical laboratory measures were also assessed. Holter monitoring was carried out at screening in all studies and at week 12 in a subpopulation of subjects (n=153) in GOLDEN 3.
Results
Patient characteristics
Overall, 2,379 randomized subjects in the three Phase III studies were stratified into subgroups based on background LABA use (LABA-yes: n=861 [36%]; LABA-no: n=1,518 [64%]). Of 1,293 randomized subjects who received study drug, a total of 1,111 (86%) completed the two 12-week placebo-controlled studies, and 838 (77%) of the randomized population of 1,086 subjects completed the 48-week active-controlled study. Completion rates were similar within treatment groups, irrespective of background LABA use (Tables S1 and S2).
Key demographics and baseline characteristics for LABA subgroups are shown in Table 1. Mean baseline post-bronchodilator FEV1 was numerically lower in LABA-yes subgroups, compared with LABA-no subgroups, across treatment groups. Average symptom (E-RS) scores were generally higher at baseline for LABA-yes vs LABA-no subgroups, with the exception of the GLY treatment group in the 48-week study. Across all treatment groups, a higher proportion of LABA-yes subjects exhibited post-bronchodilator FEV1 % predicted indicative of severe-to-very-severe COPD (severe airflow limitation: <50% predicted; very severe airflow limitation: <30% predicted)1 compared with LABA-no subjects. The majority (84%–95%) of LABA-yes subjects also used ICS, indicating that this group had more severe disease compared with the LABA-no group. Long-acting bronchodilator use prior to study is summarized in Table S3. The incidence of prior use of LABA/ICS aligns with the LABA-yes and ICS-yes data presented in Table 1.
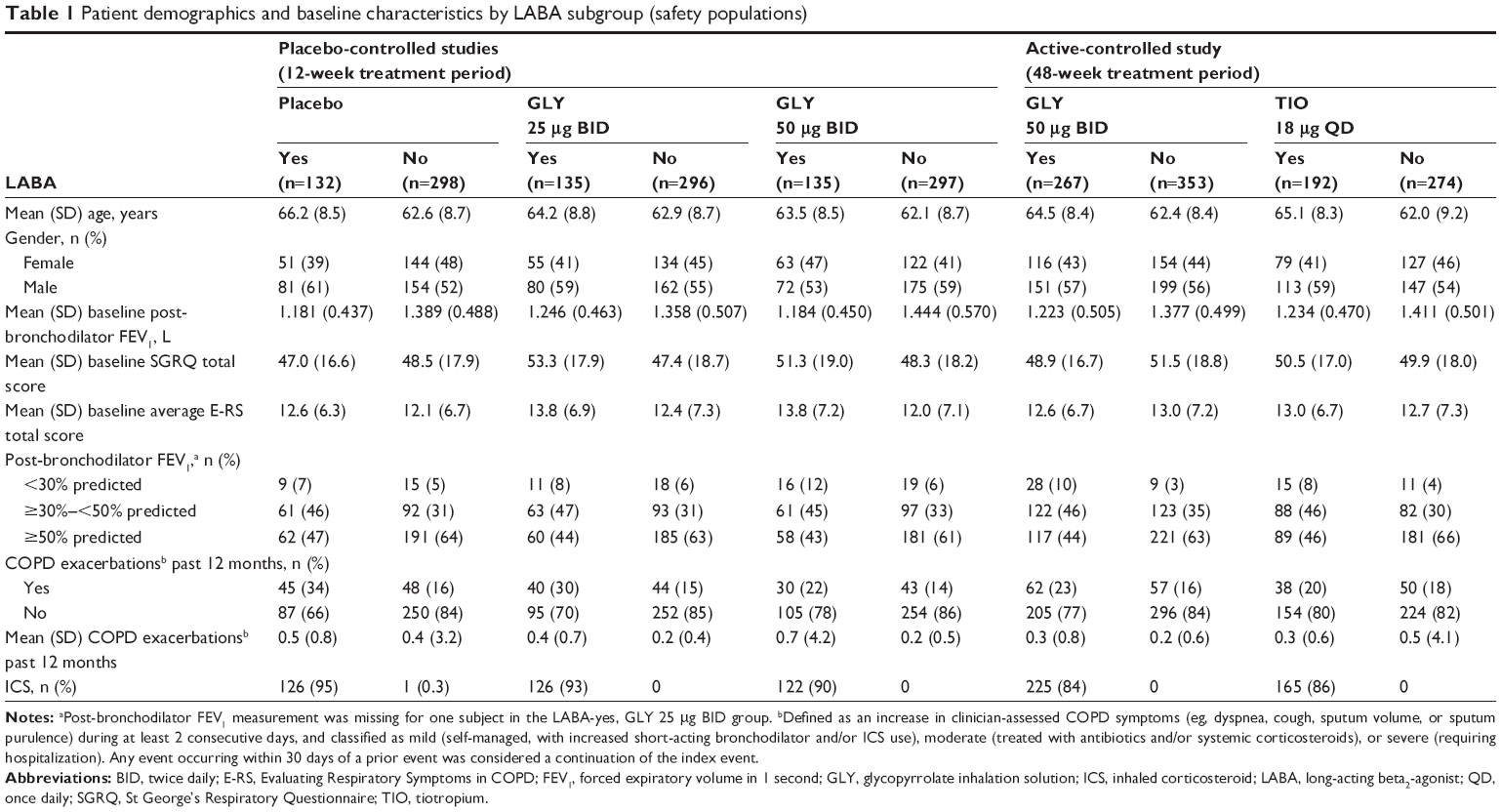 | Table 1 Patient demographics and baseline characteristics by LABA subgroup (safety populations) |
Efficacy
In the placebo-controlled studies, treatment with nebulized GLY led to significant improvements in LS mean placebo-adjusted change from baseline in trough FEV1 across LABA-yes and LABA-no subjects at week 12 (25 μg BID: 0.101 L and 0.092 L; 50 μg BID: 0.110 L and 0.101 L, respectively; Figure 2A). Both GLY doses showed highly statistically significant (P<0.001) improvements of 0.090–0.104 L in FEV1 compared to placebo at 12 weeks.
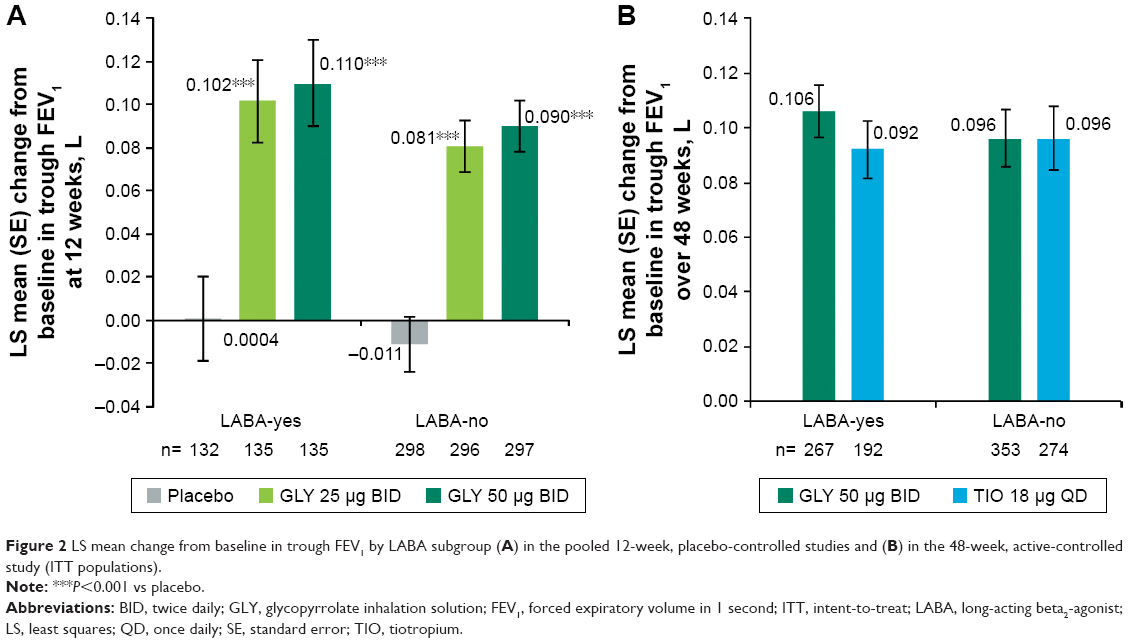 | Figure 2 LS mean change from baseline in trough FEV1 by LABA subgroup (A) in the pooled 12-week, placebo-controlled studies and (B) in the 48-week, active-controlled study (ITT populations). |
In both the LABA subgroups, overall improvements in mean trough FEV1 were similar with GLY (LABA-yes: 0.106 L; LABA-no: 0.096 L) and TIO (LABA-yes: 0.092 L; LABA-no: 0.096 L) over 48 weeks (Figure 2B).
At week 12, there was a significant improvement in placebo-adjusted SGRQ total score mean change from baseline with GLY 25 and 50 μg BID in both LABA-yes (−2.957 and −3.888, respectively) and LABA-no (−3.301 and −2.073, respectively) subgroups in the pooled placebo-controlled studies. Nebulized GLY 50 μg BID treatment led to a mean change from baseline in SGRQ total score greater than the minimum clinically important difference (MCID; defined as a reduction of ≥4.0 units) in the LABA-yes subgroup, and the GLY 25 μg BID group led to a mean change greater than the MCID in the LABA-no subgroup (Figure 3A).
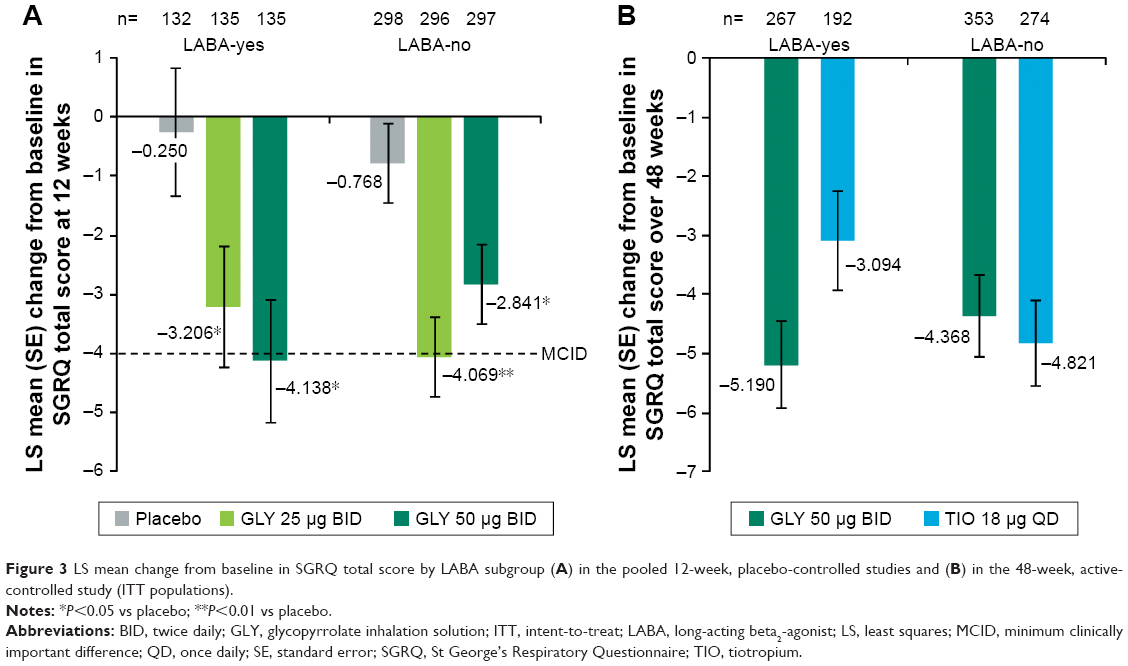 | Figure 3 LS mean change from baseline in SGRQ total score by LABA subgroup (A) in the pooled 12-week, placebo-controlled studies and (B) in the 48-week, active-controlled study (ITT populations). |
In the 48-week study, treatment with GLY or TIO reduced SGRQ total score from baseline in both the LABA subgroups (LABA-yes: −5.190 and −3.094; LABA-no: −4.368 and −4.821, respectively). Overall, there was no significant difference in SGRQ total score change from baseline between GLY and TIO (Figure 3B).
At week 12, GLY treatment resulted in significantly higher SGRQ responder rates vs placebo for LABA-yes subjects (GLY 25 μg, OR: 2.306 [95% CI: 1.275, 4.169]; GLY 50 μg BID, OR: 2.077 [95% CI: 1.141, 3.781]). In the LABA-no subgroup, only GLY 25 μg BID resulted in a significant increase in SGRQ responder rate vs placebo (OR: 1.434 [95% CI: 1.001, 2.056]) (Figure 4A).
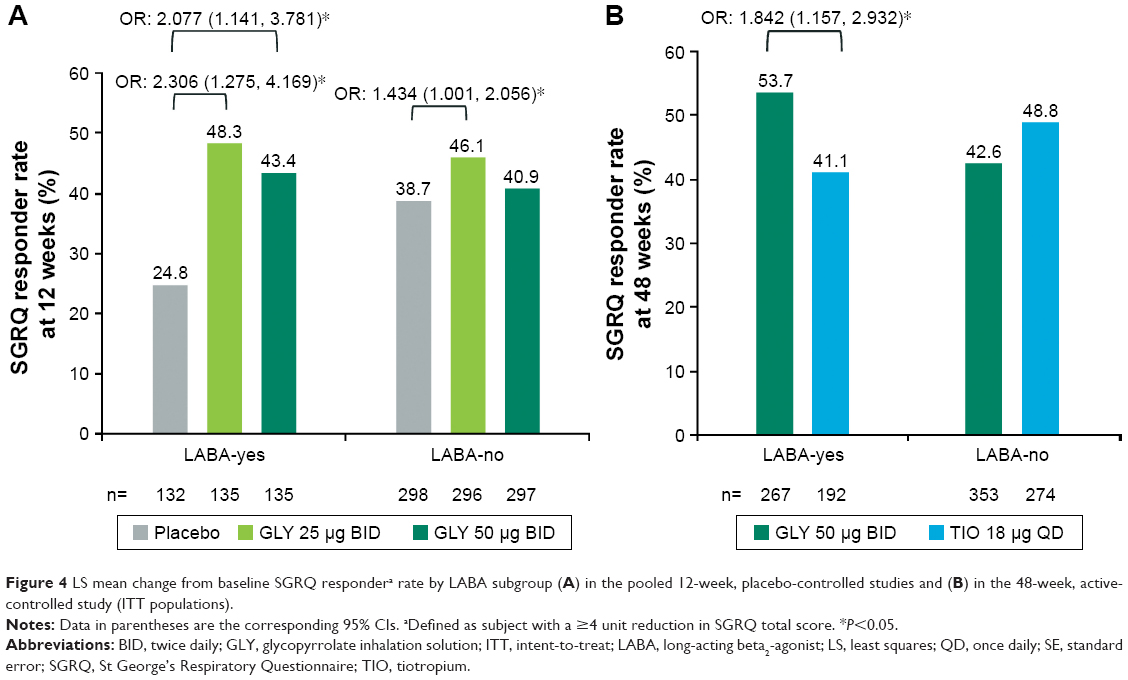 | Figure 4 LS mean change from baseline SGRQ respondera rate by LABA subgroup (A) in the pooled 12-week, placebo-controlled studies and (B) in the 48-week, active-controlled study (ITT populations). |
At week 48, SGRQ responder rate was significantly higher for GLY (53.7%) vs TIO (41.1%) in LABA-yes subjects (OR: 1.842 [95% CI: 1.157, 2.932]). SGRQ responder rates were similar for GLY and TIO groups in LABA-no subjects (Figure 4B).
Nebulized GLY produced a reduction in average E-RS total score, regardless of background LABA use (Figure 5). In the 12-week, placebo-controlled studies, GLY 25 μg BID led to a significant reduction in average E-RS total score from baseline vs placebo for LABA-no subjects (−1.165; P<0.01; Figure 5A). In the 48-week, active-controlled study, the differences in E-RS total score change from baseline for GLY and TIO were not significant. In the LABA-yes subgroup, GLY led to a larger reduction in E-RS total score compared with TIO, while in the LABA-no subgroup, subjects treated with TIO had a greater reduction in E-RS total score compared with GLY (Figure 5B).
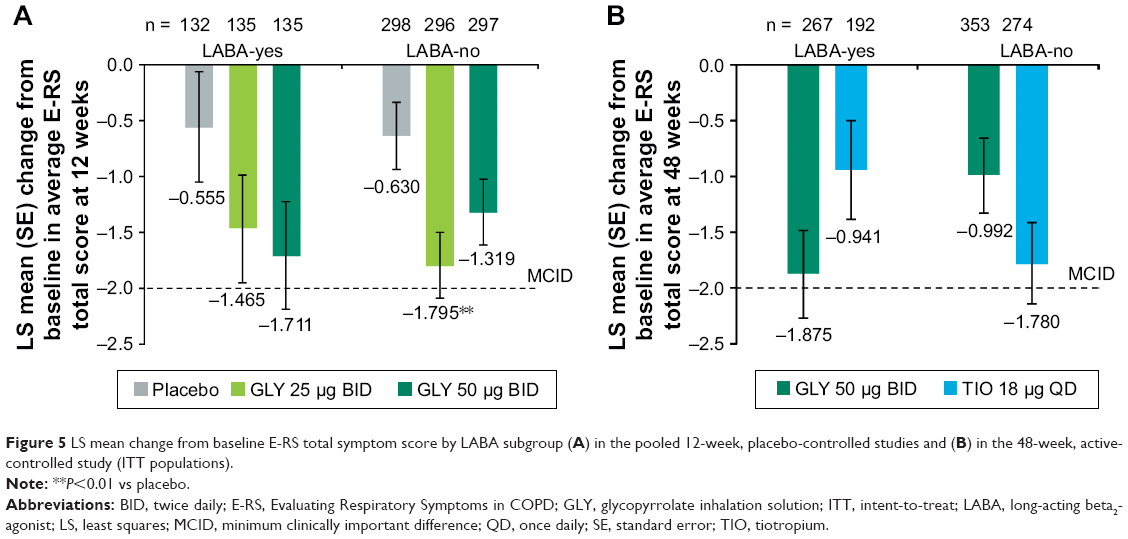 | Figure 5 LS mean change from baseline E-RS total symptom score by LABA subgroup (A) in the pooled 12-week, placebo-controlled studies and (B) in the 48-week, active-controlled study (ITT populations). |
Exacerbation rates were comparable with GLY and placebo and were numerically higher in the LABA-yes subgroup (placebo: 12.9%; GLY 25 μg: 12.6%; GLY 50 μg: 14.1%) compared with LABA-no (placebo: 8.7%; GLY 25 μg: 6.1%; GLY 50 μg: 7.7%) at 12 weeks.
Similarly, rates were comparable with GLY vs TIO at 48 weeks and were numerically higher in the LABA-yes subgroup (23.2% and 27.6%, respectively) compared with LABA-no (15.0% and 19.0%).
Safety
In the 12-week studies, nebulized GLY 25 and 50 μg BID led to a lower incidence, vs placebo, of TEAEs (43.4%, 50.7%, and 52.3%, respectively), serious TEAEs (3.0%, 4.2%, and 5.6%), and TEAEs leading to discontinuation (5.1%, 3.9%, and 9.3%).11 At week 48, the incidence of TEAEs (69.4% and 67.0%, respectively) and serious TEAEs (12.3% and 10.5%) were similar for subjects treated with GLY or TIO. However, treatment with GLY resulted in a higher incidence of TEAEs leading to discontinuation compared with TIO (10.0% vs 2.8%).12
Evaluation of safety by LABA subgroups showed that the overall incidence of TEAEs was similar by treatment group, regardless of background LABA use. In the 12-week studies, TEAE incidence was numerically lower in the GLY 25 μg group compared with placebo and GLY 50 μg. Over 48 weeks, the incidence of TEAEs was similar between GLY and TIO (Table 2).
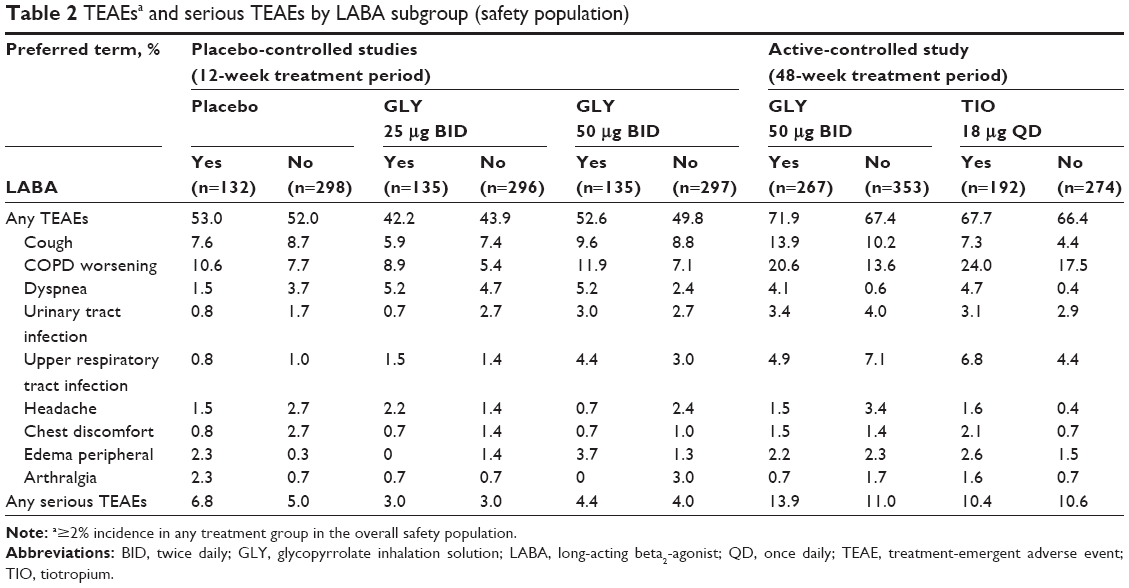 | Table 2 TEAEsa and serious TEAEs by LABA subgroup (safety population) |
In all treatment groups, incidence of COPD worsening was numerically higher in LABA-yes subgroups. Incidence of all other TEAEs was similar across LABA subgroups.
Overall incidence of TEAEs leading to discontinuation was similar regardless of background LABA use and lower with GLY 25 and 50 μg BID (LABA-yes: 4.4% and 4.4%; LABA-no: 5.4% and 3.7%, respectively) vs placebo (LABA-yes: 9.1%; LABA-no: 9.4%) in the 12-week, placebo-controlled studies (Table S4). All discontinuations due to pneumonia occurred in subjects on LABA (or LABA/ICS) in the placebo arm.
In the 48-week, active-controlled study, incidence of TEAEs leading to discontinuation was similar in the LABA subgroups and was higher with GLY (LABA-yes: 11.2%; LABA-no: 9.1%) vs TIO (LABA-yes: 2.6%; LABA-no: 2.9%) (Table S4).
Across LABA subgroups, the most common TEAEs leading to discontinuation were cough, COPD worsening, and dyspnea.
In the 12-week studies, the overall incidence rate (IR) in MACE for GLY 50 μg (IR: 23.3 per 1,000 patient-years) was similar to placebo (IR: 16.4) (Table 3).11 MACE occurred only among LABA-no subjects. In the 48-week study, the overall IR for GLY (IR: 6.4) was numerically lower than for TIO (IR: 20.3).12 MACE occurred in subjects treated with TIO, irrespective of background LABA use. Furthermore, the rates of individual CV events of special interest were ≤2.1% and showed similar incidence between placebo, GLY, and TIO and across LABA subgroups (Table S5).
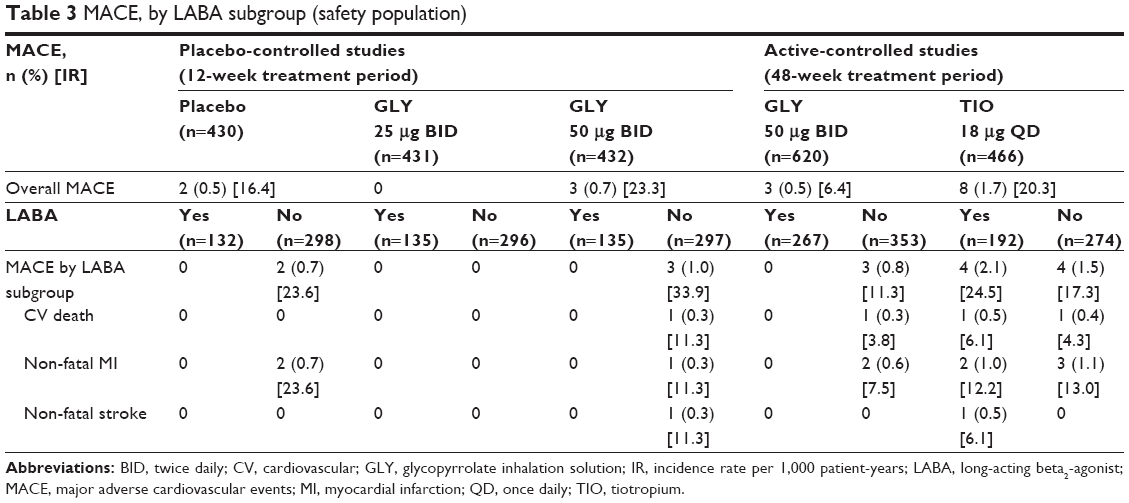 | Table 3 MACE, by LABA subgroup (safety population) |
Across the Phase III GOLDEN studies, clinical laboratory results, vital signs, Holter monitoring (GOLDEN 3 only) and ECG parameters did not show any clinically relevant changes or trends,11,12 with similar results between LABA subgroups.
Discussion
Following recent updates to treatment paradigms, a better understanding of the effects of LAMA, LAMA/LABA and LAMA/LABA/ICS combinations on efficacy and safety outcomes of COPD therapies may help clinicians to make informed decisions when tailoring a patient’s treatment. The Phase III GOLDEN studies were prospectively designed to include subjects on background LABA/ICS or LABA therapies and subjects with moderate-to-very severe COPD, in order to be representative of the general COPD population.
Substantial improvements in FEV1 were experienced in all GLY treatment groups, regardless of background LABA use. These analyses support patients using background LABA (potentially identified as a subgroup of patients with more severe COPD) in addition to nebulized LAMA, and the combination appeared to produce effects similar to those in LABA-no (ie, LAMA-only) patients with less severe COPD.
Overall, in all three studies, SGRQ total score improvements were reported in LABA-yes and LABA-no subgroups. Based on E-RS total scores, there was a general trend toward symptom reduction across all treatment groups at end of study. Although GLY 25 μg BID showed significant reduction in E-RS total score vs placebo in the LABA-no subgroup, no treatment group produced the two-unit reduction in mean score considered evidence of a response.15
These changes in health status (SGRQ and E-RS total scores) showed improvements in breathlessness and shortness of breath and were directionally aligned with, and supported, the observed improvements in lung function. Importantly, the directionality of the collective symptom changes was consistent with the treatment goals of the GOLD Report.1
The Phase III GOLDEN studies were conducted in a population that was not predominantly exacerbating, and the studies were not powered to analyze differences in exacerbation levels between subjects with and without background LABA use. Overall, in the 12 months prior to joining the studies, <20% of subjects experienced one or more exacerbation (Table 1). During treatment, LABA-yes subgroups exhibited numerically higher exacerbation rates in all treatment groups at 12 and 48 weeks compared with LABA-no subgroups.
In the Phase III GOLDEN studies, there was no evidence that adding LAMA therapy to LABA ± ICS led to any additional safety signals. Overall incidence of TEAEs was similar across LABA subgroups in the 12- and 48-week studies and similar between GLY and TIO treatment groups over 48 weeks. In all studies, incidence of COPD worsening was numerically higher in the LABA-yes subgroup compared with LABA-no. This may be due to the higher proportion of subjects with severe-to-very-severe airflow limitation at baseline in LABA-yes subjects compared with LABA-no subjects.
Discontinuations due to TEAEs at 12 weeks were higher in placebo vs GLY, and there was no difference across LABA subgroups. The 2.3% incidence of cough in the placebo group in the 12-week studies suggests a drug-independent effect. In the 48-week, open-label study, discontinuations due to TEAEs were higher for GLY compared with TIO, and there was no difference across LABA subgroups. As previously reported, respiratory-related TEAEs, particularly cough, contributed to the difference in discontinuation rates, with many of the discontinuations occurring in the first 15 days of the study. The early discontinuations due to cough and/or dyspnea may have been due to BID administration of nebulized GLY, >90% of subjects being naïve to nebulizer therapy, and hydration of airway mucus and epithelium by the aerosol potentially altering airway lumen dimensions. In addition, prior use of TIO may have contributed to the perception and reporting of TEAEs, as ~30% of subjects were treated with TIO for ≥2 years prior to randomization.12
Due to their individual mechanisms of action, LAMAs and LABAs can raise heart rate, which may lead to incidences of arrhythmia, MI, stroke, or sudden death in patients with underlying CV comorbidity. Thus, combination LAMA/LABA therapy may result in increased rate of CV AEs in subjects with COPD compared with each component alone.16 Very few CV events of special interest occurred in the Phase III GOLDEN studies, and the incidence was similar between treatment groups in each study and across LABA subgroups. Treatment with nebulized GLY did not generate any new CV safety signals in the study populations, which were prospectively designed to include subjects with high CV risk.
The majority of subjects on background LABA therapy were also using ICS (range: 84%–95%), representing the subset of subjects more likely to have severe disease and airflow limitation, and with the addition of nebulized LAMA, these subjects were effectively using an open LAMA/LABA/ICS triple therapy. Recent studies have demonstrated that triple therapy plays a role in the COPD therapeutic landscape,9,17,18 and it is a recommended treatment option for patients who experience recurrent exacerbations or continuing symptoms despite current treatment with either LAMA/LABA or LABA/ICS combination.1 An important finding of this analysis is that there seems to be a treatment benefit with “free,” “open-triple,” or combination therapy in the population studied. The results from the Phase III GOLDEN studies, specifically improvements in lung function and health status, are in agreement with previously reported efficacy of triple therapies9,17–19 and add to the growing body of evidence regarding the potential role of triple therapy in the clinical management of COPD. We expect the place of triple LAMA/LABA/ICS therapy within the treatment algorithm to be more clearly defined in future updates of the GOLD Report.
Nebulized GLY could be beneficial as initial bronchodilator maintenance therapy for COPD patients with low burden of symptoms and high exacerbation risk, or low exacerbation risk and high symptoms, or as additional therapy for those already receiving LABA or LABA/ICS.
Conclusion
Lung function (trough FEV1) and SGRQ patient-reported outcomes were improved in patients with moderate-to-very-severe COPD treated with nebulized GLY with and without background LABA ± ICS medication. Patients using background LABA (± ICS) during the treatment period showed robust improvements in trough FEV1 and SGRQ with the addition of nebulized GLY. Exacerbation rates with GLY were comparable to open-label control (TIO) over 48 weeks for patients in both LABA subgroups. Nebulized GLY, with or without background LABA treatment, did not generate any additional overall or CV safety signals.
These results provide important information to clinicians managing COPD patients who require polypharmacy (ie, combination therapy) over their disease continuum.
Acknowledgments
These studies were funded by Sunovion Pharmaceuticals Inc. Medical writing support was provided by Linda Townsend, PhD, of FireKite, an Ashfield company, part of UDG Healthcare plc, and was funded by Sunovion Pharmaceuticals Inc.
Author contributions
RT contributed to study design; EMK contributed to data collection. All authors contributed toward data analysis, drafting, and revising the manuscript and agree to be accountable for all aspects of the work.
Disclosure
EMK has participated in consulting, advisory boards, speaker panels, or received travel reimbursement for Amphastar, AstraZeneca, Forest, Mylan, Novartis, Oriel, Pearl, Sunovion, Teva, and Theravance. He has conducted multicenter clinical research trials for ~40 pharmaceutical companies. RT, BP, SS, and TG are employees of Sunovion Pharmaceuticals Inc. The authors report no other conflicts of interest in this work.
References
Global Initiative for Chronic Obstructive Lung Disease Inc. Global Initiative for Chronic Obstructive Lung Disease (GOLD). Global strategy for the diagnosis, management and prevention of COPD; 2018. [Guidelines]. 2018. Available from: http://goldcopd.org/. Accessed March 28, 2018. | ||
Wheaton AG, Cunningham TJ, Ford ES, Croft JB, Centers for Disease Control and Prevention (CDC). Employment and activity limitations among adults with chronic obstructive pulmonary disease – United States, 2013. MMWR Morb Mortal Wkly Rep. 2015;64(11):289–295. | ||
Heron M. Deaths: leading causes for 2010. Natl Vital Stat Rep. 2013;62(6):1–96. | ||
Guarascio AJ, Ray SM, Finch CK, Self TH. The clinical and economic burden of chronic obstructive pulmonary disease in the USA. Clinicoecon Outcomes Res. 2013;5:235–245. | ||
Miravitlles M, Ribera A. Understanding the impact of symptoms on the burden of COPD. Respir Res. 2017;18(1):67. | ||
Agusti A, Hedner J, Marin JM, Barbé F, Cazzola M, Rennard S. Night-time symptoms: a forgotten dimension of COPD. Eur Respir Rev. 2011;20(121):183–194. | ||
Vogelmeier C, Hederer B, Glaab T, et al. Tiotropium versus salmeterol for the prevention of exacerbations of COPD. N Engl J Med. 2011;364(12):1093–1103. | ||
Decramer ML, Chapman KR, Dahl R, et al. Once-daily indacaterol versus tiotropium for patients with severe chronic obstructive pulmonary disease (INVIGORATE): a randomised, blinded, parallel-group study. Lancet Respir Med. 2013;1(7):524–533. | ||
Lipson DA, Barnhart F, Brealey N, et al. Once-daily single-inhaler Triple versus dual therapy in patients with COPD. N Engl J Med. 2018;378(18):1671–1680 Epub ahead of print. | ||
LONHALA MAGNAIR (glycopyrrolate) inhalation solution [package insert]. Marlborough, MA: Sunovion Pharmaceuticals Inc. 2018. | ||
Kerwin E, Donohue JF, Goodin T, Tosiello R, Wheeler A, Ferguson GT. Efficacy and safety of glycopyrrolate/eFlow® CS (nebulized glycopyrrolate) in moderate-to-very-severe COPD: results from the glycopyrrolate for obstructive lung disease via electronic nebulizer (GOLDEN) 3 and 4 randomized controlled trials. Respir Med. 2017;132:238–250. | ||
Ferguson GT, Goodin T, Tosiello R, Wheeler A, Kerwin E. Long-term safety of glycopyrrolate/eFlow® CS in moderate-to-very-severe COPD: Results from the Glycopyrrolate for Obstructive Lung Disease via Electronic Nebulizer (GOLDEN) 5 randomized study. Respir Med. 2017;132:251–260. | ||
Jones P, Miravitlles M, van der Molen T, Kulich K. Beyond FEV1 in COPD: a review of patient-reported outcomes and their measurement. Int J Chron Obstruct Pulmon Dis. 2012;7:697–709. | ||
Singh D, Miravitlles M, Vogelmeier C. Chronic Obstructive Pulmonary Disease Individualized Therapy: Tailored Approach to Symptom Management. Adv Ther. 2017;34(2):281–299. | ||
Leidy NK, Murray LT, Monz BU, et al. Measuring respiratory symptoms of COPD: performance of the EXACT-Respiratory Symptoms Tool (E-RS) in three clinical trials. Respir Res. 2014;15:124. | ||
Kardos P, Worsley S, Singh D, Román-Rodríguez M, Newby DE, Müllerová H. Randomized controlled trials and real-world observational studies in evaluating cardiovascular safety of inhaled bronchodilator therapy in COPD. Int J Chron Obstruct Pulmon Dis. 2016;11:2885–2895. | ||
Singh D, Corradi M, Spinola M, et al. Triple therapy in COPD: new evidence with the extrafine fixed combination of beclomethasone dipropionate, formoterol fumarate, and glycopyrronium bromide. Int J Chron Obstruct Pulmon Dis. 2017;12:2917–2928. | ||
Frith PA, Thompson PJ, Ratnavadivel R, et al. Glycopyrronium once-daily significantly improves lung function and health status when combined with salmeterol/fluticasone in patients with COPD: the GLISTEN study, a randomised controlled trial. Thorax. 2015;70(6):519–527. | ||
Bremner PR, Birk R, Brealey N, Ismaila AS, Zhu CQ, Lipson DA. Single-inhaler fluticasone furoate/umeclidinium/vilanterol versus fluticasone furoate/vilanterol plus umeclidinium using two inhalers for chronic obstructive pulmonary disease: a randomized non-inferiority study. Respir Res. 2018;19(1):19. |
Supplementary materials
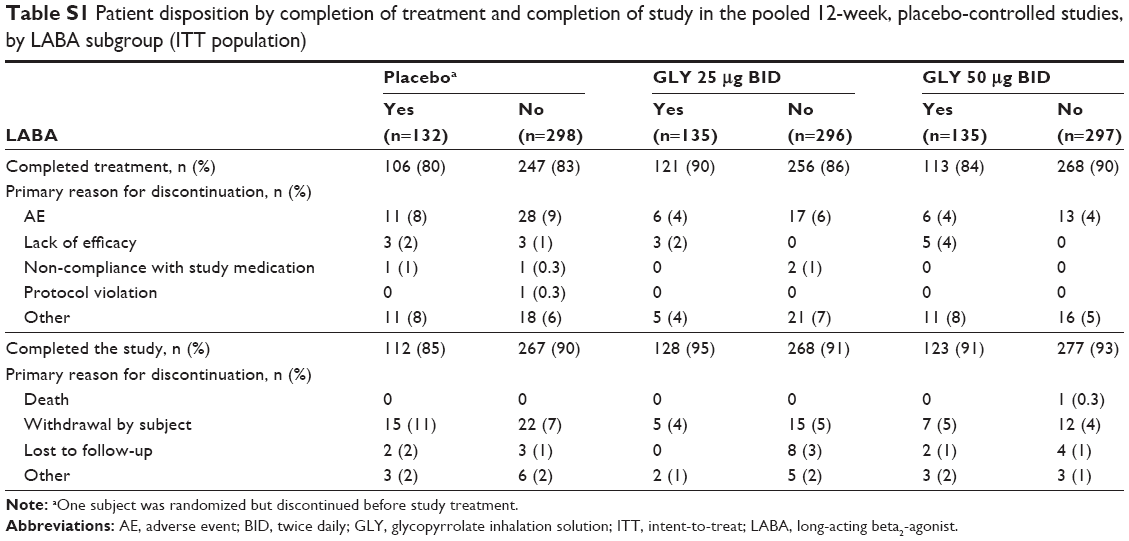 | Table S1 Patient disposition by completion of treatment and completion of study in the pooled 12-week, placebo-controlled studies, by LABA subgroup (ITT population) |
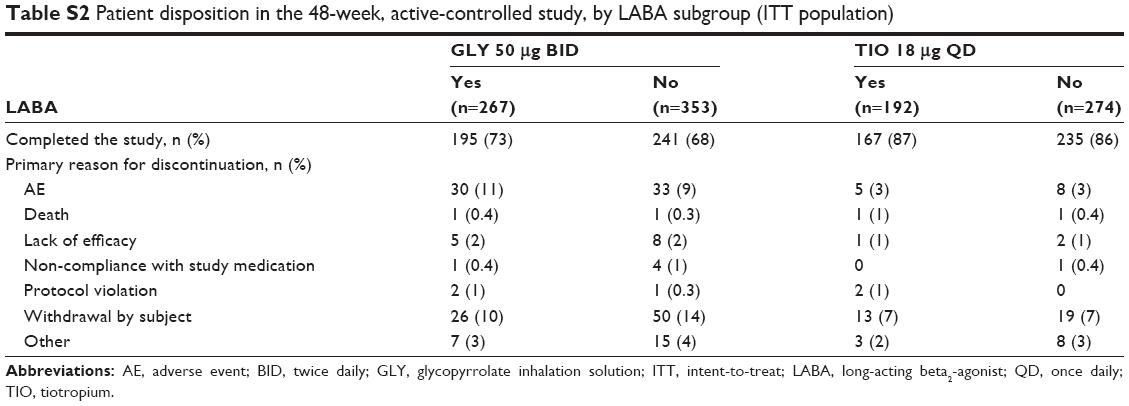 | Table S2 Patient disposition in the 48-week, active-controlled study, by LABA subgroup (ITT population) |
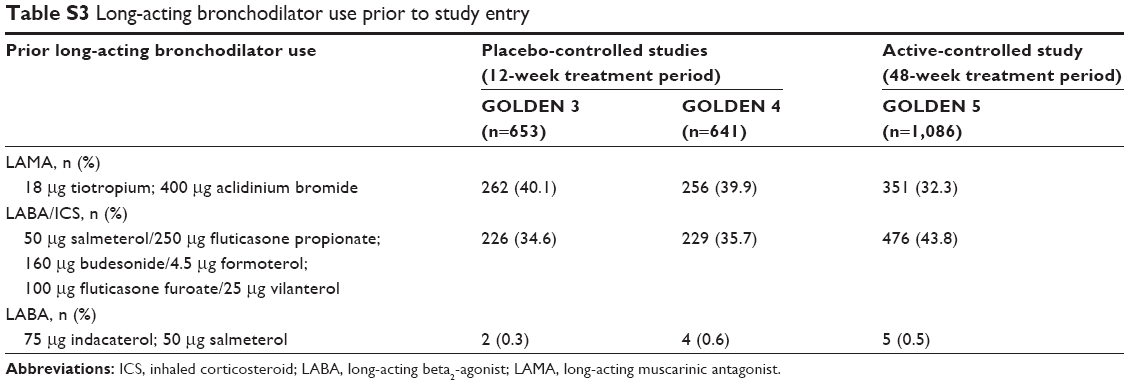 | Table S3 Long-acting bronchodilator use prior to study entry |
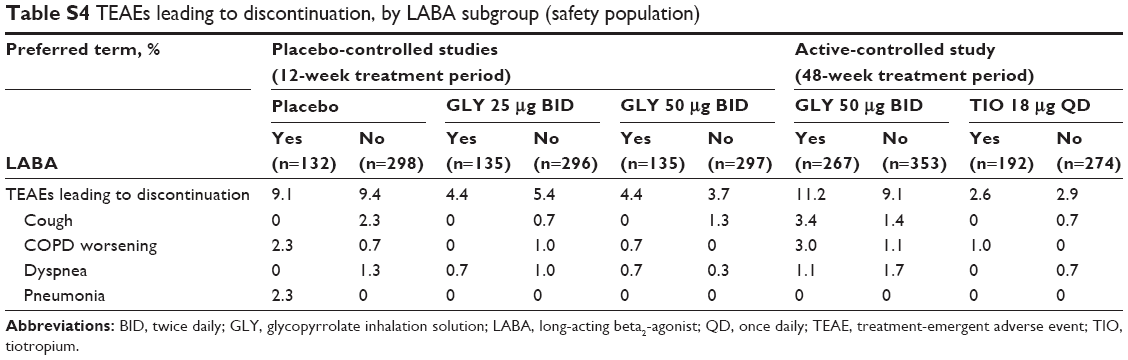 | Table S4 TEAEs leading to discontinuation, by LABA subgroup (safety population) |
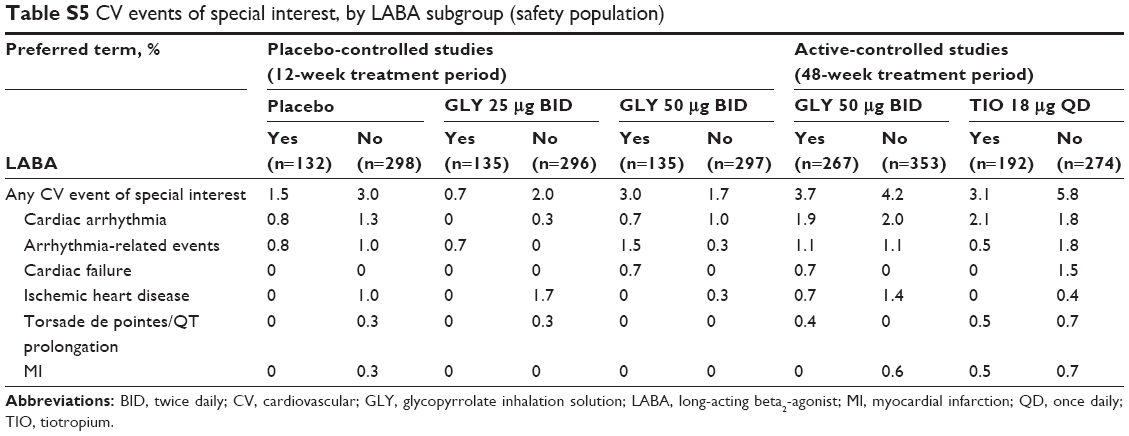 | Table S5 CV events of special interest, by LABA subgroup (safety population) |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.